Neuroprotective Effects of Rhynchophylline Against Ischemic Brain Injury via Regulation of the Akt/mTOR and TLRs Signaling Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
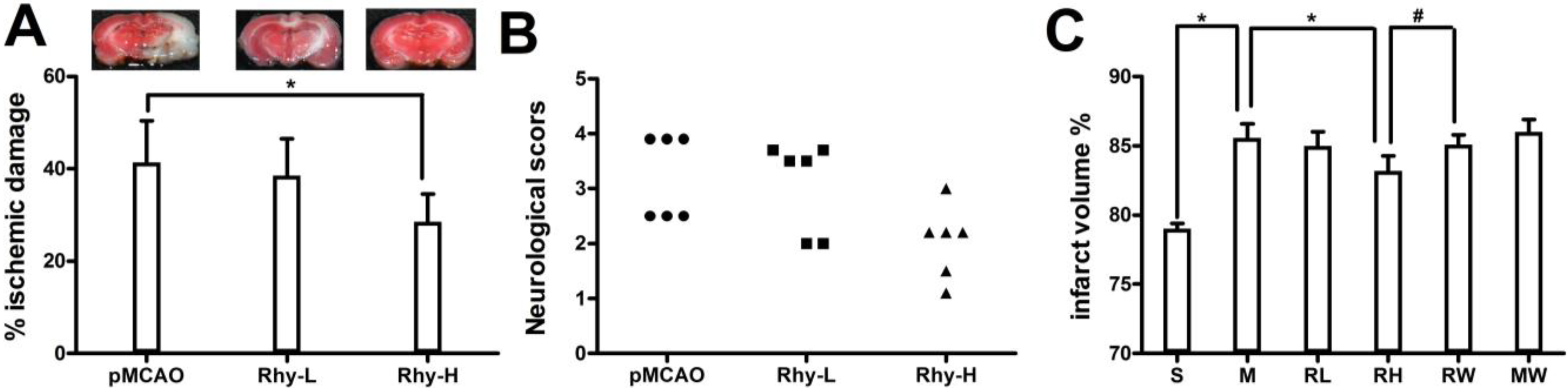
2.1. Effect of Rhy on Cerebral Infarction, Neurological Deficit, and Brain Edema

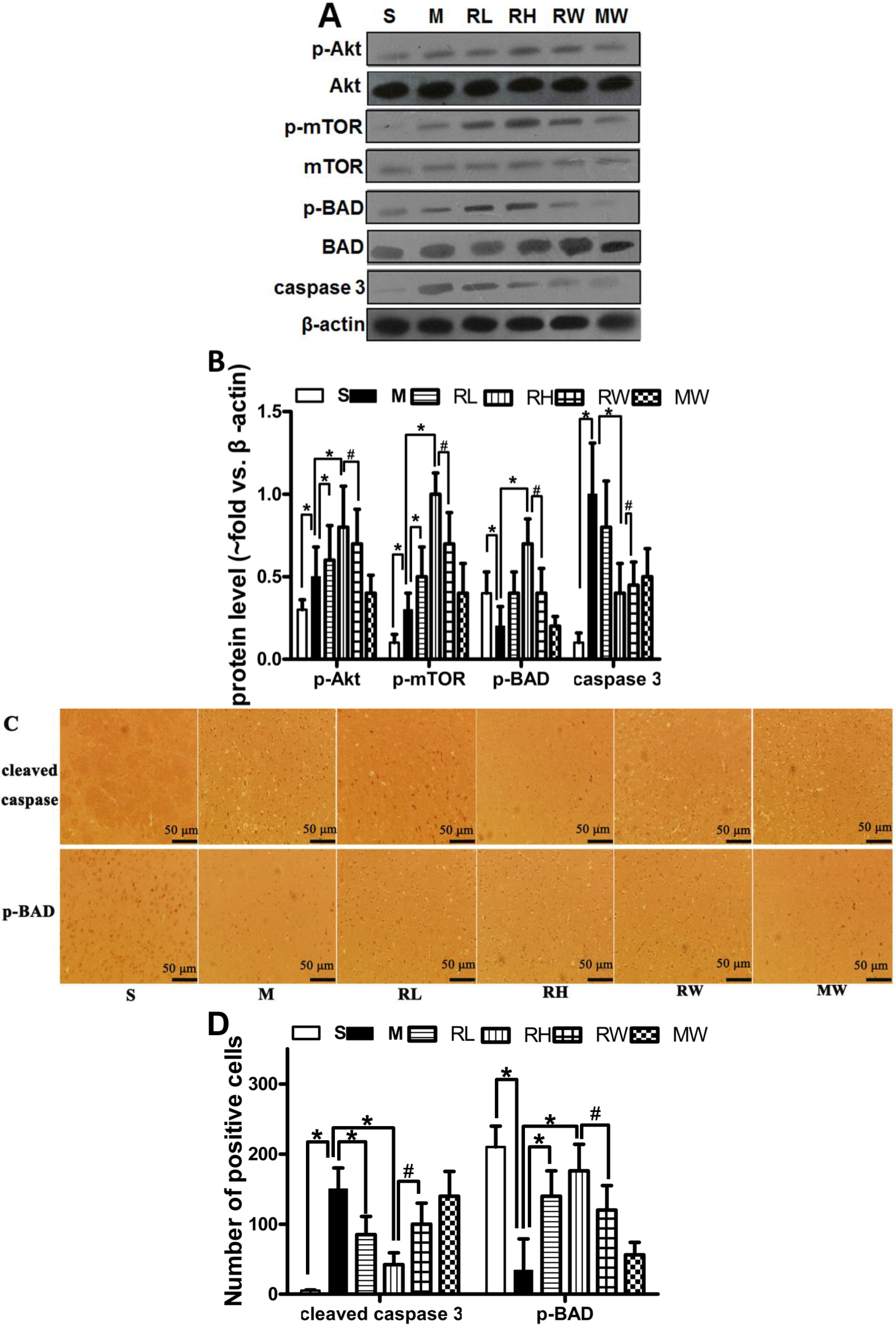
2.2. Effects of Rhy on p-Akt, p-mTOR, p-BAD and Caspase-3 Expressions

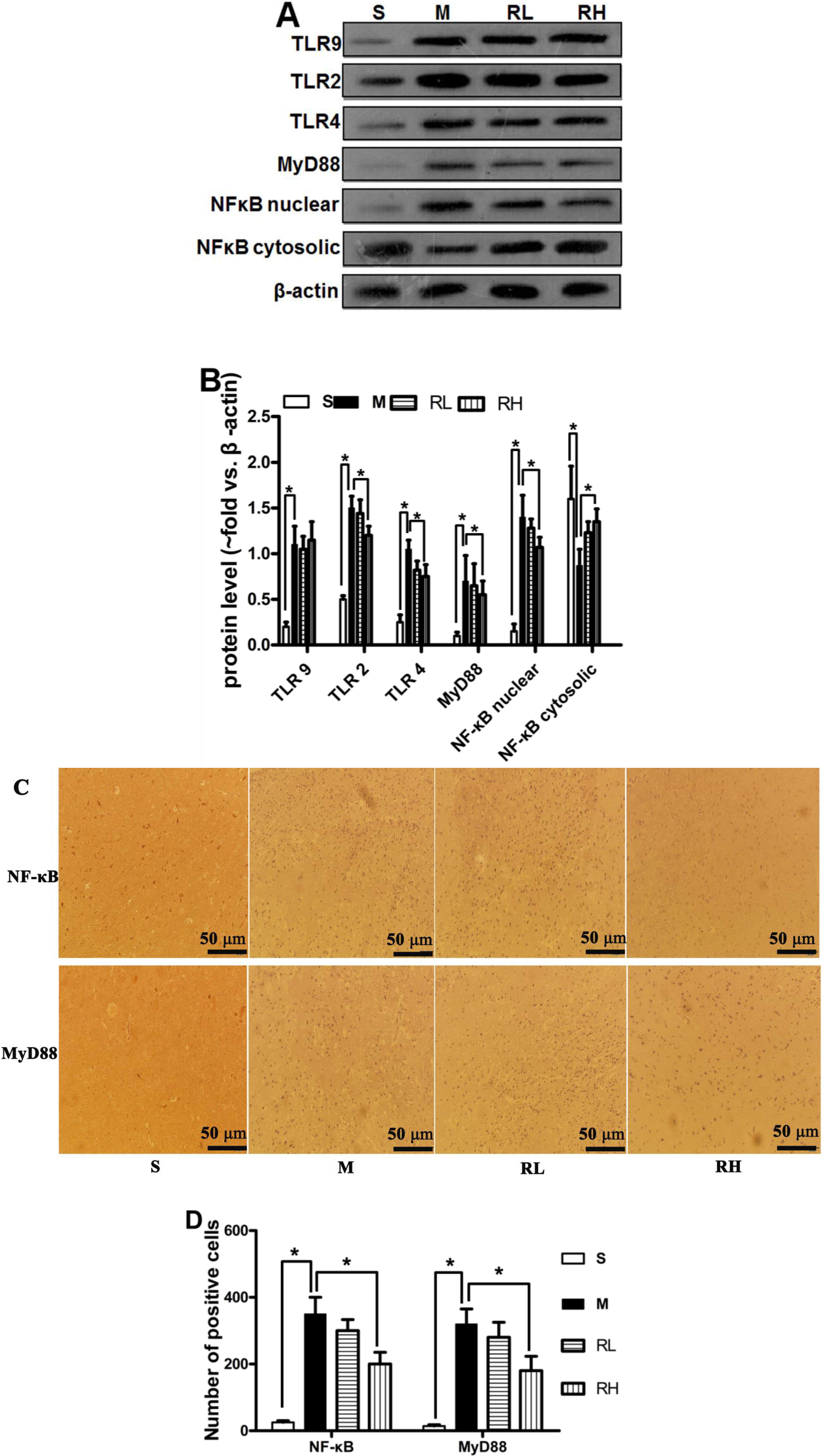
2.3. Effect of Rhy on the Activation of TLRs/NF-κB Signaling

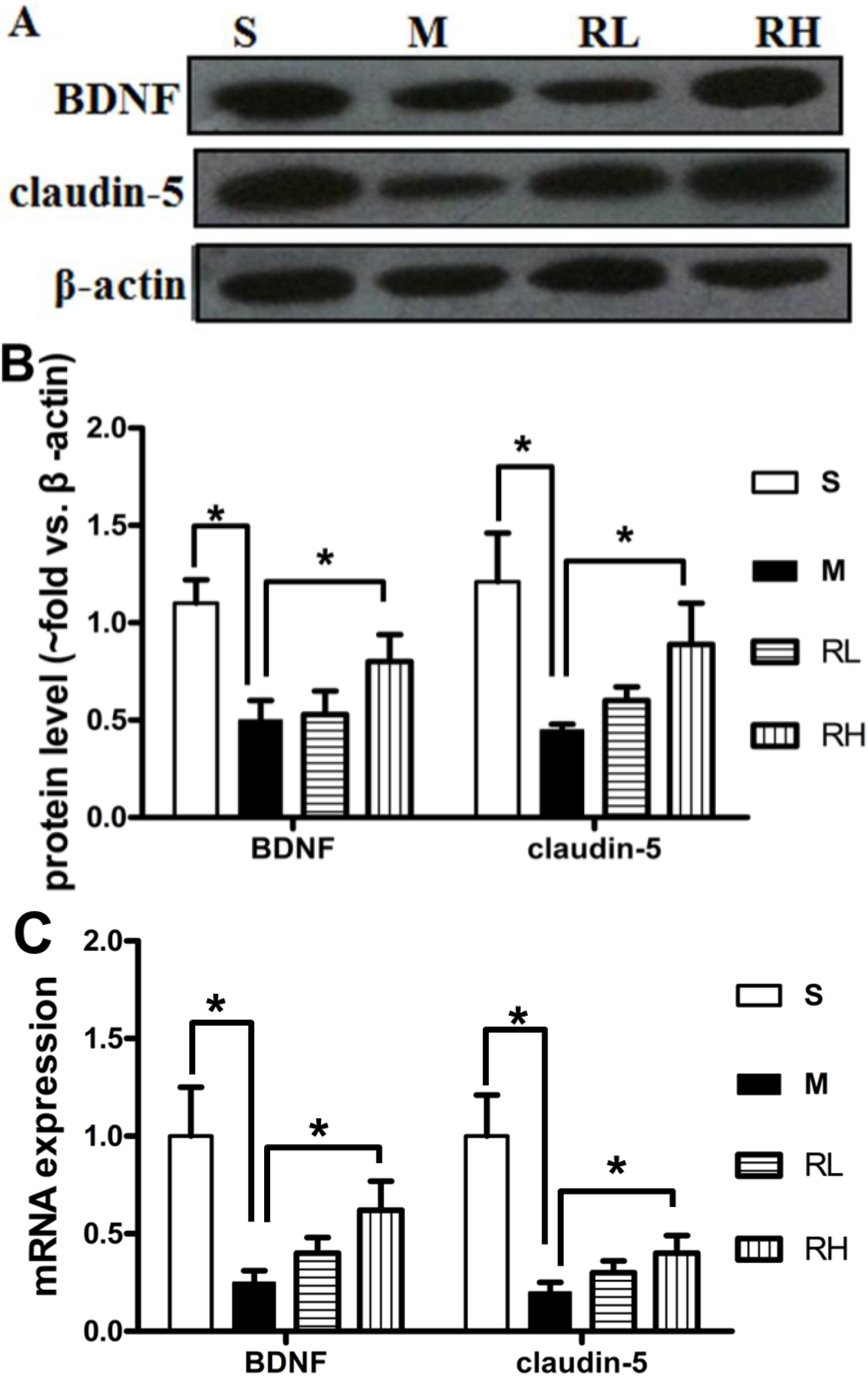
2.4. Effect of Rhy on the Expression of Claudin-5 and BDNF

2.5. Discussion
3. Experimental Section
3.1. Animals
3.2. Surgery
3.3. Experimental Design
3.4. Neurobehavioral Evaluation
3.5. Infarct Volumes
3.6. Brain Edema
3.7. Western Blot Analysis
3.8. Immunohistochemical Rtudies
3.9. Real-Time Reverse Ranscription-Quantitative PCR (RT-qPCR)
3.10. Statistics
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lu, C.; Ha, T.; Wang, X.; Liu, L.; Zhang, X.; Kimbrough, E.O.; Sha, Z.; Guan, M.; Schweitzer, J.; Kalbfleisch, J.; et al. The TLR9 ligand, CpG-ODN, induces protection against cerebral ischemia/reperfusion injury via activation of PI3K/Akt signaling. J. Am. Heart Assoc. 2014, 3, e000629. [Google Scholar] [CrossRef]
- Marder, V.J.; Jahan, R.; Gruber, T.; Goyal, A.; Arora, V. Thrombolysis with plasmin: Implications for stroke treatment. Stroke 2010, 41, S45–S49. [Google Scholar] [CrossRef]
- Wang, Q.; Tang, X.N.; Yenari, M.A. The inflammatory response in stroke. J. Neuroimmunol. 2007, 184, 53–68. [Google Scholar] [CrossRef]
- Zhang, X.; Li, H.; Hu, S.; Zhang, L.; Liu, C.; Zhu, C.; Liu, R.; Li, C. Brain edema after intracerebral hemorrhage in rats: The role of inflammation. Neurol. India 2006, 54, 402–407. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Liu, H.; Liu, X.; Wei, X.; Chen, L.; Xiang, Y.; Yi, F.; Zhang, X. Losartan, an angiotensin II type 1 receptor blocker, ameliorates cerebral ischemia-reperfusion injury via PI3K/Akt-mediated eNOS phosphorylation. Brain Res. Bull. 2012, 89, 65–70. [Google Scholar] [CrossRef]
- Del Zoppo, G.J. Acute anti-inflammatory approaches to ischemic stroke. Ann. NY Acad. Sci. 2010, 1207, 143–148. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, X.; Liu, L.; Cui, L.; Yang, R.; Li, M.; Du, W. Tanshinone II A down-regulates HMGB1, RAGE, TLR4, NF-kappaB expression, ameliorates BBB permeability and endothelial cell function, and protects rat brains against focal ischemia. Brain Res. 2010, 1321, 143–151. [Google Scholar]
- Tsung, A.; Sahai, R.; Tanaka, H.; Nakao, A.; Fink, M.P.; Lotze, M.T.; Yang, H.; Li, J.; Tracey, K.J.; Geller, D.A.; et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 2005, 201, 1135–1143. [Google Scholar] [CrossRef]
- Lu, C.; Liu, L.; Chen, Y.; Ha, T.; Kelley, J.; Schweitzer, J.; Kalbfleisch, J.H.; Kao, R.L.; Williams, D.L.; Li, C. TLR2 ligand induces protection against cerebral ischemia/reperfusion injury via activation of phosphoinositide 3-kinase/Akt signaling. J. Immunol. 2011, 187, 1458–1466. [Google Scholar] [CrossRef]
- Zhou, J.; Zhou, S. Antihypertensive and neuroprotective activities of rhynchophylline: the role of rhynchophylline in neurotransmission and ion channel activity. J. Ethnopharmacol. 2010, 132, 15–27. [Google Scholar] [CrossRef]
- Kang, T.H.; Murakami, Y.; Matsumoto, K.; Takayama, H.; Kitajima, M.; Aimi, N.; Watanabe, H. Rhynchophylline and isorhynchophylline inhibit NMDA receptors expressed in Xenopus oocytes. Eur. J. Pharmacol. 2002, 455, 27–34. [Google Scholar] [CrossRef]
- Kang, T.H.; Murakami, Y.; Takayama, H.; Kitajima, M.; Aimi, N.; Watanabe, H.; Matsumoto, K. Protective effect of rhynchophylline and isorhynchophylline on in vitro ischemia-induced neuronal damage in the hippocampus: putative neurotransmitter receptors involved in their action. Life Sci. 2004, 76, 331–343. [Google Scholar] [CrossRef]
- Mohamed, A.F.; Matsumoto, K.; Tabata, K.; Takayama, H.; Kitajima, M.; Watanabe, H. Effects of Uncaria tomentosa total alkaloid and its components on experimental amnesia in mice: Elucidation using the passive avoidance test. J. Pharm. Pharmacol. 2000, 52, 1553–1561. [Google Scholar] [CrossRef]
- Xu, D.D.; Hoeven, R.; Rong, R.; Cho, W.C. Rhynchophylline protects cultured rat neurons against methamphetamine cytotoxicity. Evid. Based Complement. Alternat. Med. 2012, 2012, 636091. [Google Scholar]
- Shi, J.S.; Kenneth, H.G. Effect of rhynchophylline on apoptosis induced by dopamine in NT2 cells. Acta Pharm. Sin. 2002, 23, 445–449. [Google Scholar]
- Yuan, D.; Ma, B.; Yang, J.Y.; Xie, Y.Y.; Wang, L.; Zhang, L.J.; Kano, Y.; Wu, C.F. Anti-inflammatory effects of rhynchophylline and isorhynchophylline in mouse N9 microglial cells and the molecular mechanism. Int. Immunopharmacol. 2009, 9, 1549–1554. [Google Scholar] [CrossRef]
- Greenberg, M.E.; Xu, B.; Lu, B.; Hempstead, B.L. New insights in the biology of BDNF synthesis and release: implications in CNS function. J. Neurosci. 2009, 29, 12764–12767. [Google Scholar] [CrossRef]
- Kitagawa, K. CREB and cAMP response element-mediated gene expression in the ischemic brain. FEBS J. 2007, 274, 3210–3217. [Google Scholar]
- Han, B.H.; D’Costa, A.; Back, S.A.; Parsadanian, M.; Patel, S.; Shah, A.R.; Gidday, J.M.; Srinivasan, A.; Deshmukh, M.; Holtzman, D.M. BDNF blocks caspase-3 activation in neonatal hypoxia-ischemia. Neurobiol. Dis. 2000, 7, 38–53. [Google Scholar] [CrossRef]
- Chen, L.; Wang, L.; Zhang, X.; Cui, L.; Xing, Y.; Dong, L.; Liu, Z.; Li, Y.; Zhang, X.; Wang, C.; et al. The protection by octreotide against experimental ischemic stroke: Up-regulated transcription factor Nrf2, HO-1 and down-regulated NF-kappaB expression. Brain Res. 2012, 1475, 80–87. [Google Scholar] [CrossRef]
- Liesz, A.; Zhou, W.; Na, S.Y.; Hammerling, G.J.; Garbi, N.; Karcher, S.; Mracsko, E.; Backs, J.; Rivest, S.; Veltkamp, R. Boosting regulatory T cells limits neuroinflammation in permanent cortical stroke. J. Neurosci. 2013, 33, 17350–17362. [Google Scholar] [CrossRef]
- Hua, F.; Ma, J.; Ha, T.; Kelley, J.L.; Kao, R.L.; Schweitzer, J.B.; Kalbfleisch, J.H.; Williams, D.L.; Li, C. Differential roles of TLR2 and TLR4 in acute focal cerebral ischemia/reperfusion injury in mice. Brain Res. 2009, 1262, 100–108. [Google Scholar]
- Hua, F.; Ma, J.; Ha, T.; Kelley, J.; Williams, D.L.; Kao, R.L.; Kalbfleisch, J.H.; Browder, I.W.; Li, C. Preconditioning with a TLR2 specific ligand increases resistance to cerebral ischemia/reperfusion injury. J. Neuroimmunol. 2008, 199, 75–82. [Google Scholar] [CrossRef]
- He, W.; Zhang, Y.; Zhang, J.; Yu, Q.; Wang, P.; Wang, Z.; Smith, A.J. Cytidine-phosphate-guanosine oligonucleotides induce interleukin-8 production through activation of TLR9, MyD88, NF-kappaB, and ERK pathways in odontoblast cells. J. Endod. 2012, 38, 780–785. [Google Scholar]
- Hua, F.; Ma, J.; Ha, T.; Xia, Y.; Kelley, J.; Williams, D.L.; Kao, R.L.; Browder, I.W.; Schweitzer, J.B.; Kalbfleisch, J.H.; et al. Activation of Toll-like receptor 4 signaling contributes to hippocampal neuronal death following global cerebral ischemia/reperfusion. J. Neuroimmunol. 2007, 190, 101–111. [Google Scholar] [CrossRef]
- Ziegler, G.; Harhausen, D.; Schepers, C.; Hoffmann, O.; Rohr, C.; Prinz, V.; Konig, J.; Lehrach, H.; Nietfeld, W.; Trendelenburg, G. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem. Biophys. Res. Commun. 2007, 359, 574–579. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nature reviews Immunology 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Tang, S.C.; Arumugam, T.V.; Xu, X.; Cheng, A.; Mughal, M.R.; Jo, D.G.; Lathia, J.D.; Siler, D.A.; Chigurupati, S.; Ouyang, X.; et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 13798–13803. [Google Scholar] [CrossRef]
- Barakat, W.; Safwet, N.; El-Maraghy, N.N.; Zakaria, M.N. Candesartan and glycyrrhizin ameliorate ischemic brain damage through downregulation of the TLR signaling cascade. Eur. J. Pharm. 2014, 724, 43–50. [Google Scholar] [CrossRef]
- Bamboat, Z.M.; Balachandran, V.P.; Ocuin, L.M.; Obaid, H.; Plitas, G.; DeMatteo, R.P. Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury. Hepatology 2010, 51, 621–632. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Z.; Zhang, X.; Zhang, X.; Dong, L.; Xing, Y.; Li, Y.; Liu, Z.; Chen, L.; Qiao, H.; et al. Protection by silibinin against experimental ischemic stroke: up-regulated pAkt, pmTOR, HIF-1alpha and Bcl-2, down-regulated Bax, NF-kappaB expression. Neurosci. Lett. 2012, 529, 45–50. [Google Scholar] [CrossRef]
- Zhang, Z.; Yu, B.; Tao, G.Z. Apelin protects against cardiomyocyte apoptosis induced by glucose deprivation. Chin. Med. J. 2009, 122, 2360–2365. [Google Scholar]
- Zhang, Y.; Ren, J. Autophagy in ALDH2-elicited cardioprotection against ischemic heart disease: Slayer or savior? Autophagy 2010, 6, 1212–1213. [Google Scholar] [CrossRef]
- Osuka, K.; Watanabe, Y.; Usuda, N.; Nakazawa, A.; Tokuda, M.; Yoshida, J. Modification of endothelial NO synthase through protein phosphorylation after forebrain cerebral ischemia/reperfusion. Stroke 2004, 35, 2582–2586. [Google Scholar]
- Noshita, N.; Lewen, A.; Sugawara, T.; Chan, P.H. Evidence of phosphorylation of Akt and neuronal survival after transient focal cerebral ischemia in mice. J. Cerebr. Blood Flow Metabol. 2001, 21, 1442–1450. [Google Scholar]
- Shibata, M.; Yamawaki, T.; Sasaki, T.; Hattori, H.; Hamada, J.; Fukuuchi, Y.; Okano, H.; Miura, M. Upregulation of Akt phosphorylation at the early stage of middle cerebral artery occlusion in mice. Brain Res. 2002, 942, 1–10. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, H.; Takahashi, T.; Hsieh, J.; Liao, J.; Steinberg, G.K.; Zhao, H. The Akt signaling pathway contributes to postconditioning’s protection against stroke; the protection is associated with the MAPK and PKC pathways. J. Neurochem. 2008, 105, 943–955. [Google Scholar] [CrossRef]
- Ishrat, T.; Sayeed, I.; Atif, F.; Hua, F.; Stein, D.G. Progesterone is neuroprotective against ischemic brain injury through its effects on the phosphoinositide 3-kinase/protein kinase B signaling pathway. Neuroscience 2012, 210, 442–450. [Google Scholar]
- Yano, S.; Morioka, M.; Fukunaga, K.; Kawano, T.; Hara, T.; Kai, Y.; Hamada, J.; Miyamoto, E.; Ushio, Y. Activation of Akt/protein kinase B contributes to induction of ischemic tolerance in the CA1 subfield of gerbil hippocampus. J. Cerebr. Blood Flow Metabol. 2001, 21, 351–360. [Google Scholar]
- Perez-Alvarez, M.J.; Maza Mdel, C.; Anton, M.; Ordonez, L.; Wandosell, F. Post-ischemic estradiol treatment reduced glial response and triggers distinct cortical and hippocampal signaling in a rat model of cerebral ischemia. J. Neuroinflamm. 2012, 9, 157. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Sapolsky, R.M.; Steinberg, G.K. Phosphoinositide-3-kinase/akt survival signal pathways are implicated in neuronal survival after stroke. Mol. Neurobiol. 2006, 34, 249–270. [Google Scholar] [CrossRef]
- Yang, E.; Zha, J.; Jockel, J.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 1995, 80, 285–291. [Google Scholar] [CrossRef]
- Gardiner, J.; Barton, D.; Overall, R.; Marc, J. Neurotrophic support and oxidative stress: converging effects in the normal and diseased nervous system. Neuroscientist 2009, 15, 47–61. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, X.; Wang, L.; Yang, R.; Cui, L.; Li, M.; Du, W.; Wang, S. The neuroprotective effects of Tanshinone IIA are associated with induced nuclear translocation of TORC1 and upregulated expression of TORC1, pCREB and BDNF in the acute stage of ischemic stroke. Brain Res. Bull. 2010, 82, 228–233. [Google Scholar] [CrossRef]
- Lan, X.; Zhang, M.; Yang, W.; Zheng, Z.; Wu, Y.; Zeng, Q.; Liu, S.; Liu, K.; Li, G. Effect of treadmill exercise on 5-HT, 5-HT1A receptor and brain derived neurophic factor in rats after permanent middle cerebral artery occlusion. Neurol. Sci. 2014, 35, 761–766. [Google Scholar] [CrossRef]
- Zhang, Z.; Peng, D.; Zhu, H.; Wang, X. Experimental evidence of Ginkgo biloba extract EGB as a neuroprotective agent in ischemia stroke rats. Brain Res. Bull. 2012, 87, 193–198. [Google Scholar] [CrossRef]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989, 20, 84–91. [Google Scholar]
- Yang, C.; Zhang, X.; Fan, H.; Liu, Y. Curcumin upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res. 2009, 1282, 133–141. [Google Scholar] [CrossRef]
- Garcia, J.H.; Wagner, S.; Liu, K.F.; Hu, X.J. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Stroke 1995, 26, 627–634. [Google Scholar] [CrossRef]
- Xing, Y.; Zhang, X.; Zhao, K.; Cui, L.; Wang, L.; Dong, L.; Li, Y.; Liu, Z.; Wang, C.; Zhang, X.; et al. Beneficial effects of sulindac in focal cerebral ischemia: a positive role in Wnt/beta-catenin pathway. Brain Res. 2012, 1482, 71–80. [Google Scholar]
- Zhang, J.; Fu, B.; Zhang, X.; Chen, L.; Zhang, L.; Zhao, X.; Bai, X.; Zhu, C.; Cui, L.; Wang, L. Neuroprotective effect of bicyclol in rat ischemic stroke: down-regulates TLR4, TLR9, TRAF6, NF-kappaB, MMP-9 and up-regulates claudin-5 expression. Brain Res. 2013, 1528, 80–88. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Huang, H.; Zhong, R.; Xia, Z.; Song, J.; Feng, L. Neuroprotective Effects of Rhynchophylline Against Ischemic Brain Injury via Regulation of the Akt/mTOR and TLRs Signaling Pathways. Molecules 2014, 19, 11196-11210. https://doi.org/10.3390/molecules190811196
Huang H, Zhong R, Xia Z, Song J, Feng L. Neuroprotective Effects of Rhynchophylline Against Ischemic Brain Injury via Regulation of the Akt/mTOR and TLRs Signaling Pathways. Molecules. 2014; 19(8):11196-11210. https://doi.org/10.3390/molecules190811196
Chicago/Turabian StyleHuang, Houcai, Rongling Zhong, Zhi Xia, Jie Song, and Liang Feng. 2014. "Neuroprotective Effects of Rhynchophylline Against Ischemic Brain Injury via Regulation of the Akt/mTOR and TLRs Signaling Pathways" Molecules 19, no. 8: 11196-11210. https://doi.org/10.3390/molecules190811196