Stable Hemiaminals with a Cyano Group and a Triazole Ring

Abstract
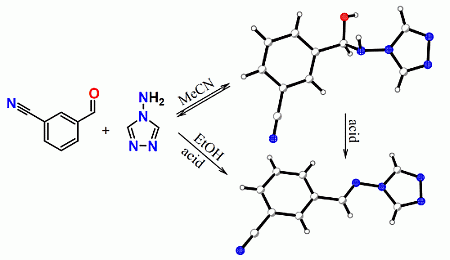
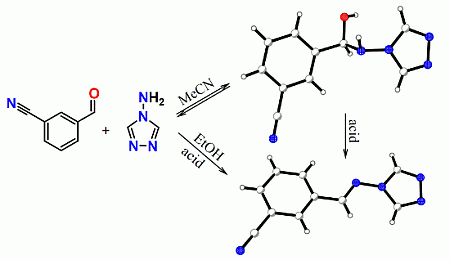
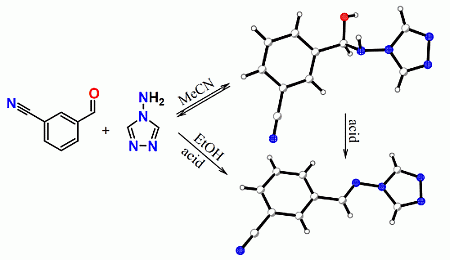
:
1. Introduction

2. Results and Discussion
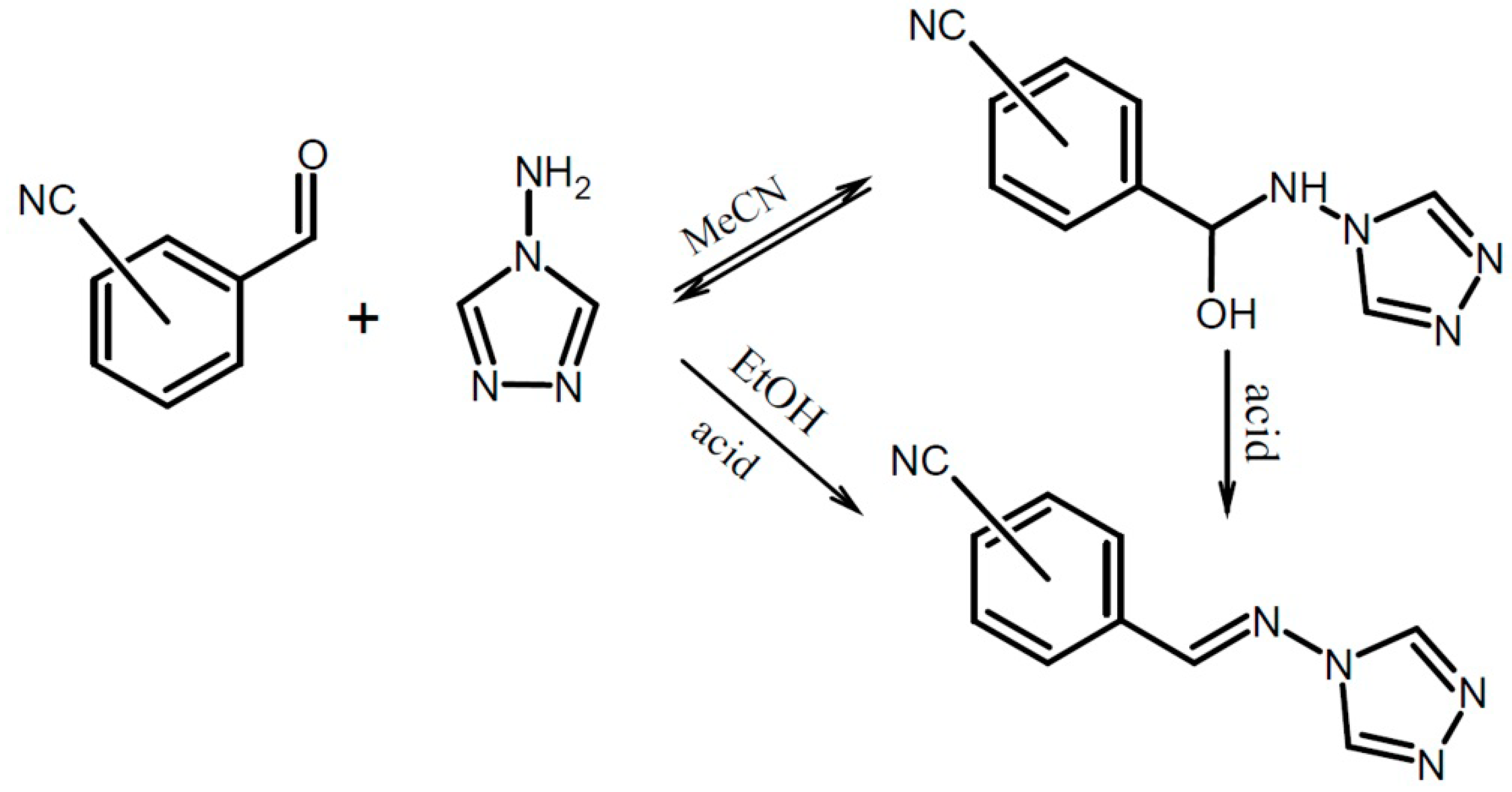
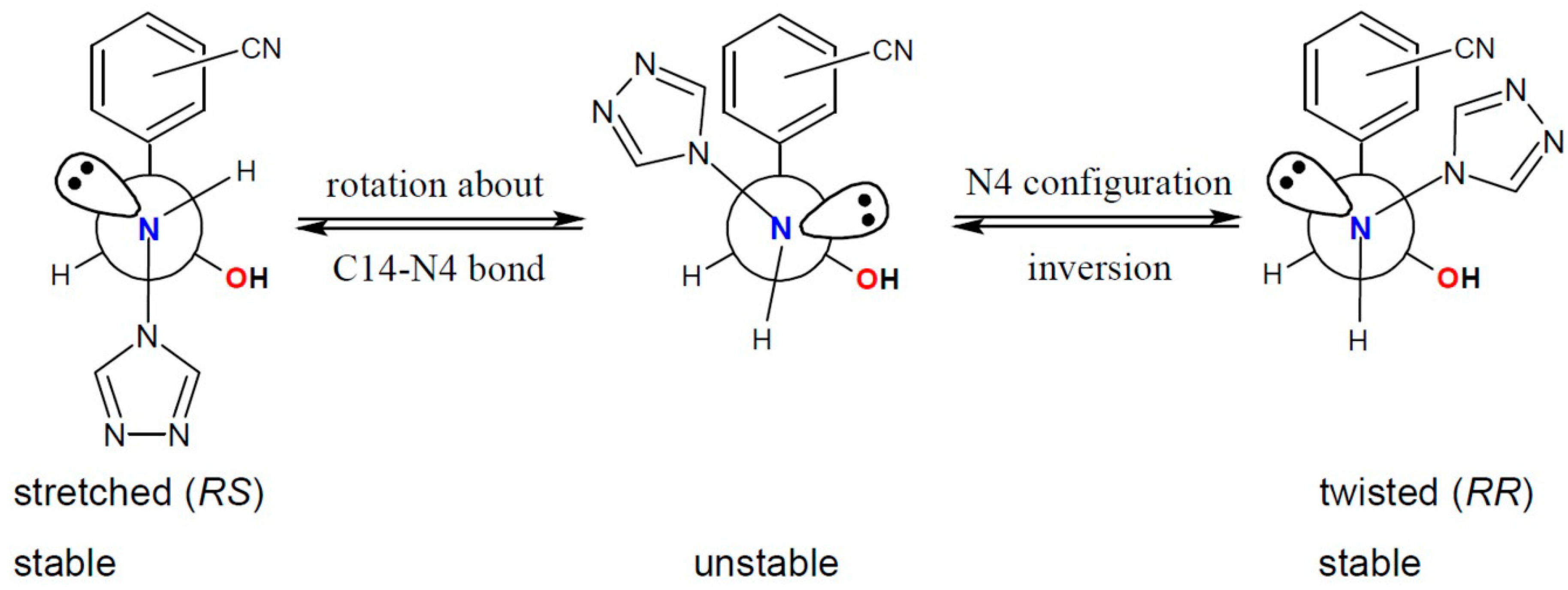
2.1. General Remarks

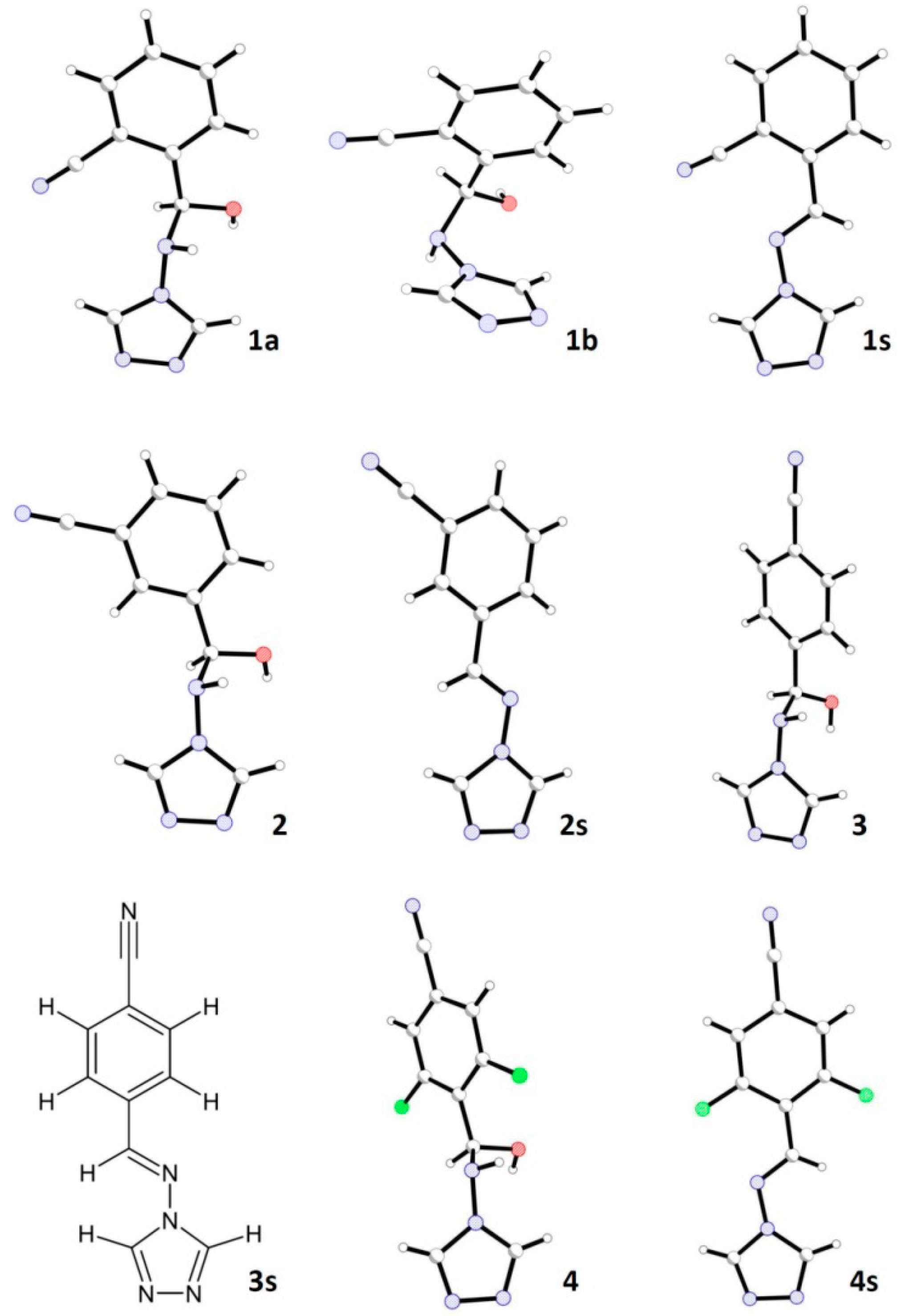
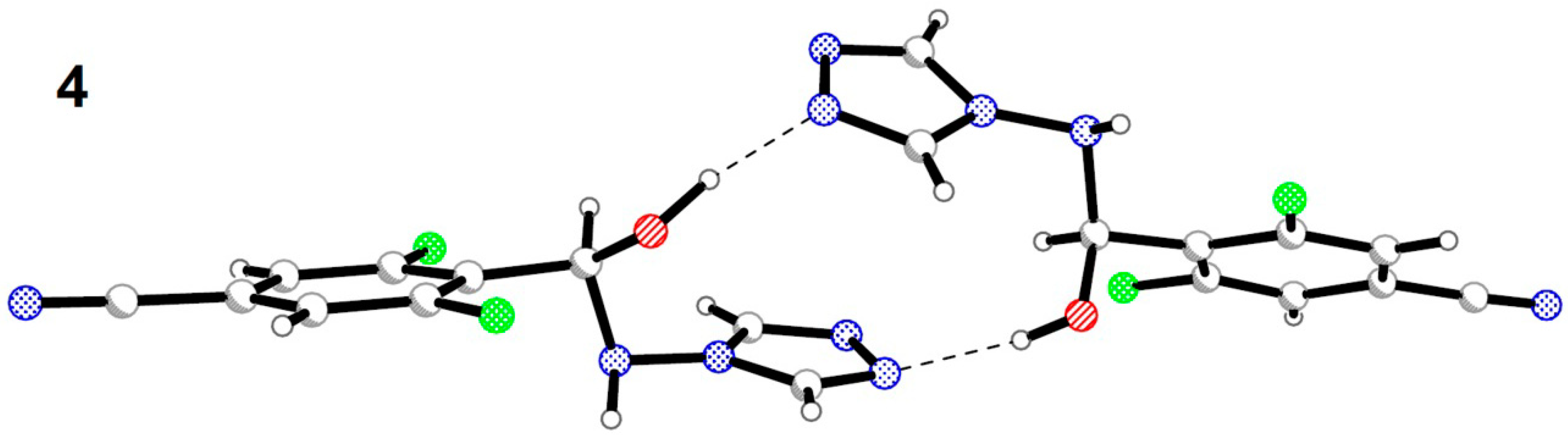
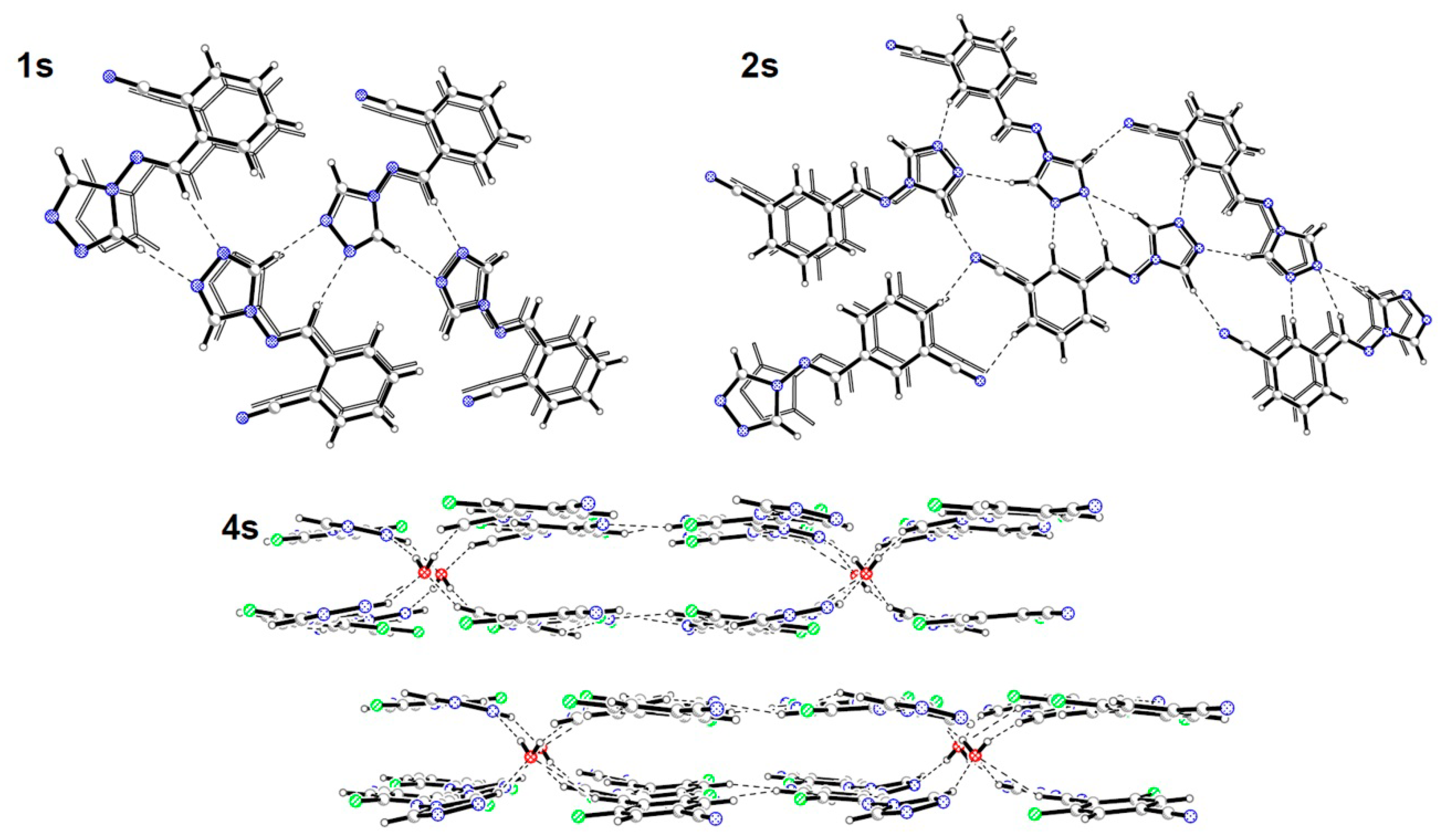
2.2. Crystal Structures Description


{kind=link}
{kind=link}
{kind=link}
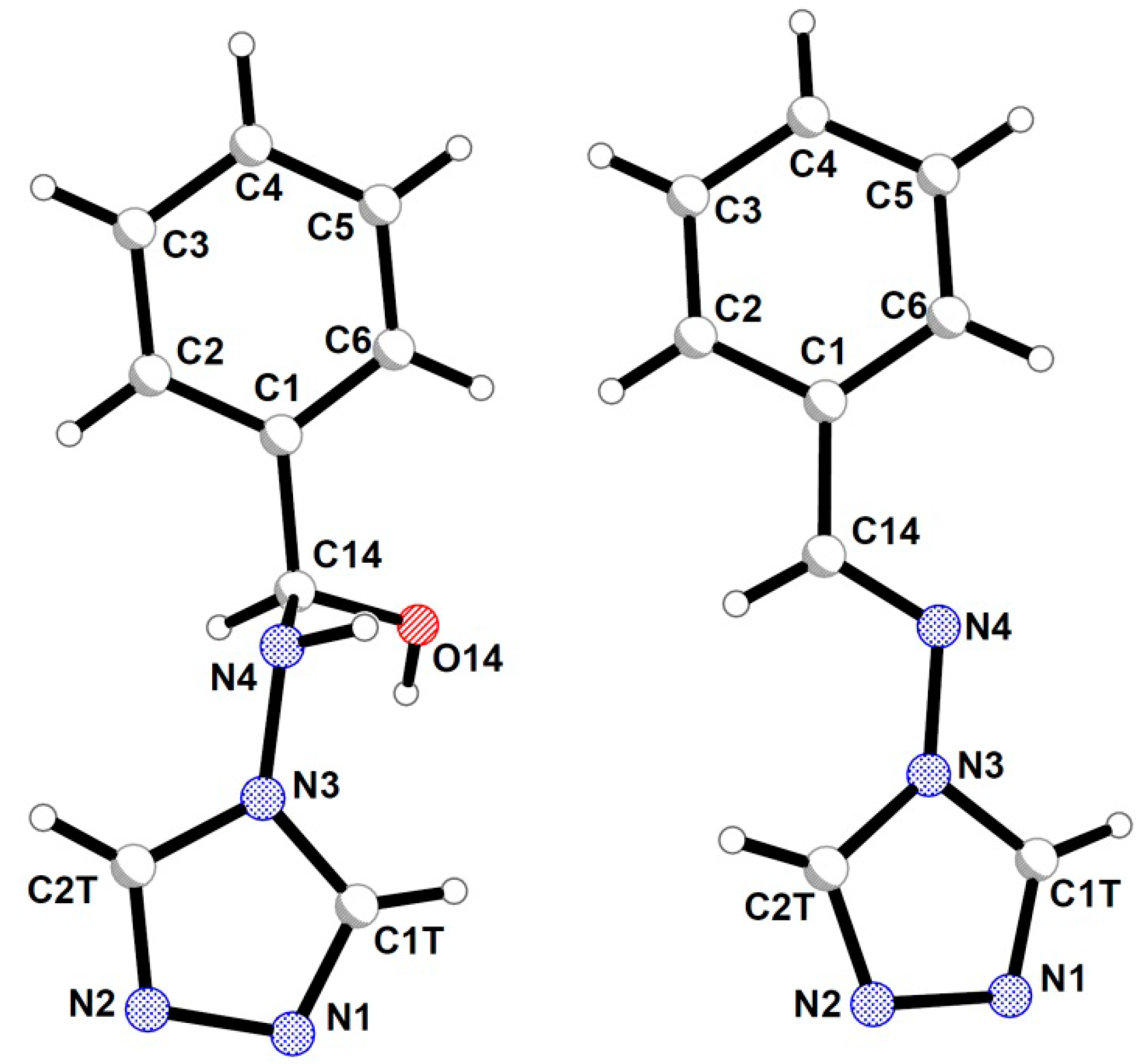{kind=link}
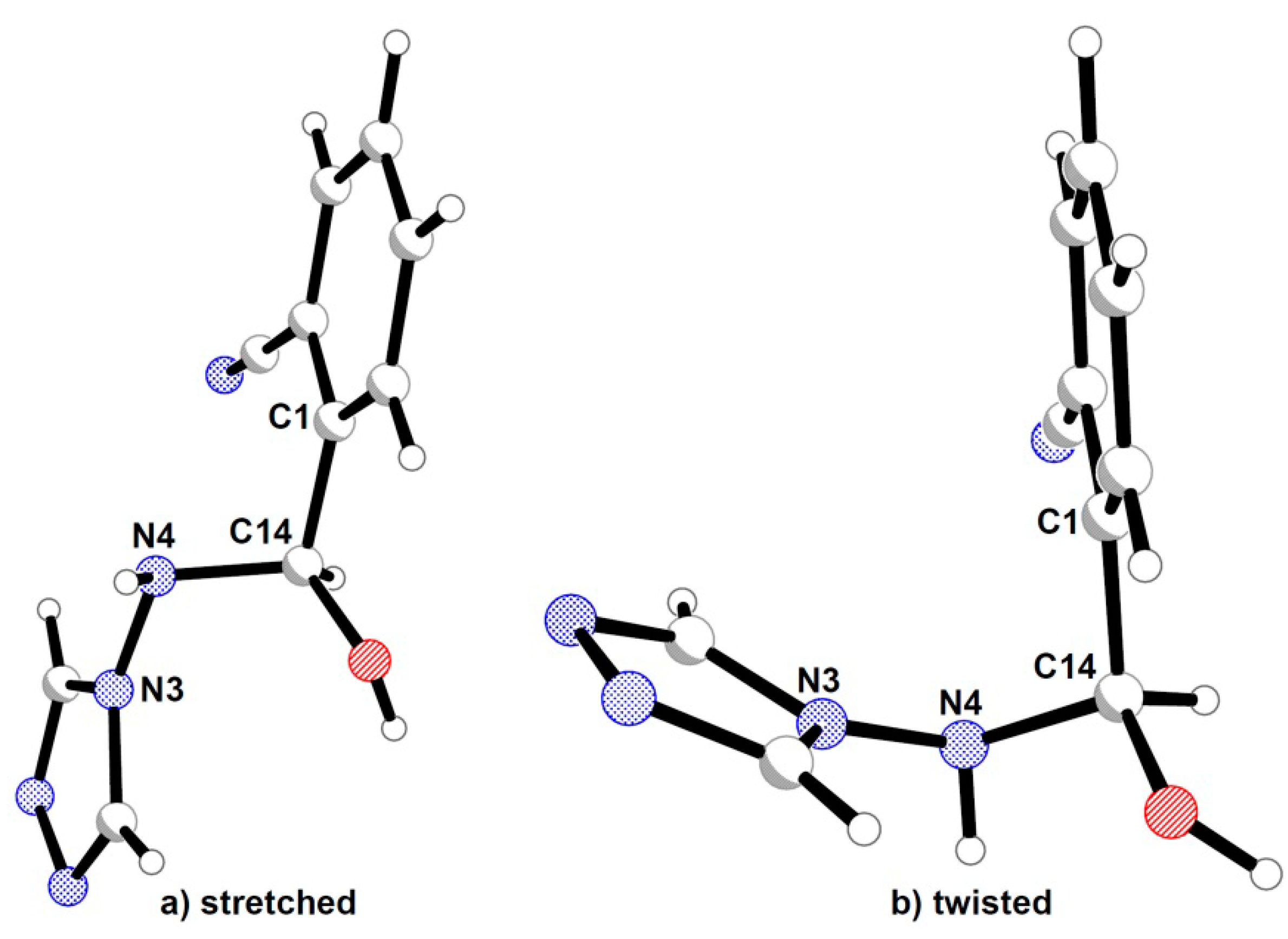{kind=link}
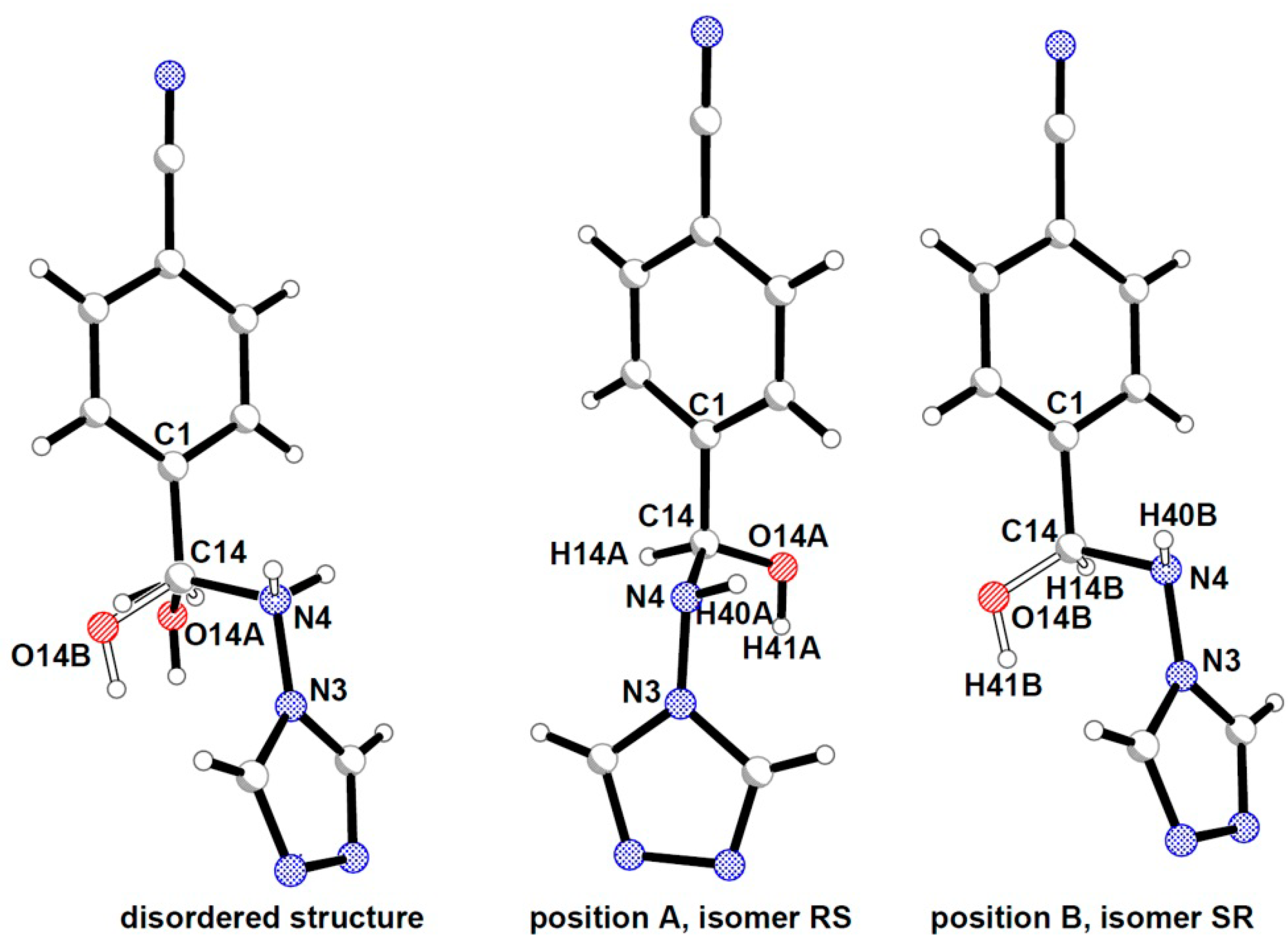{kind=link}
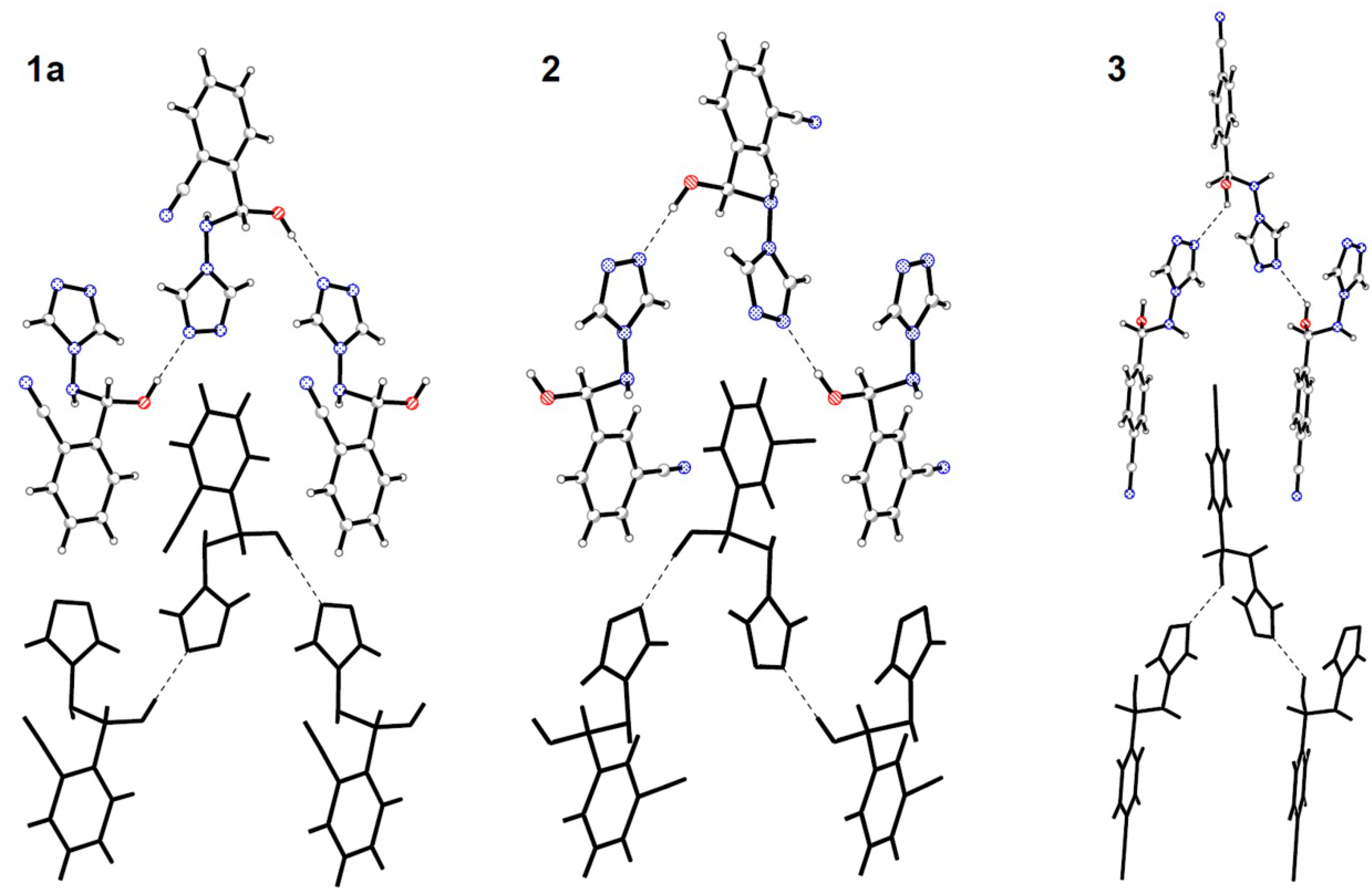{kind=link}
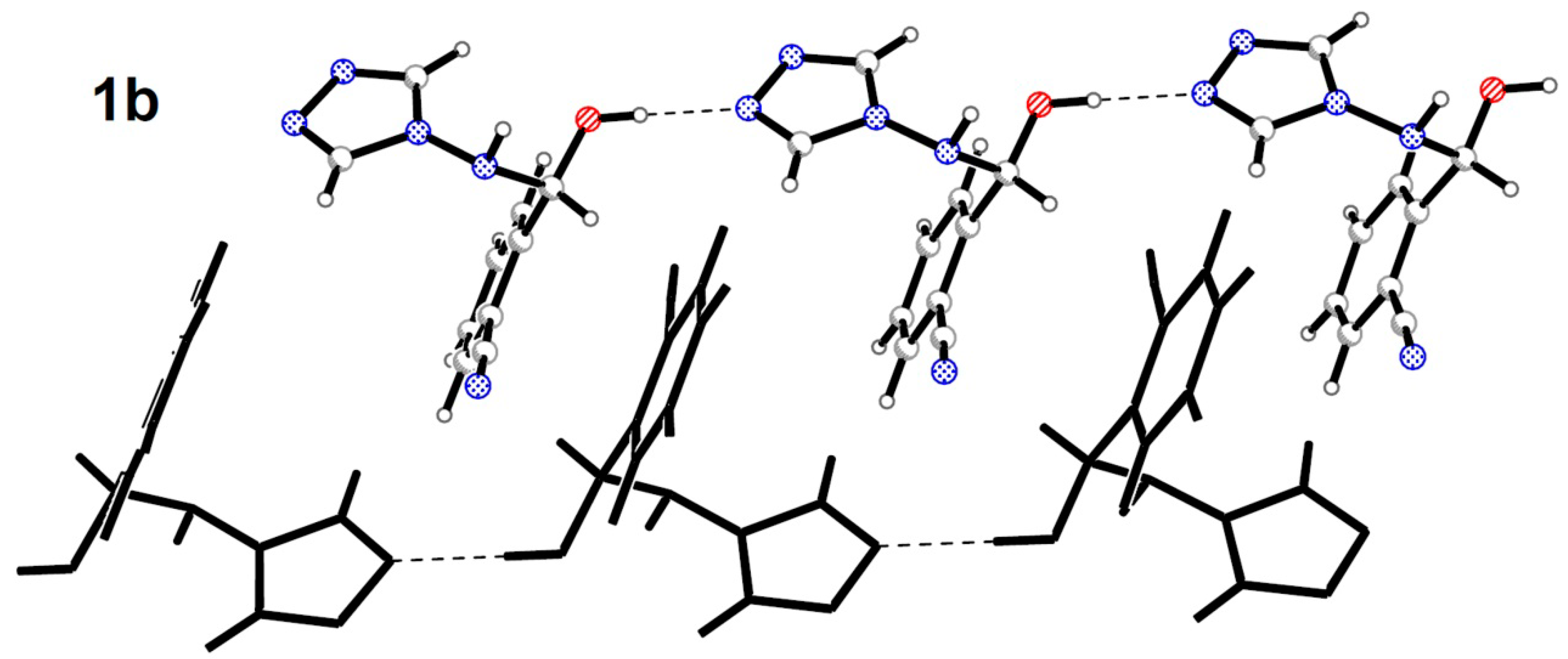{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Geometrical | Bond lengths [Å] | Torsion angles [°] | Phenyl/triazole dihedral angle [°] | |||||
|---|---|---|---|---|---|---|---|---|
| parameter | ||||||||
| C1-C14 | C14-N4 | N4-N3 | C14-O14 | N3-N4-C14-C1 | N3-N4-C14-O14 | |||
| Compound | ||||||||
| 1a | 1.516(1) | 1.474(1) | 1.408(1) | 1.399(1) | 167.4(1) | −71.9(1) | 8.2(1) | |
| 1b | 1.520(2) | 1.468(2) | 1.412(2) | 1.418(2) | −54.0(1) | 68.8(1) | 60.4(1) | |
| 2 | 1.518(2) | 1.470(2) | 1.416(2) | 1.394(2) | 166.1(2) | −72.4(2) | 3.9(1) | |
| 3 / OH group disordered in two positions | ||||||||
| position A | 1.511(3) | 1.469(3) | 1.409(2) | 1.302(3) | 178.0(2) | −59.5(3) | 20.4(1) | |
| position B | 1.246(3) | 39.7(3) | ||||||
| 4 | ||||||||
| molecule 1 | 1.525(2) | 1.459(3) | 1.410(2) | 1.394(2) | 179.1(2) | −55.8(2) | 11.6(2) | |
| molecule 2 | 1.517(2) | 1.468(2) | 1.408(2) | 1.410(2) | 168.0(2) | −71.4(2) | 54.3(8) | |
| 1s | 1.465(2) | 1.272(2) | 1.393(2) | – | 179.6(1) | – | 27.9(1) | |
| 2s | ||||||||
| molecule 1 | 1.464(2) | 1.284(2) | 1.391(2) | – | 178.0(2) | – | 11.4(1) | |
| molecule 2 | 1.469(2) | 1.278(2) | 1.386(2) | – | 177.9(2) | – | 6.5(1) | |
| 4s | ||||||||
| molecule 1 | 1.463(3) | 1.276(3) | 1.390(3) | – | 179.7(2) | – | 18.4(1) | |
| molecule 2 | 1.465(3) | 1.278(3) | 1.397(3) | – | −177.9(2) | – | 16.3(1) | |






2.3. Discussion

3. Experimental Section
3.1. Materials and Methods
3.2. Crystallography
3.3. Synthetic Procedures
3.3.1. General Procedure for Synthesis of Hemiaminals
3.3.2. General Procedure for Synthesis of Schiff Bases.
3.3.3. 2-[Hydroxy(4H-1,2,4-triazol-4-ylamino)methyl]benzonitriles (1a and 1b)
3.3.4. 2-[(E)-(4H-1,2,4-Triazol-4-ylimino)methyl]benzonitrile (1s)
3.3.5. 3-[Hydroxy(4H-1,2,4-triazol-4-ylamino)methyl]benzonitrile (2)
3.3.6. 3-[(E)-(4H-1,2,4-Triazol-4-ylimino)methyl]benzonitrile (2s)
3.3.7. 4-[Hydroxy(4H-1,2,4-triazol-4-ylamino)methyl]benzonitrile (3)
3.3.8. 4-[(4H-1,2,4-Triazol-4-ylimino)methyl]benzonitrile (3s)
3.3.9. 3,5-Difluoro-4-[hydroxy(4H-1,2,4-triazol-4-ylamino)methyl]benzonitrile (4)
3.3.10. 3,5-Difluoro-4-[(E)-(4H-1,2,4-triazol-4-ylimino)methyl]benzonitrile (4s)
4. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Conflicts of Interest
References
- McNaught, A.D.; Wilkinson, A. Compendium of Chemical Terminology, 2nd ed.; Blackwell Scientific Publications: Oxford, UK, 1997. [Google Scholar]
- Smith, J.M.B.; March, J. March’s Advanced Organic Chemistry, 6th ed.; Wiley Interscience: Hoboken, NJ, USA, 2007; pp. 1281–1284. [Google Scholar]
- Baymak, M.S.; Zuman, P. Equilibria of formation and dehydration of the carbinolamine intermediate in the reaction of benzaldehyde with hydrazine. Tetrahedron 2007, 63, 5450–5454. [Google Scholar] [CrossRef]
- Namli, H.; Turhan, O. Simultaneous observation of reagent consumption and product formation with the kinetics of benzaldehyde and aniline reaction in FTIR liquid cell. Vib. Spectrosc. 2007, 43, 274–283. [Google Scholar] [CrossRef]
- Evans, D.A.; Borg, G.; Scheidt, K.A. Remarkably stable tetrahedral intermediates: Carbinols from nucleophilic additions to N-Acylpyrroles. Angew. Chem. Int. Ed. 2002, 41, 3188–3191. [Google Scholar] [CrossRef]
- Hooley, R.J.; Iwasawa, T.; Rebek, J., Jr. Detection of reactive tetrahedral intermediates in a deep cavitand with an introverted functionality. J. Am. Chem. Soc. 2007, 129, 15330–15339. [Google Scholar] [CrossRef] [PubMed]
- Iwasawa, T.; Hooley, R.J.; Rebek, J., Jr. Stabilization of Labile Carbonyl Addition Intermediates by a Synthetic Receptor. Science 2007, 317, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hua, S.; Li, S. Insight into the reaction between a primary amine and a cavitand with an introverted aldehyde group: An enzyme-like mechanism. Chem. Commun. 2013, 49, 1542–1544. [Google Scholar] [CrossRef]
- Kawamichi, T.; Haneda, T.; Kawano, M.; Fujita, M. X-ray observation of a transient hemiaminal trapped in a porous network. Nature 2009, 461, 633–635. [Google Scholar] [CrossRef] [PubMed]
- Morris, W.; Doonan, C.J.; Yaghi, O.M. Postsynthetic modification of a metal–organic framework for stabilization of a hemiaminal and ammonia uptake. Inorg. Chem. 2011, 50, 6853–6855. [Google Scholar] [CrossRef] [PubMed]
- Dolotko, O.; Wiench, J.W.; Dennis, K.W.; Pecharsky, V.K.; Balema, V.P. Mechanically induced reactions in organic solids: Liquid eutectics or solid-state processes? New J. Chem. 2010, 34, 25–28. [Google Scholar]
- Yufit, D.S.; Howard, J.A.K. Structure of hemiaminal intermediate of the reaction of diethylamine with cyclobutanone. J. Mol. Struct. 2010, 984, 182–185. [Google Scholar] [CrossRef]
- Suni, V.; Kurup, M.R.P.; Nethaji, M. Unusual isolation of a hemiaminal product from 4-cyclohexyl-3-thiosemicarbazide and di-2-pyridyl ketone: Structural and spectral investigations. J. Mol. Struct. 2005, 749, 177–182. [Google Scholar] [CrossRef]
- Bouffard, F.A.; Hammond, M.L.; Arison, B.H. Pneumocandin Bo acid degradate. Tetrahedron Lett. 1995, 36, 1405–1408. [Google Scholar] [CrossRef]
- Lal, B.; Gund, V.G. Approaches towards the stabilization of hemiaminal function at ornithine unit of mulundocandin. Bioorg. Med. Chem. Lett. 2004, 14, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- LaLonde, R.T.; Wong, C. Hemiaminal Derivatives of Neothiobinupharidine. J. Org. Chem. 1976, 41, 291–294. [Google Scholar] [CrossRef]
- Graff von Stosch, A. Aspartate Aminotransferase Complexed with erythro-β-Hydroxyaspartate: Crystallographic and Spectroscopic Identification of the Carbinolamine Intermediate. Biochemistry 1996, 35, 15260–15268. [Google Scholar] [CrossRef] [PubMed]
- Salvà, A.; Donoso, J.; Frau, J.; Muñoz, F. Theoretical studies on Schiff base formation of vitamin b6 analogues. J. Mol. Struc.-Theochem. 2002, 577, 229–238. [Google Scholar]
- Erdtman, E.; Bushnell, E.A.C.; Gauld, J.W.; Eriksson, L.A. Computational studies on Schiff-base formation: Implications for the catalytic mechanism of porphobilinogen synthase. Comp. Theor. Chem. 2011, 963, 479–489. [Google Scholar] [CrossRef]
- Matsui, M.; Yamada, K.; Funabiki, K. Hemiacetal and hemiaminal formation at fluoroacyl moiety. Tetrahedron 2005, 61, 4671–4677. [Google Scholar] [CrossRef]
- Liu, X.; Banister, S.D.; Christie, M.J.; Banati, R.; Meikle, S.; Coster, M.J.; Kassiou, M. Trishomocubanes: Novel σ ligands modulate cocaine-induced behavioural effects. Eur. J. Pharmacol. 2007, 555, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Prins, L.H.A.; de Vries, A.; Caira, M.R.; Oliver, D.W.; van Dyk, S.; Malan, S.F. Crystal Structure of a Rearranged Cage Compound, 3-Hydroxy-4-aza-8-oxoheptacyclo [9.4.1.02,10.03,14.04,9.09,13.012,15]tetradecane. J. Chem. Crystallogr. 2008, 38, 705–709. [Google Scholar]
- Prins, L.H.A.; du Preez, J.L.; van Dyk, S.; Malan, S.F. Polycyclic cage structures as carrier molecules for neuroprotective non-steroidal anti-inflammatory drugs. Eur. J. Med. Chem. 2009, 44, 2577–2582. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, D.K.; de Vries, A.; Oliver, D.W.; Malan, S.F. Nitric Oxide Synthase Inhibition by Pentacycloundecane Conjugates of Aminoguanidine and Tryptamine. Arch. Pharm. Chem. Life Sci. 2009, 342, 73–79. [Google Scholar] [CrossRef]
- Banister, S.D.; Moussa, I.A.; Jordan, M.J.T.; Coster, M.J.; Kassiou, M. Oxo-bridged isomers of aza-trishomocubane sigma (r) receptor ligands: Synthesis, in vitro binding, and molecular modeling. Bioorg. Med. Chem. Lett. 2010, 20, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Banister, S.D.; Manoli, M.; Doddareddy, M.R.; Hibbs, D.E.; Kassiou, M. A σ1 receptor pharmacophore derived from a series of N-substituted 4-azahexacyclo[5.4.1.02,6.03,10.05,9.08,11]dodecan-3-ols (AHDs). Bioorg. Med. Chem. Lett. 2012, 22, 6053–6058. [Google Scholar]
- El Mail, R.; Garralda, M.A.; Hernández, R.; Ibarlucea, L.; Pinilla, E.; Torres, M.R. Hydroxyalkyl Complexes and Hemiaminal Formation in the Reaction of o-Diphenylphosphinobenzaldehyde with Rhodium(I) Dihydrazone Complexes. Organometallics 2000, 19, 5310–5317. [Google Scholar] [CrossRef]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. 2002, B58, 380–388. [Google Scholar] [CrossRef]
- Barys, M.; Ciunik, Z.; Drabent, K.; Kwiecień, A. Stable hemiaminals containing a triazole ring. New J. Chem. 2010, 34, 2605–2611. [Google Scholar]
- Sayer, J.M.; Jencks, W.P. Imine-Forming Elimination Reactions. 2. Imbalance of Charge Distribution in the Transition State for Carbinolamine Dehydration. J. Am. Chem. Soc. 1977, 99, 464–474. [Google Scholar]
- Hansch, C.; Leo, A.; Taft, R.W. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Ciunik, Z.; Drabent, K.; Szterenberg, L. Molecular conformation versus C–H···Ph weak hydrogen bonds in 4-(4-H-1,2,4-triazol-4-yl)-2-X-phenylmethanimine (X = CH3, Cl, Br) crystals. J. Mol. Struct. 2002, 641, 175–182. [Google Scholar] [CrossRef]
- Ciunik, Z.; Drabent, K. The C-H···[FBF3]− hydrogen bonds as origin of the linear polytetrameric self-organisation of Schiff base containing substituted 1,2,4-triazole. Pol. J. Chem. 2001, 75, 1475–1482. [Google Scholar]
- Berski, S.; Ciunik, Z.; Drabent, K.; Latajka, Z.; Panek, J. Dominant role of C-Br···N halogen bond in molecular self-organization. Crystallographic and quantum-chemical study of Schiff-base-containing triazoles. J. Phys. Chem. B 2004, 108, 12327–12332. [Google Scholar]
- Kitaev, Y.P.; Savin, V.I.; Zverev, V.V.; Popova, G.V. Structures and reactivities of nitrogen-containing derivatives of carbonyl compounds. Chem. Heterocycl. Compd. 1971, 7, 522–526, (translated from Khim. Geterotsikl. Soedin. 1971, 7, 559–564. [Google Scholar]
- Carey, F.A.; Sundberg, R.J. Advanced Organic Chemistry: Part A: Structure and Mechanisms, 5th ed.; Publisher: Springer, New York, NY, USA, 2007; pp. 81–85. [Google Scholar]
- Kirby, A.J. The Anomeric Effect and Related Stereoelectronic Effects at Oxygen, 1st ed.; Springer-Verlag: Berlin, Germany, 1983. [Google Scholar]
- Cosier, J.; Glazer, A.M. A nitrogen-gas-stream cryostat for general X-ray diffraction studies. J. Appl. Cryst. 1986, 19, 105–107. [Google Scholar] [CrossRef]
- CrysAlis CCD, CrysAlis RED, CrysAlisPRO; Oxford Diffraction /Agilent Technologies UK Ltd: Yarnton, UK, 2009.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A: Found. Crystallogr. 2008, 64, 112–122. [Google Scholar]
- XP—Interactive Molecular Graphics, Bruker Analytical X-ray System, Version 5.1; Bruker AXS Inc.: Madison, WI, USA, 1998.
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kwiecień, A.; Barys, M.; Ciunik, Z. Stable Hemiaminals with a Cyano Group and a Triazole Ring. Molecules 2014, 19, 11160-11177. https://doi.org/10.3390/molecules190811160
Kwiecień A, Barys M, Ciunik Z. Stable Hemiaminals with a Cyano Group and a Triazole Ring. Molecules. 2014; 19(8):11160-11177. https://doi.org/10.3390/molecules190811160
Chicago/Turabian StyleKwiecień, Anna, Maciej Barys, and Zbigniew Ciunik. 2014. "Stable Hemiaminals with a Cyano Group and a Triazole Ring" Molecules 19, no. 8: 11160-11177. https://doi.org/10.3390/molecules190811160