Chemistry of Phosphorylated Formaldehyde Derivatives. Part I

Laboratory of Organophosphorus Compounds, Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, Vavilova str., 28, Moscow 119991, Russia
Molecules 2014, 19(9), 12949-13009; https://doi.org/10.3390/molecules190912949
Submission received: 8 July 2014
/
Revised: 8 August 2014
/
Accepted: 15 August 2014
/
Published: 25 August 2014
(This article belongs to the Special Issue Organophosphorus Chemistry)
Abstract
:The underinvestigated derivatives of unstable phosphorylated formaldehyde acetals and some of the structurally related compounds, such as thioacetals, aminonitriles, aminomethylphosphinoyl compounds, are considered. Separately considered are halogen aminals of phosphorylated formaldehyde, acetals of phosphorylated formaldehyde of H-phosphinate-type and a phosphorylated gem-diol of formaldehyde. Synthetic methods, chemical properties and examples of practical applications are given.
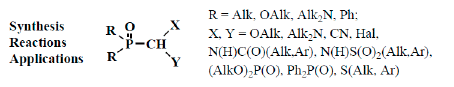
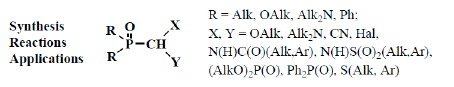{kind=link}
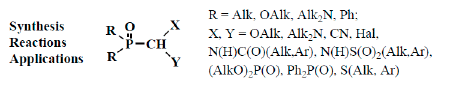{kind=link}
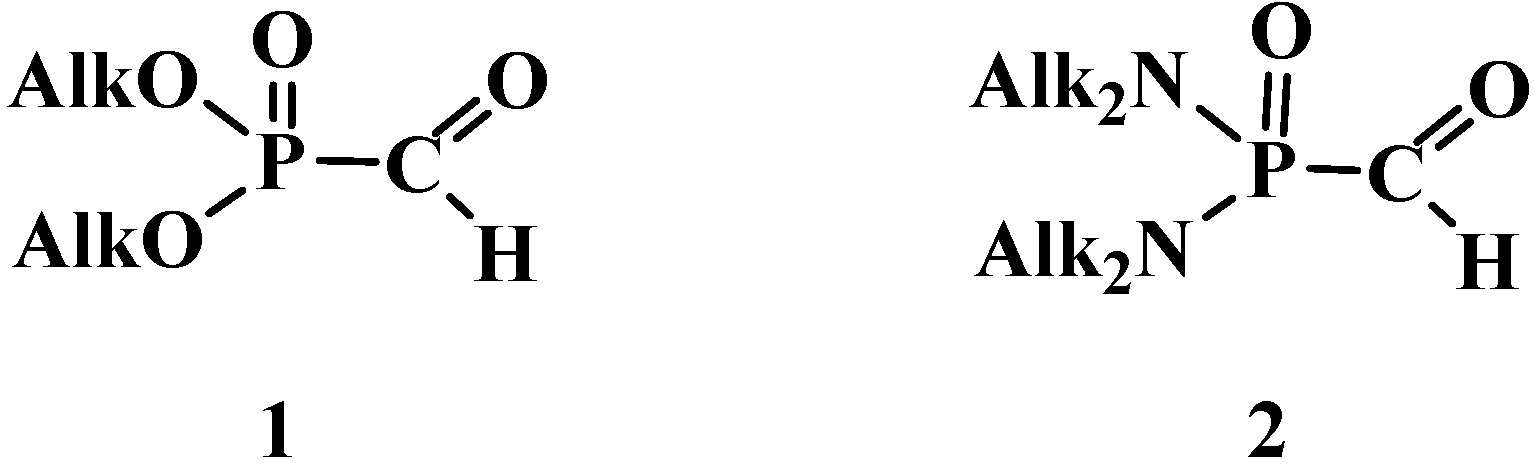{kind=link}
{kind=link}
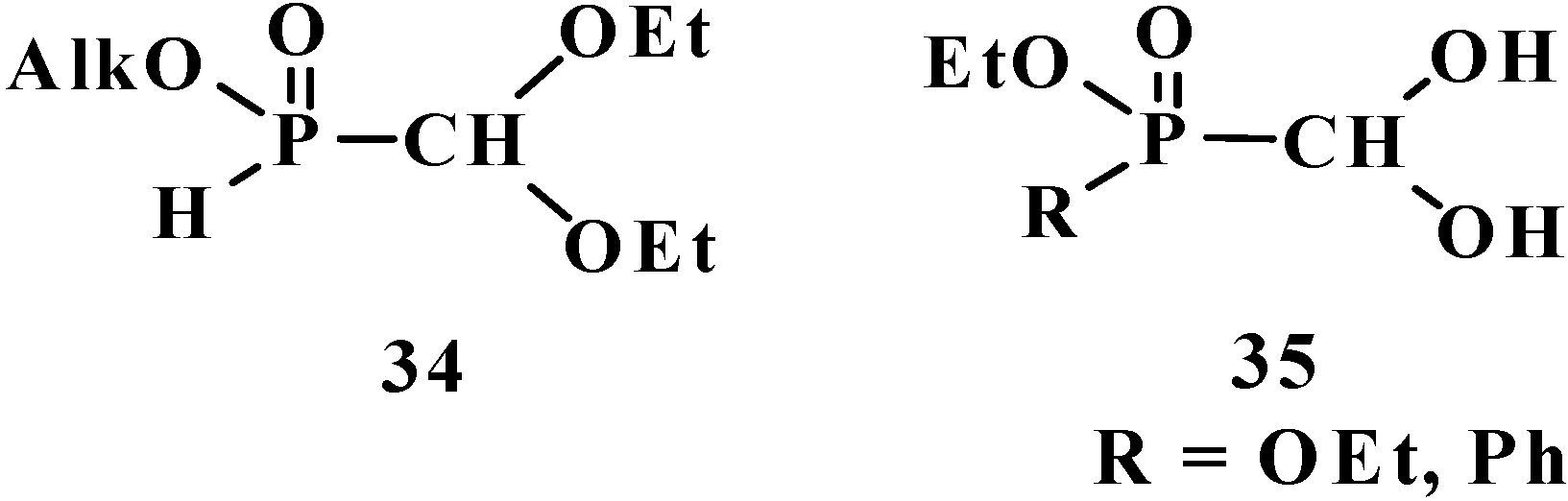{kind=link}
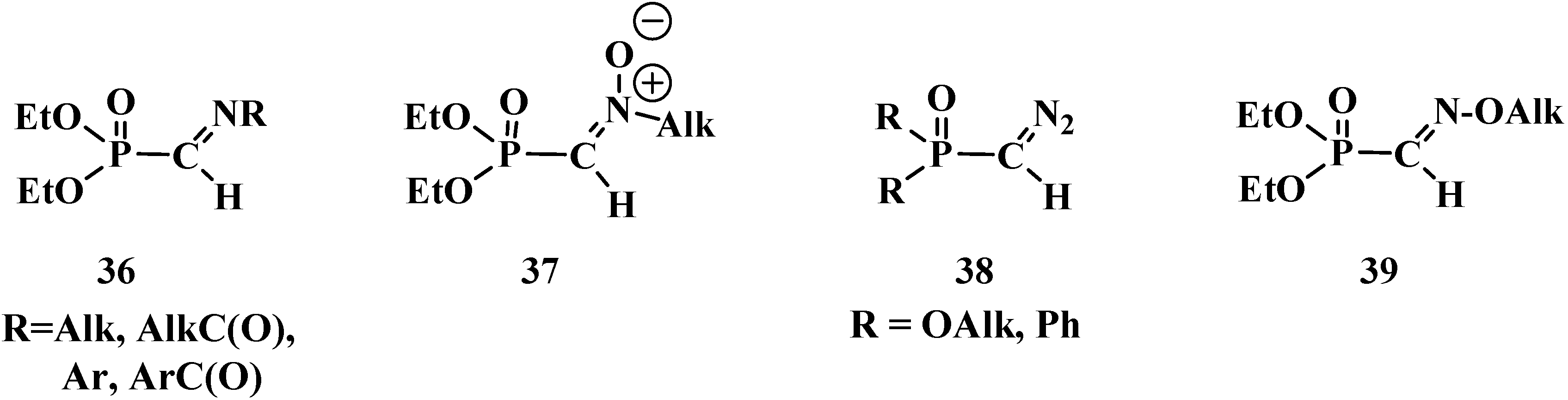{kind=link}
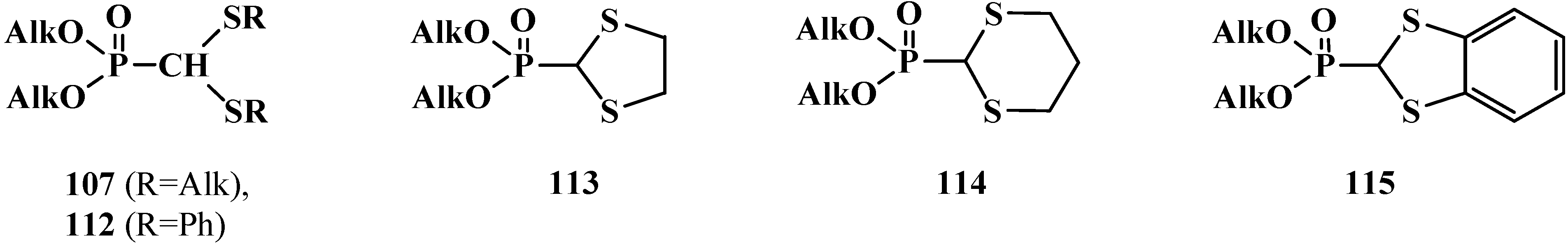{kind=link}
{kind=link}
{kind=link}
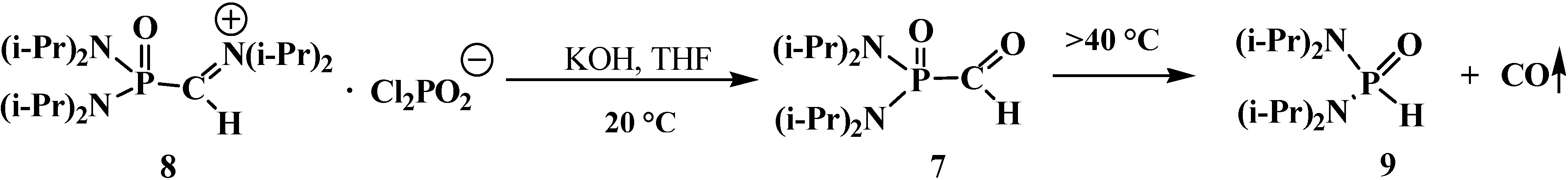{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
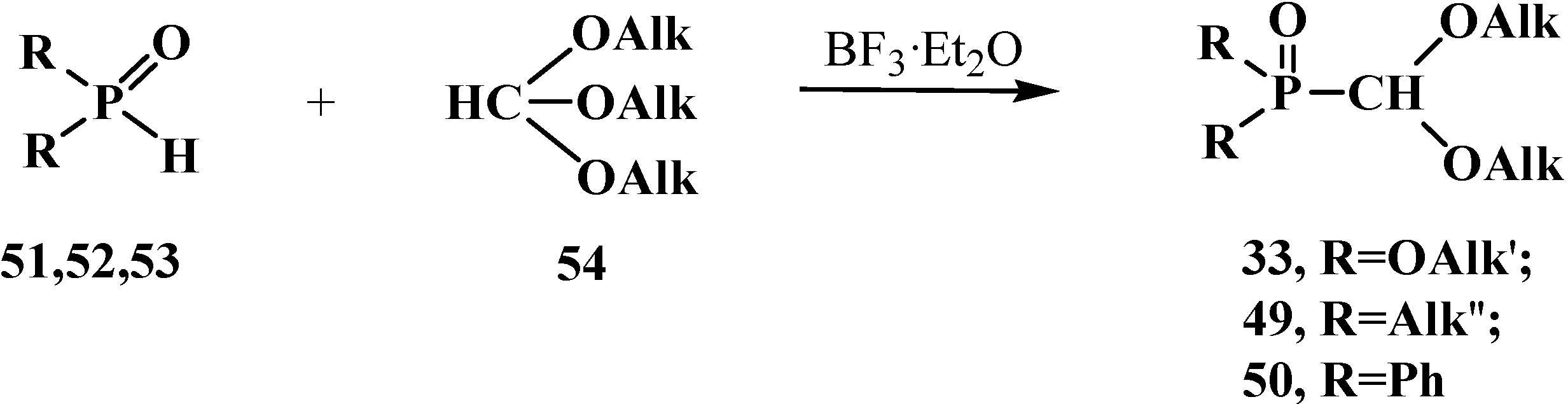{kind=link}
{kind=link}
{kind=link}
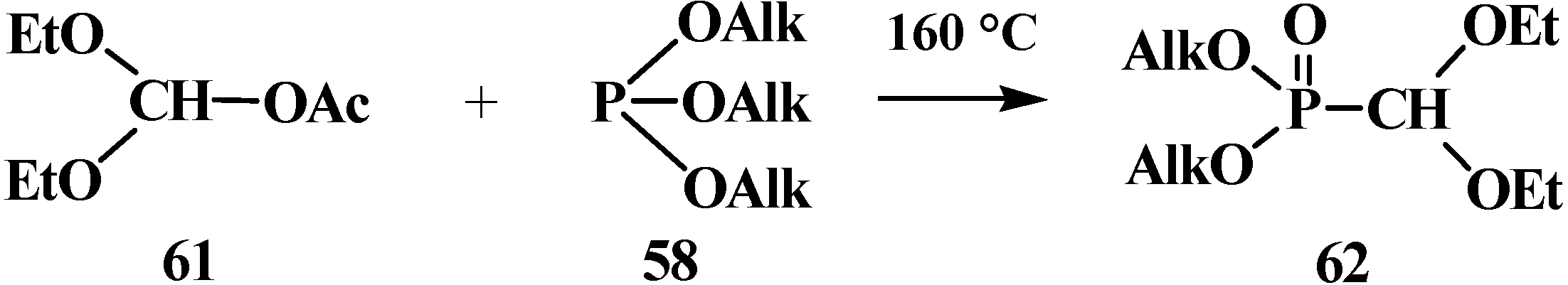{kind=link}
{kind=link}
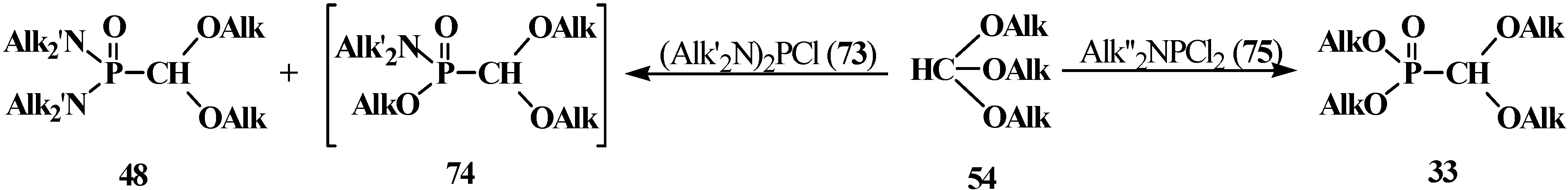{kind=link}
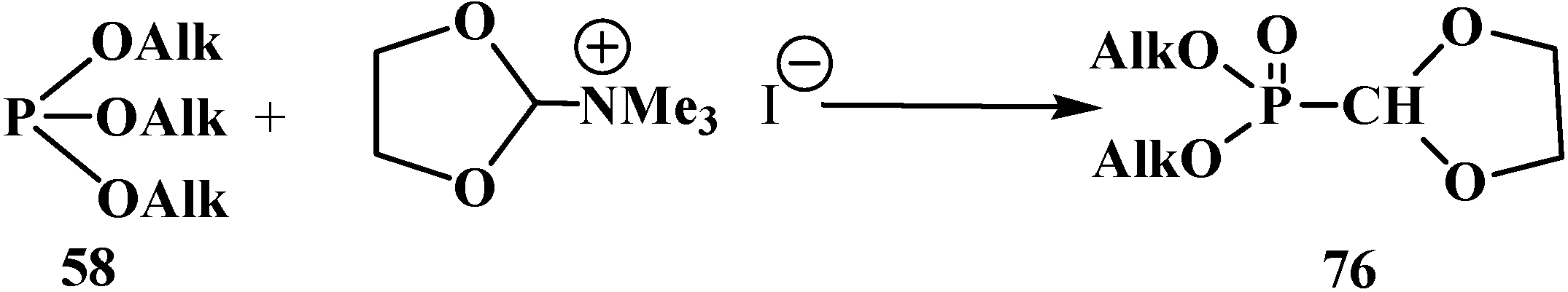{kind=link}
{kind=link}
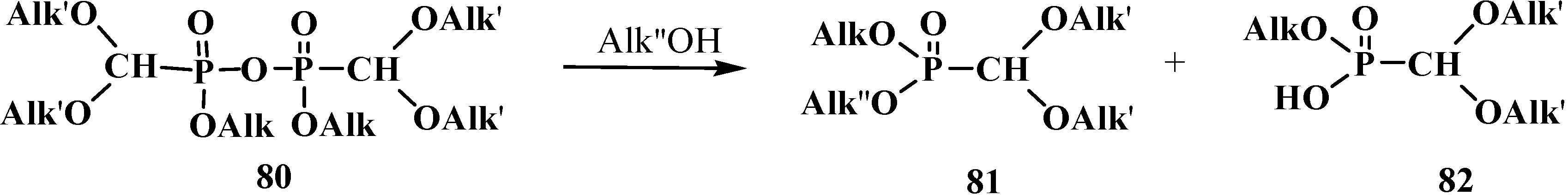{kind=link}
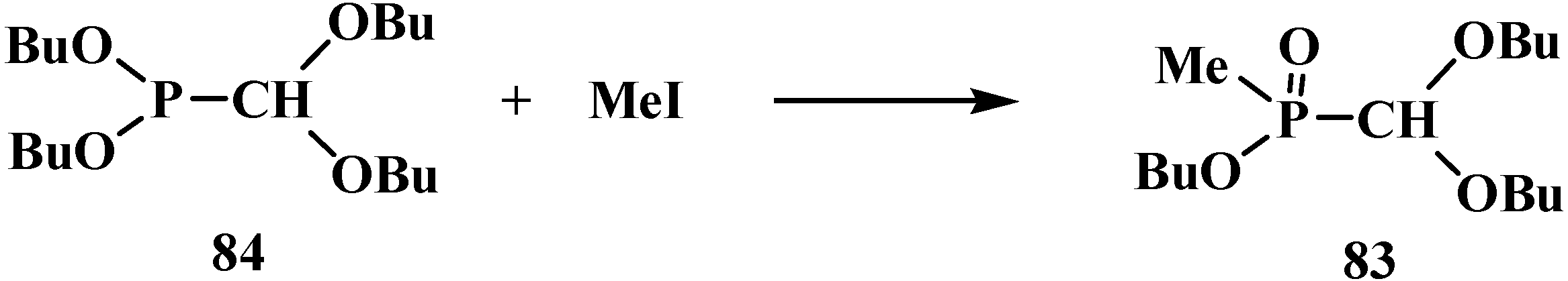{kind=link}
{kind=link}
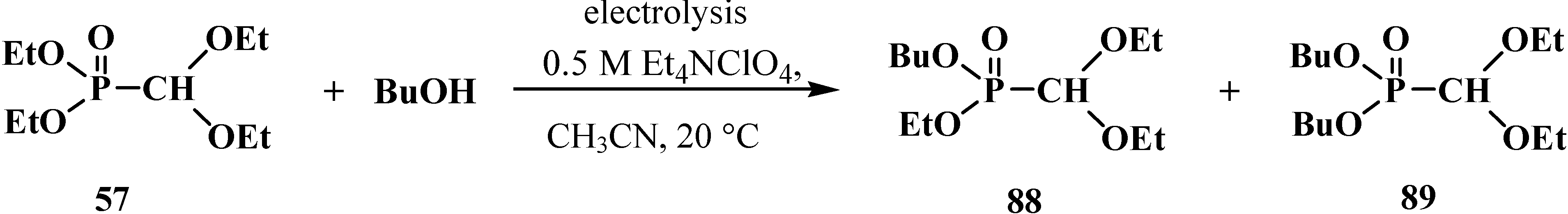{kind=link}
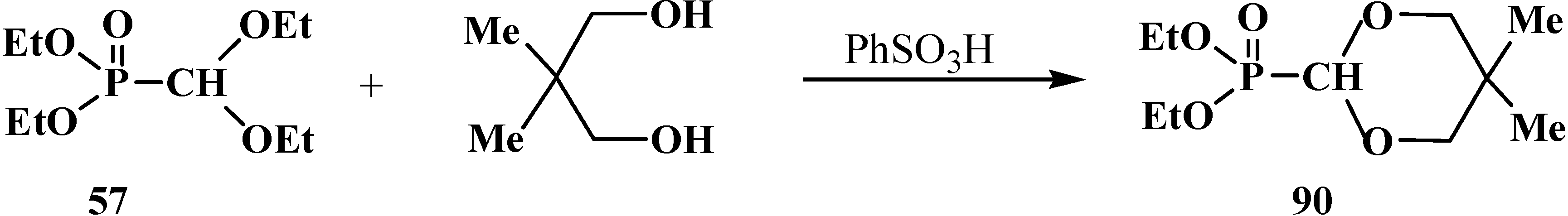{kind=link}
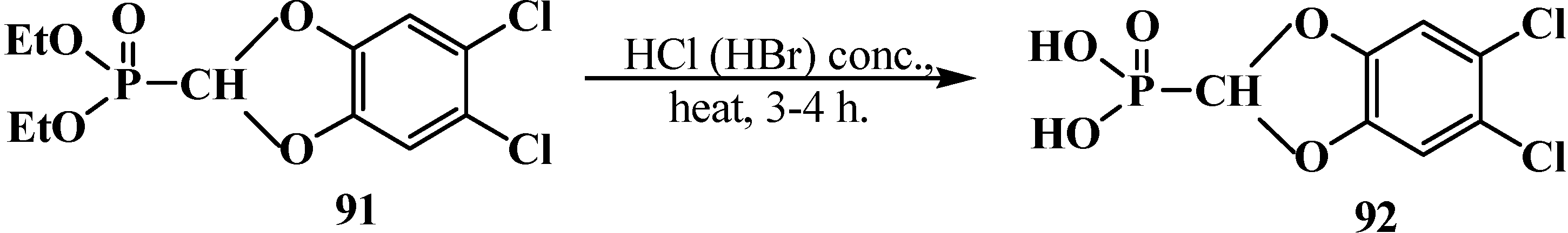{kind=link}
{kind=link}
{kind=link}
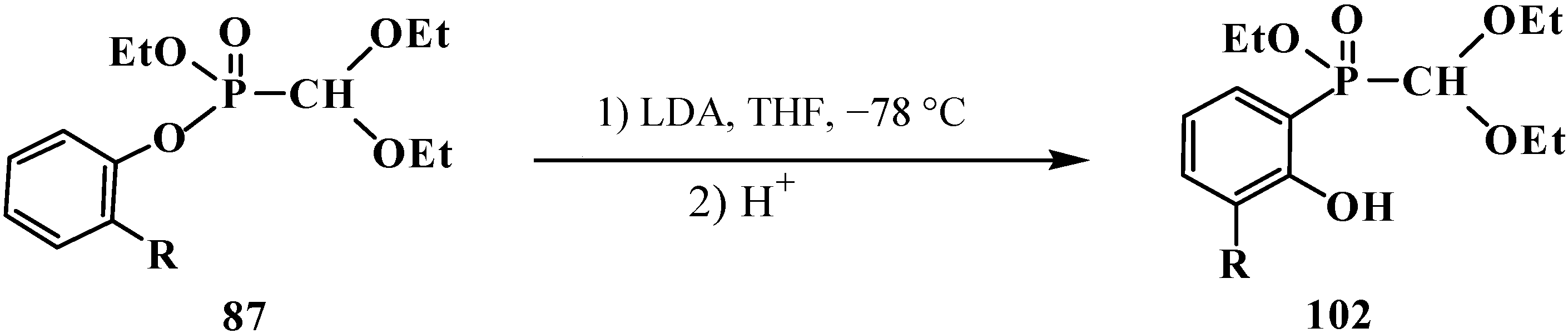{kind=link}
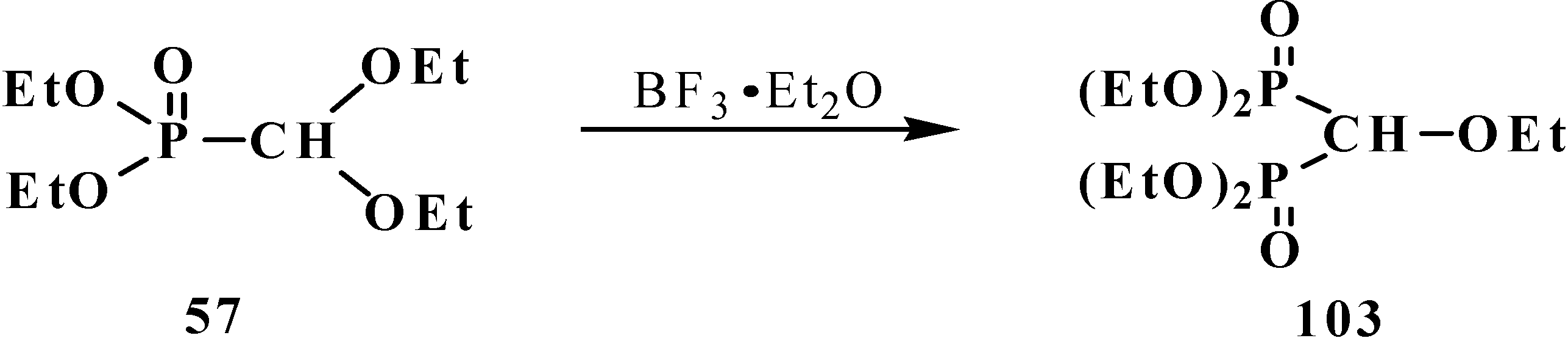{kind=link}
{kind=link}
{kind=link}
{kind=link}
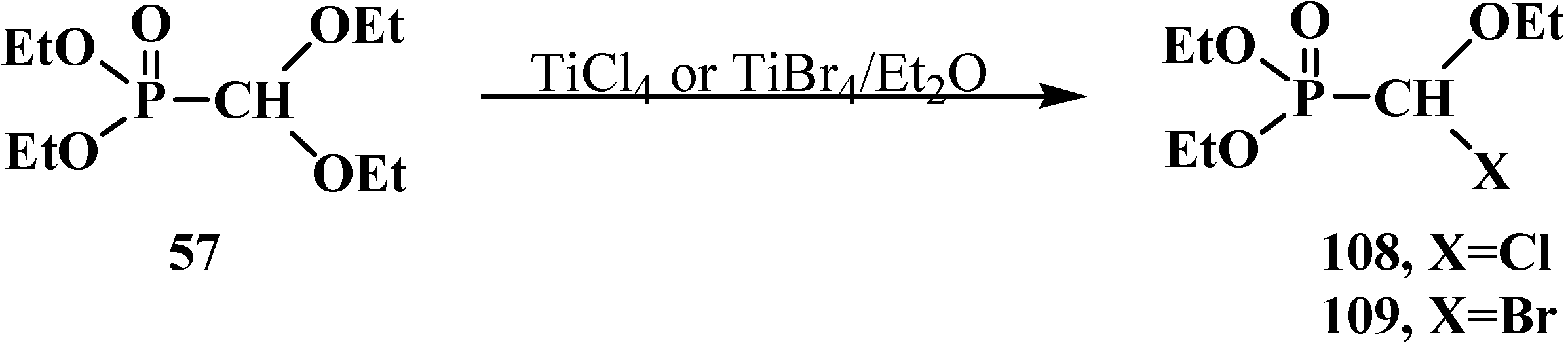{kind=link}
{kind=link}
{kind=link}
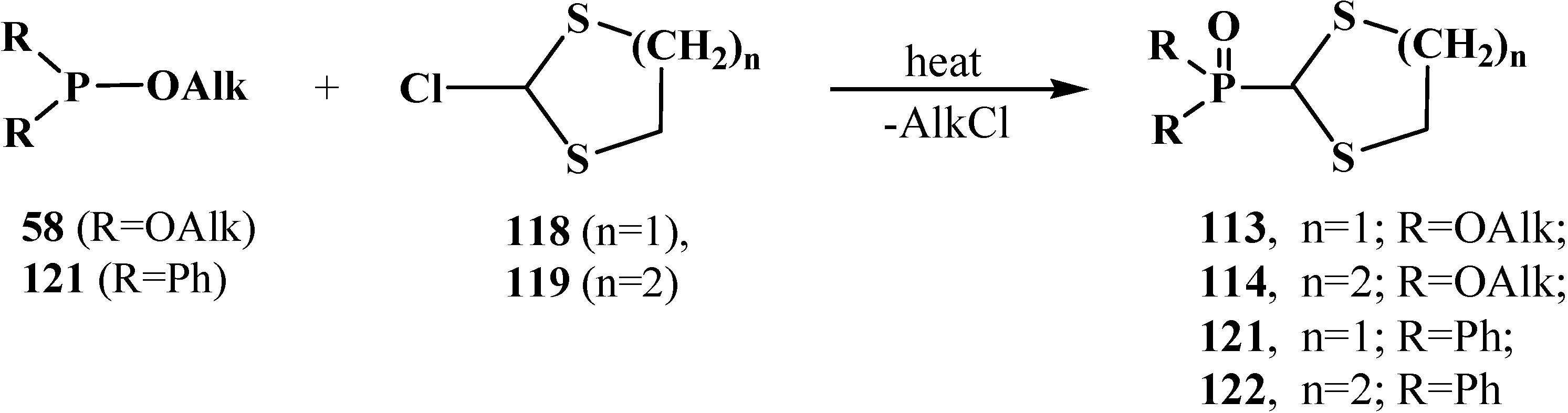{kind=link}
{kind=link}
{kind=link}
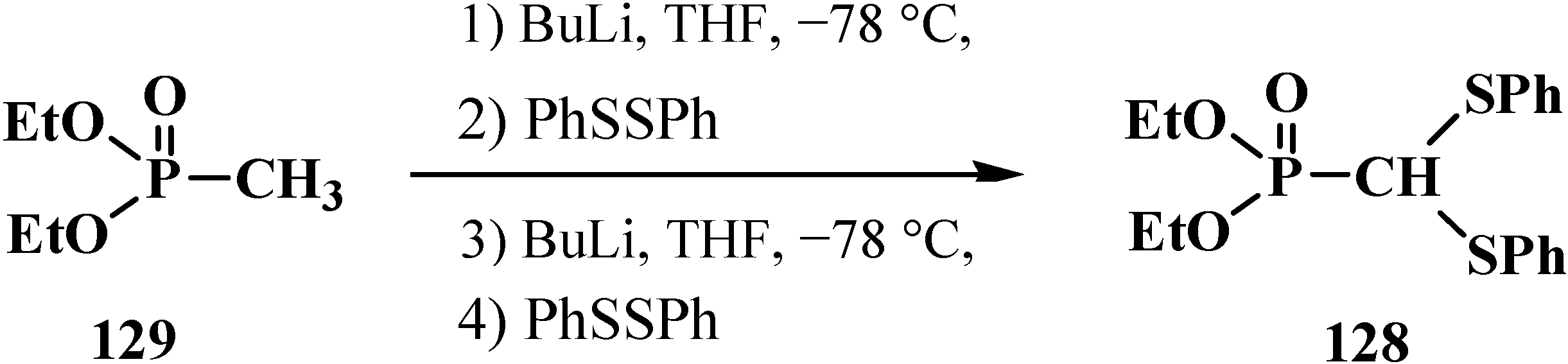{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
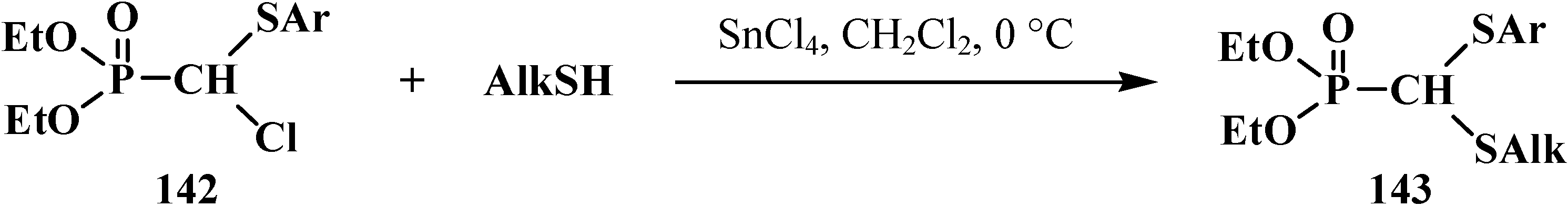{kind=link}
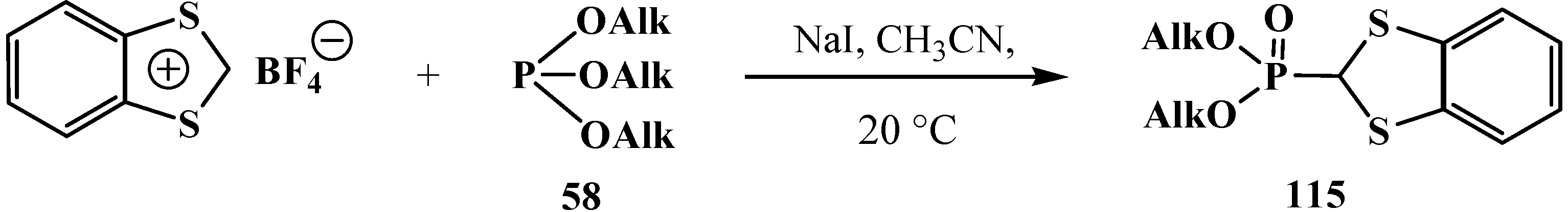{kind=link}
{kind=link}
{kind=link}
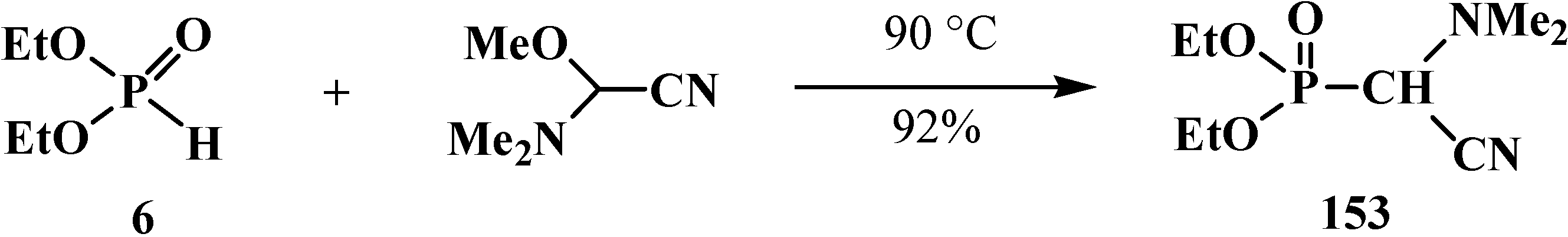{kind=link}
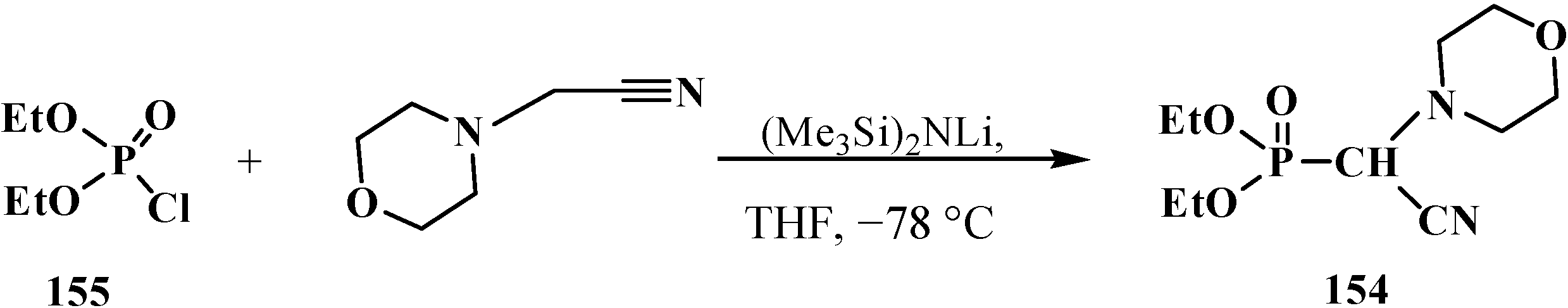{kind=link}
{kind=link}
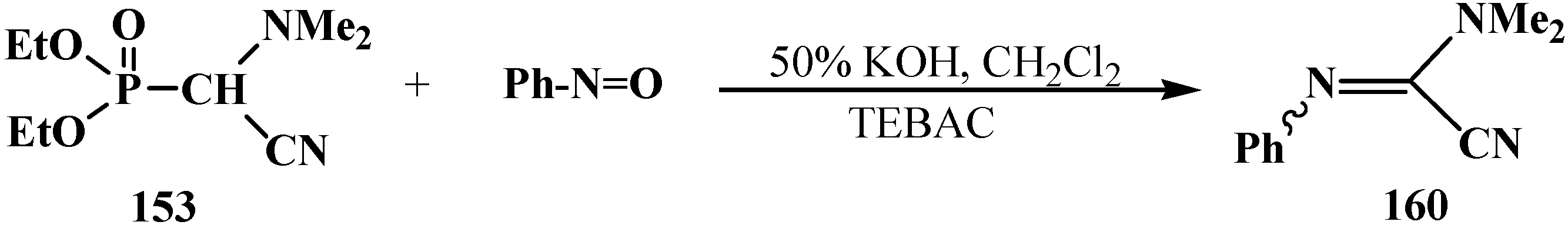{kind=link}
{kind=link}
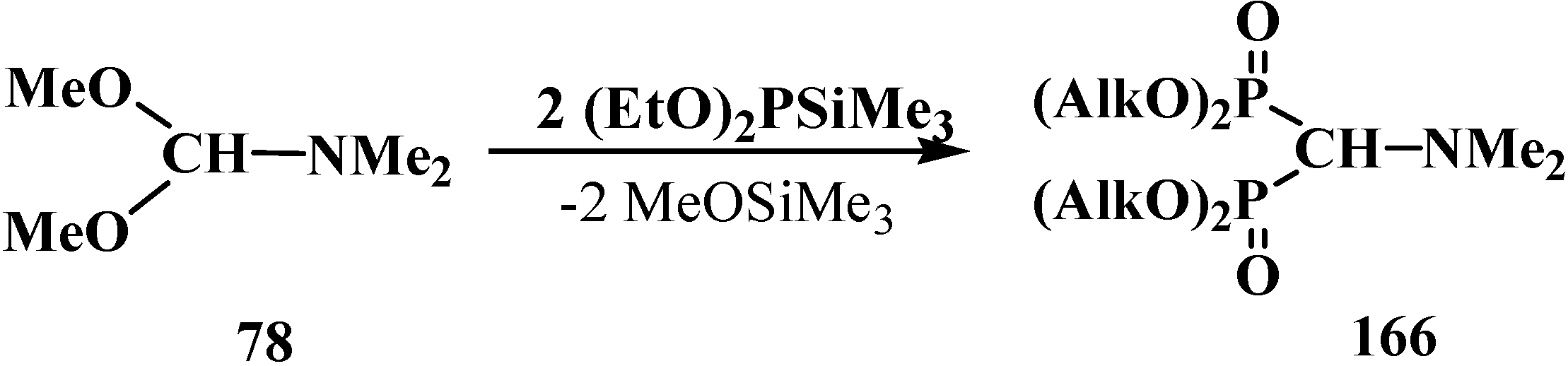{kind=link}
{kind=link}
{kind=link}
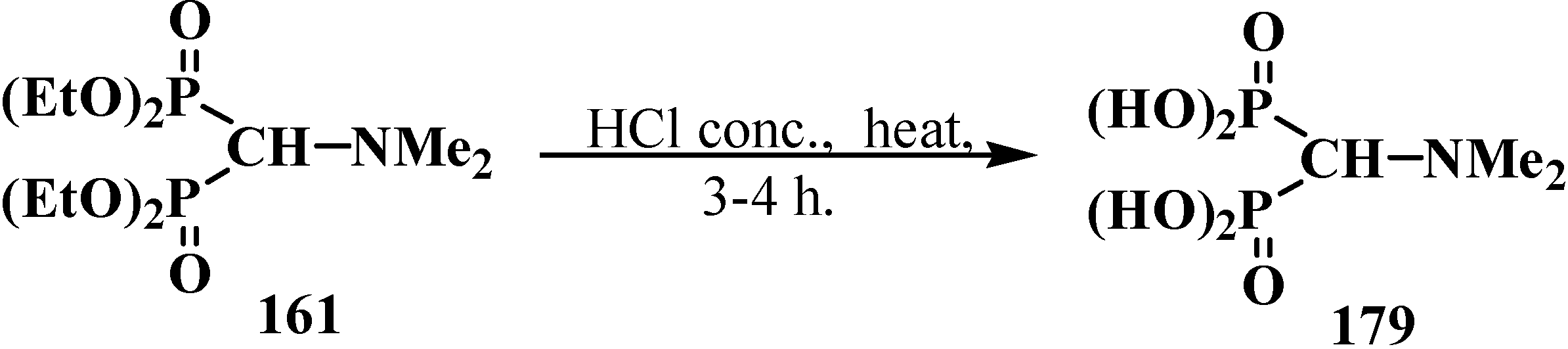{kind=link}
{kind=link}
{kind=link}
{kind=link}
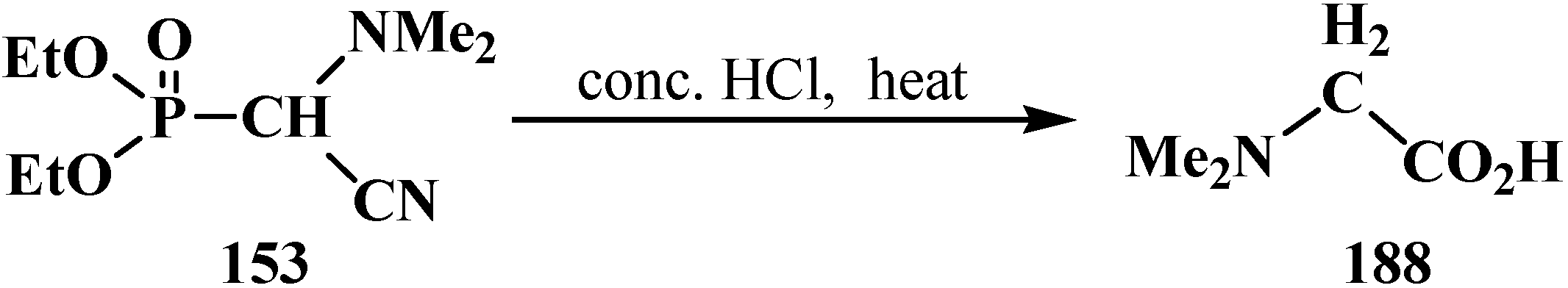{kind=link}
{kind=link}
{kind=link}
{kind=link}
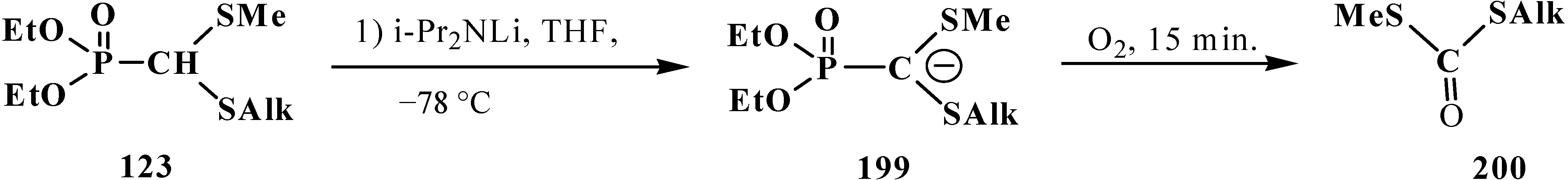{kind=link}
{kind=link}
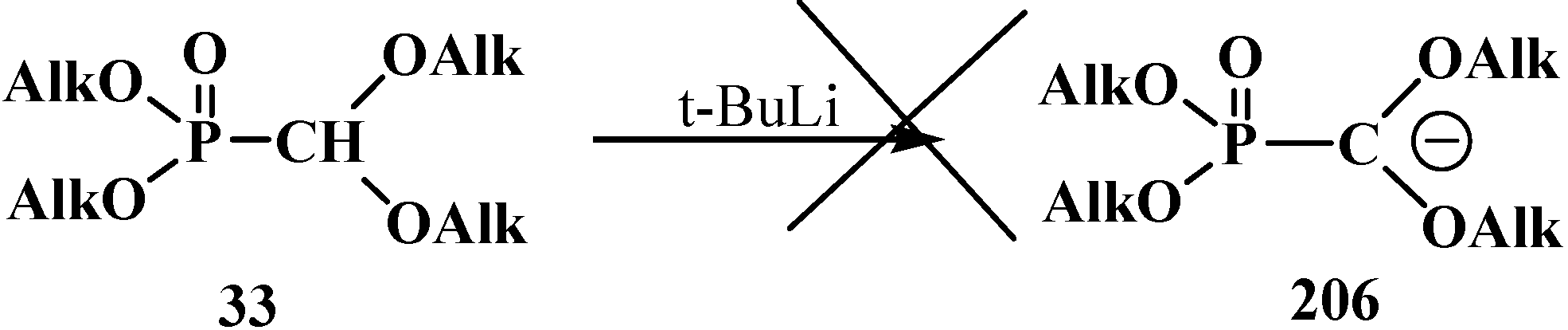{kind=link}
{kind=link}
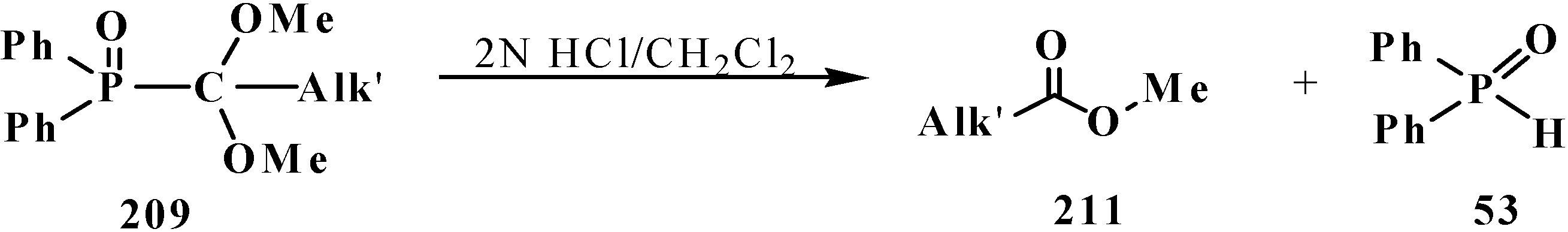{kind=link}
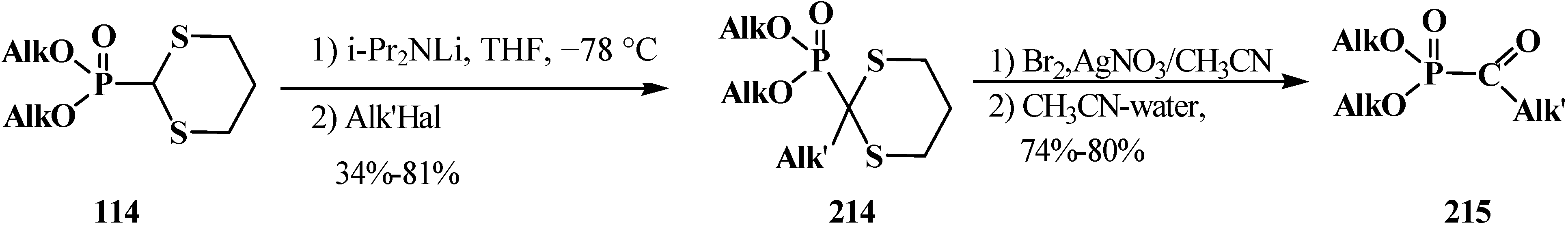{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
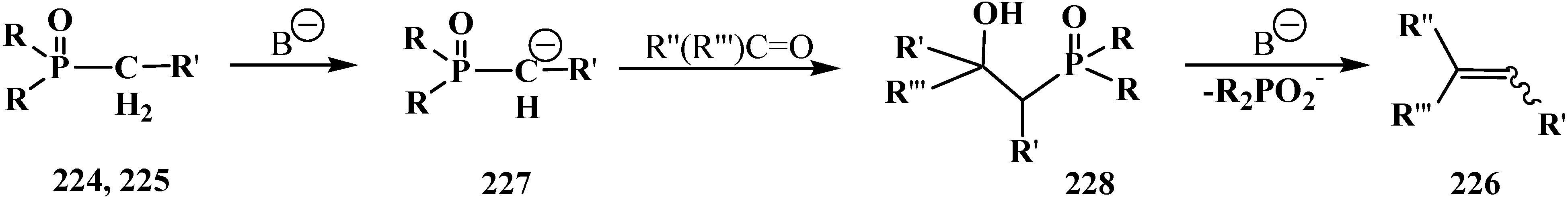{kind=link}
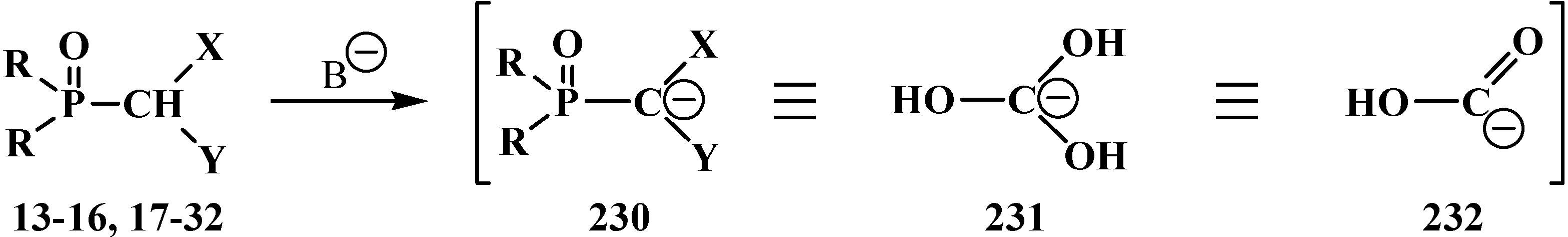{kind=link}
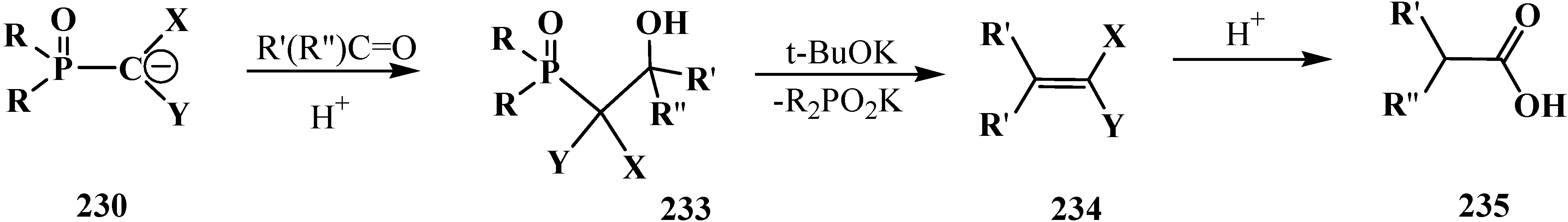{kind=link}
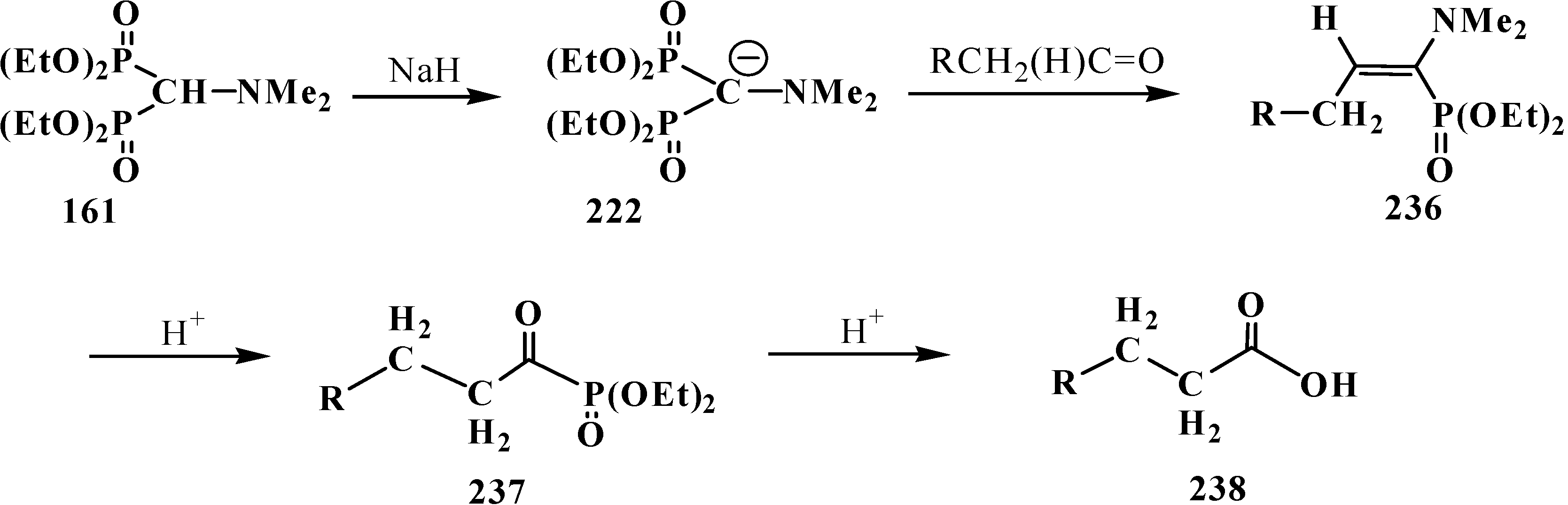{kind=link}
{kind=link}
{kind=link}
{kind=link}
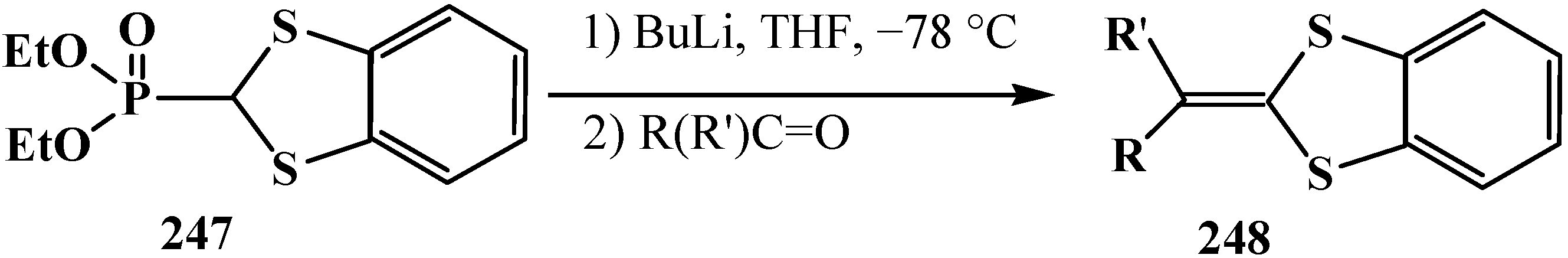{kind=link}
{kind=link}
{kind=link}
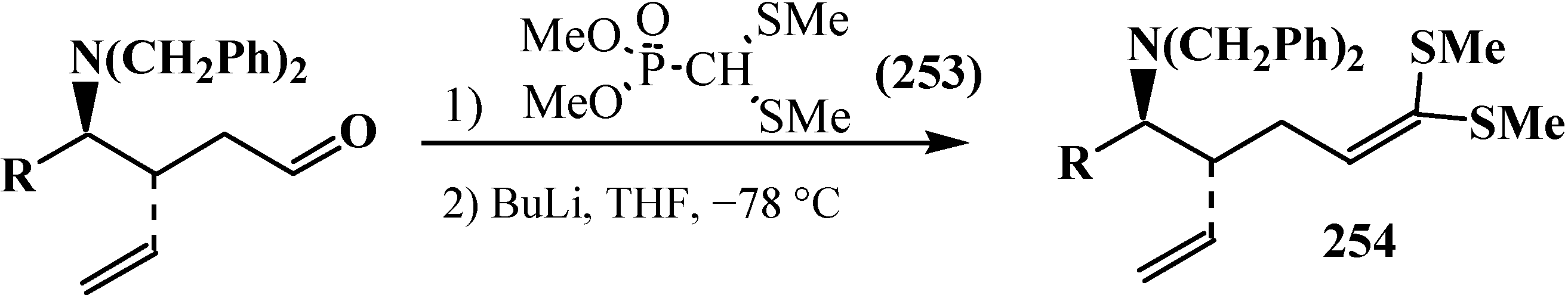{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
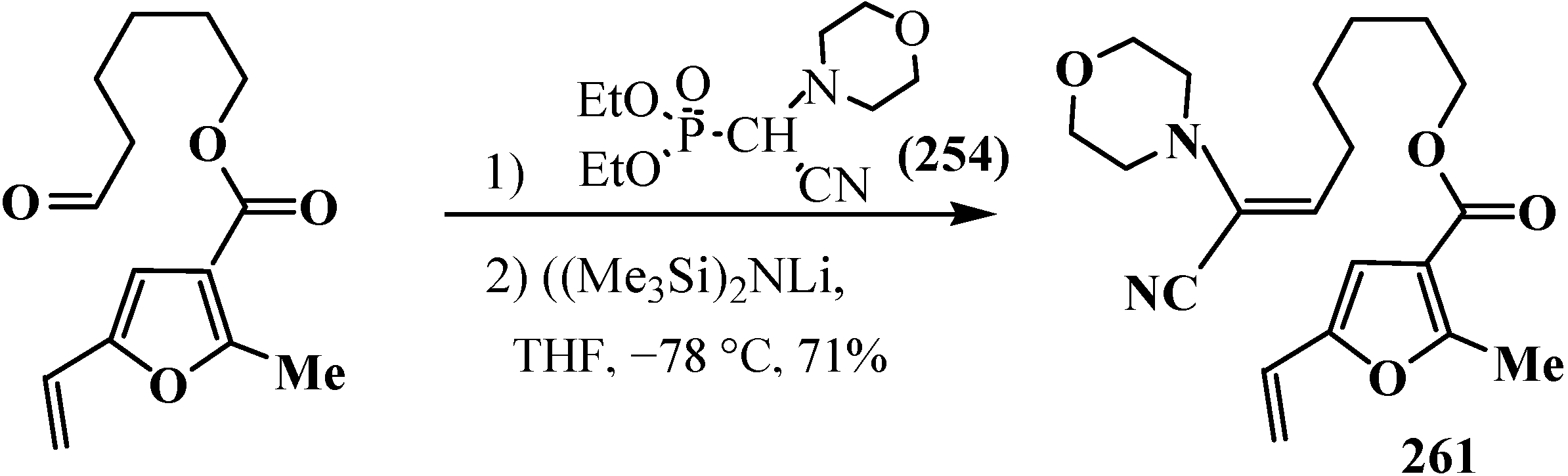{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
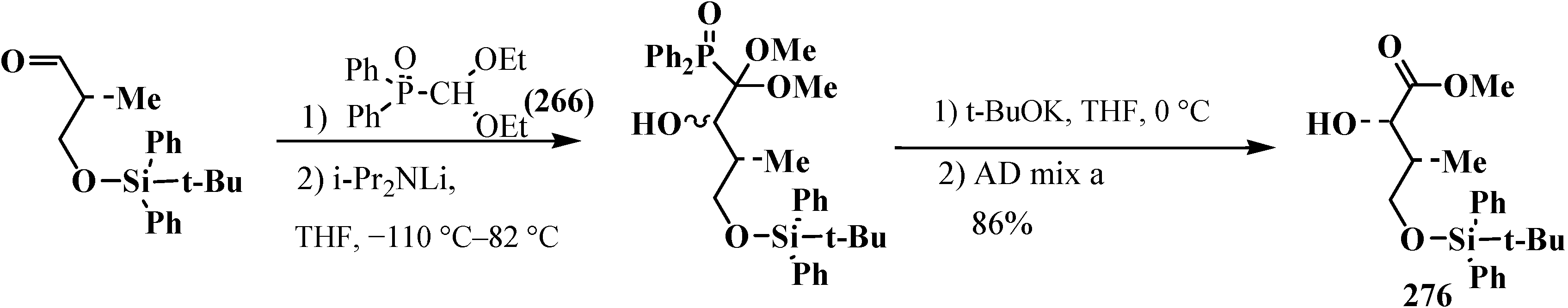{kind=link}
{kind=link}
{kind=link}
{kind=link}
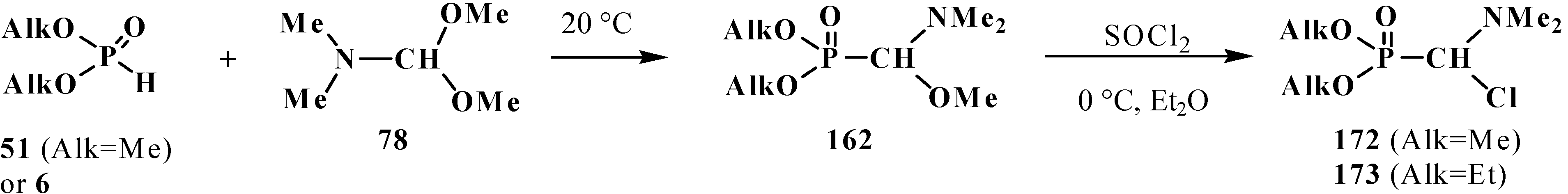{kind=link}
{kind=link}
{kind=link}
{kind=link}
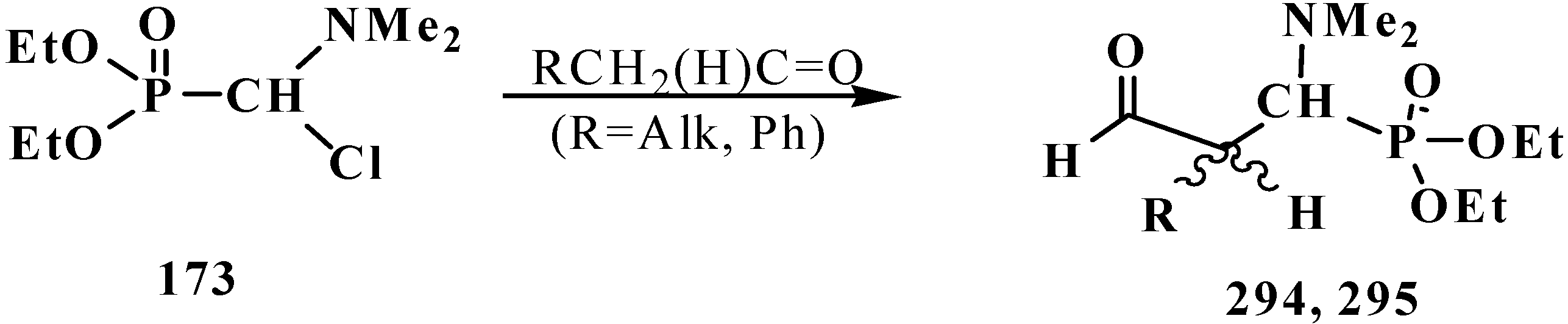{kind=link}
{kind=link}
{kind=link}
{kind=link}
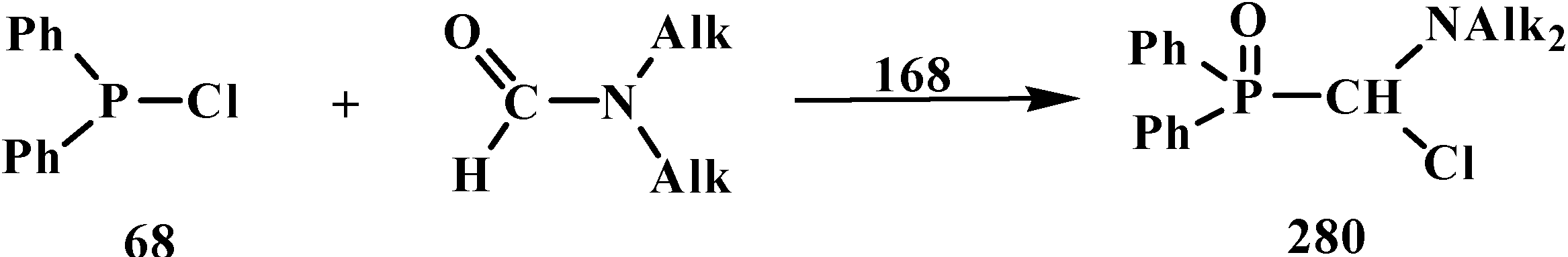{kind=link}
{kind=link}
{kind=link}
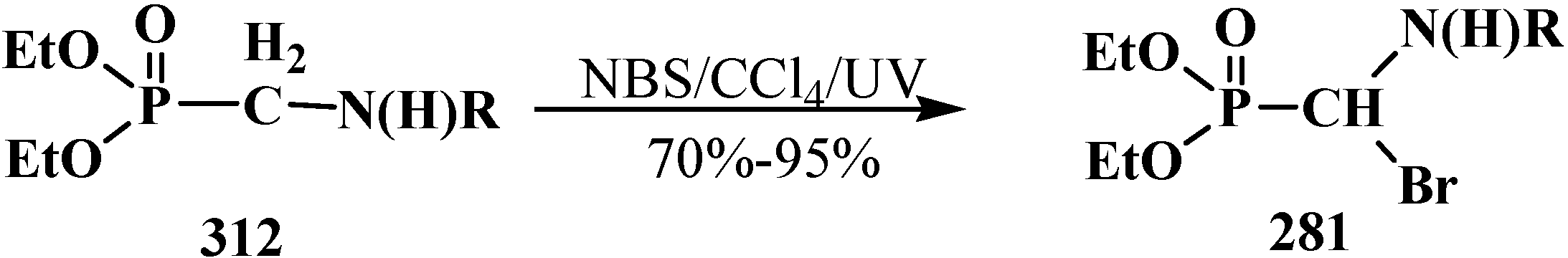{kind=link}
{kind=link}
{kind=link}
{kind=link}
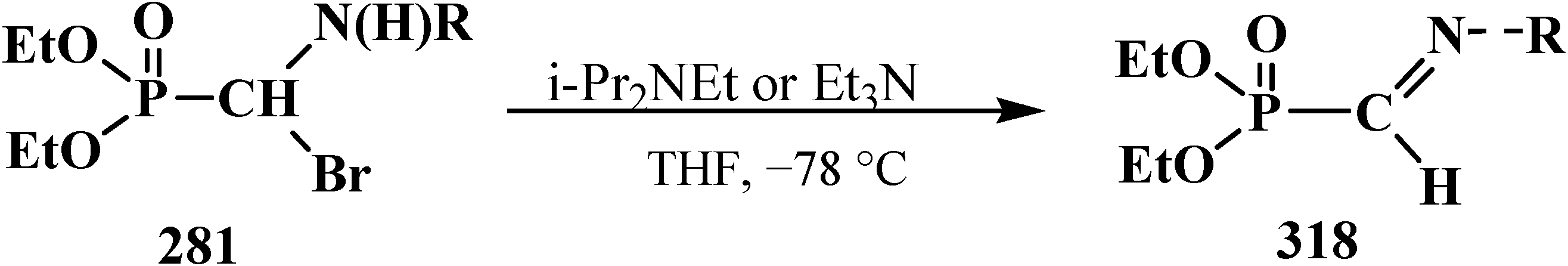{kind=link}
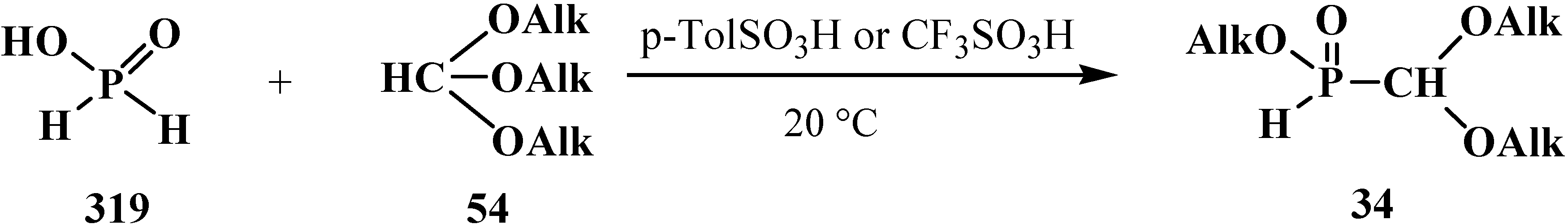{kind=link}
{kind=link}
{kind=link}
{kind=link}
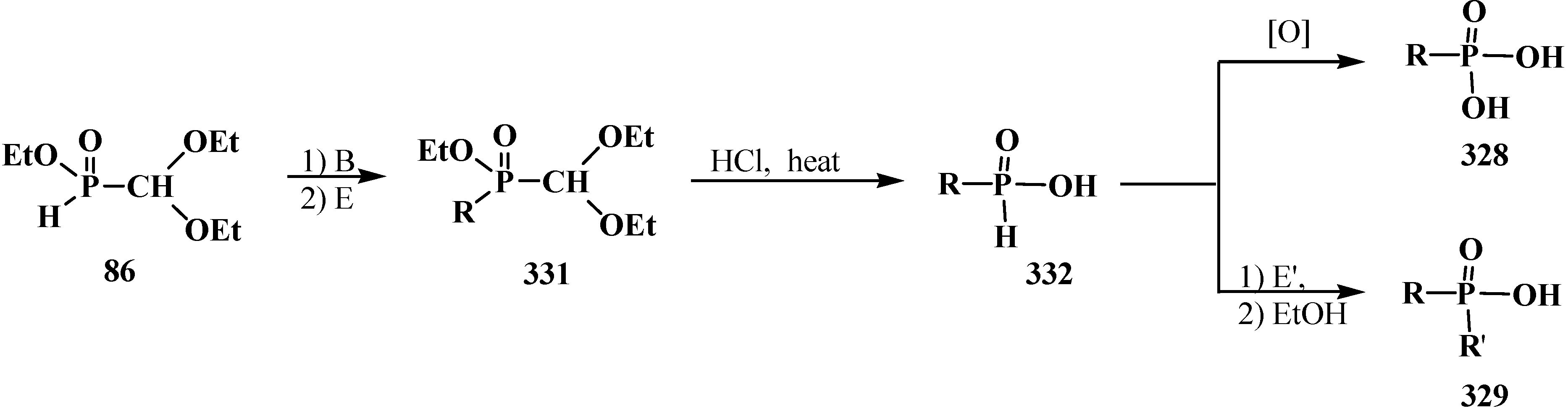{kind=link}
{kind=link}
{kind=link}
{kind=link}
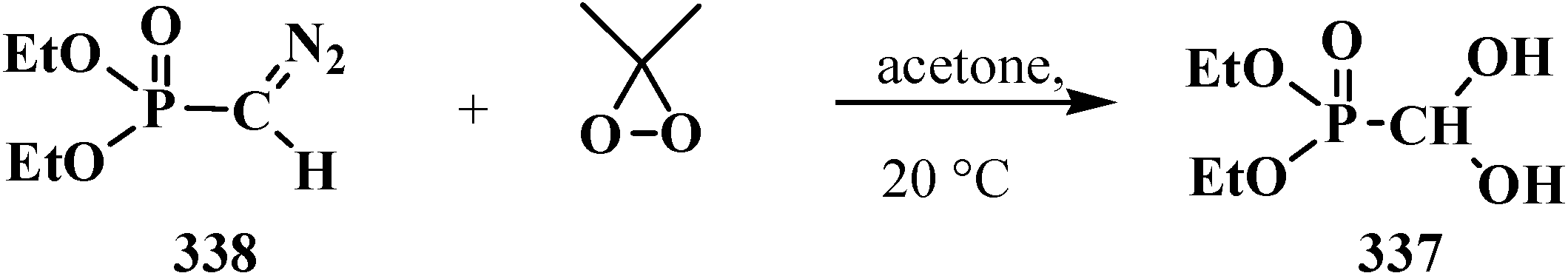{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Among organophosphorus compounds, α-phosphorylated carbonyl compounds stand out by the capacity of cleavage of phosphorus–carbon bond under mild conditions when reacted with nucleophiles [1,2,3,4,5]. The cleavage of phosphorus–carbon bond may proceed spontaneously as well. At the same time, α-oxoalkylphosphinoyl compounds also retain properties inherent in carbonyl compounds, for example, they undergo cross aldol condensation.
The least stable compounds among them are phosphorylated formaldehyde derivatives, dialkyl formylphosphonates (1) [6,7,8,9,10,11] and N,N,N',N'-tetraalkyl formylphosphondiamides (2) [12] (Figure 1), whose existence was even disputed in the first half of the 1970s [13,14].
Figure 1.
Structures of dialkyl formylphosphonates (1) and N,N,N',N'-tetraalkyl formylphosphondiamides (2).
Figure 1.
Structures of dialkyl formylphosphonates (1) and N,N,N',N'-tetraalkyl formylphosphondiamides (2).

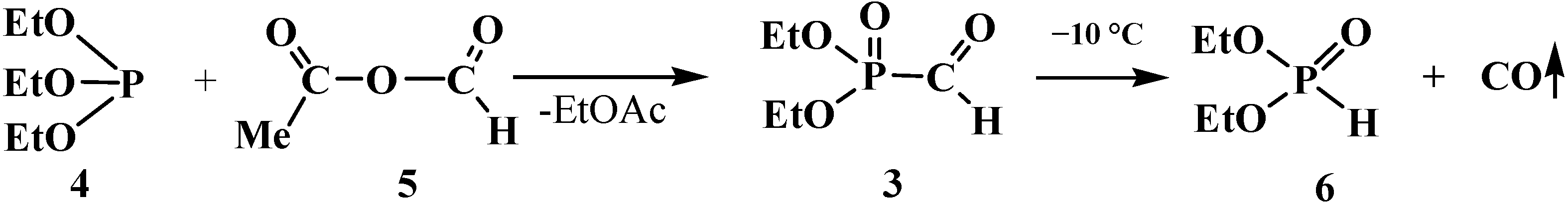
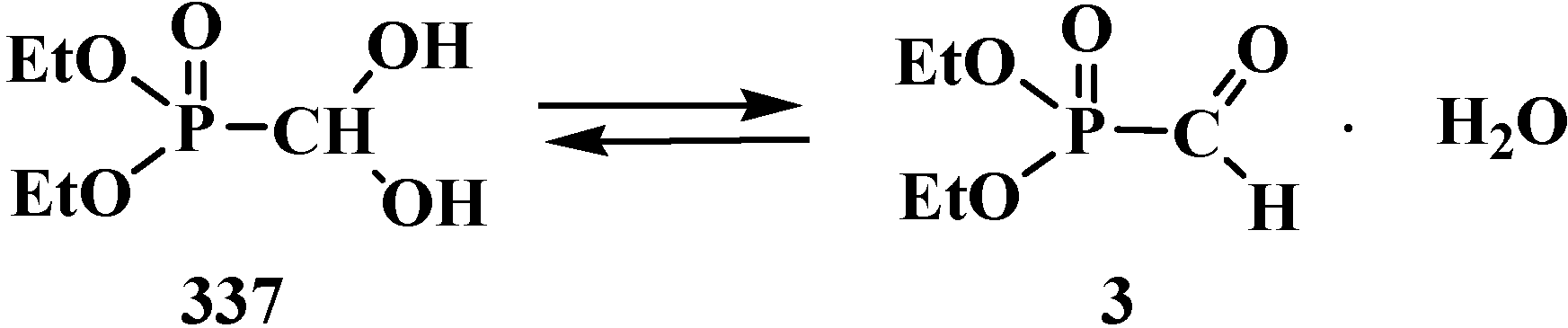
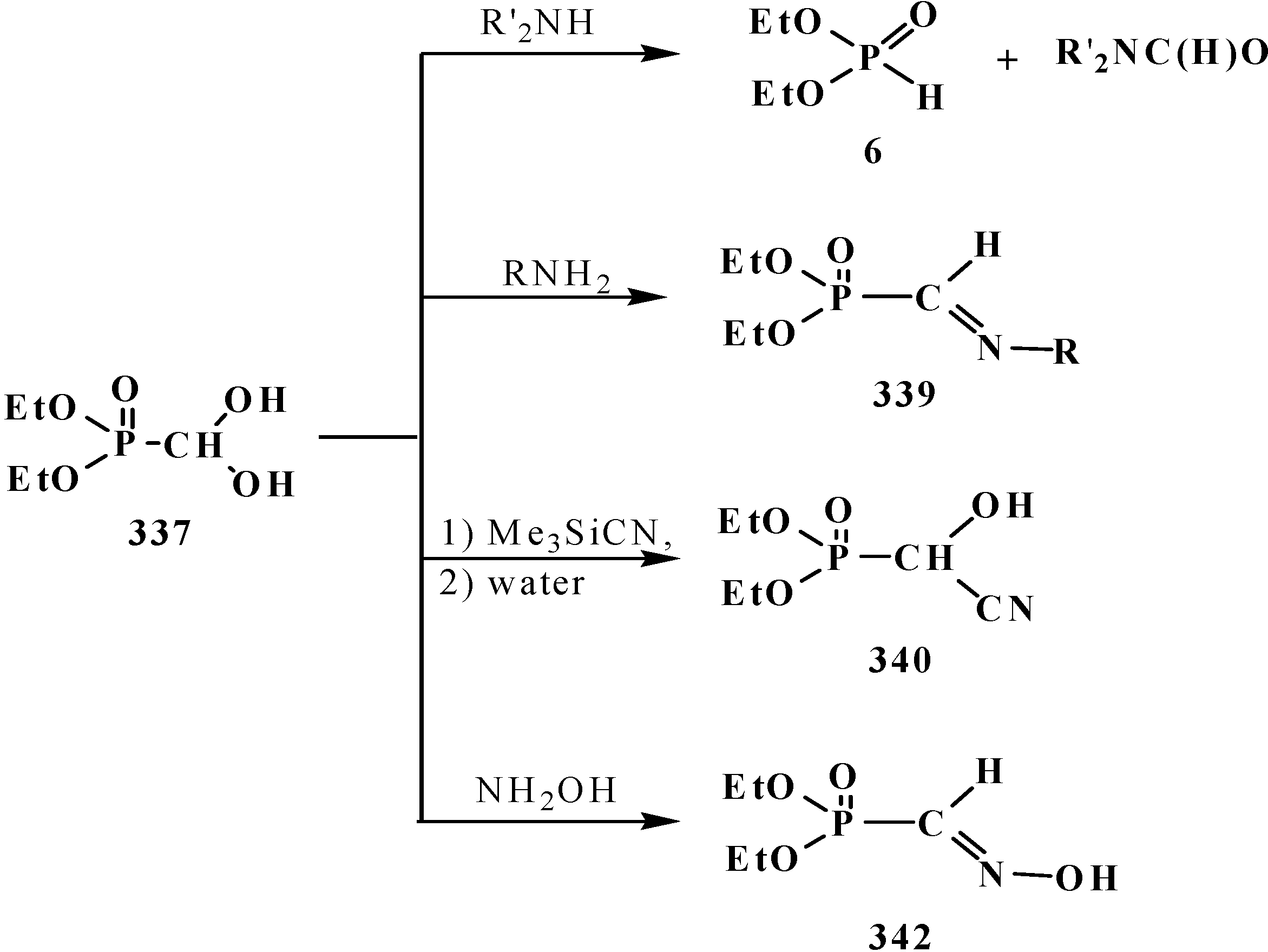
However, the first synthesis of 1 by reaction of sodium derivatives of dialkyl phosphites with acetic formic anhydride was described in 1974 [15]. Compounds 1 were shown to be unstable and prone to spontaneous degradation [9,10,16,17]; thus, diethyl formylphosphonate (3) prepared by the reaction of triethyl phosphite (4) with acetic formic anhydride (5) at low temperature begins to undergo decarbonylation even at −10 °C [9,10] to give diethyl phosphite (6) (Scheme 1).
Scheme 1.
Syntheses and destruction of diethyl formylphosphonate (3).

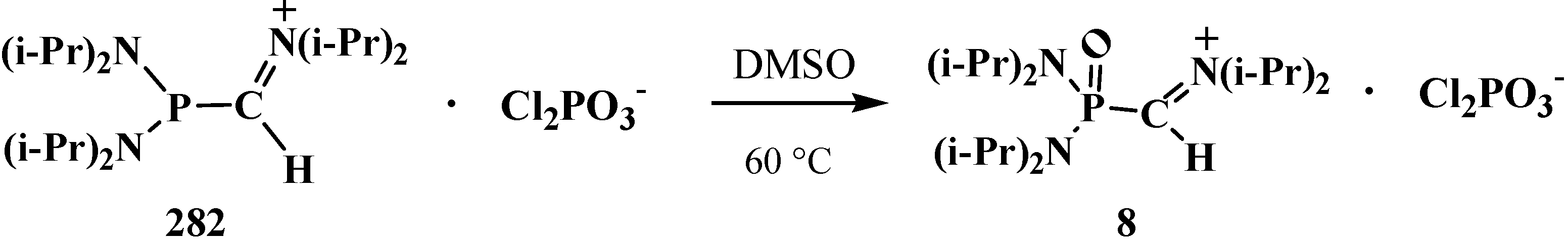
N,N,N',N'-Tetraisopropylformylphosphondiamide (7) obtained by saponification of N,N,N′,N′- tetraisopropyl[(N'',N''-diisopropylamino)methylydeniminium]phosphondiamide dichlorophosphate (8) with potassium hydroxide in tetrahydrofuran at 20 °C proved to be slightly more stable; it undergoes decarbonylation only above 40 °C [12] to give N,N,N',N'-tetraisopropylphosphonicdiamide (9) (Scheme 2).
Scheme 2.
Syntheses and destruction of N,N,N',N'-tetraisopropyl formylphosphondiamide (7).

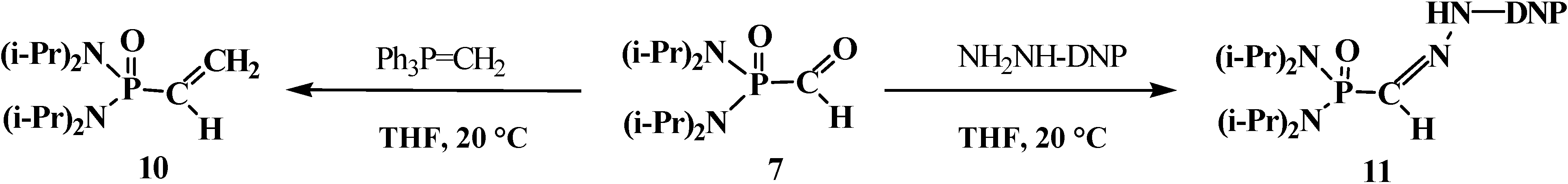
The stability of the formylphosphondiamide 7 allowed the study some of its chemical properties. It was shown that in its reactions with methylene(triphenyl)phosphorane (Ph3P=CH2) and 2,4-dinitrophenylhydrazine (NH2NH-DNP) 7 behaves as a typical aldehyde, yielding the corresponding α-phosphorylated olefin 10 and hydrazone 11 [12] (Scheme 3).
Scheme 3.
Reactions N,N,N',N'-tetraisopropylformylphosphondiamide (7) as an aldehyde (DNP means a 2,4-dinitrophenyl moiety).
Scheme 3.
Reactions N,N,N',N'-tetraisopropylformylphosphondiamide (7) as an aldehyde (DNP means a 2,4-dinitrophenyl moiety).

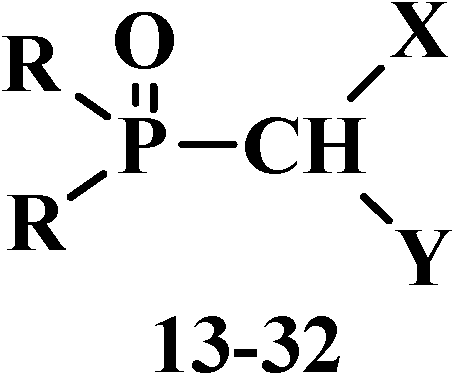
However, many authors showed that formylphosphinoyl compounds are also stable in the form of formylphosphonic acid (12) [12,14,15], its disodium salt [15], aldehyde group derivatives such as acetals 13 [18,19,20] and structurally related compounds. These compounds are thioacetals 14 [21,22,23], aminonitriles 15 [24,25,26], diphosphinoyl (N,N-dialkylaminomethyl)methanes 16 [27,28,29], chloro(or bromo)aminals 17 [30,31,32,33,34], mixed S,O-thioacetals 18 [35,36,37], aminals 19 [30,38], aminoacetals 20 [31,32,38,39], aminothioacetals 21 [32], chloroacetals 22 [40,41], chloro- (or bromo-) thioacetals 23 [40,42,43], α-chlorosulfinyl derivatives 24 [44,45], α-alkoxynitriles 25 [46,47,48], α-thionitriles 26 [46], α-dihalo derivatives 27 [49,50,51], α-alkoxydiphosphoryl compounds 28 [41], α-mercaptodiphosphoryl compounds 29 [40,52,53], α-alkoxysilyl derivatives 30 [54], α-aminosilyl derivatives 31 [29,55], or α-mercaptosilyl derivatives 32 [56] (Figure 2):
Figure 2.
Molecular structure of phosphorylated formaldehyde acetals (13) and related compounds (14–32), where R = OAlk, Alk2N, Ph; X, Y are the combinations of OAlk, Alk2N, CN, (AlkO)2P(O), S(Alk, Ar), S(O)(Alk, Ar), N(H)C(O)Alk, C(O)Ar, N(H)S(O)2Alk, S(O)2Ar, Hal, Me3Si [18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,4142,43,44,45,46,47,48,49,50,51,52,53,54,55,56].
Figure 2.
Molecular structure of phosphorylated formaldehyde acetals (13) and related compounds (14–32), where R = OAlk, Alk2N, Ph; X, Y are the combinations of OAlk, Alk2N, CN, (AlkO)2P(O), S(Alk, Ar), S(O)(Alk, Ar), N(H)C(O)Alk, C(O)Ar, N(H)S(O)2Alk, S(O)2Ar, Hal, Me3Si [18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,4142,43,44,45,46,47,48,49,50,51,52,53,54,55,56].

Among these compounds, phosphorylated acetals 13, thioacetals 14, α-dimethylaminonitriles 15, aminodiphosphinoyl compounds 16 and chloro- (or bromo-) aminals 17 are used in contemporary organic synthesis. Nonetheless, the phosphorylated acetals of formaldehyde and structurally related compounds remain poorly studied types of organophosphorus compounds until now. The chemical properties of this type of compounds were most studied on an example of dialkyl(dialkoxymethyl)phosphonates 33. The properties of other compounds 18–32 have been studied in much less detail, and the majority of them are examined in one-sided manner, only as precursors for the synthesis of ketene acetals and similar compounds by the Horner reaction.
To date, it is known that—except for formylphosphonic acid (12), phosphorylated acetals 13, and similar compounds 14–32—the phosphorylated formaldehyde derivatives are stable in the form of H-phosphinate acetals: alkyl (diethoxymethyl)phosphinate 34 [57,58,59], and geminal dioles, phosphorylated formaldehyde hydrates (hydrates of phosphorylated formaldehyde) 35 [60,61,62] (Figure 3).
Figure 3.
Molecular structure of alkyl (diethoxymethyl)phosphinate 34 and phosphorylated formaldehyde hydrates 35.
Figure 3.
Molecular structure of alkyl (diethoxymethyl)phosphinate 34 and phosphorylated formaldehyde hydrates 35.

Noteworthy are nitrogen-containing analogs of phosphorylated formaldehyde: N-substituted imines (36) [16,63,64], N-alkylnitrones 37 [8,65,66], phosphorylated diazomethane (38 [67,68,69] and O-alkylated oximes of diethyl formylphosphonates 39 [7,70], used in contemporary organic synthesis (Figure 4).
Figure 4.
Molecular structures of formylphosphonates of N-substituted imines 36, N-alkylnitrones 37, phosphorylated diazomethane 38 and alkylated oximes 39.
Figure 4.
Molecular structures of formylphosphonates of N-substituted imines 36, N-alkylnitrones 37, phosphorylated diazomethane 38 and alkylated oximes 39.

However, the properties of compounds 36–39 differ significantly from those of compounds 13–32. Therefore, compounds 36–39 will be considered in a separate publication.
2. Chemistry of Phosphorylated Formaldehyde Derivatives
2.1. Syntheses and Chemical Properties of Formylphosphonic Acid (12)
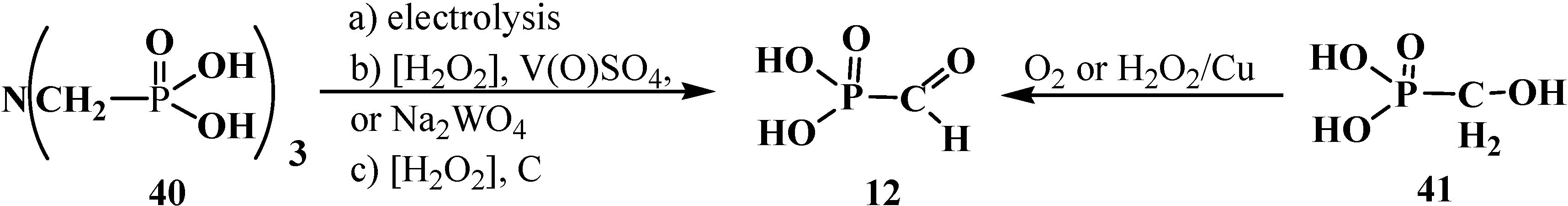
Formylphosphonic acid (12), which shows distinct antiviral activity [71] was obtained for the first time in 1974 [14] as a byproduct of the electrolysis of an aqueous solution of nitrilotrimethylphosphonic acid (40) (Scheme 4). Later it was shown that 12 might be also obtained in high yields by the catalytic oxidation of nitrilotrimethylphosphonic acid (40) with hydrogen peroxide in the presence of vanadyl sulfate or potassium tungstate (88%–93% yield) [17] or activated carbon (up to 82% yield) [72]. Compound 12 can be also prepared by the oxidation of hydroxymethylphosphonic acid (41) in aqueous solution with air oxygen or hydrogen peroxide in the presence of Raney copper in a yield up to 65% [73] (Scheme 4).
Scheme 4.
Possible routes of syntheses of formylphosphonic acid (12).

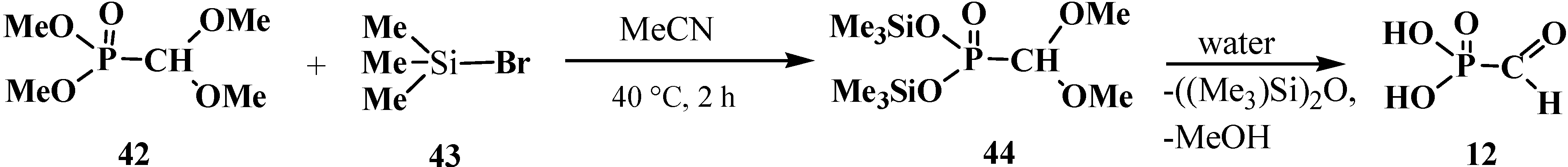
In a third method of synthesis, 12 was obtained via a two-stage synthesis starting from dimethyl (dimethoxymethyl)phosphonate (42), which was converted by treatment with bromotrimethylsilane (43) into bis(trimethylsilyl)(dimethoxymethyl)phosphonate (44). Hydrolysis of the latter resulted in formylphosphonic acid (12) [74] (Scheme 5). See also Section 3.2.—Synthesis of formacetalphosphonic acids.
Scheme 5.
Synthesis of formylphosphonic acid (12) from dimethyl (dimethoxymethyl)phosphonate (42).

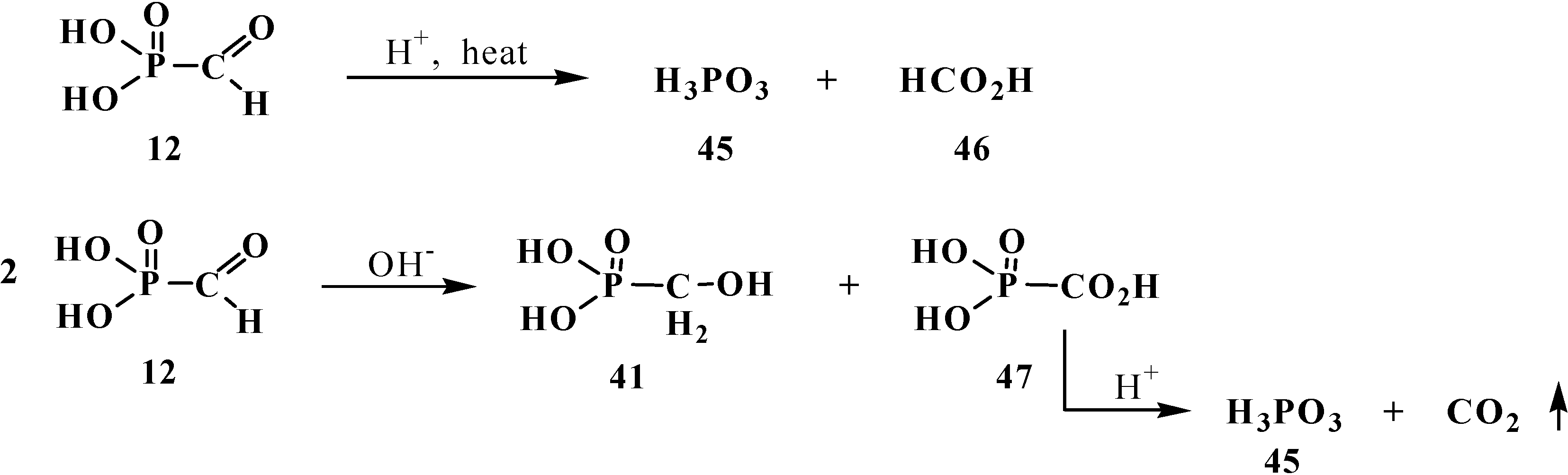
However, the chemical properties of formylphosphonic acid (12) have been poorly studied until now. It is known that heating 12 in acidic medium leads to the cleavage of the P–C bond to form phosphoric and formic acids 45 and 46. In the presence of bases 12 undergoes disproportionation (Cannizzaro reaction) to give hydroxymethylphosphonic 41 and carboxyphosphonic acids 47. Acid 47 undergoes rapid decarboxylation on acidification of the reaction medium [14] (Scheme 6).
Scheme 6.
Degradation of formylphosphonic acid (12) to form phosphoric 45 and formic acids 46 and disproportionation of 12 in aqueous solutions.
Scheme 6.
Degradation of formylphosphonic acid (12) to form phosphoric 45 and formic acids 46 and disproportionation of 12 in aqueous solutions.

2.2. Chemistry of Phosphorylated Formaldehyde Acetals 13
Chemistry of phosphorylated formaldehyde acetals 13 started as a chemistry of dialkyl (dialkoxymethyl)phosphonates 33 due to their more ready availability as compared with the analogs—N,N,N',N'-tetraalkyl(dialkoxymethyl)phosphondiamides 48, dialkyl (dialkoxymethyl)-phosphine oxides 49 or diphenyl(dialkoxymethyl)phosphine oxide 50. Many types of acetals are known to date, but their chemical properties are still insufficiently studied, although they are more studied than the other derivatives of phosphorylated formaldehyde.
2.2.1. Methods of Synthesis of Phosphorylated Formaldehyde Acetals 13
First phosphorylated formaldehyde acetals 13 were obtained by the reaction of hydrophosphinoyl compounds 51–53 with orthoformate esters 54 on heating. The reaction with dialkyl phosphites 51 [19,75] is conducted by heating to 182 °C [76,77] or at 60 °C in the presence of BF3·Et2O as catalyst (without the catalyst the yields decrease from 69%–90% to 25% [75]). In the case of sec-phosphine oxides—dialkylphosphine oxides 52 [77] and diphenylphosphine oxide 53 [77,78], the reaction proceeds at 100 °C [19,77]. This method provides high yields of compounds 33, 49 and 50 (Scheme 7).
Scheme 7.
Synthesis of phosphorylated formaldehyde acetals 33, 49, 50 from dialkyl phosphites 51 or sec-phosphine oxides 52, 53 by means of orthoformate esters 54.
Scheme 7.
Synthesis of phosphorylated formaldehyde acetals 33, 49, 50 from dialkyl phosphites 51 or sec-phosphine oxides 52, 53 by means of orthoformate esters 54.

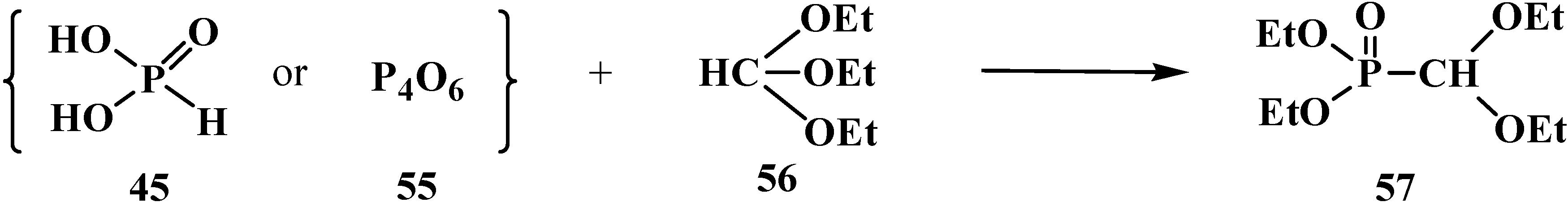
A variant of this method consists in the reaction of phosphoric acid (H3PO3, 45) or phosphoric anhydride (55) with triethyl orthoformate (56) in 1:3 ratio that results in diethyl (diethoxymethyl)phosphonate (57) with minimal effort [79] (Scheme 8).
Scheme 8.
Synthesis of diethyl (diethoxymethyl)phosphonate (57) from phosphoric acid (45) or phosphorous anhydride (55).
Scheme 8.
Synthesis of diethyl (diethoxymethyl)phosphonate (57) from phosphoric acid (45) or phosphorous anhydride (55).

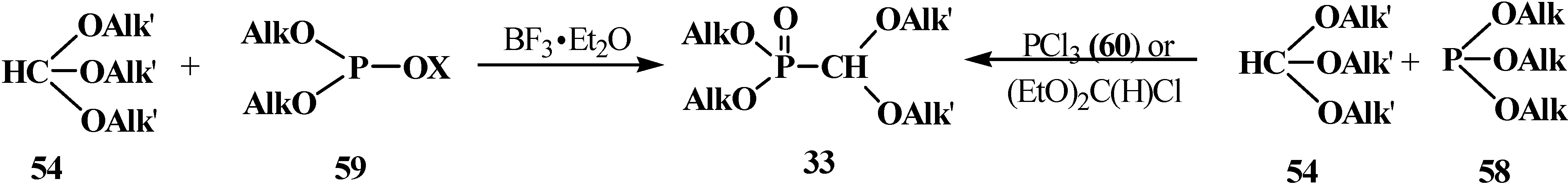
Compounds 33 can be also prepared by the reaction of trialkyl phosphites 58 with orthoformate esters 54. It was shown that phosphites 58 do not react directly with 54 even on heating [19]. However, as in the case of dialkyl phosphites 51, orthoformates 54 react with trialkyl phosphites 58 and their analogs (AlkO)2POX 59, where X = Me3Si, (AlkO)2P, (AlkO2)P(O) on heating in the presence of catalytic amounts of boron trifluoride etherate BF3·Et2O [19]. The reaction of 58 with orthoformate esters 54 is also possible in the presence of phosphorus trichloride (PCl3, 60) or diethoxychloromethane (EtO)2C(H)Cl [80,81] (Scheme 9).
Scheme 9.
Synthesis of dialkyl (dialkoxymethyl)phosphonates 33 from orthoformate esters 54, trialkyl phosphites 58 and their analogs (AlkO)2POX 59.
Scheme 9.
Synthesis of dialkyl (dialkoxymethyl)phosphonates 33 from orthoformate esters 54, trialkyl phosphites 58 and their analogs (AlkO)2POX 59.

It was reported that the reaction of trialkyl phosphites 58 with acetoxy(diethoxy)methane (61), which is more reactive derivative than orthoformate esters 54 [19,82], results in dialkyl (diethoxymethyl)phosphonates 62 (Scheme 10).
Scheme 10.
Synthesis of dialkyl (diethoxymethyl)phosphonates 62 from orthoformate esters 54 and acetoxy(diethoxy)methane (61).
Scheme 10.
Synthesis of dialkyl (diethoxymethyl)phosphonates 62 from orthoformate esters 54 and acetoxy(diethoxy)methane (61).

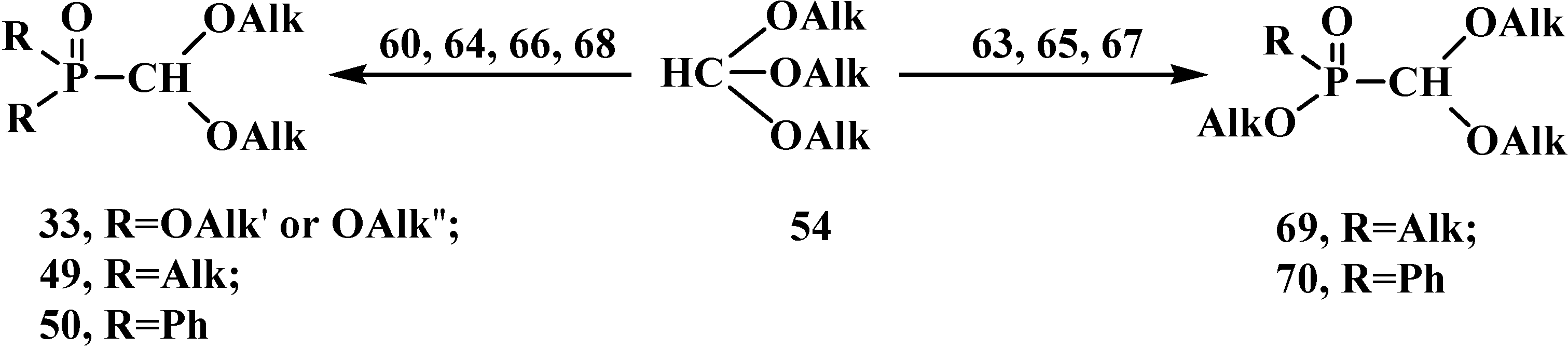
The most general method of synthesis of phosphorylated formaldehyde acetals 13 is the reaction of phosphorus trichloride derivatives 60 with orthoformate esters 54. Excess of 54 reacts on heating with 60 [79,81,83,84,85] and mono- and dialkyl AlkPCl263, Alk2PCl 64, alkoxy (AlkO)PCl265, (AlkO)2PCl 66, and phenyl PhPCl267, Ph2PCl 68 [80,84,86,87,88,89] derivatives of 60 (Scheme 11), to form symmetrical and unsymmetrical acetals 33, 49, 50 and 69, 70, respectively. It was noted that the reaction of 2-chloro-1,2,3-dioxaphospholanes 71 with 54 leads to the opening of the dioxaphospholane ring to form ethyl (β-chloroethyl) ([1,3]-dioxolan-2-yl)phosphonates 72 [90].
Scheme 11.
Syntheses of phosphorylated formaldehyde acetals 33, 49, 50, 69, 70 from phosphorus trichloride derivatives 63–68 with orthoformate esters 54. See the text above.
Scheme 11.
Syntheses of phosphorylated formaldehyde acetals 33, 49, 50, 69, 70 from phosphorus trichloride derivatives 63–68 with orthoformate esters 54. See the text above.

The reaction of N,N,N',N'-tetraalkyl(chloro)phosphindiamides 73 with orthoformates 54 proceeds with a partial exchange of dialkylamino groups at phosphorus atom of 73 to give both N,N,N',N'-tetraalkyl (dialkoxymethyl)phosphondiamides 48 and mixed phosphonamidates 74 [19,91,92]. In the case of N,N-dialkyl(dichloro)phosphinamides 75, a total exchange of dialkylamino groups at the phosphorus atom for alkoxy groups takes place to yield dialkyl (dialkoxymethyl)phosphonates 33 [85] (Scheme 12).
Scheme 12.
Interaction of N,N,N',N'-tetraalkyl(chloro)phosphindiamides 73 and N,N-dialkyl(dichloro)phosphinamides 75 with orthoformates 54.
Scheme 12.
Interaction of N,N,N',N'-tetraalkyl(chloro)phosphindiamides 73 and N,N-dialkyl(dichloro)phosphinamides 75 with orthoformates 54.

Quaternary ammonium salts of dimethylformamide acetals can be used instead of orthoformates 54 [18,93]. The method only allows preparation of cyclic phosphorylated formaldehyde dialkyl acetals 76 since the quaternary ammonium salts of dialkylformamide acetals of linear structure are unstable and undergo fast degradation [93] (Scheme 13).
Scheme 13.
Synthesis of cyclic acetals 76 by interaction of trialkyl phosphites 58 with the quaternary ammonium salts of dimethylformamide acetals.
Scheme 13.
Synthesis of cyclic acetals 76 by interaction of trialkyl phosphites 58 with the quaternary ammonium salts of dimethylformamide acetals.

Nonetheless, linear compounds 33 can be obtained by this method in low yield (15%–25%) using a one-pot method from methyl iodide, dialkylformamide acetals 77, and trialkyl phosphites 58 [18].
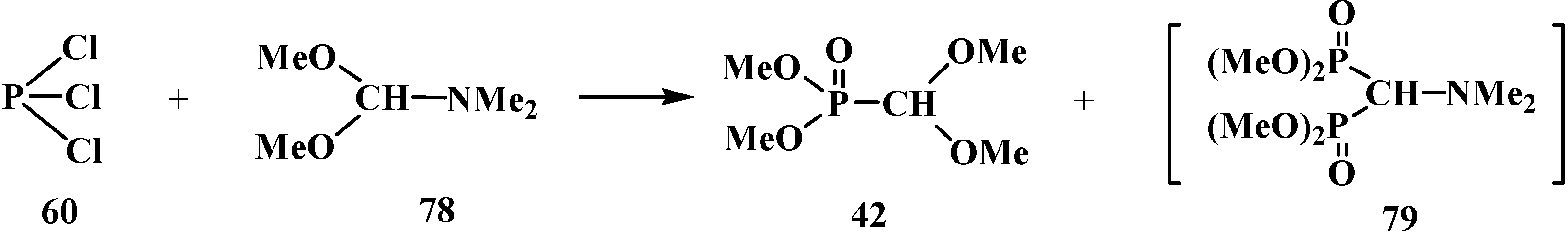
The reaction of phosphorus trichloride (60) with dimethylformamide dimethylacetal (78) also leads to the formation of dimethyl (dimethoxymethyl)phosphonate (42) (along with a certain amount of tetramethyl (N,N-dimethylaminomethyl)diphosphonate (79)) [83] (Scheme 14).
Scheme 14.
Syntheses of dimethyl (dimethoxymethyl)phosphonate (42) from phosphorus trichloride (60) and dimethylformamide dimethylacetal (78).
Scheme 14.
Syntheses of dimethyl (dimethoxymethyl)phosphonate (42) from phosphorus trichloride (60) and dimethylformamide dimethylacetal (78).

Dialkyl acetals 33 can be also prepared by the reaction of alcohols with tetraalkyl bis(dialkoxymethyl)pyrophosphonates 80. The method provides a possibility to obtain formaldehyde dialkyl acetals with different alkoxy substituents at phosphorus atom 81 [89] that are difficult to prepare from trivalent phosphorus derivatives and orthoformates 54 or their derivatives because of competitive exchange of substituents at phosphorus atom [19,89] (Scheme 15). Alkyl (dialkoxymethyl)phosphonic acid 82 is also formed at the same time. See also Scheme 22 and Scheme 23.
Scheme 15.
Preparation of acetals 81 with different alkoxy substituents at phosphorus atom by means of alcoholysis of tetraalkyl bis(dialkoxymethyl)pyrophosphonates 80.
Scheme 15.
Preparation of acetals 81 with different alkoxy substituents at phosphorus atom by means of alcoholysis of tetraalkyl bis(dialkoxymethyl)pyrophosphonates 80.

Alkyl alkyl(dialkoxymethyl)phosphinates 83 unsymmetrically substituted at the phosphorus atom were synthesized by the Arbuzov reaction of dialkyl (dialkoxymethyl)phosphonites 84 with alkyl iodides [82,84], for example see Scheme 16.
Scheme 16.
Example synthesis of acetals with different substitutients at phosphorus atom by the Arbuzov reaction. See also Scheme 11, Scheme 15, Scheme 17, Scheme 21, Scheme 22, Scheme 23, Scheme 111, Scheme 112, Scheme 113 and Scheme 114.
Scheme 16.
Example synthesis of acetals with different substitutients at phosphorus atom by the Arbuzov reaction. See also Scheme 11, Scheme 15, Scheme 17, Scheme 21, Scheme 22, Scheme 23, Scheme 111, Scheme 112, Scheme 113 and Scheme 114.

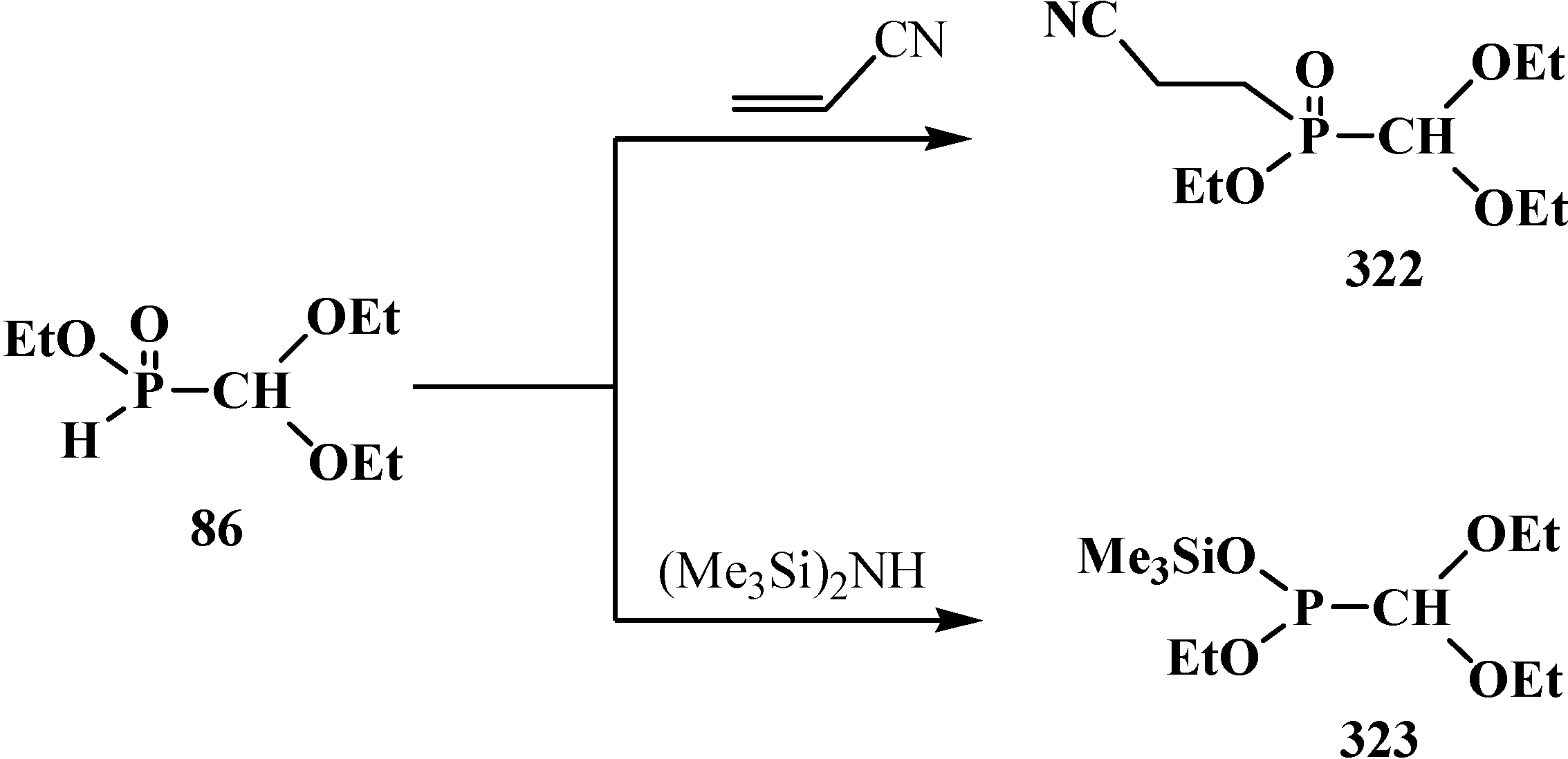
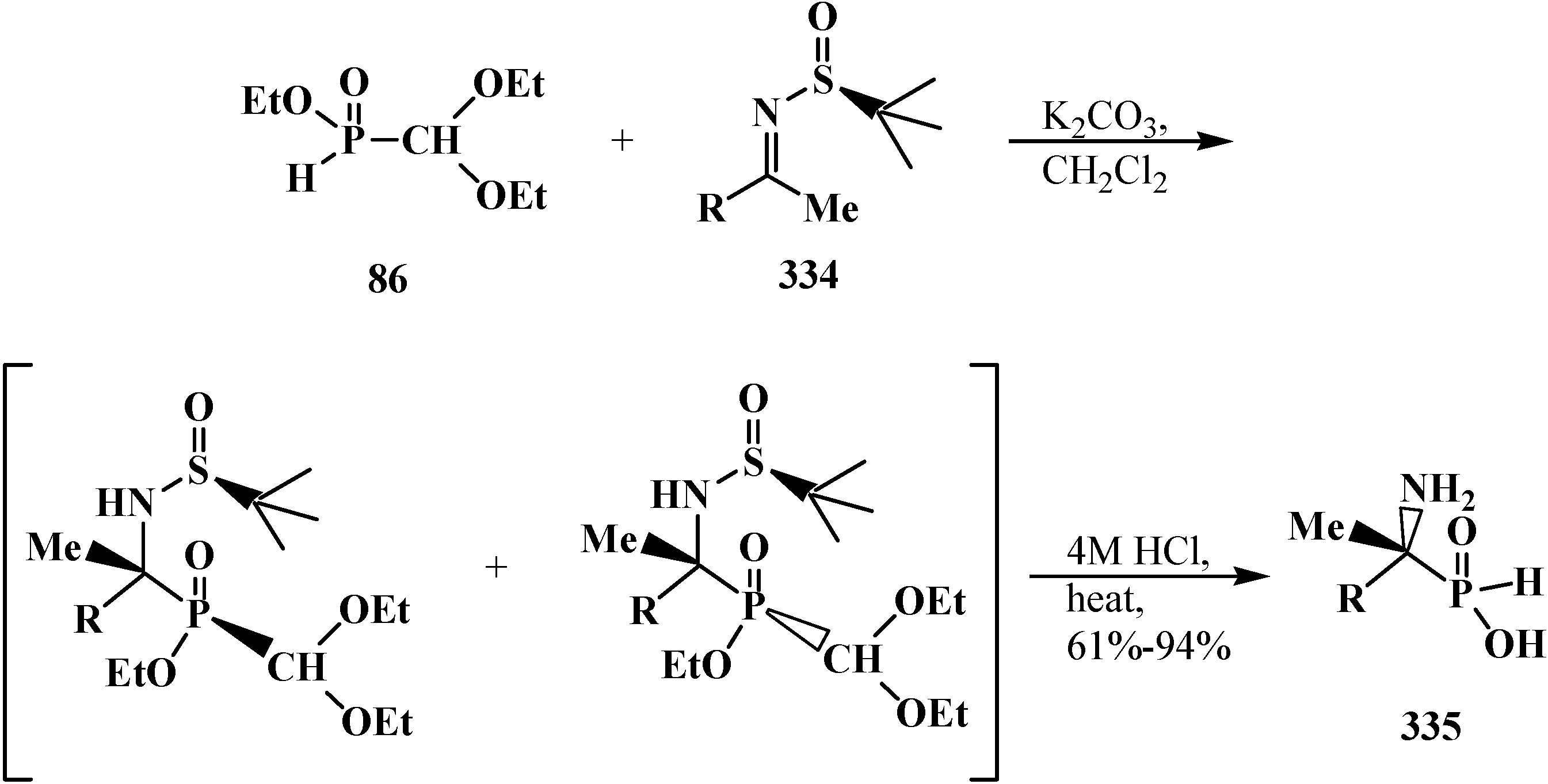
The catalytic synthesis of ethyl aryl(diethoxymethyl)phosphinates 85 by the reaction of ethyl (diethoxymethyl)phosphinate (86) with ortho-substituted aryl bromides in the presence of tetrakis(triphenylphosphine)palladium(0) Pd(Ph3P)4 in 75%–90% yields was reported in 1995 [94]. The Todd-Atherton reaction of 86 with ortho-substituted phenols in the presence of triethylamine at 0–23 °C leads to ethyl aryl (diethoxymethyl)phosphonates 87 [94] (Scheme 17).
Scheme 17.
Synthesis of ethyl aryl(diethoxymethyl)phosphinates 85 and ethyl aryl (diethoxymethyl)phosphonates 87 from ethyl (diethoxymethyl)phosphinate (86), R are Alk or Hal.
Scheme 17.
Synthesis of ethyl aryl(diethoxymethyl)phosphinates 85 and ethyl aryl (diethoxymethyl)phosphonates 87 from ethyl (diethoxymethyl)phosphinate (86), R are Alk or Hal.

The attempted preparations of novel acetales 33 by the transesterification of phosphorus ester groups failed because they gave rise to intractable mixtures of compounds [95]. Ethoxyphosphoryl groups in acetal 57 were replaced by butoxyphosphoryl groups only under cathode electrolysis conditions. As a result, butyl ethyl (diethoxymethyl)phosphonate (88) and dibutyl (diethoxymethyl)phosphonate (89) were obtained in low yields—11% and 5%, respectively [96] (Scheme 18).
Scheme 18.
Synthesis of acetales 88 and 89 by transesterification under the conditions of electrolysis.
Scheme 18.
Synthesis of acetales 88 and 89 by transesterification under the conditions of electrolysis.

However, the heating of 57 with 1,3-dimethylpropanediols in the presence of a catalytic amount of benzenesulfonic acid (PhSO3H) results in the replacement of ethoxy groups of the acetal fragment to give cyclic diethyl (5-dimethyl-[1,3]-dioxan-2-yl)phosphonates 90 [97], for example, see Scheme 19.
Scheme 19.
Preparation of cyclic diethyl (5-dimethyl-[1,3]-dioxan-2-yl)phosphonates 90 by the transesterification under acid catalysis.
Scheme 19.
Preparation of cyclic diethyl (5-dimethyl-[1,3]-dioxan-2-yl)phosphonates 90 by the transesterification under acid catalysis.

2.2.2. Chemical Properties of Phosphorylated Formaldehyde Acetals 13
The chemistry of phosphorylated formaldehyde acetals 13 was initially developed for the most part as a chemistry of available dialkyl (dialkoxymethyl)phosphonates 33. Therefore the properties of acetals as a separate type of organophosphorus compounds were studied mainly by the examples of compounds 33 whose reactivity is affected by the presence of both phosphorus ester and acetal groups.
Hydrolysis of acetals 33 was studied by the example of compound 57 and a compound with aromatic substituents in the acetal group, diethyl (5,6-dichloro-1,3-benzodioxomethyl)phosphonate (91). However, the attempted acid hydrolysis of acetal 57 on heating lead to the cleavage of phosphorus–carbon bond [77,95,98]. Compound 91 underwent acid hydrolysis on heating to give (5,6-dichloro-1,3-benzodioxomethyl)phosphonic acid (92) (Scheme 20). See also the section “Cleavage of Phosphorus–Carbon Bond under the Action of Acids and Acidic Reagents”, Scheme 54.
Scheme 20.
Acid hydrolysis of diethyl (5,6-dichloro-1,3-benzodioxomethyl)phosphonate (91).

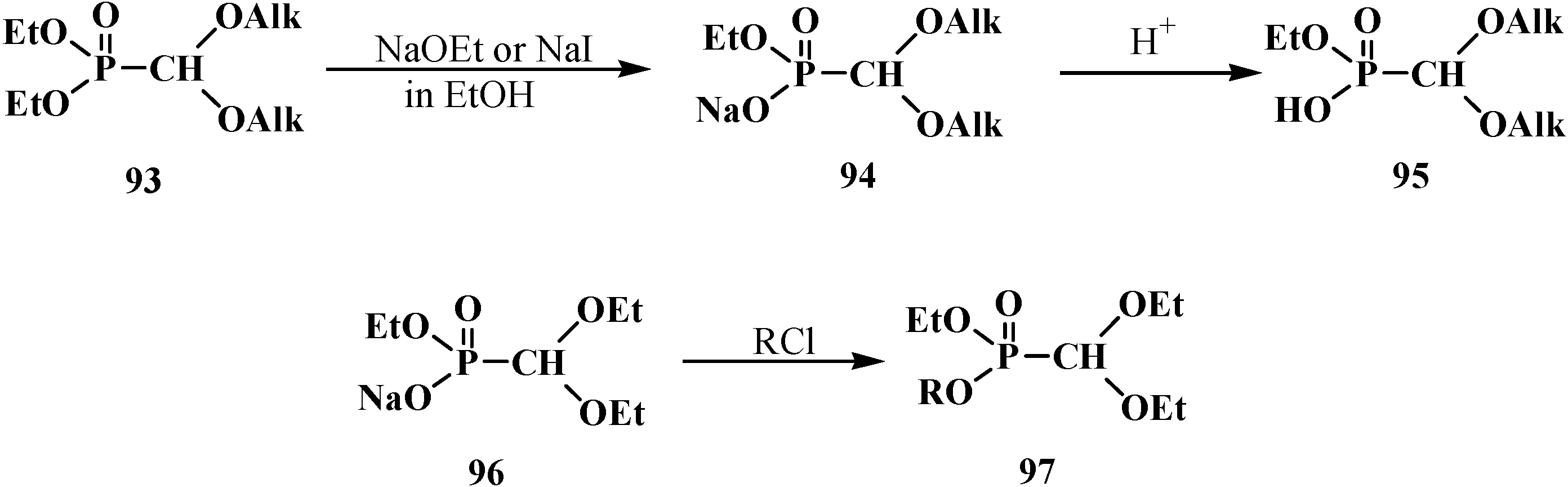
The heating of a solution of diethyl (dialkoxymethyl)phosphonate 93 in absolute ethanol with sodium ethoxide (NaOEt) leads to dealkylation of one of the ethoxy groups by phosphorus atom to form ethyl sodium (dialkoxymethyl)phosphonate 94, which produces the free acid 95 on acidification [95]. Heating of 93 with sodium iodide NaI leads to the same result [93]. The reaction of ethyl sodium (diethoxymethyl)phosphonate (96) with electrophilic reagents brings about the formation of phosphonates 97 with different substituents at the phosphorus atom [95] (Scheme 21).
Scheme 21.
Acetals 93 dealkylation at their interaction with sodium ethoxide or sodium iodide. Synthesis of acetals 97 with different substituents at phosphorus atom, where R = Alk, Ac, Me3Si, MeOCH2, ArOCH2.
Scheme 21.
Acetals 93 dealkylation at their interaction with sodium ethoxide or sodium iodide. Synthesis of acetals 97 with different substituents at phosphorus atom, where R = Alk, Ac, Me3Si, MeOCH2, ArOCH2.

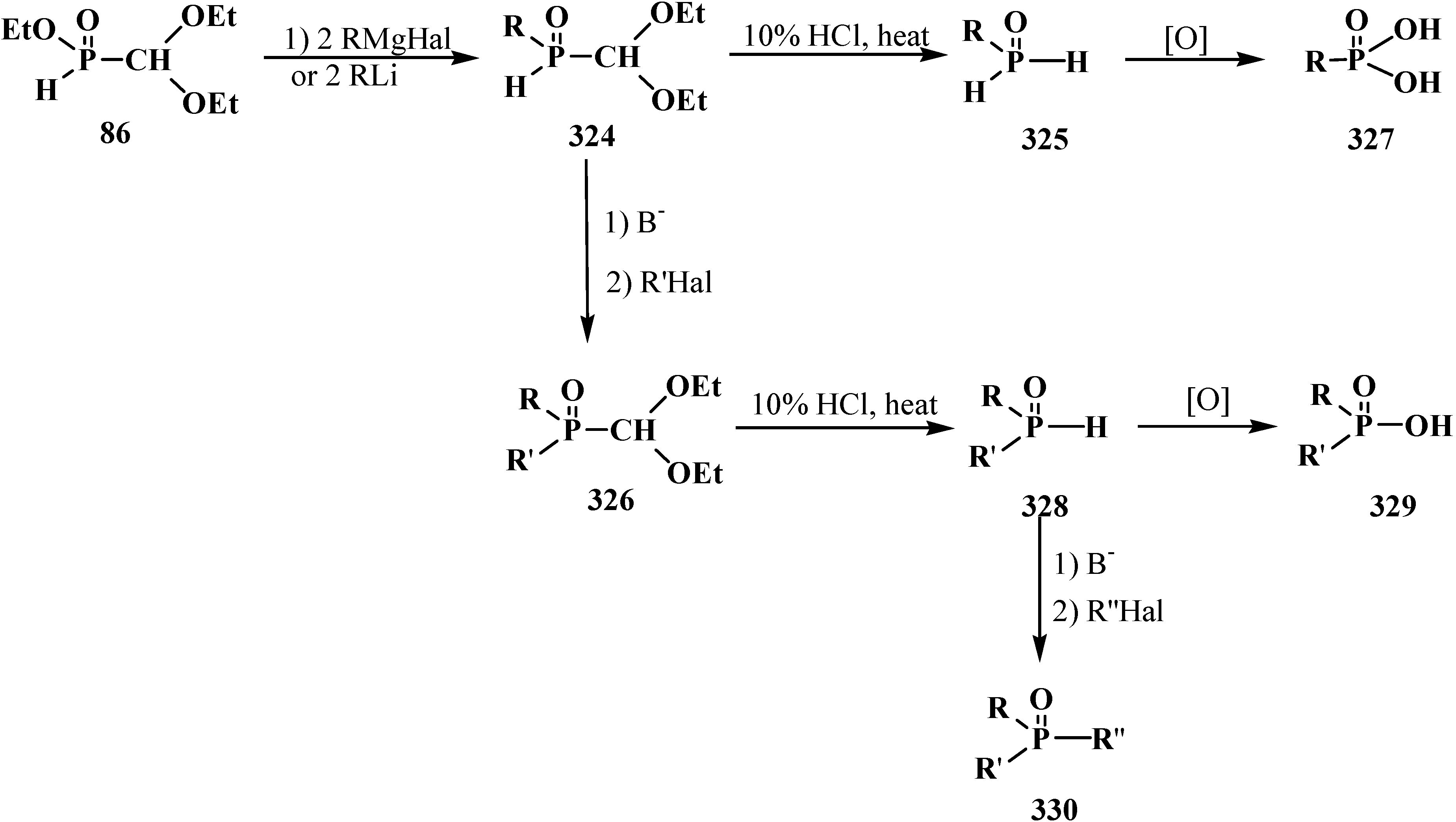
Acids 95 show typical properties of hydroxy compounds. Using as example ethyl (diethoxymethyl)phosphonic acid (98) it is shown that they react with diazomethane and thionyl chloride. The reaction products are ethyl methyl (diethoxymethyl)phosphonate (99) and ethyl (diethoxymethyl)phosphonic chloride (100) [95], which reacts with phenylmagnesium bromide in tetrahydrofuran to produce ethyl phenyl(diethoxymethyl)phosphinate (101) (Scheme 22).
Scheme 22.
Ethyl (diethoxymethyl)phosphonic acid (98) transformations.

Unsymmetrical ethyl aryl (diethoxymethyl)phosphonates 87 containing aryloxy substituents at the phosphorus atom [94] undergo rearrangement in the presence of equimolar amount of lithium diisopropylamide LDA in tetrahydrofuran at −70 °C to yield ethyl (2-hydroxyaryl)- (diethoxymethyl)phosphinates 102 (Scheme 23).
Scheme 23.
Rearrangement of compounds 87, R = Alk, Hal.

Heating of diethyl (diethoxymethyl)phosphonate (57) with a catalytic amount of BF3·Et2O gives rise to formation of tetraethyl (ethoxymethyl)diphosphonate (103) in a 14% yield [19] (Scheme 24).
Scheme 24.
Formation of tetraethyl (ethoxymethyl)diphosphonate (103) from acetal 57 under a catalysis by BF3·Et2O.
Scheme 24.
Formation of tetraethyl (ethoxymethyl)diphosphonate (103) from acetal 57 under a catalysis by BF3·Et2O.

The acetal group of compounds 13 is rather stable to the action of co-reactants. Nonetheless, a series of transformations of phosphorylated formaldehyde acetals (33) that involves dialkoxyacetal group is described.
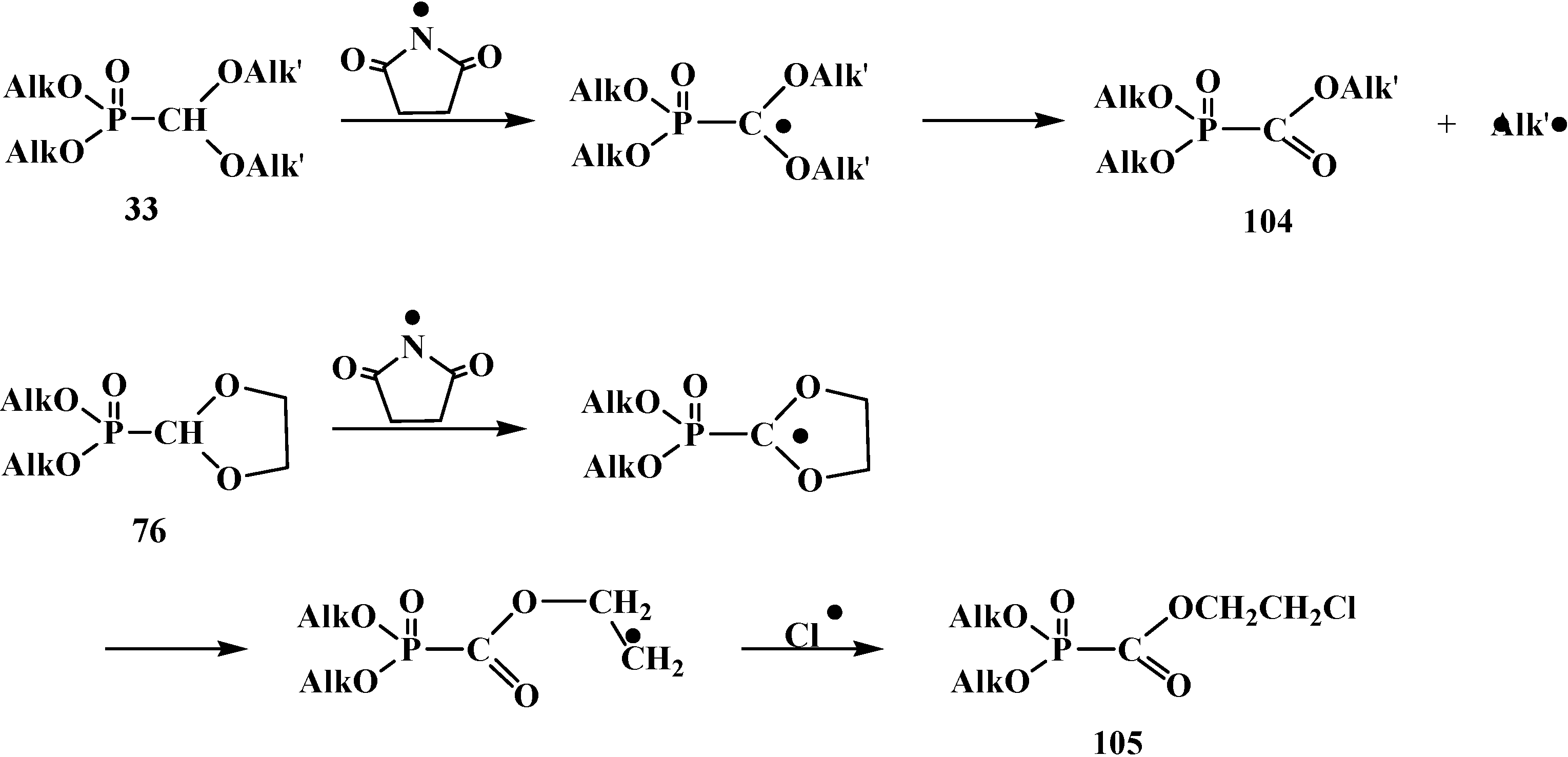
The halogenation of compounds 33 with N-bromosuccinimide leads to alcoxycarbonylphosphonates 104. Five-membered cyclic acetals 76 by halogenation with N-chlorosuccinimide and azodiisobutyro-nitrile mix produce β-haloethoxycarbonylphosphonates 105 [99]. Reactions proceed via a radical mechanism (Scheme 25).
Scheme 25.
Interaction of compounds 33 and 76 with N-bromosuccinimide or N-chlorosuccinimide.

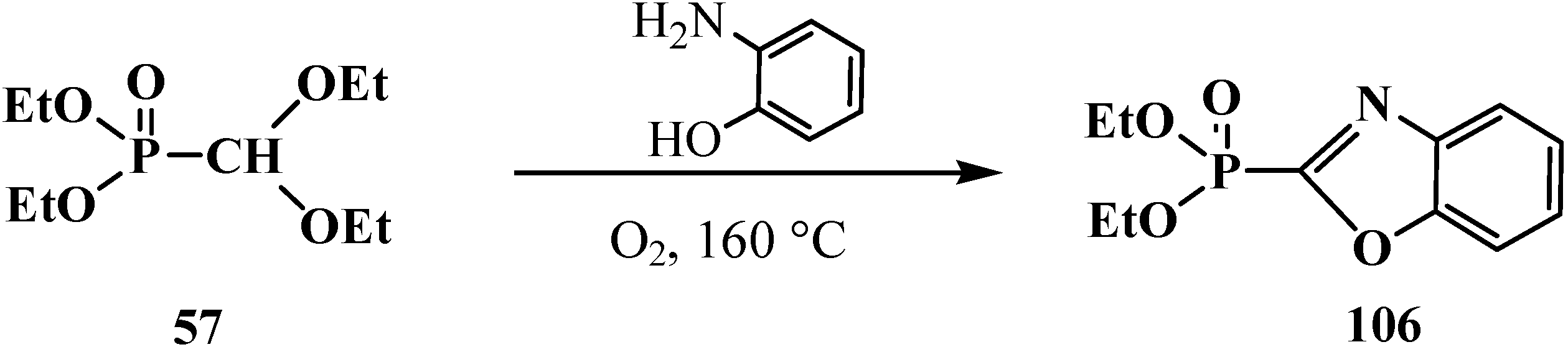
The reaction of 57 with o-aminophenol in oxygen flow at 160 °C results in diethyl (2-benzoxazolyl)phosphonate (106) [100] (Scheme 26).
Scheme 26.
The reaction of diethyl (diethoxymethyl)phosphonate (57) with o-aminophenol.

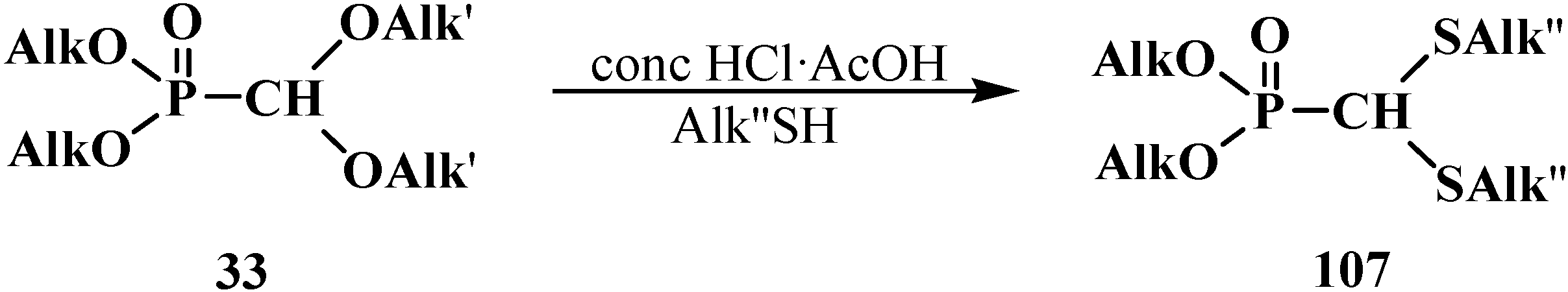
When compound 33 reacted with thiols in a 1:1 mixture of acetic and hydrochloric acid at 0 °C, dialkyl (dialkylthiomethyl)phosphonates 107 were obtained in 61%–64% yield [21] (Scheme 27).
Scheme 27.
Transformation of compounds 33 into dialkyl (dialkylthiomethyl)phosphonates 107.

Diethyl (diethoxymethyl)phosphonate (57) reacts with titanium tetrachloride TiCl4 or tetrabromide TiBr4 in diethyl ether to give diethyl [(ethoxy)chloromethyl]phosphonate (108) or diethyl [(ethoxy)bromomethyl]phosphonate (109) [61] (Scheme 28).
Scheme 28.
Syntheses of phosphorylated halogenacetals 108, 109 from diethyl (diethoxymethyl)phosphonate (57).
Scheme 28.
Syntheses of phosphorylated halogenacetals 108, 109 from diethyl (diethoxymethyl)phosphonate (57).

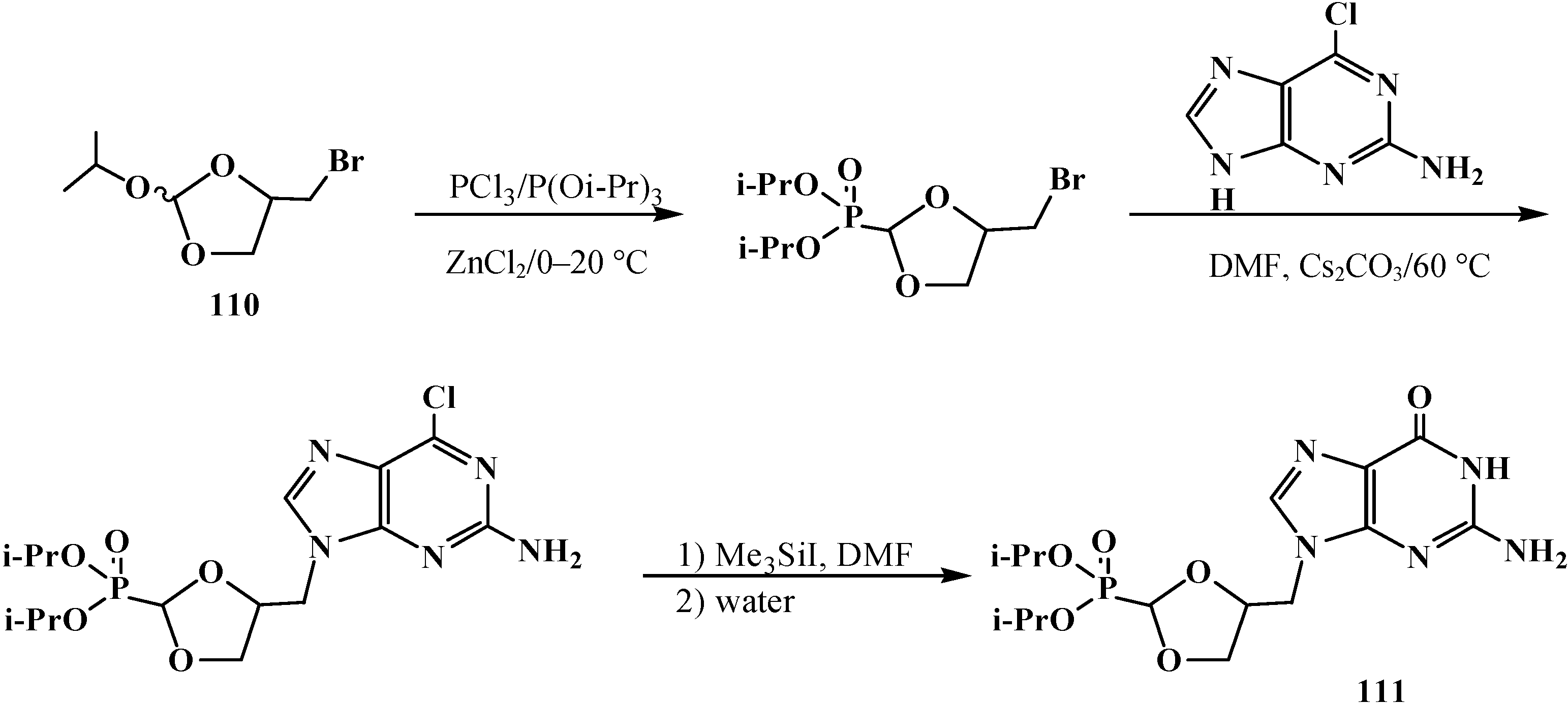
Cyclic diisopropyl [(4-bromomethyl-[1,3]-dioxolan)-2-yl]phosphonates 110 were used in the synthesis of analogs of natural purine and pyrimidine nucleotides [101]. Guanine analog 111 was prepared by the scheme (Scheme 29).
Scheme 29.
Synthesis of [1,3]-dioxolane analog guanine nucleotide 111.

The corresponding uracil analog was obtained in a similar manner. Both compounds showed moderate activity in vitro toward human cytomegalovirus [101]. See also Scheme 16, Scheme 18, Scheme 19, Scheme 54, Scheme 57, Scheme 58, Scheme 60, Scheme 62, Scheme 86, Scheme 87, Scheme 88, Scheme 90, Scheme 91 and Scheme 92.
2.3. Phosphorylated Formaldehyde Thioacetals 14
The first syntheses of compounds 14, linear dialkyl (dialkylthiomethyl)phosphonates 107 or dialkyl (diphenylthiomethyl)phosphonates 112 [21,22], cyclic dialkyl ([1,3]-dithiolan-2-yl)phosphonates 113, dialkyl ([1,3]-dithian-2-yl)phosphonates 114 [102] and dialkyl (1,3-benzodithiolylmethyl)phosphonates 115 [103] (Figure 5) were reported almost simultaneously in 1976–1977.
Figure 5.
First representatives of the phosphorylated formaldehyde thioacetals 107, 112–115.

2.3.1. Methods of Synthesis of Phosphorylated Formaldehyde Thioacetals 14
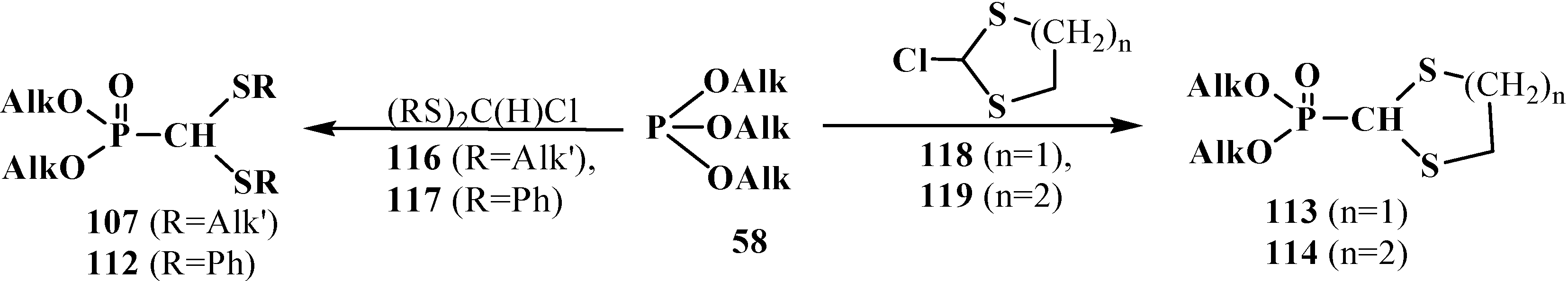
The first syntheses of linear and cyclic dialkyl (dialkylthiomethyl)phosphonates 107, 112–114 were performed by the analogy with the syntheses of dialkyl (dialkoxymethyl)phosphonates 33 (Scheme 9 and Scheme 13), namely, by the Arbuzov reaction of trialkyl phosphites 58 with linear (dialkylthio)chloromethanes 116 or (diphenylthio)chloromethane (117) [21,22], and cyclic 2-chloro-1,3-dithiolane [22,102] (118) or 2-chloro-1,3-dithiane (119) with 93%–95% yields (or quaternary ammonium salts of dimethylformamide thioacetal (only for 113), 50%–85%) [21,22] (Scheme 30).
Scheme 30.
The first syntheses of linear 107, 112 and cyclic 113, 114 phosphorylated formaldehyde thioacetals.
Scheme 30.
The first syntheses of linear 107, 112 and cyclic 113, 114 phosphorylated formaldehyde thioacetals.

At present, the method is used for the synthesis of cyclic dialkyl (dialkylthiomethyl)phosphonates 113, 114, because initial 2-chloro-1,3-dithiolane (118) and 2-chloro-1,3-dithiane (119) are readily prepared by the reaction of 1,3-dithiolane and 1,3-dithiane with N-chlorosuccinimide [102,104,105,106]. The method allows one to obtain in good yields both cyclic dialkyl (dialkylthiomethyl)phosphonates 113, 114 (55%–96%), [102,104,106]) and diphenyl(dialkylthiomethyl)phosphine oxides 121, 122 (57%–85%), [56,104,105]) (Scheme 31).
Scheme 31.
Syntheses of cyclic dialkyl (dialkylthiomethyl)phosphonates 113, 114 and diphenyl(dialkylthiomethyl)phosphine oxides 121, 122.
Scheme 31.
Syntheses of cyclic dialkyl (dialkylthiomethyl)phosphonates 113, 114 and diphenyl(dialkylthiomethyl)phosphine oxides 121, 122.

However, the poor availability of linear dialkylthiochloromethanes [21] confined the use of this method of synthesis of phosphorylated thioacetals 14 and stimulated the search for alternative methods for their synthesis.
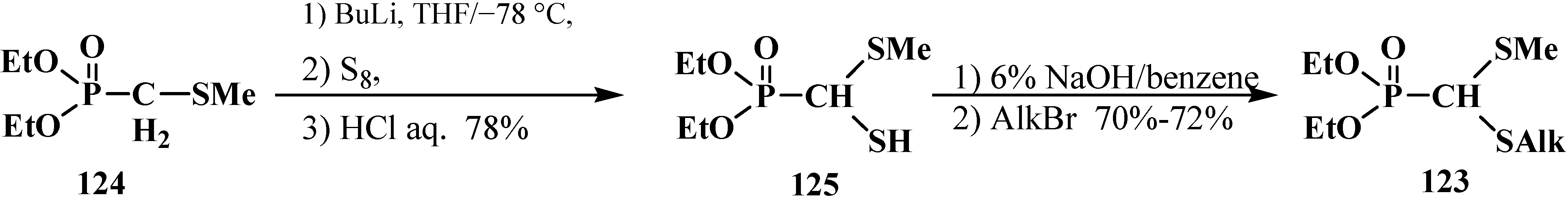
In 1979, a new two-step method was proposed for the synthesis of unsymmetrical diethyl (dialkylthiomethyl)phosphonates 123 starting from diethyl (methylthiomethyl)phosphonate (124). After one-pot treatment with butyllithium and elemental sulfur (sulfenylation) followed by aqueous treatment, 124 was converted into diethyl [(methylthio)mercaptomethyl]phosphonate (125), which under phase transfer catalysis conditions was further alkylated to give final unsymmetrical thioacetals 123 [107] (Scheme 32). In further work, alkyl halides (AlkHal) were introduced in the reaction medium immediately after sulfenylation, which enabled the preparation of thioacetals 123 by a one-pot method [108].
Scheme 32.
The synthesis of unsymmetrical diethyl (dialkylthiomethyl)phosphonates (123).

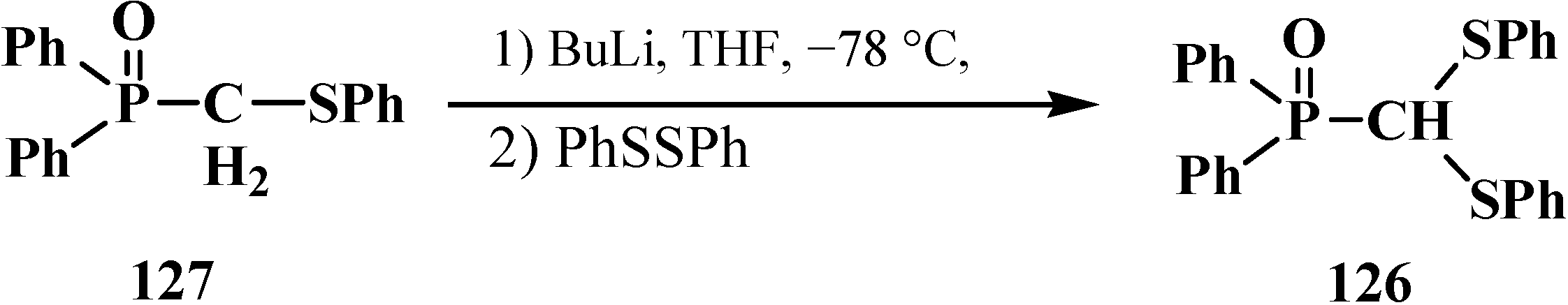
Diphenyl(diphenylthiomethyl)phosphine oxide (126) [109] was previously obtained in similar manner in 75% yield from diphenyl(phenylthiomethyl)phosphine oxide (127) by its interaction with diphenyldisulfide (Scheme 33).
Scheme 33.
Syntheses of diphenyl(diphenylthiomethyl)phosphine oxide (126) from diphenyl(phenylthiomethyl)phosphine oxide (127).
Scheme 33.
Syntheses of diphenyl(diphenylthiomethyl)phosphine oxide (126) from diphenyl(phenylthiomethyl)phosphine oxide (127).

It was shown later that diethyl (diphenylthiomethyl)phosphonate (128) can be prepared from dialkyl methylphosphonate (129) [108] by a one-pot technique in 84% yield (Scheme 34).
Scheme 34.
Syntheses of diethyl (diphenylthiomethyl)phosphonate (128) from dialkyl methylphosphonate (129) by a one-pot technique.
Scheme 34.
Syntheses of diethyl (diphenylthiomethyl)phosphonate (128) from dialkyl methylphosphonate (129) by a one-pot technique.

Diethyl [(methylthio)(trimethylsilyl)methyl]phosphonate (130) undergoes a similar transformation with diethyldisulfide to form diethyl (diphenylthiomethyl)phosphonate (131) (yield 62%–75%) [110] (Scheme 35).
Scheme 35.
Interaction of diethyl [(methylthio)(trimethylsilyl)methyl]phosphonate (130) with diethyldisulfide.
Scheme 35.
Interaction of diethyl [(methylthio)(trimethylsilyl)methyl]phosphonate (130) with diethyldisulfide.

This method [107] and its variations [108,109,110] provide a possibility to synthesize linear dialkyl (dialkylthiomethyl)phosphonates 107 containing different substituents in the thioacetal group.
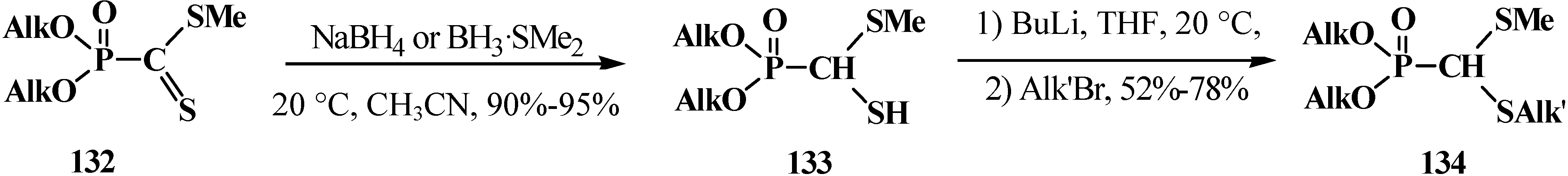
The reduction of phosphonodithioformates 132 with sodium borohydride (NaBH4) or borane–dimethyl sulfide adduct BH3·SMe2 [111] followed by alkylation of the resulted dialkyl [(methylthio)mercaptomethyl]phosphonates 133 finally results in unsymmetrical dialkyl [(methylthio)(alkylthio)methyl]phosphonates 134 (Scheme 36).
Scheme 36.
Transformation of phosphonodithioformates 132 into dialkyl [(methylthio)(alkylthio)methyl]phosphonates 134.
Scheme 36.
Transformation of phosphonodithioformates 132 into dialkyl [(methylthio)(alkylthio)methyl]phosphonates 134.

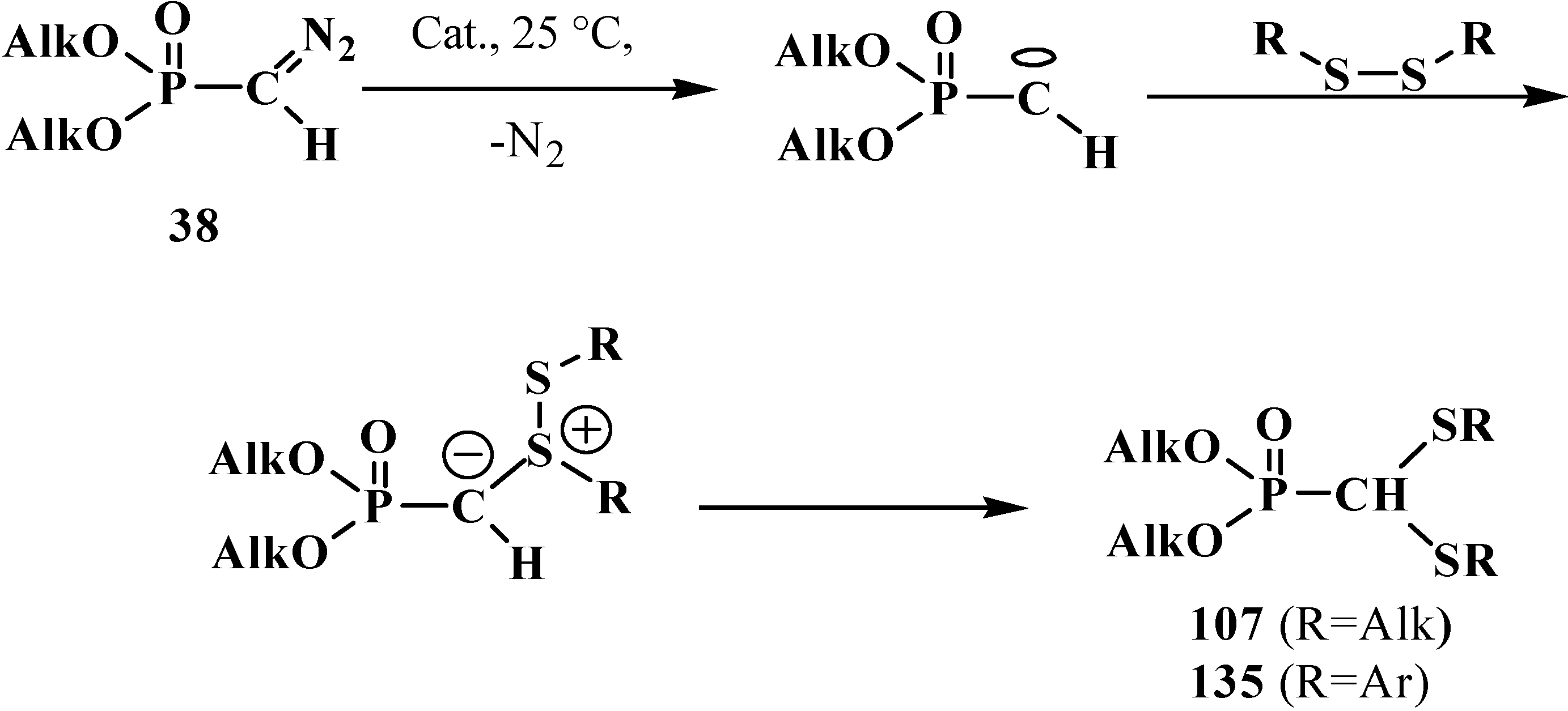
It was shown later that dialkyl and diaryl disulfides in the presence of catalysts (Cat.) like BF3·Et2O, rhodium(II) tetraacetate Rh2(OAc)4, or copper(II) sulfate CuSO4, can react with diazomethanephosphonates 38, where R=OAlk. Phosphorylated carbenes produced in the reaction undergo insertion into S–S bond to afford dialkyl (dialkylthiomethyl)phosphonates 107 or dialkyl (diarylthiomethyl)phosphonates 135 in 42%–93% yields [56,112]. Reactions proceed by the carbene mechanism (Scheme 37) [112].
Scheme 37.
Diazomethanephosphonates 38 reaction with organic disulfides with formation of dialkyl (dialkylthiomethyl)phosphonates 107 or dialkyl (diarylthiomethyl)-phosphonates 135.
Scheme 37.
Diazomethanephosphonates 38 reaction with organic disulfides with formation of dialkyl (dialkylthiomethyl)phosphonates 107 or dialkyl (diarylthiomethyl)-phosphonates 135.

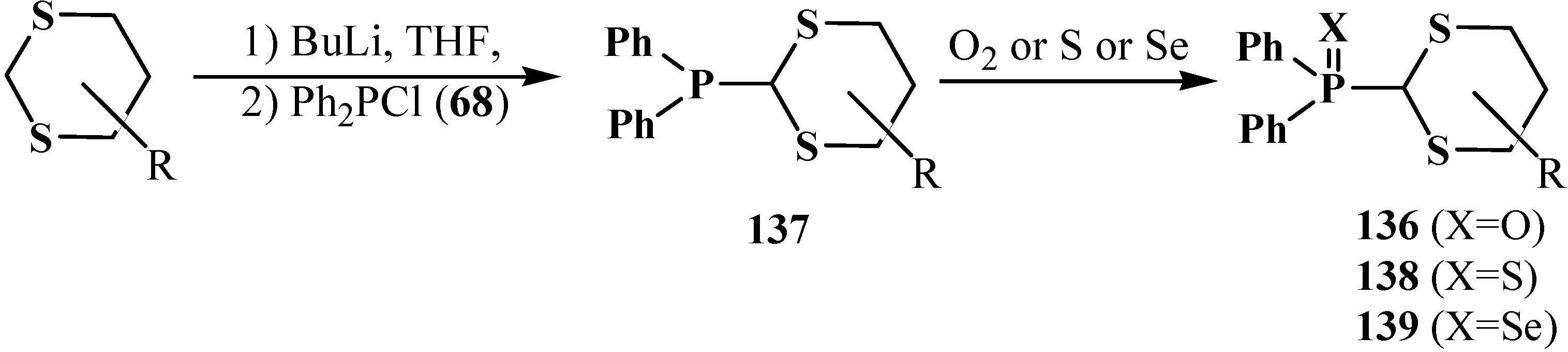
A method of synthesis of substituted diphenyl([1,3]-dithian-2-yl)phosphine oxides 136 (yields 34%–85%) starting from substituted 1,3-dithianes and chlorodiphenylphosphine (68) was also suggested [104,113,114]. Initially formed diphenyl([1,3]-dithian-2-yl)phosphines 137 undergo further oxidation with molecular oxygen to final 136 (Scheme 38). Diphenyl([1,3]-dithian-2-yl)phosphine sulfides 138 or selenides 139 can be obtained by this method when elemental sulfur or selenium are used as oxidants for 137 [104,113].
Scheme 38.
Obtaining of substituted diphenyl([1,3]-dithian-2-yl)phosphine oxides 136 and corresponding phosphine sulfides 138 and phosphine selenides 139 from dithianes.
Scheme 38.
Obtaining of substituted diphenyl([1,3]-dithian-2-yl)phosphine oxides 136 and corresponding phosphine sulfides 138 and phosphine selenides 139 from dithianes.

The attempted preparation of 107 and 114 in one step from dialkylthiomethanes or 1,3-dithianes and dialkyl chlorophosphate in the presence of strong bases failed, as evidenced by the negligible yield of final products [21,115]. The syntheses of 107 by the interaction of thioles with dialkyl [(N,N-dimethylamino)alkoxymethyl]phosphonates 140 or dialkyl [(alkylthio)chloromethyl]phosphonates 141 also failed [21].
Hovewer a method of synthesis of linear phosphorylated formaldehyde thioacetals 14 by the reaction of dialkyl [(arylthio)chloromethyl]phosphonates 142 with thiols at 0 °C in the presence of equimolar amount of tin(IV) tetrachloride SnCl4 was succesful. This route provided a preparation of unsymmetrical dialkyl [(alkylthio)(arylthio)methyl]phosphonates 143 in 73%–84% yields [43] (Scheme 39).
Scheme 39.
Obtaining of dialkyl [(alkylthio)(arylthio)methyl]phosphonates 143 by the reaction of dialkyl [(arylthio)chloromethyl]phosphonates 142 with thiols.
Scheme 39.
Obtaining of dialkyl [(alkylthio)(arylthio)methyl]phosphonates 143 by the reaction of dialkyl [(arylthio)chloromethyl]phosphonates 142 with thiols.

Dialkyl (dialkylthiomethyl)phosphonates 107 were also obtained by the reaction of dialkyl (dialkoxymethyl)phosphonates 33 with thiols in the presence of acids [21] (see Scheme 27).
Dialkyl (1,3-benzodithiolylmethyl)phosphonates 115 were prepared for the first time by the reaction of 1,3-benzodithiolyl tetrafluoroborate with 58 in the presence of NaI [103]. The method is used at present without changes [116] (Scheme 40).
Scheme 40.
The synthesis of dialkyl (1,3-benzodithiolylmethyl)phosphonates 115 from 1,3-benzodithiolyl tetrafluoroborate.
Scheme 40.
The synthesis of dialkyl (1,3-benzodithiolylmethyl)phosphonates 115 from 1,3-benzodithiolyl tetrafluoroborate.

2.3.2. Chemical Properties of Phosphorylated Formaldehyde Thioacetals 14
The chemical properties of phosphorylated formaldehyde thioacetals 14 have been studied in much less detail compared with the corresponding acetals 13. Their properties are determined by the presence of sulfur atoms and a disubstituted phosphoryl group. Both sulfur atoms are oxidized when diethyl ([1,3]-dithian-2-yl)phosphonate (144) is treated with sodium periodate NaIO4 in aqueous methanol at 20 °C [106] to yield the dioxo form 145 (Scheme 41).
Scheme 41.
Diethyl ([1,3]-dithian-2-yl)phosphonate (144) oxidation by periodate.

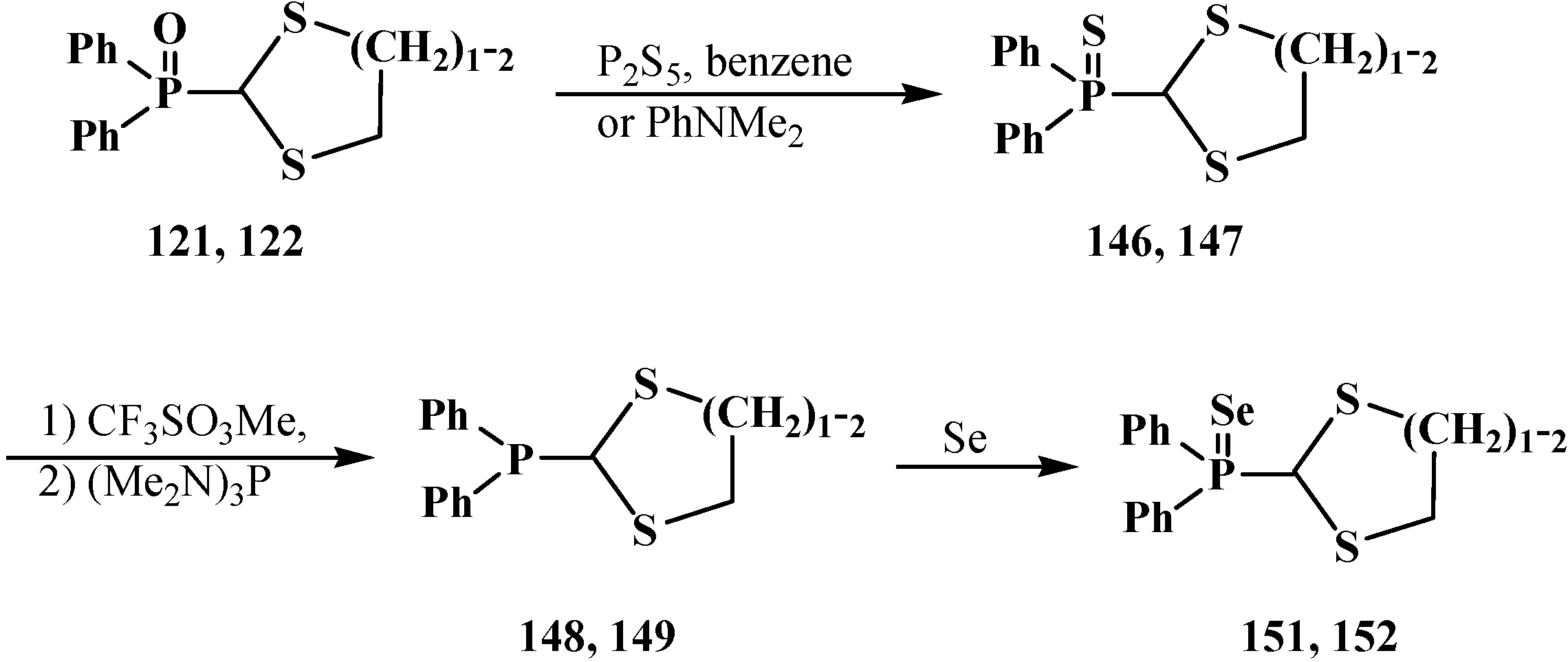
The heating of diphenyl([1,3]-dithiolan-2-yl)phosphine oxides 121 [115] and diphenyl([1,3]-dithian-2-yl)phosphine oxides 122 [56,104] with phosphorus pentasulfide P2S5 in benzene or N,N-diethylaniline PhNMe2 leads to the replacement of phosphinoyl oxygen by sulfur to yield diphenyl([1,3]-dithiolan-2-yl)phosphine sulfide (146) and diphenyl([1,3]-dithian-2-yl)phosphine sulfide, respectively (147) (Scheme 42). See also Scheme 38.
Scheme 42.
Transformation of phosphorylated cyclic thioacetals 122, 123 into corresponding phosphines 148, 149, phosphine sulfides 146, 147 and phosphine selenides 151, 152.
Scheme 42.
Transformation of phosphorylated cyclic thioacetals 122, 123 into corresponding phosphines 148, 149, phosphine sulfides 146, 147 and phosphine selenides 151, 152.

The sequential treatment of the prepared phosphine sulfides 146 and 147 with trifluoromethylsulfonate CF3SO3Me and tris(dimethylamino)phosphite (Me2N)3P finally affords diphenyl([1,3]-dithiolan-2-yl)phosphine (148) [105] and diphenyl([1,3]-dithian-2-yl)phosphines 149 [56,104]. The reaction of diphenyl(5-tert-butyl-[1,3]-dithian-2-yl)phosphine sulfide (150) with trichlorosilane leads to the same result [56]. The obtained phosphines 146, 147 combine with elemental selenium to yield diphenyl([1,3]-dithiolan-2-yl)phosphine selenide (151) [105] and diphenyl([1,3]-dithian-2-yl)phosphine selenide (152) [56,104]. See also Scheme 59, Scheme 60, Scheme 64, Scheme 65, Scheme 74, Scheme 75, Scheme 76, Scheme 77, Scheme 78, Scheme 79, Scheme 80 and Scheme 81.
2.4. (N,N-dialkylamino)cyanomethyl Derivatives of Phosphorylated Formaldehyde (α-dialkylamino-nitriles) 15
For 1982 till now, only two derivatives of this type of organophosphorus compounds of phosphonate series were obtained, diethyl [(N,N-dimethylamino)cyanomethyl]phosphonate (153) [24] and diethyl [(N-morpholino)cyanomethyl]phosphonate (154) [26].
2.4.1. Methods of Synthesis of Diethyl [(N,N-Dialkylamino)cyanomethyl]phosphonates 15
Diethyl [(N,N-dimethylamino)cyanomethyl]phosphonate (153) was obtained for the first time by a reaction similar to the preparation of phosphorylated acetals 13 (Scheme 7) from diethyl phosphite (6) and (N,N-dimethylamino)(methoxy)cyanomethane [24,25] (Scheme 43).
Scheme 43.
Syntheses of diethyl [(N,N-dimethylamino)cyanomethyl]phosphonate (153).

Diethyl [(N-morpholino)cyanomethyl]phosphonate (154) was prepared by the second method from diethyl chlorophosphate (155) and (N-cyanomethyl)morpholine [26] (Scheme 44) and was used in a subsequent reaction without isolation.
Scheme 44.
Syntheses of diethyl [(N-morpholino)cyanomethyl]phosphonate (154) from diethyl chlorophosphate (155).
Scheme 44.
Syntheses of diethyl [(N-morpholino)cyanomethyl]phosphonate (154) from diethyl chlorophosphate (155).

2.4.2. Chemical Properties of Diethyl [(N,N-Dialkylamino)cyanomethyl]phosphonates 15
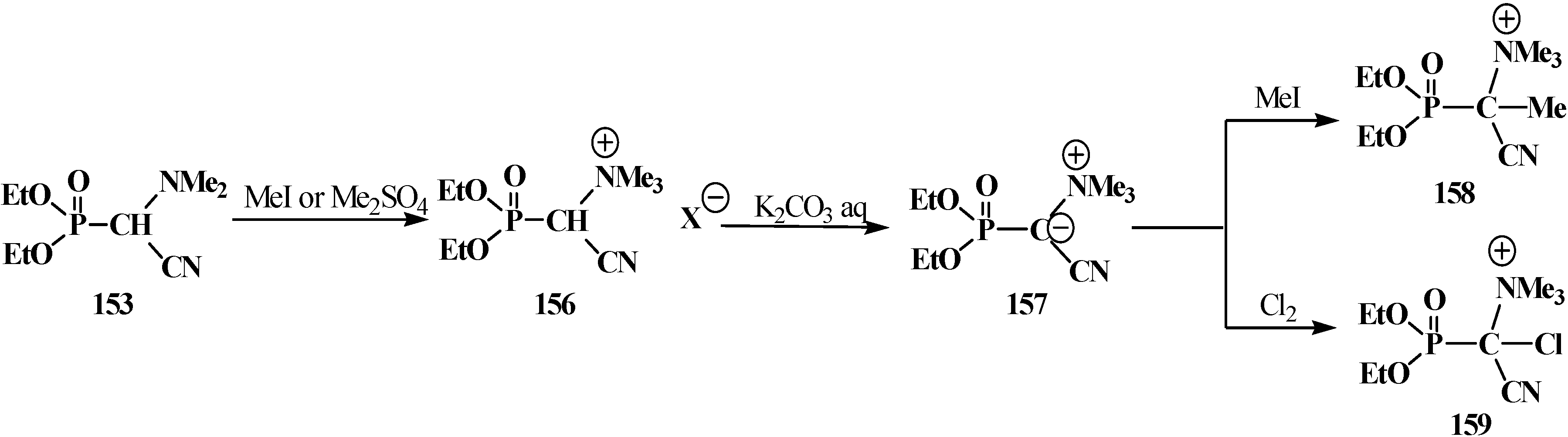
The chemical properties of this type of compounds have been studied almost completely using the example of diethyl [(N,N-dimethylamino)cyanomethyl]phosphonate (153). The treatment of 153 with methyl iodide (MeI) or dimethyl sulfate Me2SO4 [117,118] gives rise to methylation of nitrogen atom (quaternization) of the dimethylamino group and formation of diethyl [(N,N,N-trimethylammonio)cyanomethyl]phosphonate cation (156), whose α proton shows enhanced acidity and breaks off under the action of aqueous potassium carbonate solution K2CO3. The resultant diethoxy [(N,N,N-trimethylammonio)cyanomethylidium]phosphonate (157) can react with electrophiles—MeI and molecular chlorine Cl2 [117], to give diethyl [(1-(N,N,N-trimethylammonio))(1-cyano)ethan-1-yl]phosphonate cation (158) and diethyl [chloro(1-N,N,N-trimethylammonio)cyanomethyl]phosphonate cation (159), respectively (Scheme 45).
Scheme 45.
Transformation of diethyl [(N,N-dimethylamino)cyanomethyl]phosphonate (153).

Under phase transfer catalysis conditions—50% KOH solution, [Et3NCH2Ph]+Cl− (TEBAC) [24] phosphonate 153 combines with nitrosobenzene PhN=O similarly to the Horner reaction to afford [(N,N-dimethyl)(N'-phenyl)amidinoyl]oxalnitrile (160) in 58% yield (Scheme 46). See also Scheme 55, Scheme 60, Scheme 66, Scheme 67, Scheme 82, Scheme 83, Scheme 84 and Scheme 85.
Scheme 46.
Horner—analog reaction of phosphonate 153 with nitrosobenzene.

2.5. Diphosphinoyl N,N-Dialkylaminomethanes 16
2.5.1. Methods of Synthesis of Diphosphinoyl N,N-Dialkylaminomethanes 16
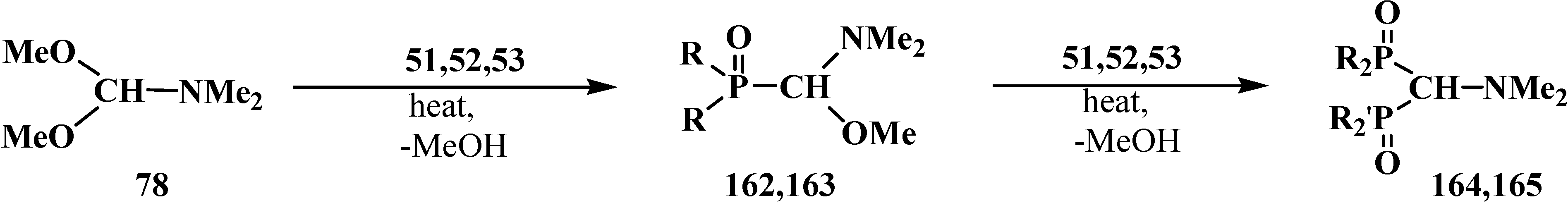
The first report on the synthesis of tetraethyl (N,N-dimethylaminomethyl)diphosphonate (161) was published in 1968. Compound 161 was obtained in 62% yield by heating a 2:1 mixture of diethyl phosphite (6) and dimethylformamide dimethylacetal (78) [27]. Dialkyl phosphites 51, dialkyl- and diphenylphosphine oxides 52, 53 can be also involved in this reaction [31,32,119]. Intermediate dialkyl [(N,N-dimethylamino)methoxymethyl]phosphonates (162) or dialkyl[(N,N-dimethylamino)-methoxymethyl]phosphine oxides 163 may be isolated in many cases [32,119]. This allows preparation of symmetrical 164 and unsymmetrical diphosphinoyl N,N-dimethylaminomethanes 165 [31,32,119,120] (Scheme 47).
Scheme 47.
Synthesis of diphosphinoyl N,N-dimethylaminomethanes 164 and 165 from dimethylformamide dimethylacetal (78), where R, R' = OAlk, Alk, Ph. See text above.
Scheme 47.
Synthesis of diphosphinoyl N,N-dimethylaminomethanes 164 and 165 from dimethylformamide dimethylacetal (78), where R, R' = OAlk, Alk, Ph. See text above.

Other methods of synthesis of symmetrical tetraalkyl (N,N-dimethylaminomethyl)diphosphonates 166 were proposed later. Since trialkyl phosphites 58 do not react with dimethylformamide dimethylacetal (78) [18], mixed dialkyl trimethylsilyl phosphites (EtO)2PSiMe3 were successfully employed in the reaction. The reaction proceeds spontaneously at 20 °C [121] in 36%–66% yield or upon heating in the presence of zinc chloride in 72%–77% yield [122,123] (Scheme 48).
Scheme 48.
Use mixed dialkyl trimethylsilyl phosphites for the syntheses of tetraalkyl diphosphonates 166 from dimethylformamide dimethylacetal (78).
Scheme 48.
Use mixed dialkyl trimethylsilyl phosphites for the syntheses of tetraalkyl diphosphonates 166 from dimethylformamide dimethylacetal (78).

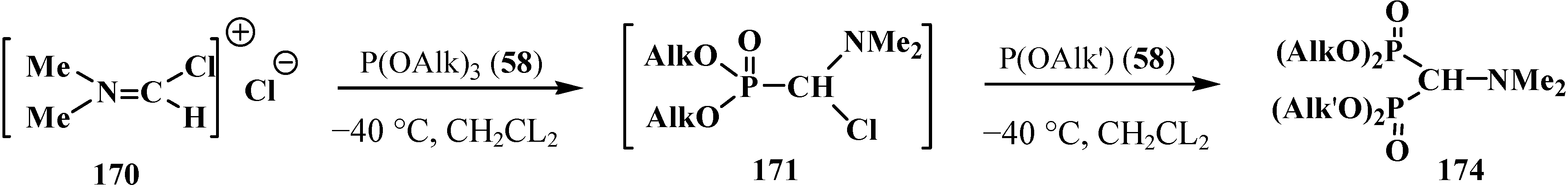
A convenient method of synthesis of unsymmetrical tetraalkyl (N,N-dialkylaminomethyl)- diphosphonates 167 by the reaction of trialkyl phosphites (58) with N,N-dialkylhalo-methylideneiminium halides [HalC(H)=NAlk2]+Hal−, where Hal = Cl (compounds 168) [28,31,32,120] or Br (compounds 169) [29] in 2:1 ratio was proposed in 1969.
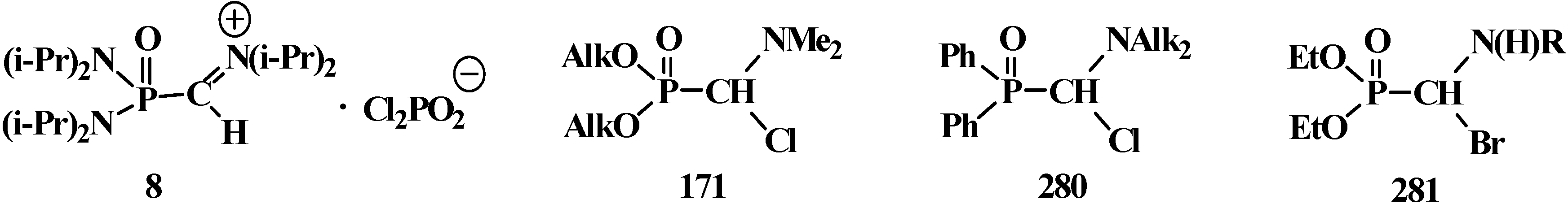
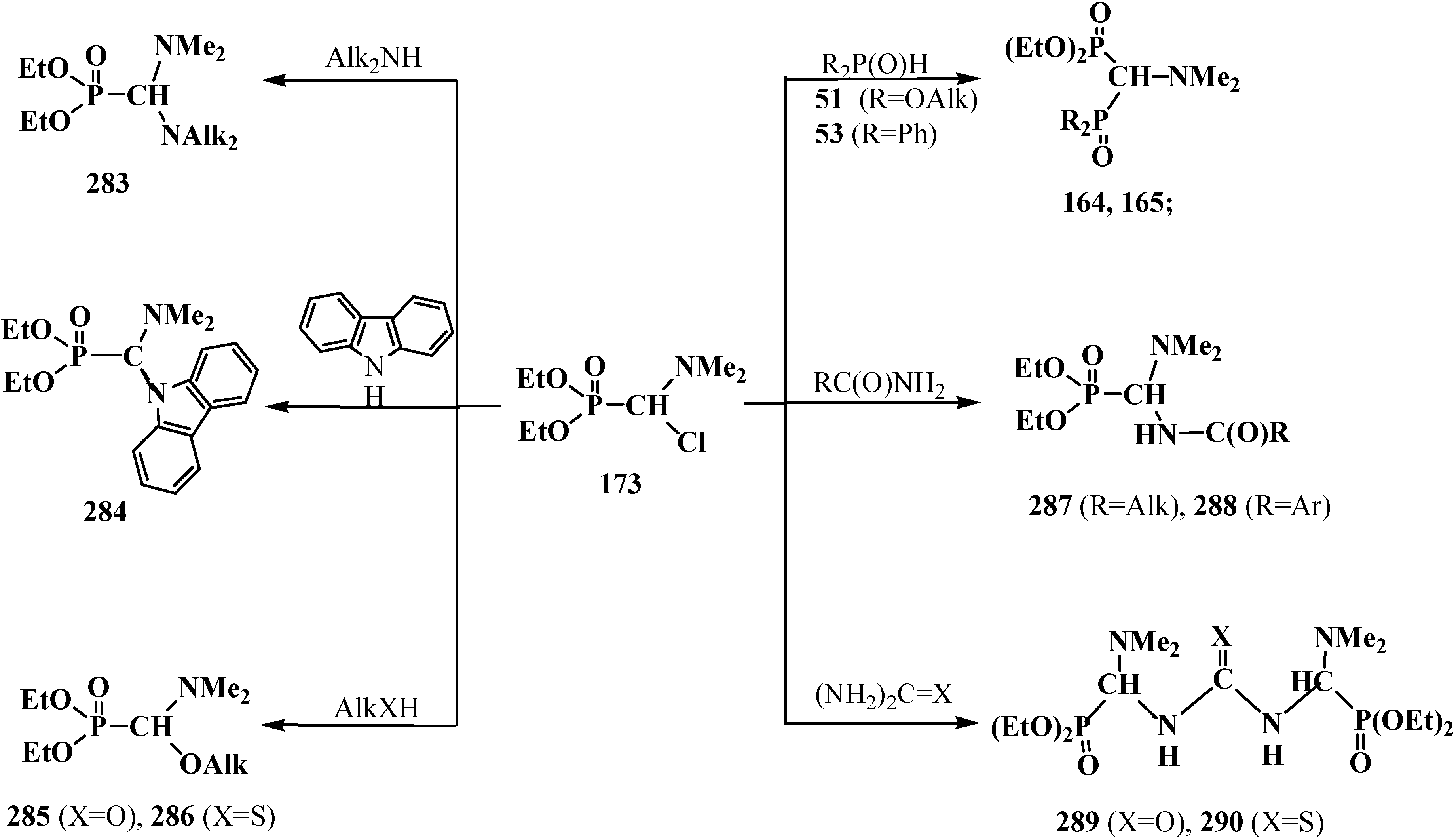
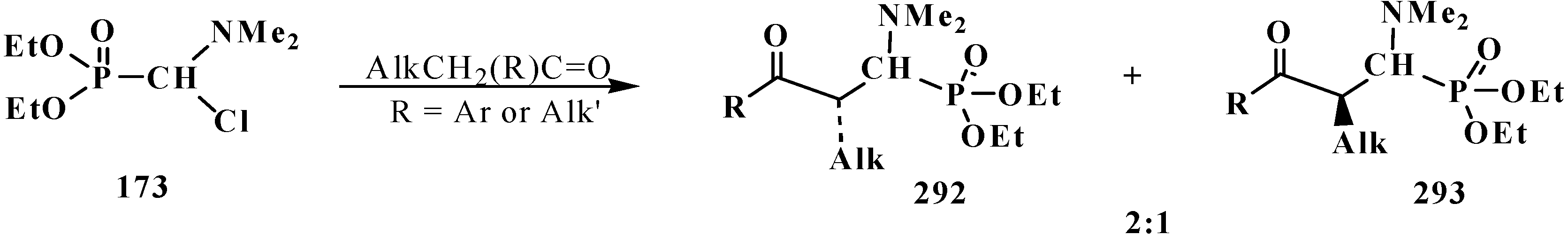
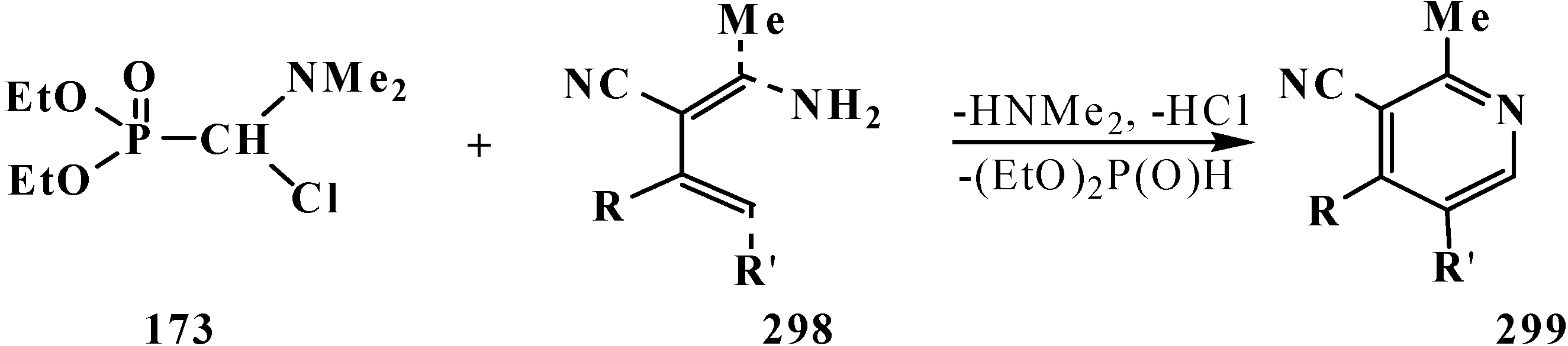
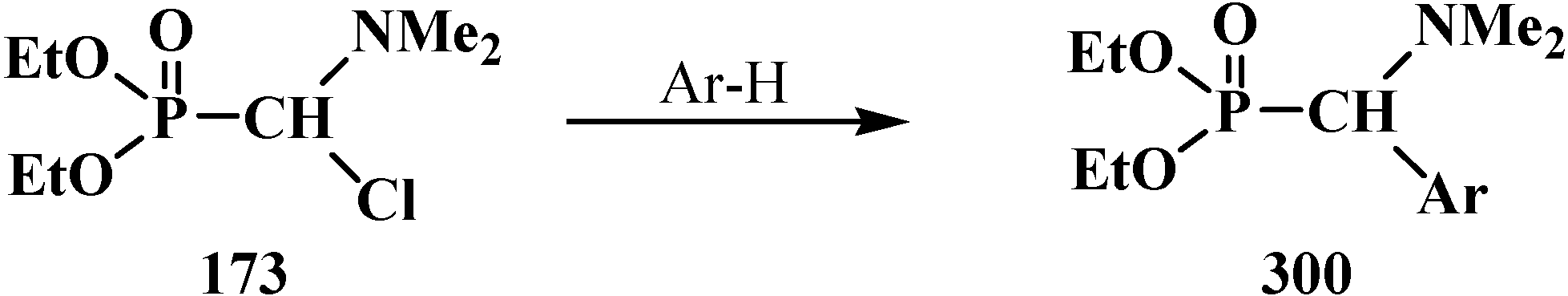
It was shown that the reaction of trialkyl phosphites 58 with N,N-dimethylchloro- methylideneiminium chloride (170) proceeds via intermediate formation of dialkyl [(N,N-dimethylamino)chloromethyl]phosphonate 171 [31,32], specially prepared dimethyl [(N,N-dimethylamino)chloromethyl]phosphonate (172) and diethyl [(N,N-dimethylamino)chloromethyl]- phosphonate (173) may be also involved in the reaction [30,31,124]. This method enables preparation of symmetrical and unsymmetrical tetraalkyl phosphonates 174 as well (Scheme 49).
Scheme 49.
Method of synthesis of symmetrical and unsymmetrical dialkyl (N,N-dimethylaminochloromethyl)phosphonates 174 from N,N-dimethylchloromethylideneiminium chloride (170) and trialkyl phosphites (58).
Scheme 49.
Method of synthesis of symmetrical and unsymmetrical dialkyl (N,N-dimethylaminochloromethyl)phosphonates 174 from N,N-dimethylchloromethylideneiminium chloride (170) and trialkyl phosphites (58).

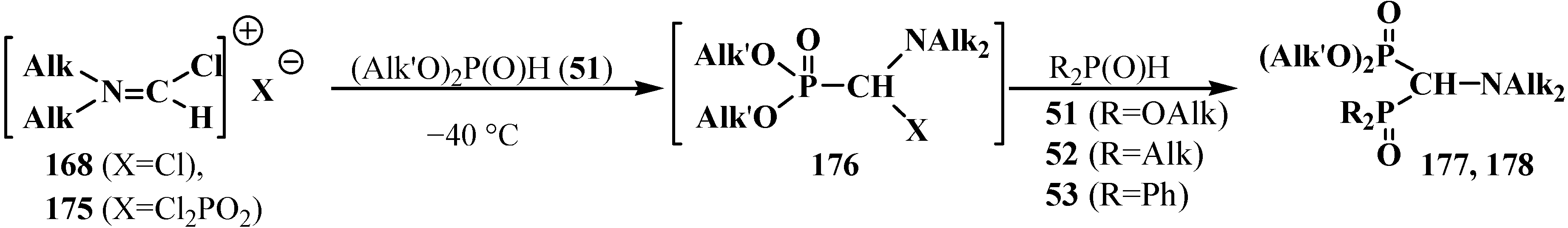
Dialkyl phosphites 51, dialkylphosphine oxides 52 and diphenylphosphine oxide (53) also react with N,N-dialkylchloromethylideneiminium chlorides 168 [31,120] and their analogs 175 obtained from N,N-dialkylformamides and phosphorus oxychloride [28] (Scheme 50). The reaction also proceeds via intermediate compounds 176, where X=Cl or Cl2PO2. This allows also the preparation of symmetrical or unsymmetrical diphosphinoyl compounds 177 and 178, respectively.
Scheme 50.
Synthesis of diphosphinoyl N,N-dialkylaminomethanes 177 and 178 from N,N-dimethylchloromethylideneiminium chloride (170) and hydrophosphorylic compounds 51, 52, 53. See text above.
Scheme 50.
Synthesis of diphosphinoyl N,N-dialkylaminomethanes 177 and 178 from N,N-dimethylchloromethylideneiminium chloride (170) and hydrophosphorylic compounds 51, 52, 53. See text above.

See also Scheme 14.
2.5.2. Chemical Properties of Diphosphinoyl N,N-Dialkylaminomethanes 16
The chemical properties of this type of organophosphorus compounds are studied insufficiently and almost exclusively by the example of tetraethyl (N,N-dimethylaminomethyl)diphosphonate (161). Their properties are attributable to the presence of both an amino group and disubstituted phosphoryl groups.
Hydrolysis of tetraalkyl (N,N-dimethylaminomethyl)diphosphonates 174 was studied using the example of compound 161. Boiling diphosphonate 161 with concentrated hydrochloric acid leads to (N,N-dimethylaminomethyl)diphosphonic acid (179) [98,120] in almost quantitative yield (Scheme 51).
Scheme 51.
Acid hydrolysis of tetraethyl (N,N-dimethylaminomethyl)diphosphonate (161).

The amino group of diphosphonate 161 undergoes methylation (quaternization) when reacted with methyl iodide (MeI) or dimethyl sulfate (Me2SO4) to give tetraethyl (N,N,N-trimethylammoniomethyl)diphosphonate cation (180) in 76 and 85% yields, respectively [118,125]. The α-proton of the latter shows enhanced acidity and undergoes elimination under the action of aqueous solution of potassium carbonate (K2CO3) to afford tetraethyl [(N,N,N-trimethylammonio)methylidium]diphosphonate (181) [120,125] (Scheme 52).
Scheme 52.
Transformation of tetraethyl (N,N-dimethylaminomethyl)diphosphonate (161) into methylidiumdiphosphonate 181, where X = I, MeSO3.
Scheme 52.
Transformation of tetraethyl (N,N-dimethylaminomethyl)diphosphonate (161) into methylidiumdiphosphonate 181, where X = I, MeSO3.

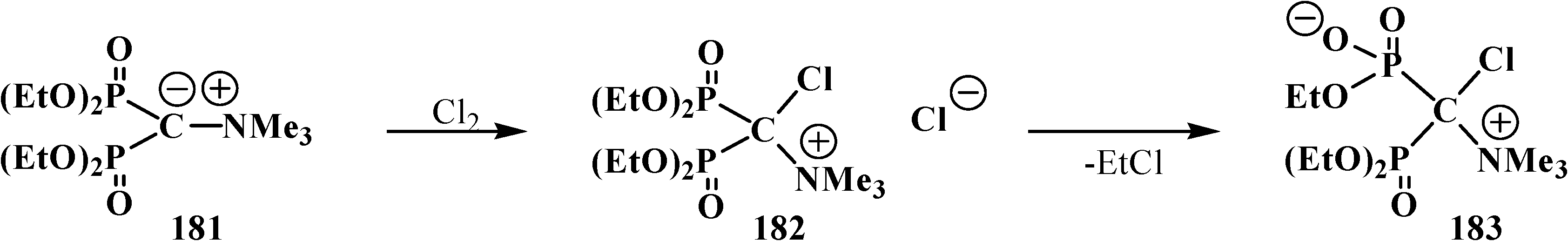
Ylide 181 exhibits an enhanced stability: it can be stored for a long time in air, it is thermally stable and does not react with methyl iodide [126]. However, ylide 181, like cyanomethylide 154, reacts with molecular chlorine to yield tetraethyl [(N,N,N-trimethylammonio)chloromethyl)diphosphonate chloride (182) that undergoes fast dealkylation on storage to afford the corresponding betaine 183 in 85% yield [120] (Scheme 53).
Scheme 53.
Interaction of methylidiumdiphosphonate 181 with chlorine with the subsequent formation of betaine 183.
Scheme 53.
Interaction of methylidiumdiphosphonate 181 with chlorine with the subsequent formation of betaine 183.

3. General Chemical Properties of Phosphorylated Formaldehyde Acetals 13 and Structurally Related Compounds 14–16
3.1. Phosphorus–Carbon Bond Cleavage
3.1.1. Cleavage of Phosphorus–Carbon Bond under the Action of Acids and Acidic Reagents
The general property of phosphorylated formaldehyde acetals 13 and structurally related compounds 14–16 is the possibility of phosphorus–carbon bond cleavage under the action of acids and acidic reagents [24,77,95,98], organic reagents [19,119], and bases [37,127,128].
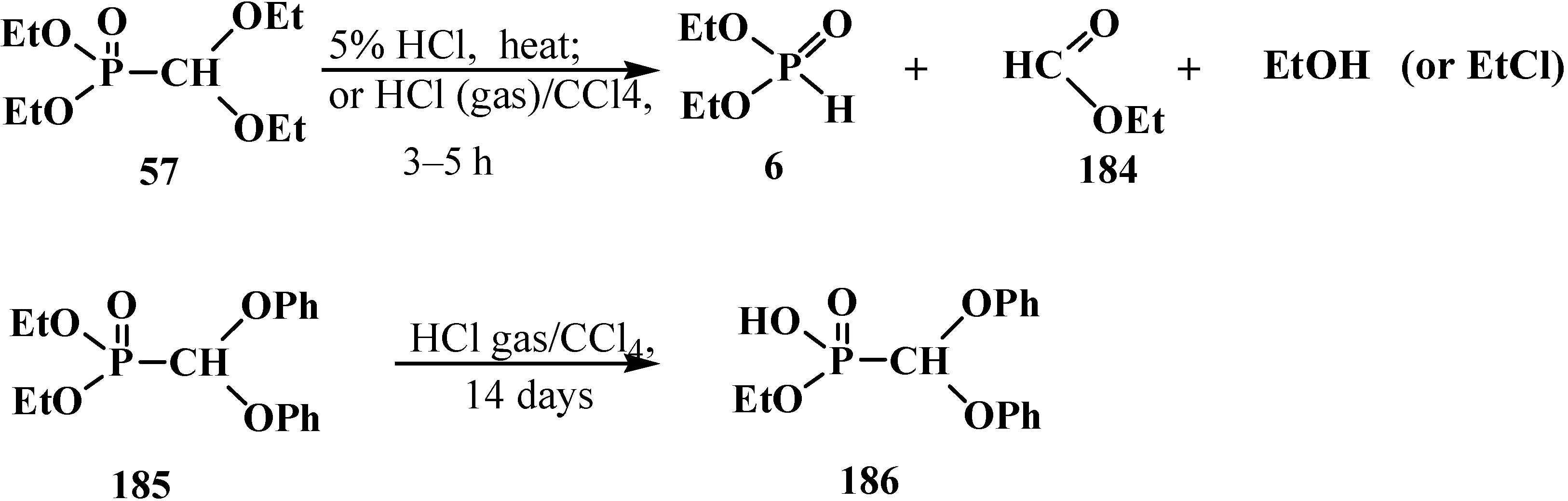
The cleavage of phosphorus–carbon bond under acidic conditions compounds 33 is studied on examples of diethyl (diethoxymethyl)phosphonate (57). Phosphonate 57 was shown to undergo cleavage of the phosphorus–carbon bond on heating with 5% hydrochloric acid [77,98] to form diethyl phosphites (6), ethyl formate (184) and ethanol. Similar degradation of compound 57 occurs in a flow of hydrogen chloride at 20 °C [95] (ethyl chloride is formed as a byproduct) (Scheme 54). Unlike 57, interaction of diethyl (diphenoxymethyl)phosphonate (185) with dry hydrogen chloride does not lead to the cleavage of phosphorus–carbon bond, but leads to the dealkylation of one ethoxy substituent at the phosphorus atom with obtaining ethyl (diphenoxymethyl)phosphonic acid (186) (Scheme 54).
Scheme 54.
Interaction of acetals 57 and 185 with hydrochloric acid (only for 57) and hydrogen chloride.
Scheme 54.
Interaction of acetals 57 and 185 with hydrochloric acid (only for 57) and hydrogen chloride.

It was shown that acid hydrolysis of diethyl (diethylthiomethyl)phosphonates (187) is accompanied by a partial cleavage of the phosphorus–carbon bond [98]. Prolonged refluxing of diethyl [(N,N-dimethylamino)cyanomethyl]phosphonate (153) in concentrated hydrochloric acid leads to N,N-dimethylaminoacetic acid (188) in 87% yield [24] (Scheme 55).
Scheme 55.
Obtaining N,N-dimethylaminoacetic acid (188) by acid hydrolysis of cyanomethylphosphonate 153.
Scheme 55.
Obtaining N,N-dimethylaminoacetic acid (188) by acid hydrolysis of cyanomethylphosphonate 153.

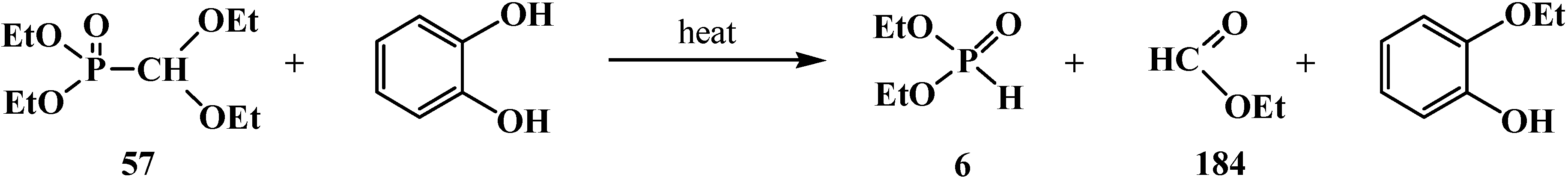
It was shown in [95] that the attempted transesterification of ethoxy substituents at the phosphorus atom of phosphonate 57 by catechol residue also leads to the cleavage of the phosphorus–carbon bond (Scheme 56).
Scheme 56.
Destruction of acetales 57 at its interaction with catechol.

The cleavage of phosphorus–carbon bond may also occur when diethyl (dialkoxymethyl)phosphonates 93 are exposed to phosphorus pentachloride [95].
3.1.2. The Cleavage of Phosphorus–Carbon Bond in Reactions with Organic Coreactants
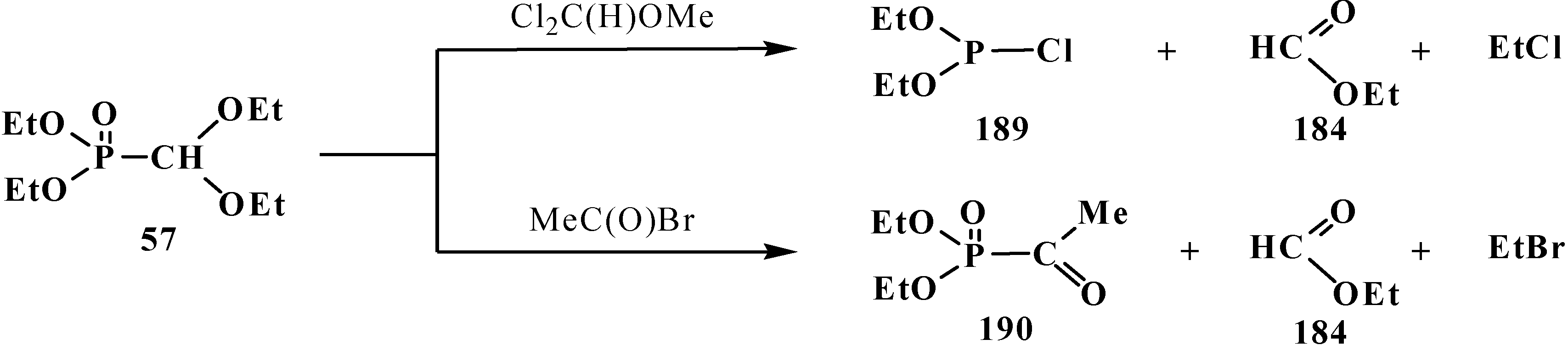
The reactions of 57 with dichloromethoxymethane Cl2C(H)OMe or acetyl bromide MeC(O)Br was shown to be accompanied by the cleavage of phosphorus–carbon bond [19] to form diethoxychlorophosphine (189) and diethyl acethylphosphonate (190) (Scheme 57).
Scheme 57.
Acetales 57 interaction with Cl2C(H)OMe and MeC(O)Br leading to the cleavage of phosphorus–carbon bond.
Scheme 57.
Acetales 57 interaction with Cl2C(H)OMe and MeC(O)Br leading to the cleavage of phosphorus–carbon bond.

The reaction of diethyl phosphite (6) with dipropyl [(N,N-dimethylamino)ethoxymethyl]phosphonate (191) is also accompanied by the partial cleavage of the phosphorus–carbon bond [119].
3.1.3. Phosphorus–Carbon Bond Cleavage under the Action of Bases
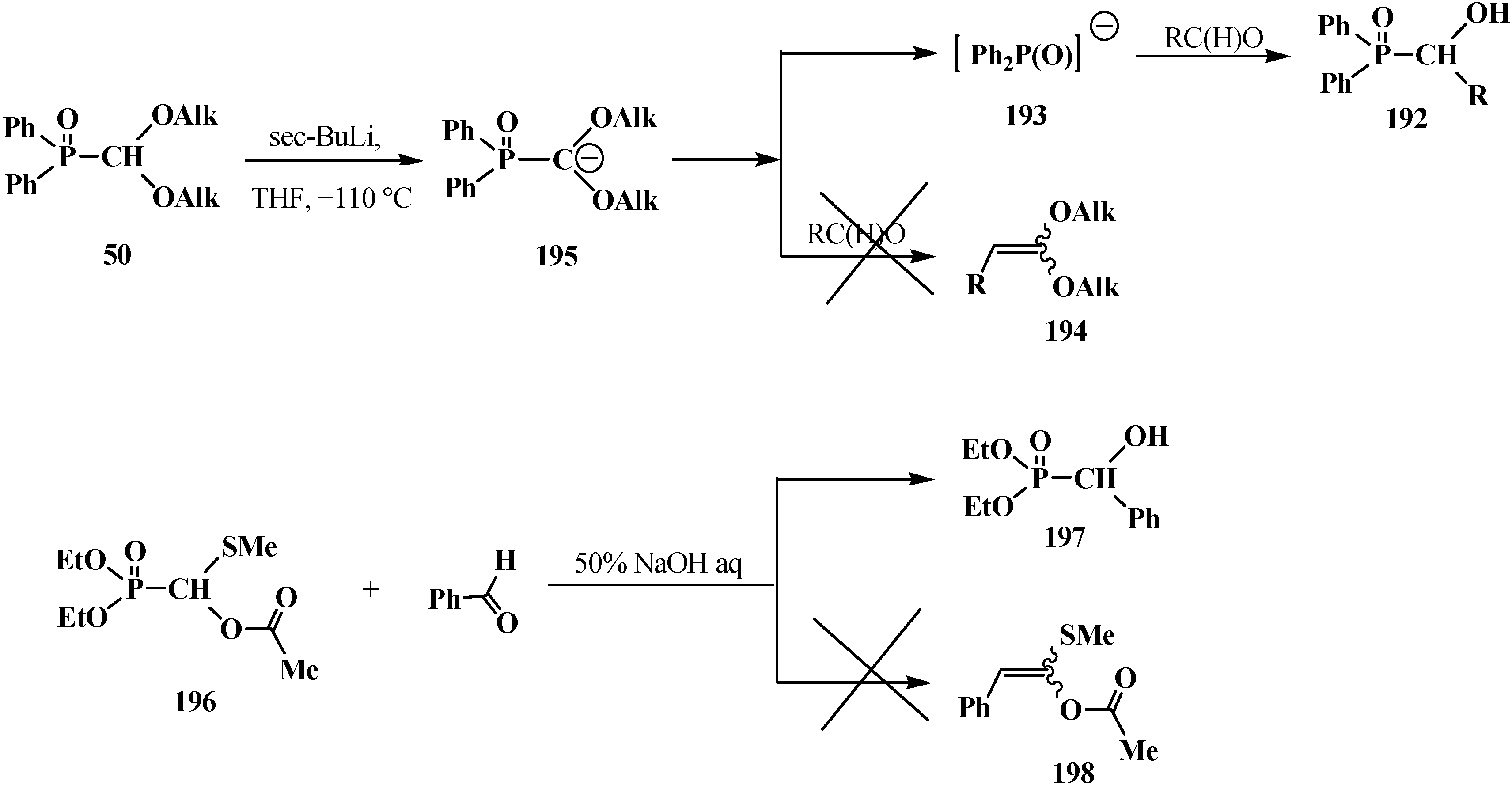
The reaction of the lithium derivatives of diphenyl(dialkoxymethyl)phosphine oxides 50 with n-octanal and p-isopropylbenzaldehyde leads to α-phosphorylated alcohols 192, the products of addition of diphenylphosphinite anion (193) to the carbonyl group (Abramov reaction) [127,128], rather than ketene O,O-acetals 194 as expected products of the Horner reaction [9,10] (Scheme 58). The reason of this course is the instability of diphenyl (dialkoxymethyl)phosphine oxide anion (195) that compound 50 decomposes after long exposition under the reaction conditions to form anion 193, which further undergoes addition to the aldehyde carbonyl group [127,128].
Scheme 58.
Cleavage of phosphorus–carbon bond of acetal 57 and O,S-acetal 196 leading to the formation of α-phosphorylated alcohols 192 and 198.
Scheme 58.
Cleavage of phosphorus–carbon bond of acetal 57 and O,S-acetal 196 leading to the formation of α-phosphorylated alcohols 192 and 198.

Like phosphine oxides 50, the reaction of diethyl [(acetoxy)methylthiomethyl]phosphonate (196) with benzaldehyde PhC(H)O under phase transfer catalysis conditions gives rise to diethyl [(hydroxy)phenylmethyl]phosphonate (197), the Abramov reaction product, instead of the expected ketene O,S-acetal 198 resulting from the Horner reaction [37] (Scheme 58).
Lithiated anions 199 of diethyl [(methylthio)(alkylthio)methyl]phosphonates 123 react quickly with molecular oxygen with the cleavage of phosphorus–carbon bond and formation of dialkyl dithiocarbonates 200 (yields 71%–72%) [129] (Scheme 59).
Scheme 59.
Destruction of thioacetal 123 in the presence of oxygen, leading to formation of dithiocarbonates 200.
Scheme 59.
Destruction of thioacetal 123 in the presence of oxygen, leading to formation of dithiocarbonates 200.

See also Scheme 63, Scheme 96, Scheme 103, Scheme 104, Scheme 113, Scheme 114, Scheme 115, Scheme 116, Scheme 117 and Scheme 120.
3.2. Synthesis of Formacetalphosphonic Acids
Attempted preparations of formacetalphosphonic acids 201, 202 and 203 from the corresponding phosphorylated acetal 57, thioacetal 187 and aminonitrile 153 by acid hydrolysis lead to cleavage of the phosphorus–carbon bond, but in the case of compound 91 with a aromatic substituent in the acetal group, it undergoes acid hydrolysis to give the expected phosphonic acid 92 (Scheme 20). See also Section 3.1.1. “Cleavage of phosphorus–carbon bond under the action of acids and acidic reagents”.
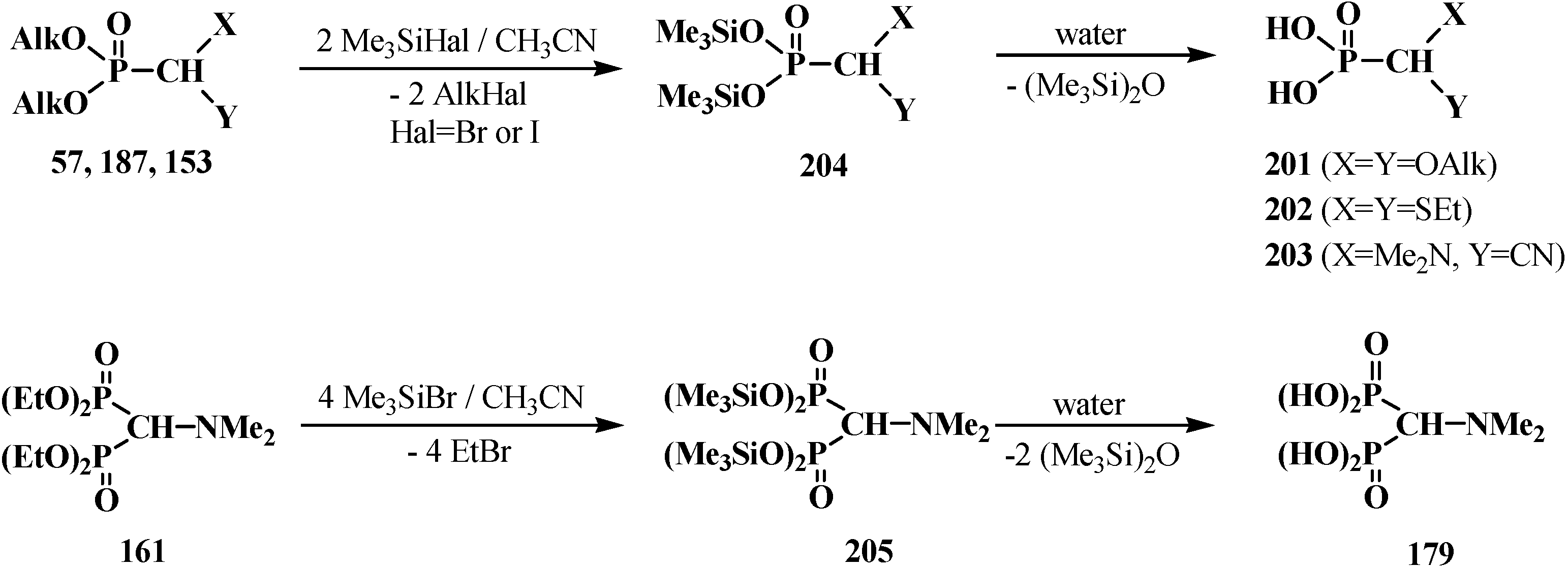
However, the target formacetalphosphonic acids 201–203 may be successfully prepared by the reaction of acetals 33, diethyl (diethylthiomethyl)phosphonate (187), diethyl [(N,N-dimethyl-amino)cyanomethyl]phosphonate (153), and tetraethyl(N,N-dimethylaminomethyl)diphosphonate (161) with trimethylsilyl bromide (Me3SiBr) in acetonitrile. Alkyl groups at the phosphorus atom are eliminated as alkyl halides to afford the corresponding intermediate bis(trimethylsilyl) phosphonates 204 (or tetrakis(trimethylsilyl) phosphonate (205) from diphosphonate 161), which are further readily hydrolyzed by water treatment to give:
Scheme 60.
Syntheses of formacetalphosphonic acids 201–203 and 179 from compounds 57, 187, 153, 161 by means of Me3SiBr or Me3SiI. See text above.
Scheme 60.
Syntheses of formacetalphosphonic acids 201–203 and 179 from compounds 57, 187, 153, 161 by means of Me3SiBr or Me3SiI. See text above.

3.3. Alkylation (Acylation) of the Formacetal Carbon Atom
Dialkyl (dialkoxymethyl)phosphonates 33 produce no stable phosphorylated carbanion 206 when reacted with bases (no metallation occurs, even under the action of tert-butyllithium (t-BuLi), which provides no possibility for further alkylation and acylation of the formacetal group [23,132] (Scheme 61).
Scheme 61.
Phosphorylated acetals 33 do not produce carbanions 206.

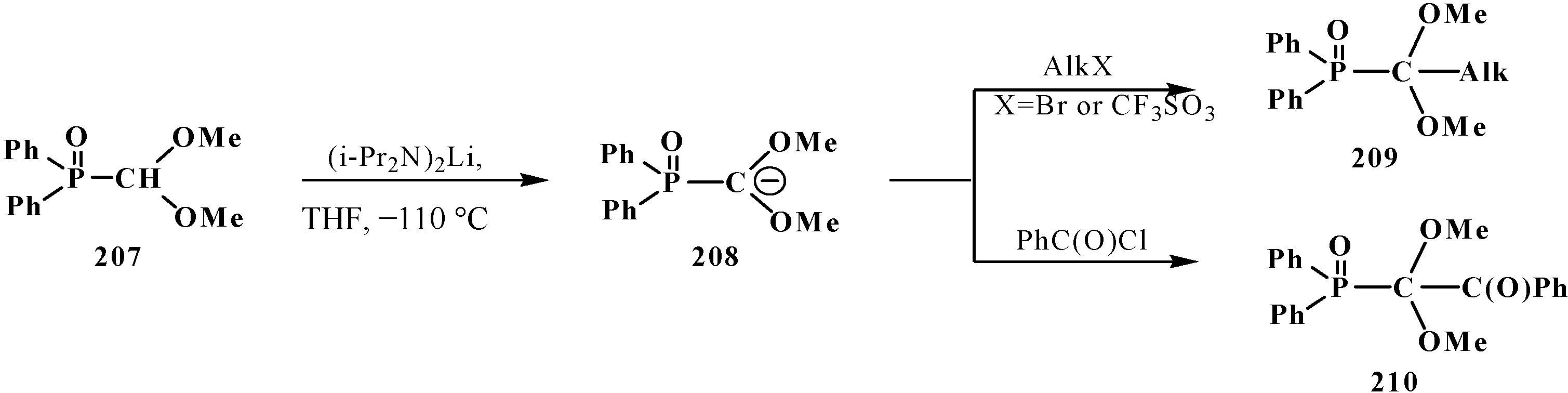
This fact was explained by insufficient stabilization of the negative charge of carbanion on the two oxygen atoms in the α-position [23]. However, it was shown in 1983 [133] that, in contrast to phosphonates 33, diphenyl(dialkoxymethyl)phosphine oxides 50 produce phosphorylated anions 195 at −110 °C that undergo metallation. The reason for the stability of the lithium derivatives of phosphine oxides 50 is the ability of diphenylphosphinoyl group to delocalize the negative charge of carbanion 195 [20,134] (Scheme 58). By the example of anion 207 of diphenyl(dimethoxy-methyl)phosphine oxide (208), it was shown that it is rather stable to subsequent alkylation with alkyl halides and acylation with benzoyl chloride [20]. The reactions afford diphenyl[(dimethoxy)alkylmethyl]phosphine oxides 209, in 30%–94% yields, and diphenyl-[(dimethoxy)benzoylmethyl]phosphine oxide (210) (60%) (Scheme 62).
Scheme 62.
Alkylation and acylation of carbanion 207.

In acidic medium at 20 °C, phosphine oxides 209 are readily decomposed with cleavage of the phosphorus–carbon bond. The resultant methyl carboxylates 211 are homologous to the initial alkyl halides—carbon chain elongation by one atom (Scheme 63).
Scheme 63.
Hydrolysis of phosphine oxide 209 leads to phosphorus–carbon bond cleavage.

Similarly, according to 1H-NMR spectroscopy, the methanolysis of diphenyl[(1,1-dialkoxy)nonan-1-yl)phosphine oxide (212) in the presence of trifluoroacetic acid leads to 1,1,1-trimethoxynonane (213) in 65% yield [20].
Nonetheless, the storage of a solution of lithiated anion 207 for two hours even at −110 °C causes the cleavage of phosphorus–carbon bond (see Scheme 58).
Distinct from acetals 33 [23,132], the two sulfur atoms of thioacetals 113, 114 stabilize well the neighboring carbanion [23], therefore the α-hydrogen atom in the thioacetal group is readily removed under the action of strong bases [23,132] in both dialkyl (dialkythiomethyl)phosphonates 113, 114 [135] and diphenyl(dialkythiomethyl)phosphine oxides 121, 122 [113]. Further, the carbanions are readily alkylated with alkyl halides [113,135]. For example, dialkyl ([1,3]-dithian-2-yl)phosphonates 114 in this reaction produce dialkyl [(2-alkyl-[1,3]-dithian)-2-yl]phosphonates 214, and their oxidative decomposition may result in α-phosphorylated carbonyl compounds 215 (Scheme 64).
Scheme 64.
Thioacetals 114 alkylation with the subsequent transformation of thioketals 214 into α-phosphorylated carbonyl compounds 215.
Scheme 64.
Thioacetals 114 alkylation with the subsequent transformation of thioketals 214 into α-phosphorylated carbonyl compounds 215.

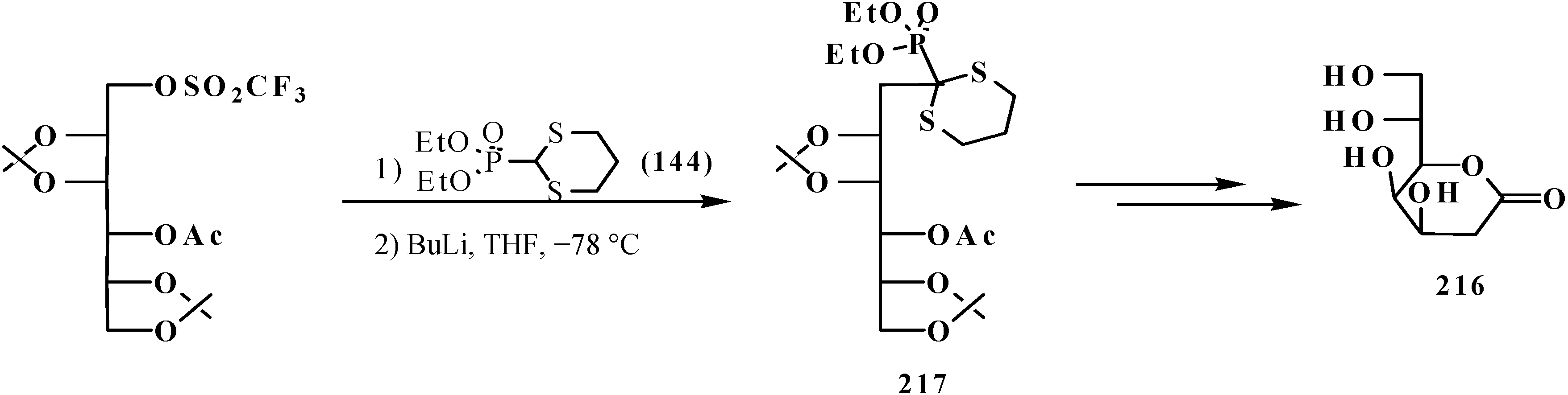
The possibility to alkylate diethyl ([1,3]-dithian-2-yl)phosphonate (144) was used for the elongation of the hydrocarbon chain in the synthesis of 3-deoxy-D-manno-octulosonic acid (216), through compound 217 as alkylated form 144 [136] (Scheme 65).
Scheme 65.
Synthesis of 3-deoxy-d-manno-octulosonic acid (216) by the alkylation of thioacetal 144.

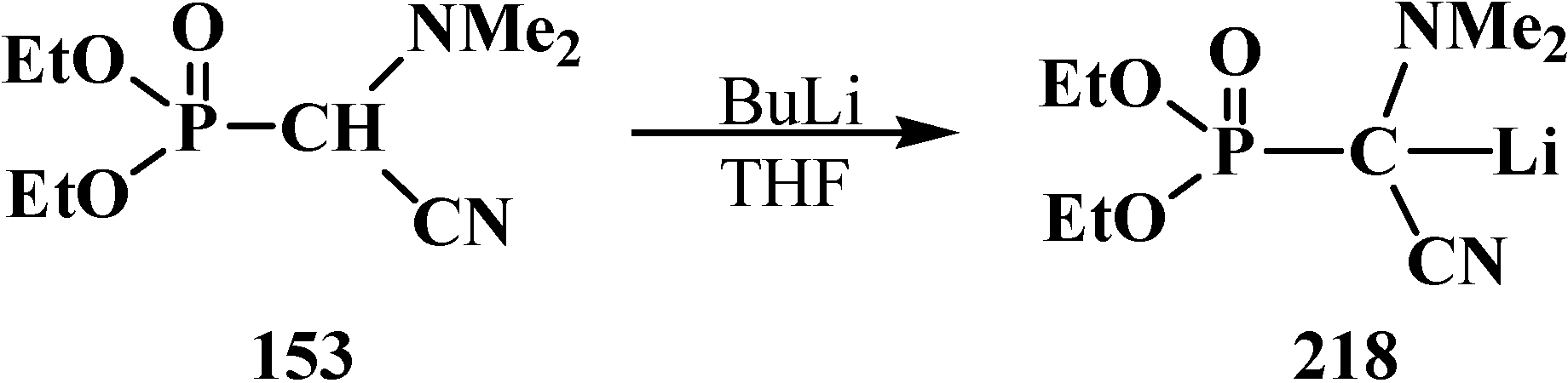
In contrast to dialkyl (dialkoxymethyl)phosphonates 33 and similarly to phosphorylated formaldehyde thioacetals 14, diethyl [(N,N-dimethylamino)cyanomethyl]phosphonate (153) is readily deprotonated under the action of sodium hydride in dioxane or dimethyl sulfoxide or 50% KOH solution under phase transfer catalysis conditions [24] as well as with butyllithium BuLi in THF [25]. Lithium derivative 218 of diethyl [(N,N-dimethylamino)cyanomethyl]phosphonate (153) proved to be so stable that it could be stored without decomposition for several months [25] (Scheme 66).
Scheme 66.
Synthesis of stable lithium derivative 218 of diethyl [(N,N-dimethylamino)- cyanomethyl]phosphonate (153).
Scheme 66.
Synthesis of stable lithium derivative 218 of diethyl [(N,N-dimethylamino)- cyanomethyl]phosphonate (153).

The anion of 218 undergoes alkylation at the carbon atom when treated with methyl iodide MeI [24,25,117], dimethyl sulfate Me2SO4 [117] or benzyl chloride PhCH2Cl [24] to give α-alkylated derivatives 219 and 220. Benzyl derivative 220 prepared by this method eliminates hydrogen cyanide on heating to give α-phosphorylated enamine 221 [24] (Scheme 67).
Scheme 67.
Anion 218 alkylation.

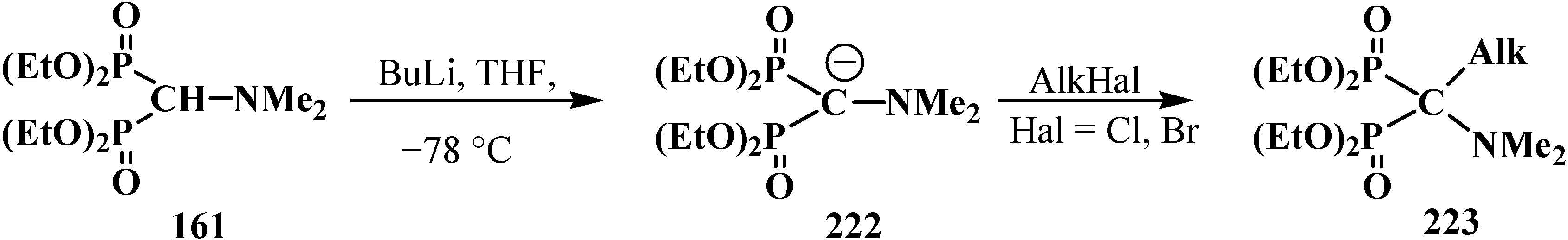
Similarly to diphenyl(dialkoxymethyl)phosphine oxides (50), cyclic dialkyl (dialkylthiomethyl)- phosphonates 113, 114, cyclic diphenyl(dialkylthiomethyl)phosphine oxides 121, 122, and diethyl [(N,N-dimethylamino)cyanomethyl]phosphonate (153), and tetraethyl (N,N-dimethylaminomethyl)- diphosphonate (161) under the action of strong bases readily eliminate a proton from the acetal carbon atom to give anion 222. This provides an opportunity for its further alkylation (compounds 223) [28], which has been used in the synthesis of pesticides (Scheme 68).
Scheme 68.
Alkylation of methyldiphosphonate 161, (Hal = Cl, Br).

3.4. Horner Reaction
In 1958 and 1959 L. Horner and co-authors reported their discovery of a new reaction [137,138] that they named as “P=O-activated olefination”. The authors showed that the reaction of alkyl(diphenyl)phosphine oxides 224 and dialkyl alkylphosphonates 225 with aldehydes and ketones in the presence of strong bases produce olefins 226 (Scheme 69). Key reaction intermediates—lithium derivatives of carbanion of the initial phosphinoyl compounds 227 and β-phosphorylated hydroxy derivatives 228 were identified on the example reaction of benzyl(diphenyl)phosphine oxide (229) with benzaldehyde in the presence of phenyllithium [138].
Scheme 69.
Syntheses of olefins 226 from aldehydes and ketones by means of Horner’s reaction, R = OAlk, Ph; R' = Alk; R'', R''' = H, Alk, Ar.
Scheme 69.
Syntheses of olefins 226 from aldehydes and ketones by means of Horner’s reaction, R = OAlk, Ph; R' = Alk; R'', R''' = H, Alk, Ar.

The reaction has a number of advantages in comparison with similar reaction of phosphorus ylides previously described by L. Wittig [139] where ketones are difficult to react, whereas both aldehydes and ketones undergo the Horner reaction. It was further shown that Horner reaction has a larger synthetic potential and is applicable for the synthesis of other types of organic compounds, for example, allenes, cyclopropanes, terminal [140] and disubstituted alkynes [132]. The involvement of phosphonates functionalized at the α-position with dialkylamino, alkoxy or alkylthio groups in the reaction leads to enamines, vinyl ethers [132,141,142,143] and vinyl thioethers [141,143]. Their subsequent hydrolysis affords aldehydes and ketones with elongated hydrocarbon chain in high yields (homologation).
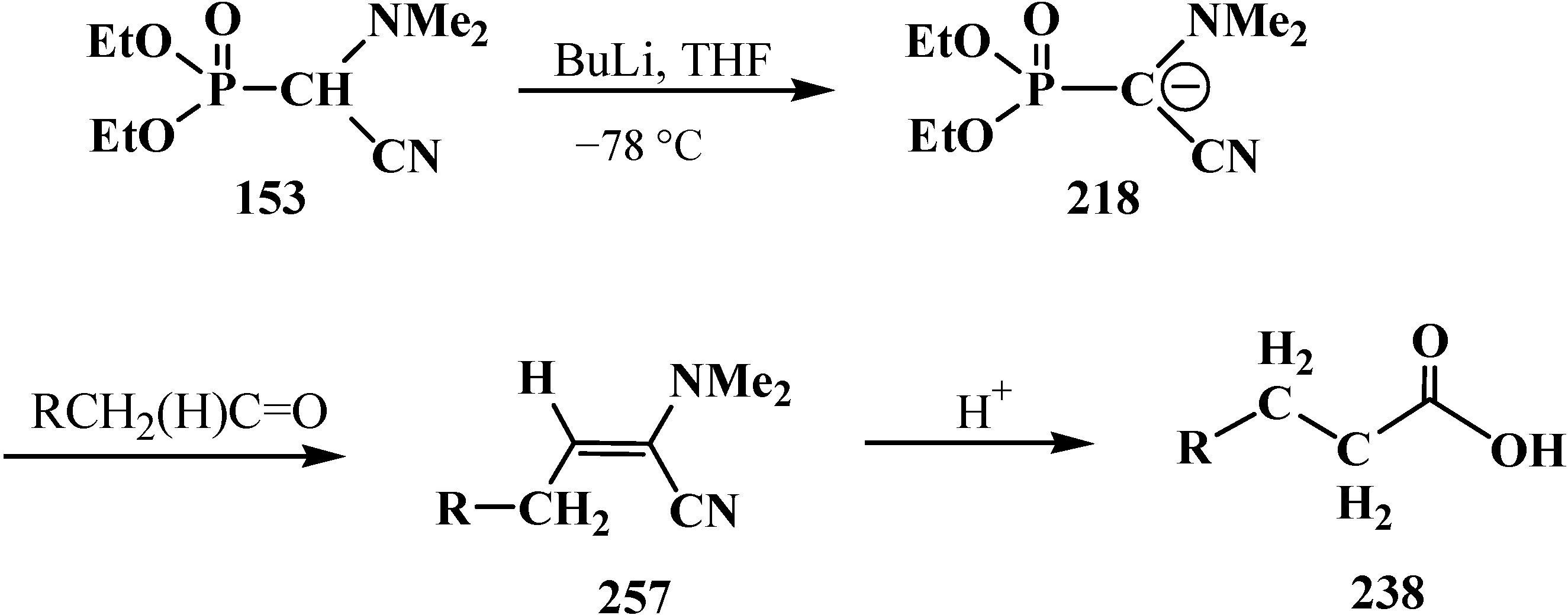
The Horner reaction also provides the possibility to prepare carboxylic acids homologized by one carbon atom via the shortest route starting from phosphinoyl compounds functionalized at the α-position with two heteroatoms, namely, phosphorylated formaldehyde acetals and structurally related compounds [132,141]. In this case, carbanions 230 of phosphorylated formaldehyde acetals and structurally related compounds 13–16, 18–32 behave as a masked form of triply functionalized carbanions 231 that may be considered as a synthetic equivalent or carrier of reversed-polarity formate carbanion [O=C–OH]− 232 [20,141] (Scheme 70).
Scheme 70.
Compounds 13–16, 18–32 as hidden form of reversed-polarity formate carbanion 232, where R = OAlk, Ph; X, Y = AlkO, AlkS, Alk2N, CN, R2P(O), Hal, AlkS(O).
Scheme 70.
Compounds 13–16, 18–32 as hidden form of reversed-polarity formate carbanion 232, where R = OAlk, Ph; X, Y = AlkO, AlkS, Alk2N, CN, R2P(O), Hal, AlkS(O).

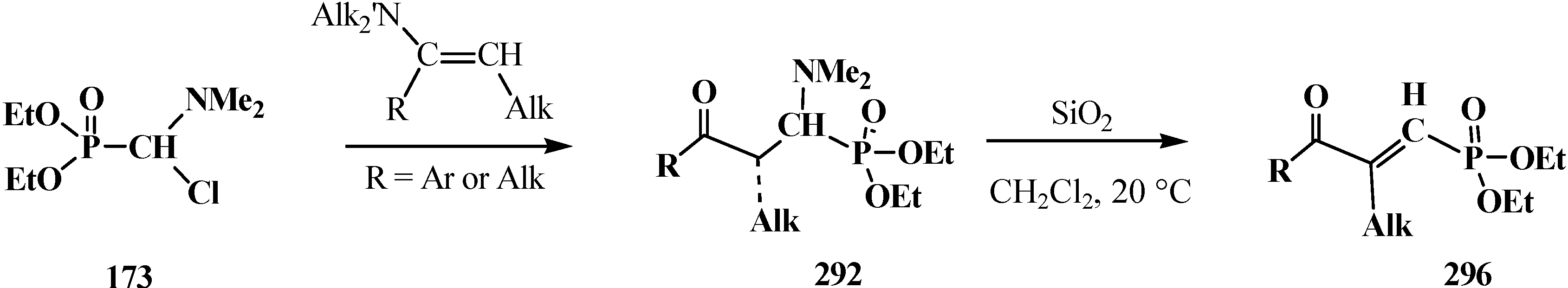
Carbanions 230, prepared by the deprotonation of the initial phosphoryl compound, react with carbonyl compounds to afford β-phosphorylated alcohols 233, which can be isolated. The subsequent treatment of alcohols 233 with strong bases, usually potassium tert-butoxide, leads to ketene acetals and structurally related compounds 234 that are valuable precursors in the synthesis of organic compounds of different kinds [23,24,132,133,143]. Further acid hydrolysis of compounds 234 produces carboxylic acids 235 (Scheme 71) or their derivatives, for example esters 236, or thioesters 237, depending on the conditions.
Scheme 71.
Syntheses of carboxylic acids 235 by means of Horner's reaction, R',R'' = H, Alk, Ar.

However, the simplest and most available phosphorylated formaldehyde acetals, dialkyl (dialkoxymethyl)phosphonates 33, do not form stable carbanions [23,132], therefore the attempted synthesis of carboxylic acids and their derivatives by Horner reaction failed for a long time. Among acetals of phosphonate type compounds, only diethyl (5,6-dichloro-1,3-benzodioxomethyl)phosphonate (91) participated in the reaction with ketones at 90 °C in dioxane in the presence of sodium hydride NaH to give ketene acetals in 19%–32% yields [23]. See also Section 3.3 “Alkylation of formacetal carbon atom”.
Carboxylic acids were obtained for the first time by Horner reaction in 75%–90% yields in 1968 by reacting tetraethyl (N,N-dimethylaminomethyl)diphosphonate (161) with aliphatic and aromatic aldehydes [27]. After formation of the carbanion 222, the reaction proceeds through the sequential formation of 1-dimethylaminoalkenylphosphonates—α-phosphorylated enamines 236, then α-phosphinoylacyl derivatives 237, and finally yields free linear acids 238 (Scheme 72).
Scheme 72.
Synthesis of carboxylic acids 238 by reacting tetraethyl (N,N-dimethylaminomethyl)diphosphonate (161) with aliphatic and aromatic aldehydes.
Scheme 72.
Synthesis of carboxylic acids 238 by reacting tetraethyl (N,N-dimethylaminomethyl)diphosphonate (161) with aliphatic and aromatic aldehydes.

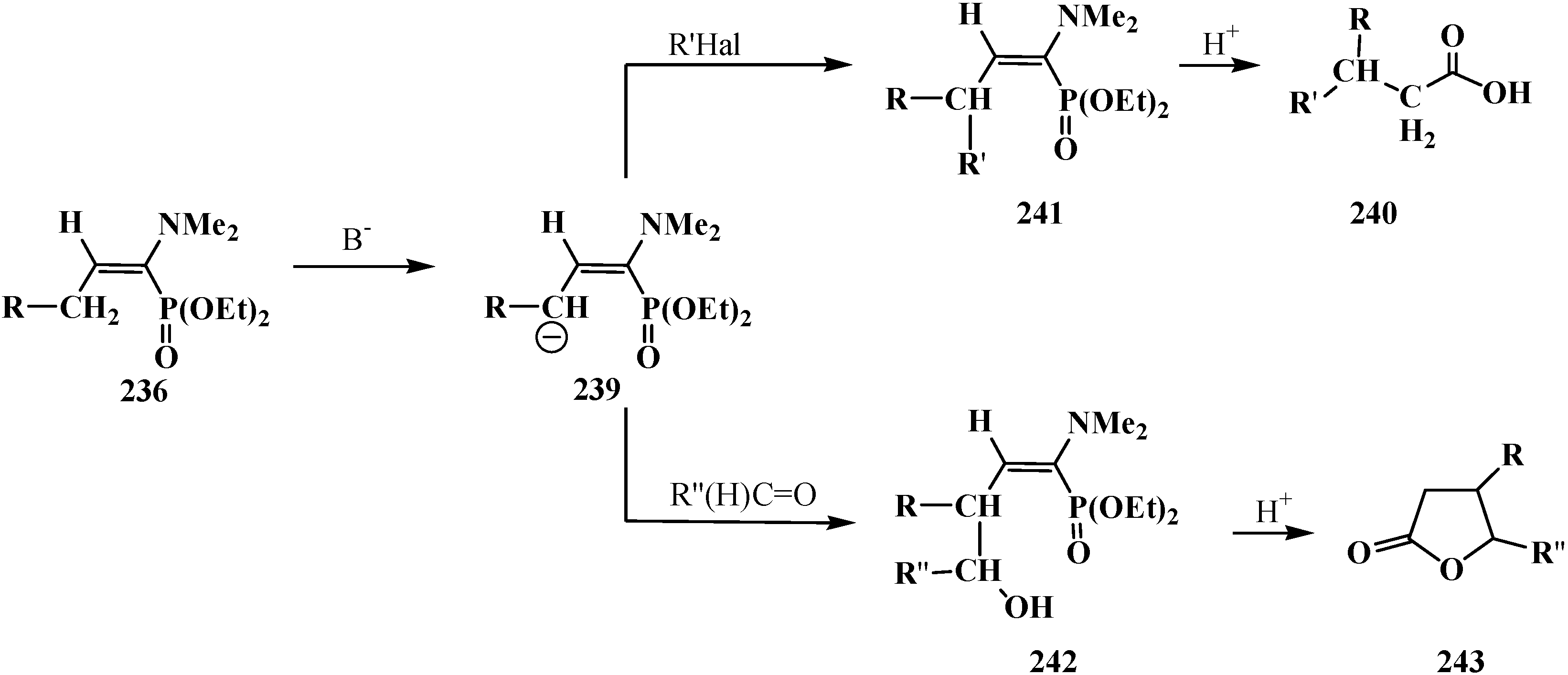
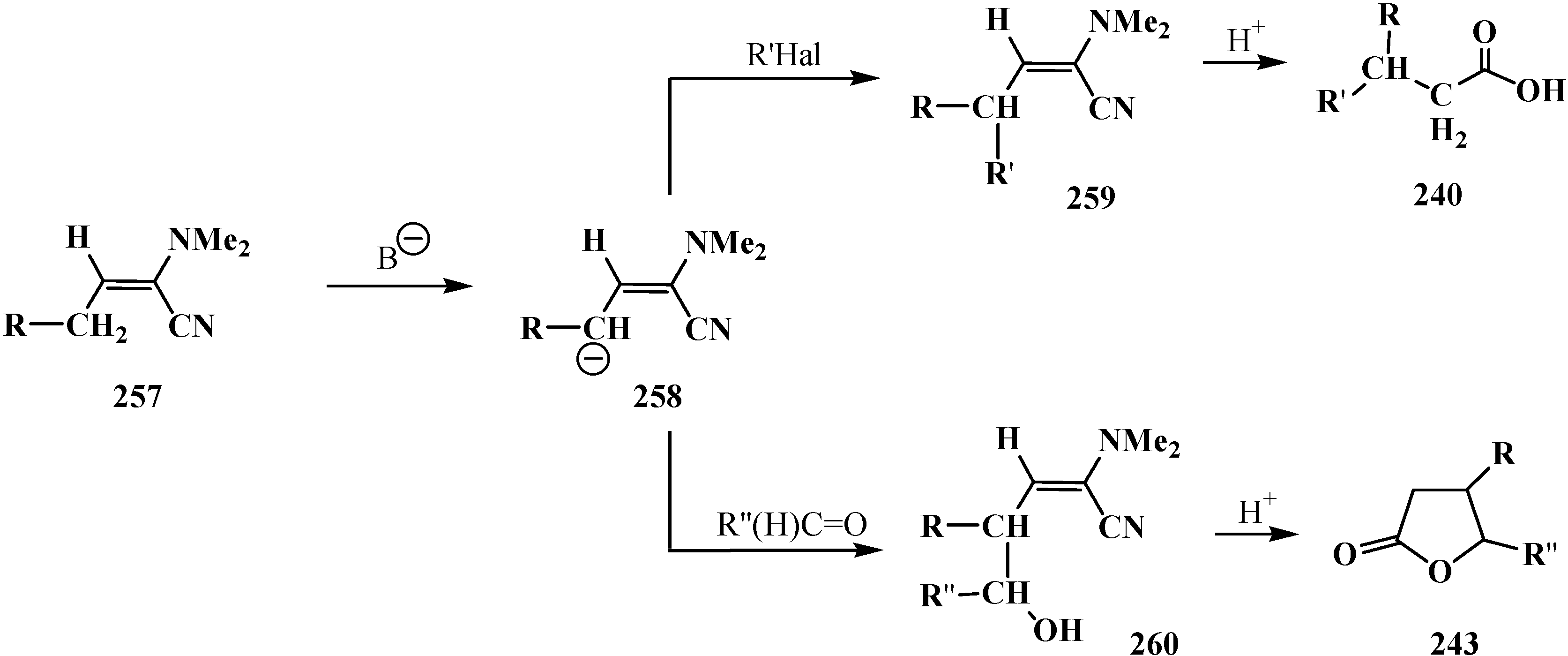
Since phosphorylated enamines 236 (synthesized from aliphatic aldehydes only) contain an anion-stabilizing diethoxyphosphinoyl group in the α-position, the methylene group in the γ-position is readily deprotonated under the action of strong bases. The resulting phosphorylated aminoallyl anions 239 react with alkyl halides and finally form carboxylic acids 240 branched at the β-position via the phosphorylated enamines 241. The reaction of anions 239 with aldehydes gives rise to hydroxy compounds (242) and then to β,γ-disubstituted γ-butyrolactones 243 over unisolated γ-hydroxy carboxylic acids that undergo fast cyclization under the reaction conditions [144] (Scheme 73).
Scheme 73.
Syntheses of carboxylic acids 240 and γ-butyrolactones 243 from phosphorylated enamines 236.
Scheme 73.
Syntheses of carboxylic acids 240 and γ-butyrolactones 243 from phosphorylated enamines 236.

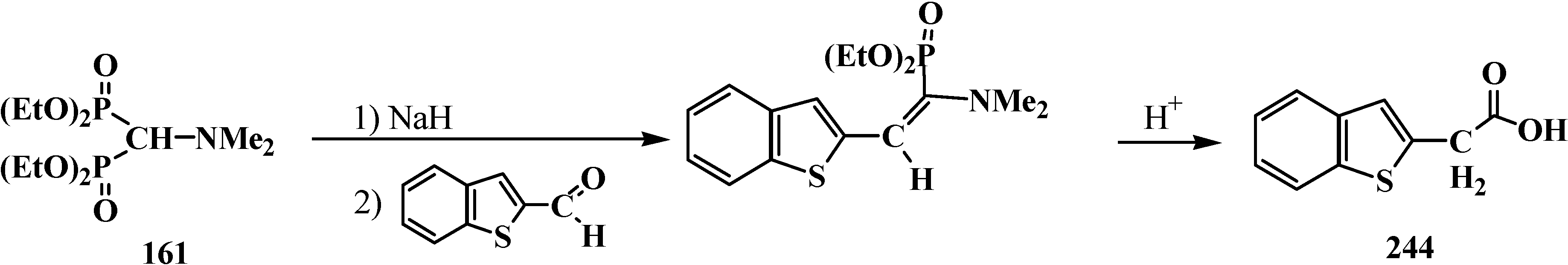
Because only aldehydes react with compound 161 [27,132], this method of synthesis of carboxylic acids is not widely used. However, compound 161 is employed for the preparation of substituted acetic acids as intermediate stages in the synthesis of potential pharmaceuticals [124,145] and pesticides [28], for example, acid 244 (Scheme 74).
Scheme 74.
Synthesis of 2-benzothienylacetic acid 244 by Horner’s reaction.

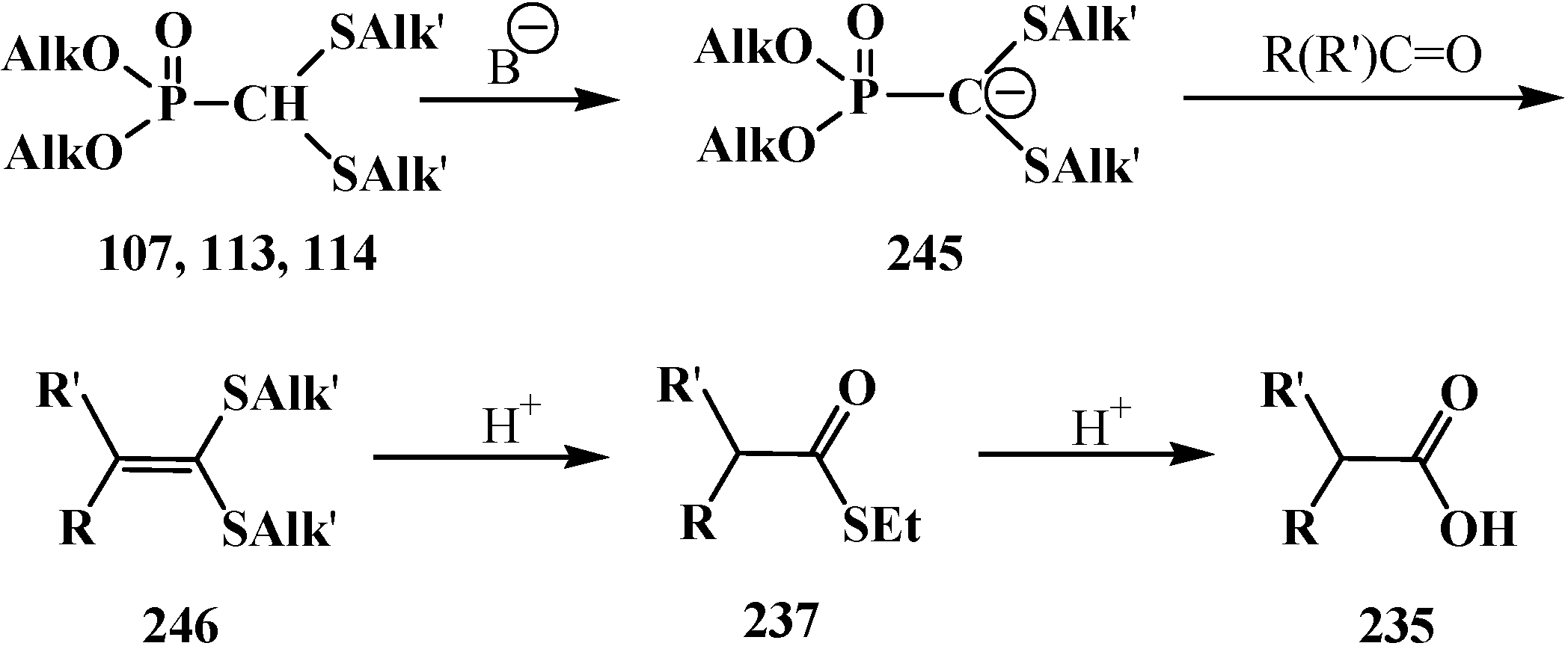
The successful homologation of aldehydes with the use of diphosphonate 161 stimulated further search for the synthetic equivalents of formate carbanion 232 among organophosphorus compounds. In 1976–1977, linear (107) and cyclic dialkyl (dialkythiomethyl)phosphonates 113, 114 were proposed [21,146]. These compounds can form a stable carbanion 245, and react with both aldehydes and ketones to form ketene thioacetals 246 and further under subsequent hydrolysis (over thioesters 237) produce homologous carboxylic acids 235 (Scheme 75), see also section “Alkylation of formacetal carbon atom”. Ketene thioacetals 246 were obtained from ketones and aldehydes [102,146], including those unsaturated, in 66%–82% and 80%–96% yields, respectively, as mixtures of Z/E isomers [23].
Scheme 75.
Syntheses of carboxylic acids 235 by reacting thiomethylphosphonates 107, 113, 114 with aldehydes and ketones.
Scheme 75.
Syntheses of carboxylic acids 235 by reacting thiomethylphosphonates 107, 113, 114 with aldehydes and ketones.

The reaction is observed also for diphenyl([1,3]-dithian-2-yl)phosphine oxide (122) [123,124] and diethyl (1,3-benzodithiolylmethyl)phosphonate (247) [113,147]. Phosphonate 247 produces benzo-analogs of ketene thioacetals, 1,4-benzodithiafulvenes 248, in 92%–98% yields when reacted with carbonyl compounds (Scheme 76).
Scheme 76.
Syntheses 1,4-benzodithiafulvenes 248 from diphenyl([1,3]-dithian-2-yl)phosphine oxide (122) by the reaction with carbonyl compounds.
Scheme 76.
Syntheses 1,4-benzodithiafulvenes 248 from diphenyl([1,3]-dithian-2-yl)phosphine oxide (122) by the reaction with carbonyl compounds.

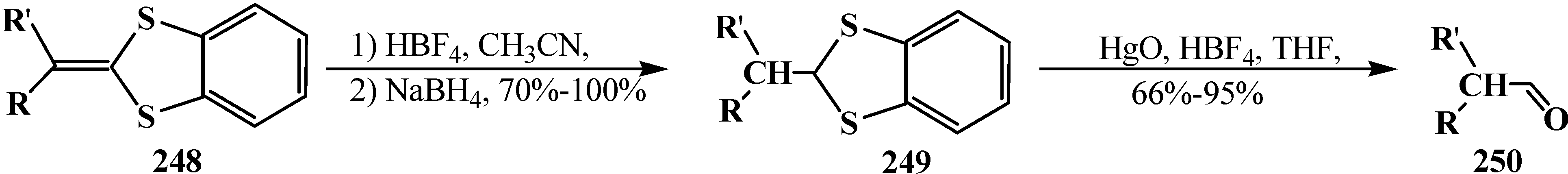
Phosphorylated thioacetals 107, 113, 114 and thioacetals produced from its ketene 246, 248 have a synthetic importance because, along with carboxylic acid synthesis, they undergo numerous reactions to afford various products [23]. For example, aldehydes 250, branched out in α-position are formed in the reduction of 1,4-benzodithiafulvenes 248 followed by hydrolysis (over the stage of reduced compounds 249) [148] (Scheme 77).
Scheme 77.
Obtaining branched aldehydes 250 from 1,4-benzodithiafulvenes 248.

Scheme 78.
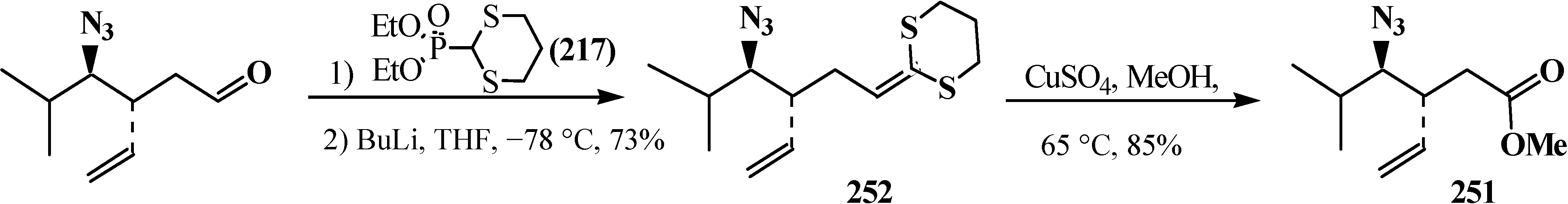
Synthesis of methyl esters 251 by methanolysis of thioacetal 252.

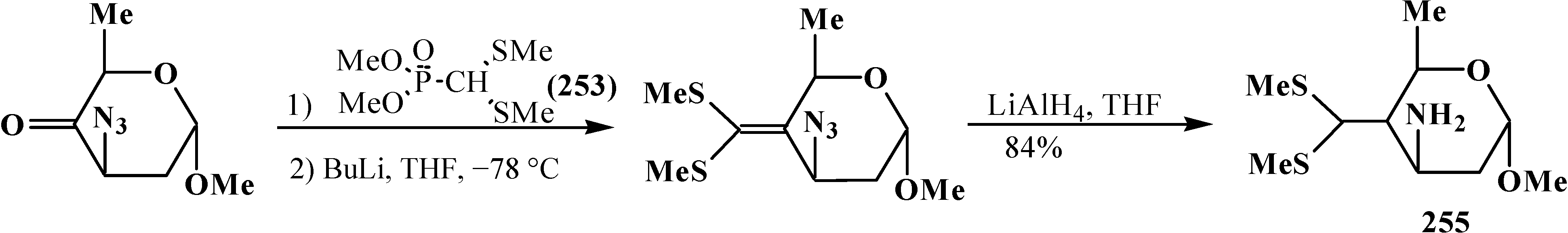
Dimethyl (dimethylthiomethyl)phosphonate 253 is used in the practice of contemporary organic chemistry, for example, in the intermediate stages of synthesis of biologically active dipeptide mimetics 254 [149] (Scheme 79), the antibiotic thienamycin (255) [150] (Scheme 80), and “organic metals” 256 [116,151,152,153,154], for example (Scheme 81).
Scheme 79.
Synthesis of compound 254—an intermediate stage of synthesis of dipeptide mimetics.

Scheme 80.
Synthesis of the antibiotic thienamycin (255) by means of thiomethylphosphonate 253.

Scheme 81.
Example of synthesis of “organic metals” 256.

However, the long duration of the two-stage conversion of ketene thioacetals 246 into acids 235, often in the presence of mercury Hg2+ [141] or copper Cu2+ salts [149] and the necessity of working with mercaptans [23,141,154] limits the application of phosphorylated formaldehyde thioacetals 14 in the Horner reaction.
Therefore, the search for efficient precursors for the synthesis of carboxylic acids 235 from carbonyl compounds by the Horner reaction has continued. From the mid-1970s to the early 1980s, many acetal-like derivatives of diethyl formylphosphonates 13–32 [18,19,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56], where the negative charge of the carbanion was stabilized by two heteroatoms of the “acetal” group, were obtained [54]. See also Figure 2, where R = OEt. In the case of X = Me3Si, Peterson olefination prevails over the Horner reaction [54,55,56,110] and trimethylsyloxy fragment is a leaving group.
Because the majority of compounds 18–32 have no substantial advantages over the phosphorylated formaldehyde thioacetals 14, the study of Horner reaction with their participation was confined mainly to academic interest. More detailed studies of reactivity of the majority of these compounds were not conducted.
Among the compounds synthesized over this period, diethyl [(N,N-dialkylamino)cyano- methyl]phosphonate (153) and diphenyl(dialkoxymethyl)phosphine oxides 50 were involved in the practice of organic synthesis.
Compound 153 proposed in 1982 [24] reacts like dialkyl (dialkylthiomethyl)phosphonates 107 with aldehydes, in 50%–69% yields, and acetophenone as ketone example, in 24% yield [24,132]. The products of Horner reaction in this case are cyanoenamines 257, whose acid hydrolysis produces linear carboxylic acids 238 homologous to the initial carbonyl compounds [24,25,132] (Scheme 82). See also Scheme 72.
Scheme 82.
Carboxylic acids 238 synthesis by means of diethyl [(N,N-dialkylamino)cyanomethyl]phosphonates 153, where R = Alk, Ph.
Scheme 82.
Carboxylic acids 238 synthesis by means of diethyl [(N,N-dialkylamino)cyanomethyl]phosphonates 153, where R = Alk, Ph.

Like phosphorylated enamines 236 (Scheme 73), compound 257 contains an anion-stabilizing CN-group in the α-position. Resulting cyanoaminoallyl anions 258 combine with alkyl halides to form carboxylic acids 240 through cyanoenamines 259, while the reaction with aldehydes leads to γ-hydroxy acids that undergo cyclization to give β,γ-disubstituted γ-butyrolactones 243 through cyano aminoallylalcohols 260 [24,25,132,144] (Scheme 83).
Scheme 83.
Enamines 257 alkylation by means of alkyl halides or aldehydes leads to obtaining branched acids 240 or butyrolactones 243.
Scheme 83.
Enamines 257 alkylation by means of alkyl halides or aldehydes leads to obtaining branched acids 240 or butyrolactones 243.

Diethyl [(N-morpholino)cyanomethyl]phosphonate (154) was used in the synthesis of the colerofragarone fragment—terminator of fungi Collerotrichum fragariac [155] (Scheme 84, compound 261).
Scheme 84.
Synthesis of colerofragarone fragment 262 using diethyl [(N-morpholino)cyanomethyl]phosphonate (154).
Scheme 84.
Synthesis of colerofragarone fragment 262 using diethyl [(N-morpholino)cyanomethyl]phosphonate (154).

It was shown that compound 153 can react also with cyclic semiacetal 262, which was used in one of the stages of synthesis of prostaglandin analog cloprosterol PGF2 [155,156] (Scheme 85, compound 263).
Scheme 85.
Synthesis of 263, a semi-product of synthesis of the analog of cloprosterol PGF2 from cyclic semiacetal 262 by means of phosphonate 153.
Scheme 85.
Synthesis of 263, a semi-product of synthesis of the analog of cloprosterol PGF2 from cyclic semiacetal 262 by means of phosphonate 153.

However, compound 153 was not widely used because of the low yields in its reactions with ketones [25] and formation of hydrogen cyanide on hydrolysis of cyanoenamines. Almost simultaneously with 153, a successful synthesis of ketene acetal at −110 °C starting from diphenyl(dialkoxymethyl)phosphine oxides 50, over carbanion 195 and both aldehydes and ketones was reported in 1983 [133]. Intermediate β-hydroxydiphenylphosphinoyl derivatives 264 were isolated in almost quantitative yield that further react with potassium tert-butoxide (t-BuOK) to give ketene acetals 265 in 45%–85% yields [133] (Scheme 86).
Scheme 86.
Synthesis of ketene acetals 265 from diphenyl(dialkoxymethyl)phosphine oxides 50 through conpounds 264 as intermediates.
Scheme 86.
Synthesis of ketene acetals 265 from diphenyl(dialkoxymethyl)phosphine oxides 50 through conpounds 264 as intermediates.

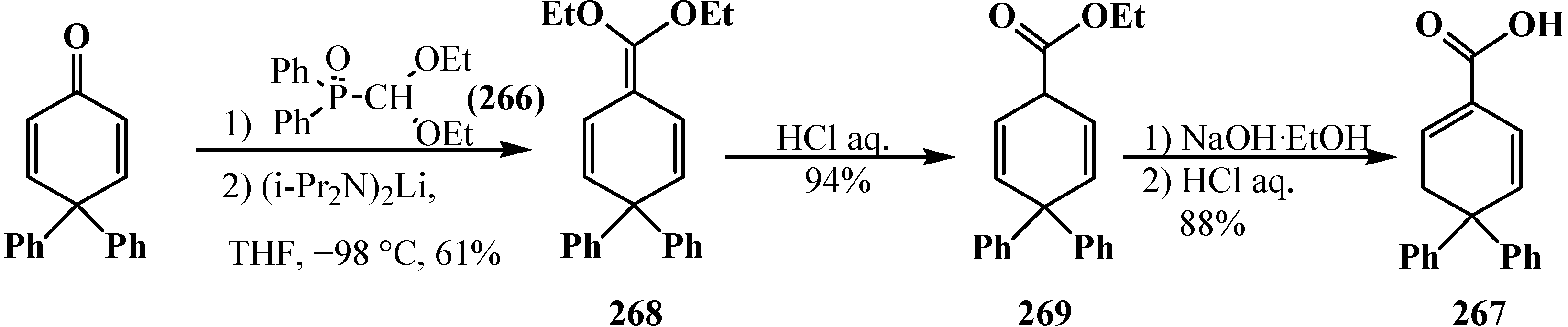
This publication attracted no attention for a long time, although in 1993 it was shown that the reaction of diphenyl(diethoxymethyl)phosphine oxide (266) with substituted cyclohexadienone in the presence of lithium diisopropylamide leads to the formation of carboxylic acid 267 through the stages of ketene acetal 268 and ethyl ester 269 formation [20,157] (Scheme 87).
Scheme 87.
Synthesis of carboxylic acid 267 from (diethoxymethyl)phosphine oxide (266) through ketene acetal 268 and ethyl ester 269.
Scheme 87.
Synthesis of carboxylic acid 267 from (diethoxymethyl)phosphine oxide (266) through ketene acetal 268 and ethyl ester 269.

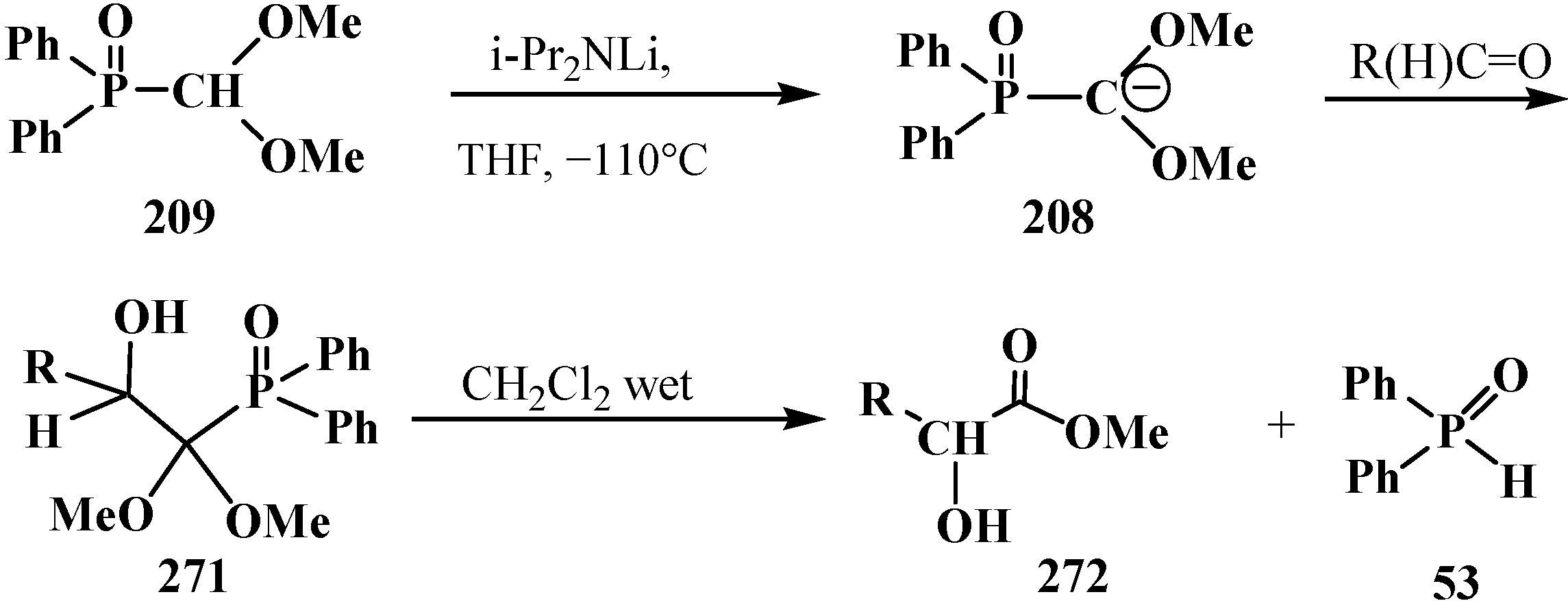
It was found in 2002, however, that the reaction of diphenyl(dimethoxymethyl)phosphine oxide (209) with aliphatic aldehydes, in contrast to other phosphorylated equivalents of formate anion—compounds 107, 112–115, 153 (154), 161 (166), may also result in derivatives of α-hydroxycarboxylic acids 270 [140,158,159]. Initially formed β-hydroxydiphenylphosphinoyl derivatives 271 in wet acidified dichloromethane readily decompose with the cleavage of the P–C bond to afford methyl esters of α-hydroxycarboxylic acids 272 in 41%–89% yield and diphenylphosphine oxide (53) (Scheme 88).
Scheme 88.
Synthesis and decomposition of β-hydroxydiphenylphosphinoyl compounds 271 lead to methyl esters of α-hydroxycarboxylic acids 272.
Scheme 88.
Synthesis and decomposition of β-hydroxydiphenylphosphinoyl compounds 271 lead to methyl esters of α-hydroxycarboxylic acids 272.

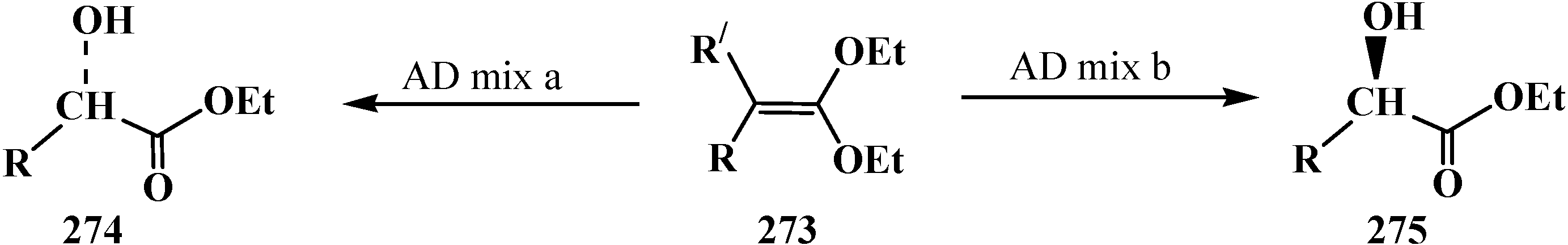
It was further shown that ketene acetals 273 resulting from aldehydes and phosphine oxide 266 may be successfully oxidized [20,128,160] in the Sharpless asymmetric dihydroxylation—AD reaction [161]. As a result, the esters of chiral α-hydroxycarboxylic acids 274, 275 were obtained in 49%–94% yields and enantiomeric excess up to 98% (Scheme 89).
Scheme 89.
Using of Sharpless asymmetric dihydroxylation (AD reaction) for synthesis of chiral α-hydroxycarboxylic acids 274, 275 from ketene acetals 273.
Scheme 89.
Using of Sharpless asymmetric dihydroxylation (AD reaction) for synthesis of chiral α-hydroxycarboxylic acids 274, 275 from ketene acetals 273.

It should be noted that ketene thioacetals 246 do not undergo the AD reaction. Mixed O,S-acetal 18 reacts but yields and enantiomeric excess of α-hydroxycarboxylic acids do not exceed 7%–37% and 80%, correspondingly [127,128]. The combination of Horner and AD reactions was successfully used on one of stages of synthesis of the diterpenoid tonantzitlolone ( 276) [162] (Scheme 90).
Scheme 90.
Using of combination of Horner and AD reactions to obtain compound 276—intermediate stage of synthesis of the diterpenoid tonantzitlolone.
Scheme 90.
Using of combination of Horner and AD reactions to obtain compound 276—intermediate stage of synthesis of the diterpenoid tonantzitlolone.

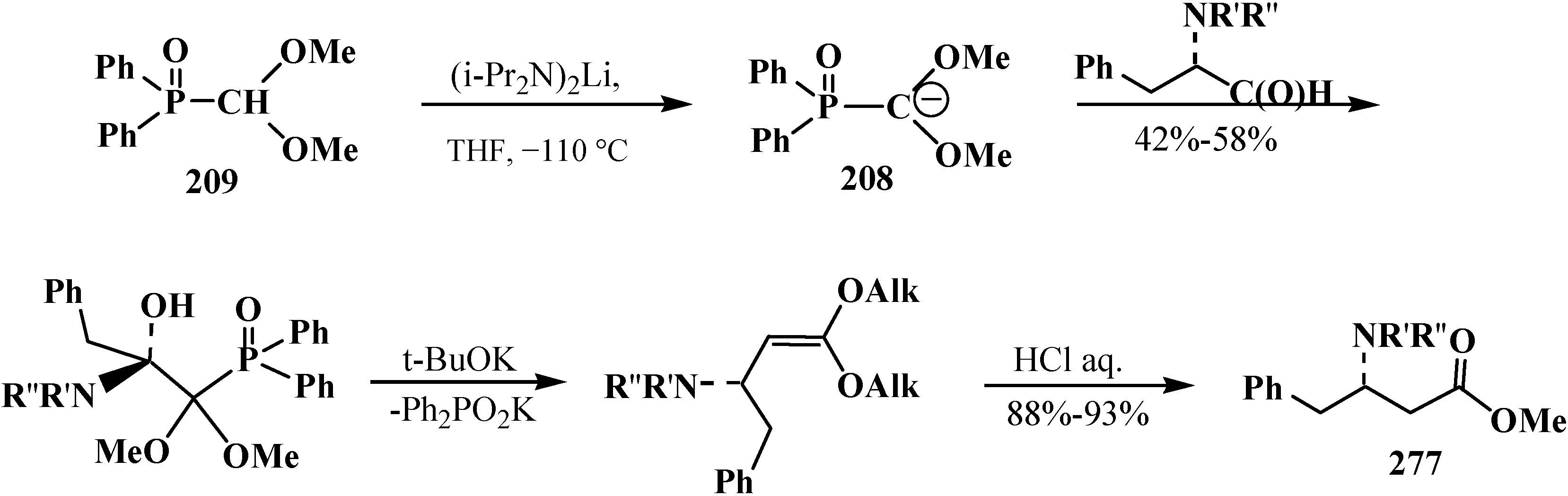
The involvement of chiral α-alkylaminoaldehydes in the Horner reaction, correspondingly, leads to the synthesis of esters of chiral β-amino acids 277 [20] (Scheme 91).
Scheme 91.
Synthesis of chiral β-amino acids 277 by the Horner reaction.

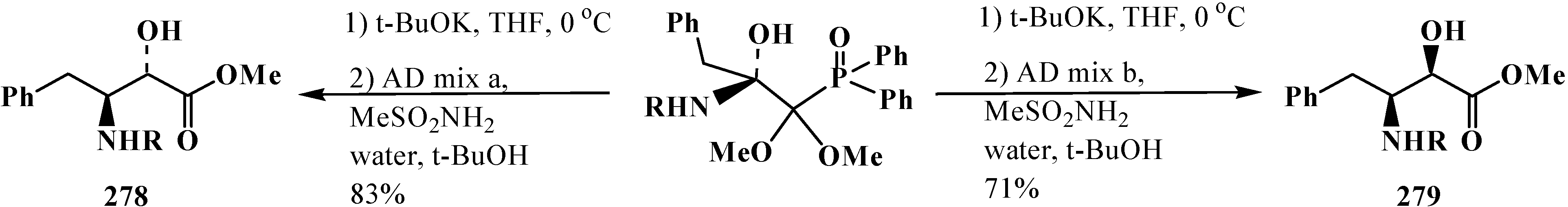
Sharpless asymmetric dihydroxylation followed of Horner reaction also provides an opportunity to synthesize α-hydroxy-β-amino acid diastereomers 278, 279 [20,128] (Scheme 92).
Scheme 92.
Using of combination of Horner and AD reactions to form α-hydroxy-β-amino acid diastereomers 278, 279 (R = t-BuOC(O)).
Scheme 92.
Using of combination of Horner and AD reactions to form α-hydroxy-β-amino acid diastereomers 278, 279 (R = t-BuOC(O)).

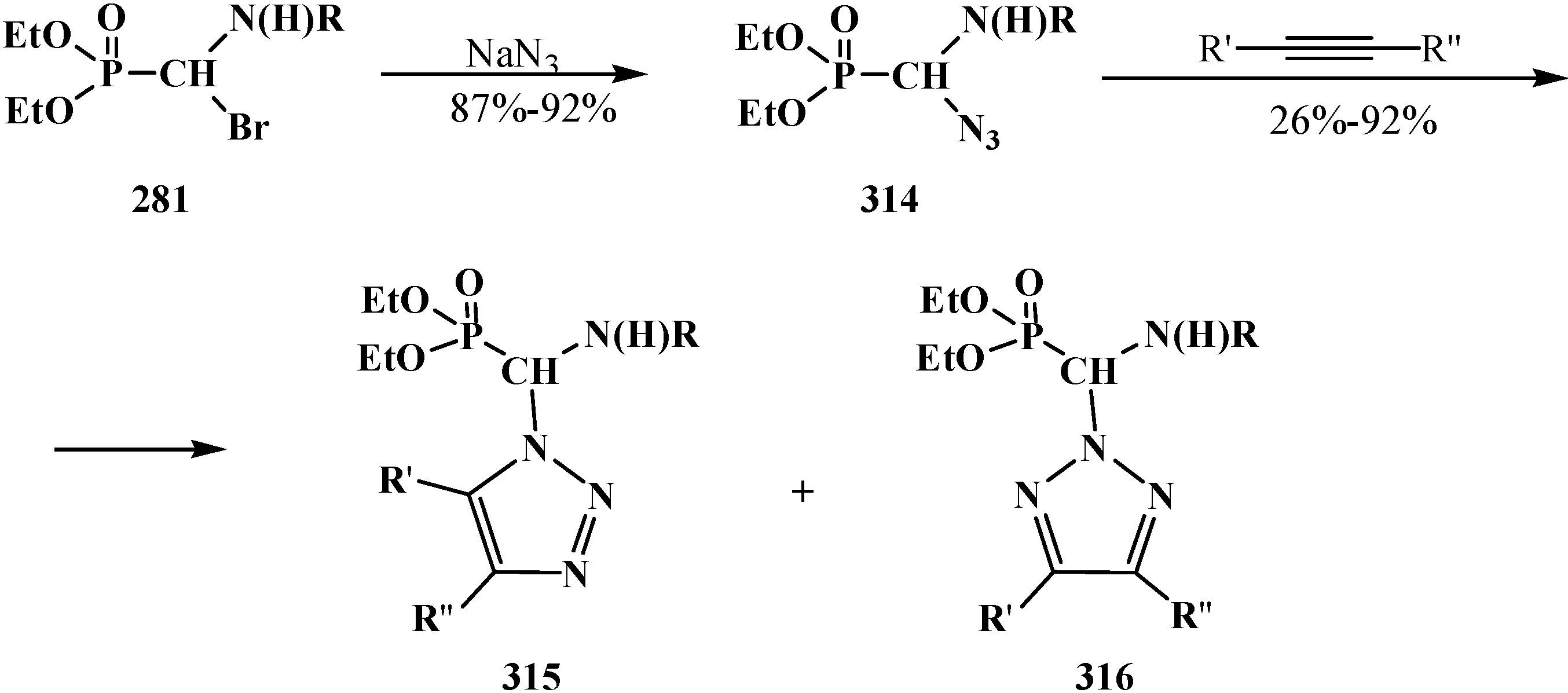
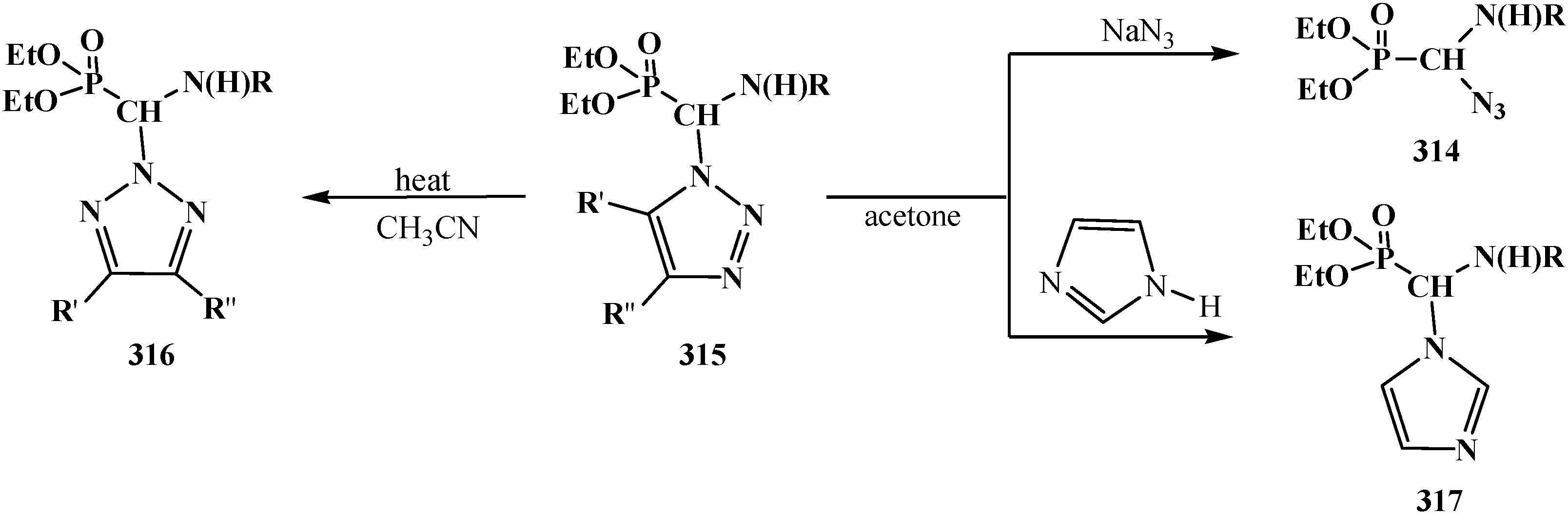
4. Phosphorylated Formaldehyde Halogenoaminals (Phosphorylated Vilsmeier–Haak Reagents) 17
At present, four types of phosphorus compounds are known that can be related to phosphorylated Vilsmeier–Haak reagents: N,N,N',N'-tetraisopropyl [(N'',N''-diisopropylamino)methylydeniminium] phosphondiamide dichlorophosphate (8) [12], dialkyl [(N,N-dimethylamino)chloromethyl]phosphonates 171 [31,32], diphenyl[(N,N-dialkylamino)chloromethyl]phosphine oxides 280 [163], and structurally related to the phosphorylated Vilsmeier–Haak reagents, diethyl [(N-acylamino)bromomethyl]phosphonates 281 [164] (Figure 6).
Figure 6.
Representatives of phosphorylated Vilsmeier–Haak reagents 8, 171, 280, 281 synthesized to date.
Figure 6.
Representatives of phosphorylated Vilsmeier–Haak reagents 8, 171, 280, 281 synthesized to date.

No general methods of synthesis have been found for the compounds of this type 8, 171, 280, 281. Their chemical properties also differ significantly from each other.
4.1. Synthesis of N,N,N',N'-Tetraisopropyl [(N'',N''-Diisopropylamino)methylydeniminium] Phosphondiamide Dichlorophosphate (8)
A sole paper was published in 1999 [12] on the synthesis of N,N,N',N'-tetraisopropyl [(N'',N''-diisopropylamino)methylydeniminium]phosphondiamide dichlorophosphate (8) by the oxidation of N,N,N',N'-tetraisopropyl [(N'',N''-diisopropylamino)methylydeniminium]phosphindiamide dichlorophosphate (282) with dimethyl sulfoxide DMSO (Scheme 93). However, the chemical properties of 8 are insufficiently studied (for some chemical properties of 8 and its derivatives, see Section 1. Introduction).
Scheme 93.
Phosphindiamide dichlorophosphate 282 oxidation by means of DMSO leads to the formation of phosphondiamide dichlorophosphate 8.
Scheme 93.
Phosphindiamide dichlorophosphate 282 oxidation by means of DMSO leads to the formation of phosphondiamide dichlorophosphate 8.

4.2. Synthesis and Chemical Properties of Dialkyl [(N,N-Dimethylamino)chloromethyl]phosphonates 171
The first compounds of this type, dimethyl [(N,N-dimethylamino)chloromethyl] phosphonate (172) and diethyl [(N,N-dimethylamino)chloromethyl]phosphonate (173) were synthesized in 1969 [31,32]. For the first time, these compounds were obtained as intermediates in the synthesis of tetraalkyl (N,N-dimethylaminomethyl)diphosphonates 174 by reaction of N,N-dimethyl(chloromethylideneiminium) chloride (171) with trialkyl phosphites 58 [32]—see Scheme 49, section “Diphosphinoyl N,N-dialkylaminomethanes”.
Preparative amounts of these compounds were prepared by a two-step scheme [30,31]. First, the reaction of dimethylformamide dimethylacetal (78) with dialkyl phosphites 51 resulted in dialkyl [(N,N-dimethylamino)methoxymethyl]phosphonates 162, which were further reacted with thionyl chloride (SOCl2) at 0 °C to yield the final products 172, 173 (Scheme 94). See also Scheme 49 and Scheme 50.
Scheme 94.
Reaction dialkyl phosphites 51 and dimethylformamide dimethylacetal (78) leads to phosphorilated halogenoaminals 172 (Alk = Me), 173 (Alk = Et).
Scheme 94.
Reaction dialkyl phosphites 51 and dimethylformamide dimethylacetal (78) leads to phosphorilated halogenoaminals 172 (Alk = Me), 173 (Alk = Et).

This scheme is currently used [33,165]; phosphorus trichloride can be used instead of thionyl chloride [83]. Dimethyl [(N,N-dimethylamino)chloromethyl]phosphonate (172) was found to undergo spontaneous dealkylation on storage [31], therefore only diethyl [(N,N-dimethylamino)chloromethyl]phosphonate (173) is used in organic synthesis. Phosphonate 173, as a phosphorylated Vilsmeier–Haak reagent where one chlorine atom is substituted by a diethoxyphosphoryl group, show electrophilic properties [166] inherent in compounds of such kind and readily reacts with nucleophiles. Thus, compound 173 reacts vigorously with secondary amines, carbazole, alcohols, thiols, and hydrophosphinoyl compounds 51, 53 in the presence of equimolar amount of triethylamine. The reaction of α-phosphono-α-aminomethylation leads to the corresponding diethoxyphosphinoylformaldehyde derivatives: asymmetric aminals 283, 284, aminoacetals 285, aminothioacetals 286, and diphosphorylated N,N-dimethylaminomethanes 164 [30], including unsymmetrical 165 [165] (Scheme 95). The reaction with aromatic and aliphatic amides RC(O)NH2, urea and thiourea (NH2)2C=X [30] proceeds in a similar manner. Products of reactions are phosphorylated amidoaminals 287, 288 and symmetrically disubstituted derivatives of urea and thiourea 289, 290 (Scheme 95). See also Scheme 53, section “Diphosphinoyl N,N-dialkylaminomethanes”.
Scheme 95.
Reactions of diethyl [(N,N-dimethylamino)chloromethyl]phosphonate (173) with nucleophilic co-reagents.
Scheme 95.
Reactions of diethyl [(N,N-dimethylamino)chloromethyl]phosphonate (173) with nucleophilic co-reagents.

Diethyl [(N,N-dimethylamino)chloromethyl]phosphonate (173) reacts with arylsulfonamides to lead to the cleavage of the phosphorus–carbon bond and give N-sulfonyl-substituted derivatives of formamidines 291 and diethyl phosphite (6) [30] (Scheme 96). See also Section 3.1. Cleavage of phosphorus–carbon bond.
Scheme 96.
Cleavage of phosphorus–carbon bond by means of the reaction of compound 173 with arylsulfonamides.
Scheme 96.
Cleavage of phosphorus–carbon bond by means of the reaction of compound 173 with arylsulfonamides.

Compound 173 reacts with methyl aryl ketones [30] and dialkyl or aryl alkyl ketones [33] to give diethyl [(N,N-dimethylamino)(aroyl(or alkanoyl)alkylmethyl)methyl]phosphonates 292 and 293 [30,33], phosphorylated analogs of natural α-amino acids potentially possessing biological activity [33] (Scheme 97). However, reaction stereoselectivity is low, and the anti/syn ratio is 2:1.
Scheme 97.
Reaction of compound 173 with ketones leads to diethyl [(N,N-dimethylamino)(aroyl(or alkanoyl)alkylmethyl)methyl]phosphonates 292 and 293.
Scheme 97.
Reaction of compound 173 with ketones leads to diethyl [(N,N-dimethylamino)(aroyl(or alkanoyl)alkylmethyl)methyl]phosphonates 292 and 293.

Like ketones, the reaction of 173 proceeds also with aldehydes branched at the α-position to the carbonyl group to give phosphorylated aminoaldehydes 294 and 295 (Scheme 98), but the reaction is not stereoselective: the stereoisomer ratio of the resulting α-formyl-α-methylaminophosphonates is 1:1 [33].
Scheme 98.
Reaction of compound 173 with aldehydes leads to phosphorylated aminoaldehydes 294 and 295.
Scheme 98.
Reaction of compound 173 with aldehydes leads to phosphorylated aminoaldehydes 294 and 295.

The involvement of enamines instead of ketones as their synthetic equivalents in the reaction with phosphonate 173 provides stereoselective synthesis of diethyl (α-methylamino-α-aroyl(or alkanoyl))methylphosphonates 292; the reaction leads to the products with only anti configuration [33] (Scheme 99).
Scheme 99.
Synthesis of diethyl (α-methylamino-α-aroyl(or alkanoyl))methylphosphonates 292 and phosphorylated vinyl ketones 296.
Scheme 99.
Synthesis of diethyl (α-methylamino-α-aroyl(or alkanoyl))methylphosphonates 292 and phosphorylated vinyl ketones 296.

Diethyl [(N,N-dimethylamino)(aroyl(or alkanoyl)alkylmethyl)methyl]phosphonates 292, 293 (Scheme 101, Scheme 102 and Scheme 103) are obtained as hydrochlorides in the above-stated syntheses. They are unstable and on storage undergo spontaneous β-elimination of dimethylamino group as dimethylammonium chloride—with conversion up to 40% over 24 h [30] to form the corresponding phosphorylated vinyl ketones 296. The same result was achieved on the heating of hydrochlorides of compounds 292 and 293 to 100 °C over 45 min [30] or stirring their solutions in methylene chloride with silica gel at 20 °C for 15 h [33]. The reaction of amino group elimination with the use of silica gel is also stereoselective: the anti stereoisomers 292 produce only E isomers of vinyl compounds 297 [33] (Scheme 99).
The reaction of β-enaminonitriles 298 with compound 173 was used in the synthesis of substituted nitriles of nicotinic acid 299 [167] (Scheme 100).
Scheme 100.
The reaction of compound 173 with β-enaminonitriles 298 leads to nitriles of nicotinic acid 299.
Scheme 100.
The reaction of compound 173 with β-enaminonitriles 298 leads to nitriles of nicotinic acid 299.

As a phosphorylated Vilsmeier–Haak reagent, phosphonate 173 undergoes typical reactions with activated aromatic compounds: N,N-dimethylaniline, triethylammonium salts of β-naphthol and p-cresol, and heteroaromatic compounds: N-methylindole, N-methylpyrrole, α-methylfuran. The synthesis of aromatic α-aminomethylphosphonates 300 is regiospecific: only one addition product forms in each case [33] (Scheme 101).
Scheme 101.
Interactions of phosphonate 173 with aromatic compounds (Ar = 4-dimethylaminophenyl; 2-hydroxynaphthyl-1; 2-hydroxy(5-methyl)phenyl; 1-methylaminoindol-3-yl; 1,5-dimethylaminopyrrol-2-yl; 5-methylfur-2-yl).
Scheme 101.
Interactions of phosphonate 173 with aromatic compounds (Ar = 4-dimethylaminophenyl; 2-hydroxynaphthyl-1; 2-hydroxy(5-methyl)phenyl; 1-methylaminoindol-3-yl; 1,5-dimethylaminopyrrol-2-yl; 5-methylfur-2-yl).

4.3. Syntheses and Chemical Properties of Diphenyl[(N,N-dialkylamino)chloromethyl]phosphine Oxides 280
Diphenyl[(N,N-dialkylamino)chloromethyl]phosphine oxides 280 are the diphenylphosphinoyl analogs of dialkyl [((N,N-dialkylamino)chloromethyl]phosphonates. They are obtained by the reaction of chlorodiphenylphosphine (68) with N,N-dialkylformamides in the presence of 10%–20% of N,N-dialkylchloromethyleniminium chloride [Alk2N=C(H)Cl]+Cl− (168)—Vilsmeier–Haak reagent [168,169] according to Scheme 102.
Scheme 102.
Synthesis of diphenyl[(N,N-dialkylamino)chloromethyl]phosphine oxides 280.

Although phosphine oxides 280 are the formal analogs of dialkyl [(N,N-dialkylamino)chloromethyl]phosphonates 171, they differ substantially in reactivity. It was shown by the example of diphenyl[(N,N-dimethylamino)chloromethyl]phosphine oxide (301) that compounds 280 can dissociate in solutions with the cleavage of C–Cl and C–P bonds. See also section “Cleavage of phosphorus–carbon bond”. The dissociation in solution results in the formation of both diphenyl[(N,N-dimethylamino)chloromethyliden]phosphine oxide cation (302) and diphenylphosphinite anion (193) [170] (Scheme 103).
Scheme 103.
Diphenyl[(N,N-dimethylamino)chloromethyl]phosphine oxide (301) dissociation in solutions with a cleavage as bond C-Cl and bond C-P.
Scheme 103.
Diphenyl[(N,N-dimethylamino)chloromethyl]phosphine oxide (301) dissociation in solutions with a cleavage as bond C-Cl and bond C-P.

Therefore there are two kinds of reactivity for 301 (and accordingly 280) [163,170]: electrophilic diphenyl[(N,N-dimethylamino)chloromethylidene]phosphine oxide cation (302) reacts with nucleophiles such as hydrophosphinoyl compounds 6, 53, mixed diethyl trimethylsilyl phosphite (EtO)2POSiMe3 and N,N-dimethylamino aniline PhNMe2, to give symmetrical 303, R=Ph and unsymmetrical 304, R=OEt diphosphorylated N,N-dimethylaminomethanes and diphenyl[(bis(4-N,N-dimethylamino)phenyl)methyl] phosphine oxide (305), the product of substitution of both chloro and amino groups at the carbon atom of 301. However, nucleophilic diphenylphosphinite anion (193) reacts with electrophiles: acetic aldehyde MeC(O)H, acetone Me2C(O), phenyl isocyanate PhNCO, triethyl orthoformate (EtO)3CH, dimethylformamide dimethylacetal (79), and bis(diethylamino)methane (Et2N)2CH2 to form addition or substitution products, α-phosphorylated compounds: alcohols 306 and 307, carbamoyl compound 308, acetal 309, aminoacetal 310 and amine 311 (Scheme 104).
Scheme 104.
Reactions of diphenyl[(N,N-dimethylamino)chloromethyl]phosphine oxide (280) with nucleophilic and electrophilic coreagents.
Scheme 104.
Reactions of diphenyl[(N,N-dimethylamino)chloromethyl]phosphine oxide (280) with nucleophilic and electrophilic coreagents.

4.4. Synthesis and Chemical Properties of Diethyl [(N-Acylamino)bromomethyl]phosphonates 281
Diethyl [(N-acylamino)bromomethyl]phosphonates 281 were prepared in high yields by the bromination of diethyl (N-acylamino)methylphosphonates 312 with N-bromosuccinimide (NBS) in carbon tetrachloride [164,171] (Scheme 105).
Scheme 105.
Synthesis of diethyl [(N-acylamino)bromomethyl]phosphonates 281 where R = AlkC(O), ArC(O), AlkS(O)2, ArS(O)2.
Scheme 105.
Synthesis of diethyl [(N-acylamino)bromomethyl]phosphonates 281 where R = AlkC(O), ArC(O), AlkS(O)2, ArS(O)2.

To date diethyl [(N-acylamino)bromomethyl]phosphonates 281 remain poorly studied, and their chemical properties are insufficiently studied. Nonetheless, compounds 281, structurally related to the phosphorylated Vilsmeier–Haak reagents, behave as electrophiles similarly to dialkyl [(N,N-dimethylamino)chloromethyl]phosphonates 171 [166]. They react readily with such nucleophiles as primary aliphatic and aromatic amines and secondary cyclic amines in the presence of ethyldiisopropylamine—Hünig’s base as well as triethylamine (Scheme 106). Reaction products are diethyl [(N-acylamino)aminomethyl]phosphonates 313, unsymmetrical amidoaminals of diethyl (formyl)phosphonate (3) [30] showing biological and pharmacological activity [172].
Scheme 106.
Transformation of bromomethylphosphonates 281 into unsymmetrical amidoaminals 313, where HNR'R'' are morpholine, piperidine, pyrrole, 2-aminomethyltetrahydrofuran, N-methylbenzylamine, and aniline.
Scheme 106.
Transformation of bromomethylphosphonates 281 into unsymmetrical amidoaminals 313, where HNR'R'' are morpholine, piperidine, pyrrole, 2-aminomethyltetrahydrofuran, N-methylbenzylamine, and aniline.

The reaction of compounds 282 with sodium azide leads to diethyl [(N-acylamino)azidomethyl]-phosphonates 314, valuable precursors in the synthesis of phosphinoyl-substituted 1,2,3-triazoles, the products of 1,3-dipolar addition to disubstituted alkynes [173,174]. A mixture of the resultant two regioisomers of diethyl [(N-acylamino)(1-(1,2,3-triazolyl))methyl]phosphonates (315) and diethyl [(N-acylamino)(2-(1,2,3-triazolyl))methyl]phosphonates (316) can be separated by chromatography on silica gel (Scheme 107).
Scheme 107.
Synthesis of (triazolylmethyl)phosphonates 315 and 316 from bromomethylphosphonates 281, where R', R'' = AlkC(O), ArC(O), AlkS(O)2, ArS(O)2.
Scheme 107.
Synthesis of (triazolylmethyl)phosphonates 315 and 316 from bromomethylphosphonates 281, where R', R'' = AlkC(O), ArC(O), AlkS(O)2, ArS(O)2.

Prolonged heating of acetonitrile solutions of 315 leads to migration of [(N-acylamino)(diethylphosphinoyl)]methyl group from the first to the second nitrogen atom (N1–N2 migration) of 1,2,3-triazole group to give 316. In the presence of nucleophiles, azide ion or imidazole, the triazole fragment is displaced to yield diethyl [(N-acylamino)azidomethyl]phosphonates 314 or diethyl [(N-acylamino)(1-imidazolyl)methyl]phosphonates 317 (Scheme 108) [174].
Scheme 108.
Reaction replacement of 1,2,3-triazole group of compound 315 with nucleophiles to give compounds 314 and 317 and isomerisation of 315 into compound 316.
Scheme 108.
Reaction replacement of 1,2,3-triazole group of compound 315 with nucleophiles to give compounds 314 and 317 and isomerisation of 315 into compound 316.

The treatment of phosphonates 281 with Hünig’s base i-Pr2NEt [164] or triethylamine Et3N [171] in tetrahydrofuran at −78 °C was shown to result in diethyl [(N-acyl)iminomethyl]phosphonates 318 in yields up to 98% (Scheme 109).
Scheme 109.
Synthesis of diethyl [(N-acyl)iminomethyl]phosphonates (318) from phosphonates 281.

5. Alkyl (dialkoxymethyl)phosphinates—H-Phosphinates 34. Syntheses and Chemical Properties
The synthesis of alkyl (dialkoxymethyl)phosphinates—H-phosphinates 34 in yields up to 96% by the reaction of hypophosphorous acid (319) with orthoformates 54 in the presence of catalytic amounts amounts of p-toluenesulfonic acid (p-TolSO3H) [57,175] or trifluoroacetic acid (CF3SO3H) [58] was reported in 1977 (Scheme 110). The method is still used at present.
Scheme 110.
Synthesis of alkyl (dialkoxymethyl)phosphinates 34.

The chemical properties of phosphinates 34 were studied almost exclusively by the example of ethyl (diethoxymethyl)phosphinate (86). Ethyl (diethoxymethyl)phosphinate (86) retains properties typical for both phosphorus esters and hydrophosphoryl compounds and retains the general ability of phosphorylated formaldehyde acetals to undergo the cleavage of the phosphorus–carbon bond.
Phosphinate 86 is readily alkylated under the action of alkyl halides in the presence of bases: Na, NaH, BuLi (B−) (via anion 320) to give ethyl alkyl(diethoxymethyl)phosphinates 321 [58,176] (Scheme 111).
Scheme 111.
Phosphinate 86 alkylation with alkylhalydes.

Compound 86 also readily undergoes addition to activated double bonds, or it can transform into three-coordinated phosphorus compounds [59,176,177], for example, in Scheme 112, compounds 322 and ethyl trimethylsilyl (diethoxymethyl)phosphinite (323), respectively.
Scheme 112.
Transformations of compound 86 into phosphinate 322 and a three-coordinated phosphorus compound (phosphinite 323).
Scheme 112.
Transformations of compound 86 into phosphinate 322 and a three-coordinated phosphorus compound (phosphinite 323).

It also undergoes Todd–Atherton reaction with phenols and cross-coupling reactions with aryl bromides [106] in the presence of tetrakis(triphenylphosphine)palladium(0), Pd(Ph3P)4, see Scheme 17. In 86, one of two P–H bonds of initial hypophosphorous acid 319 is protected by a diethoxymethyl group and can be restored in subsequent stages after deprotection [58,176]. When treated with two equivalents of organomagnesium or organolithium compounds, phosphinate 86 produces secondary phosphine oxides 324 [178], a hidden form of unstable primary phosphine oxides 325 apt to disproportionation [179], that can be easily obtained by subsequent acid hydrolysis. Further alkylation of secondary phosphine oxides 324 in the presence of a base leads to unsymmetrical tertiary phosphine oxides 326, which in turn are the hidden form of unsymmetrical secondary phosphine oxides 328. Similarly to primary phosphine oxides 325, compounds 327 can be also obtained by subsequent acid hydrolysis of tertiary phosphine oxides 326. Further oxidation leads to the corresponding phosphonic 328 and unsymmetrical phosphinic acids 329. Unsymmetrical tertiary phosphine oxides 330 can be obtained by the following alkylation of secondary phosphine oxides 327 (Scheme 113).
Scheme 113.
Using phosphinate 86 for the synthesis of phosphine oxides 325, 328, 330, phosphonic 328 and unsymmetrical phosphinic acids 329.
Scheme 113.
Using phosphinate 86 for the synthesis of phosphine oxides 325, 328, 330, phosphonic 328 and unsymmetrical phosphinic acids 329.

Phosphonic and unsymmetrical phosphinic acids 328 and 329 can also be obtained by the scheme that begins with the alkylation reaction by introducing 86 in the reaction of one or sequentially both P–H bonds, thus solving the problem of selectivity [58,59,176,178], over phosphinates 331 and H-phosphinic acids 332, respectively. The scheme allows one to avoid the use of highly toxic hypophosphorous acid in the syntheses [177] (Scheme 114).
Scheme 114.
Phosphonic 328, H-phosphinic 332 and unsymmetrical phosphinic acids 329 syntheses starting with the reaction of 86 with electrophiles, where E = RHal or olefins with the activated double bond.
Scheme 114.
Phosphonic 328, H-phosphinic 332 and unsymmetrical phosphinic acids 329 syntheses starting with the reaction of 86 with electrophiles, where E = RHal or olefins with the activated double bond.

Due to the unique combination of chemical properties, phosphinate 86 is used in modern organic synthesis and synthesis of biologically active compounds. H-phosphinic analogs of natural α-amino acids 333 were obtained with >95% enantiomeric excess by the reaction of 86 with chiral (S)-N-tert-butylsulfinylketimines (334) [180] (Scheme 115).
Scheme 115.
Synthesis of H-phosphinic analogs of natural α-amino acids 333 by means of the reaction of phosphinate 86 with chiral (S)-N-tert-butylsulfinylketimines 334.
Scheme 115.
Synthesis of H-phosphinic analogs of natural α-amino acids 333 by means of the reaction of phosphinate 86 with chiral (S)-N-tert-butylsulfinylketimines 334.

3-Amino-2-fluoropropyl-H-phosphinic acid (335), a γ-aminobutyric acid (GABA) analog, a potential pharmaceutical for the treatment of central nervous system diseases, was prepared from compound 323 as the silylated form of ethyl (diethoxymethyl)phosphinate 86 [181] (Scheme 116).
Scheme 116.
Silylated compound 323 used for the synthesis of γ-aminobutyric acid analog 335.

Disubstituted 3-aminopropyl-4-pyridylphosphinic acid (336), a potential neurotropic pharmaceutical and GABA antagonist, was obtained in a similar manner [182] (Scheme 117).
Scheme 117.
Synthesis of potential neurotropic pharmaceutical, GABA antagonist 336.

6. Phosphorylated Formaldehyde Hydrates—Geminal Diols 35. Syntheses and Chemical Properties
The chemistry of phosphorylated formaldehyde hydrates 35 began to develop since the mid 1990s when diethyl (dihydroxymethyl)phosphonate (337) was obtained in quantitative yield by the reaction of diethyl (diazomethyl)phosphonate (338) with 3,3-dimethyldioxirane—acetone peroxide at 20 °C [60,61] (Scheme 118).
Scheme 118.
Synthesis of diethyl (dihydroxymethyl)phosphonate (337) by the reaction of diazomethylphosphonate (338) with 3,3-dimethyldioxirane.
Scheme 118.
Synthesis of diethyl (dihydroxymethyl)phosphonate (337) by the reaction of diazomethylphosphonate (338) with 3,3-dimethyldioxirane.

It was shown that 337 exists in an equilibrium with the hydrated form of diethyl formylphosphonate 3 [60,61] (Scheme 119).
Scheme 119.
Equilibrium between diethyl (dihydroxymethyl)phosphonate (337) and the hydrated form of diethyl formylphosphonate 3.
Scheme 119.
Equilibrium between diethyl (dihydroxymethyl)phosphonate (337) and the hydrated form of diethyl formylphosphonate 3.

The chemical properties of phosphonate 337 provide the possibility of considering it as a hidden form of phosphorylated formaldehyde. Compound 337 reacts with secondary amines R'2NH with the cleavage of the phosphorus–carbon bond and formation of the corresponding N,N-disubstituted formamides and diethyl phosphite (6) [60,61] (it behaves as a source of formyl group [C(O)H]+ cation, synthon type a1 [183,184].
Compound 337 also shows properties of a typical aldehyde when reacted with primary amines RNH2, trimethylsilyl cyanide Me3SiCN, and hydroxylamine NH2OH to give phosphorylated formaldimines 339, formalcyanohydrin 340, and formaldoxime 341, respectively [60,61] (Scheme 120).
Scheme 120.
Reactions of compound 337 with some coreagents, where R= t-Bu or Ph.

As a hidden form of diethyl (formyl)phosphonate (3), phosphonate 337 in the presence of silver trifluoroacetate AgOTf as a catalyst undergoes a Mannich reaction with anisidine and terminal alkyl- or aryl-substituted alkynes. Reaction products, α-aminopropargylphosphonates 342, are valuable precursors in the synthesis of α-aminophosphonic acids showing high biological activity as α-amino acid mimetics [63] (Scheme 121).
Scheme 121.
Catalytical synthesis of α-aminopropargylphosphonates 342 by a Mannich reaction.

In the presence l-proline amide (343) as a catalyst, 337 undergoes an asymmetrical cross aldol condensation with aliphatic ketones. Diastereomerically pure α-hydroxyphosphonates 344, precursors of α-hydroxyphosphonic acids, result from chirality induction with diastereomeric excesses greater that 99% [185] (Scheme 122).
Scheme 122.
Synthesis of diastereomeric α-hydroxyphosphonates 344 by asymmetrical cross-aldol condensation.
Scheme 122.
Synthesis of diastereomeric α-hydroxyphosphonates 344 by asymmetrical cross-aldol condensation.

Like α-aminophosphonic acids, α-hydroxyphosphonic acids are also α-amino acid mimetics and exhibit high biological activity [185]. Similar results were obtained in the aldol condensation of ketones with racemic ethyl phenyl(dihydroxymethyl)phosphinate (345). In this case, the final products are α-hydroxyphosphinic acids 346 [62].
7. Conclusions
Phosphorylated formaldehyde derivatives, i.e., acetals and related compounds, are a group of largely underinvestigated species. The experimental data accumulated since the early 1960s confirm that these compounds can be used in a wide variety of syntheses that have not been fully realized to date. For this reason, the growth of interest to these compounds will allow investigating their chemical properties in more detail and potentially enrich organic and organoelement chemistry with new synthetic methods.
Conflicts of Interests
The author declares no conflict of interests.
References
- Mitsuo, S.; Masaki, S.; Hikaru, Y.; Tsujiaki, H. Acylphosphonates: P-C Bond Cleavage of Dialkyl Acylphosphonates by Means of Amines. Substituent and Solvent Effects for Acylation of Amines. J. Org. Chem. 1980, 45, 4162–4167. [Google Scholar]
- Miller, A.; Stewart, D. Reactions of Carbonyl Compounds with Tervalent Phosporus Reagents. Part 8.l, Acetyldiphenylphosphine Oxide. J. Chem. Soc. Perkin Trans. I 1977, 17, 1898–1901. [Google Scholar]
- Frey, G.; Lesiecki, H.; Lindner, E.; Vordermaier, G. Synthese und reaktives Verhalten von Acyldiorganylphosphanoxiden. Chem. Ber. 1979, 112, 763–772. [Google Scholar] [CrossRef]
- Sekine, M.; Kume, A.; Nakajima, M.; Hata, T. A new method for acylation of enolates by means of dialkyl acylphosphonates as acylating agents. Chem. Lett. 1981, 10, 1087–1090. [Google Scholar] [CrossRef]
- Shagidullin, R.R.; Plyamovatyi, A.K.; Mukhamadeeva, R.M.; Khairullin, V.K.; Pudovik, A.N. Fourier IR—Spectroscopic study of the mechanism of anomalous Shiff base reaction with phosphorus-containing CH—Acids. Z. Obshch. Khim. 1994, 64, 926–930. [Google Scholar]
- Conti, P.; Pinto, A.; Tamborini, L.; Rizzo, V.; de Micheli, C. A regioselective route to 5-substituted pyrazole- and pyrazoline-3-phosphonic acids and esters. Tetrahedron 2007, 63, 5554–5560. [Google Scholar] [CrossRef]
- Conti, P.; Pinto, A.; Tamborini, L.; Dunkel, P.; Gambaro, V.; Visconti, G.L.; de Micheli, C. A regioselective route to 5-substituted isoxazole- and isoxazoline-3-phosphonates. Synthesis 2009, 591–596. [Google Scholar] [CrossRef]
- Vasella, A.; Voeffray, R. Asymmetric Synthesis of α- Aminophosphonic Acids by Cycloaddition of N-Glycosyl-C- dialkoxyphosphonoylnitrones. Helv. Chim. Acta 1982, 65, 1953–1964. [Google Scholar] [CrossRef]
- Moskva, V.V.; Mavrin, V.Y. O,O-Diethylformylphosphonate. J. Gen. Chem. USSR 1988, 57, 2492. [Google Scholar]
- Iorga, B.; Eumery, F.; Mouriès, V.; Savignac, P. Phosphorylated aldehydes. Tetrahedron 1998, 54, 14637–14677. [Google Scholar] [CrossRef]
- Groß, H. Zur Existenz von Estern der Formylphosphonsäure. Z. Chem. 1977, 17, 131–132. [Google Scholar]
- Amsallem, D.; Gornitzka, H.; Baceiredo, A.; Bertrand, G. New types of stable aldehydes: Formylphosphane and formylphosphane oxide. Angew. Chem. Int. Ed. Engl. 1999, 38, 2201–2203. [Google Scholar] [CrossRef] [PubMed]
- Razumov, A.I.; Liorber, B.G.; Moskva, V.V.; Sokolov, M.P. Phosphorylated aldehydes. Russ. Chem. Rev. 1973, 42, 538–550. [Google Scholar] [CrossRef]
- Wagenknecht, J. An electrochemical method for the preparation of iminodimethylenediphosphonic Acid. Synth. React. Inorg. Met. Org. Chem. 1974, 4, 567–572. [Google Scholar] [CrossRef]
- Firestone, R.A. Hydroxy and Imino Containing Phosphonic Acid Diesters. U.S. Patent 3784590, 8 January 1974. [Google Scholar]
- Mührle, H.; Vetter, W. Reaktionsbeteiligung von Phosphoester-Nachbargruppen bei Aminodehydrierungen. Z. Naturforsch. B 1988, 43, 1663–1671. [Google Scholar]
- Franczyk, T.S., II. Preparation of Formylphosphonic Acid from Tertiary Aminomethylphosphonic Acid N-oxides. U.S. Patent 6274760, 14 August 2001. [Google Scholar]
- Costisella, B.; Gross, H. Synthese und NMR-Spektren offenkettiger und cyclischer Acetale von Formylphosphonsäureestern und Formylphosphinoxid. J. Prakt. Chem. 1977, 319, 8–16. [Google Scholar] [CrossRef]
- Livantsov, M.V.; Proskurina, M.V.; Prishchenko, A.A.; Lutsenko, I.F. Synthesis and some properties of phosphorylated formals. J. Gen. Chem. USSR 1984, 54, 2504–2517. [Google Scholar]
- Brunjes, M.; Kujat, C.; Monenschein, H.; Kirschning, A. Acylation of Alkyl Halides and Amino Aldehydes with Phosphane Oxide-Based d1-Synthon. Eur. J. Org. Chem. 2004, 5, 1149–1160. [Google Scholar] [CrossRef]
- Gross, H.; Keitel, I.; Costisella, B.; Mikołajczyk, M.; Midura, W. Zur Synthese von bis-alkylmercaptomethanphosphonsäuredialkylestern. Phosphorus Sulfur Silicon Relat. Elem. 1983, 16, 257–262. [Google Scholar] [CrossRef]
- Młotkowska, B.; Gross, H.; Costisella, B.; Mikołajczyk, M.; Grejszczak, S.; Zatorski, A. Synthese offenkettiger und cyclischer S,S-Acetale von Formyl phosphonsäurestern. J. Prakt. Chem. 1977, 319, 17–22. [Google Scholar]
- Mikołajczyk, M.; Grejszczak, S.; Zatorski, A.; Młotkowska, B.; Gross, H.; Costisella, B. A new and general synthesis of ketene S,S- and S,O-Acetals based on the Horner-Wittig reaction. Tetrahedron 1978, 34, 3081–3088. [Google Scholar]
- Costisella, B.; Gross, H. 1-Dimethylamino-1-cyano methanphosphonsдurediethylester, ein neues Edukt zur Darstellung von Carbonsäuren, 1-Cyanoenaminen und Homoenolaten. Tetrahedron 1982, 38, 139–145. [Google Scholar] [CrossRef]
- Costisella, B.; Gross, H. 1-Cyano-1-dimethylamino methanphosphonsäurediethylester-Lithium—Ein “Instant-Horner-Reagent”. Z. Chem. 1987, 27, 143–144. [Google Scholar] [CrossRef]
- Marrero, Y.; Harwood, L.M. Towards the total synthesis of colletofragaranes: Constructing the macrocyclic lactone by high pressure-mediated intramolecular Diels-Alder reaction. Tetrahedron Lett. 2009, 50, 3574–3576. [Google Scholar] [CrossRef]
- Gross, H.; Costisella, B. Synthesis of carboxylic acids via PO-activated olefination of tetraethyl dimethilamino-methylene-diphosphonate. Angew. Chem. Int. Ed. Engl. 1968, 7, 391–392. [Google Scholar] [CrossRef]
- Qian, D.Q.; Shi, X.D.; Cao, K.Z.; Liu, L.Z. The Synthesis and Reactivity of Alkylaminosubstitutedmethylenediphosphonates. Heteroat. Chem. 1999, 10, 271–276. [Google Scholar] [CrossRef]
- McNulty, J.; Das, P. Development of one-pot method for the homologation of aldehides to carboxylic acids. Tetrahedron 2009, 65, 7794–7800. [Google Scholar] [CrossRef]
- Gross, H.; Costisella, B. Über α-substituerte Phosphonate, XI. Liebiegs Ann. Chem. 1971, 750, 44–52. [Google Scholar] [CrossRef]
- Gross, H.; Costisella, B. Derivate des Formylphosphonsäurediäthylestern. J. Prakt. Chem. 1969, 311, 925–929. [Google Scholar] [CrossRef]
- Gross, H.; Costisella, B.; Haase, L. Zur Reaction von Dimethylformamidederivaten mit Phosphorigsäurediestern und Phosphinoxiden. J. Prakt. Chem. 1969, 311, 577–585. [Google Scholar] [CrossRef]
- Risch, N.; Piper, S.; Winter, A.; Lefarth-Risse, A. An Efficient Synthesis of Novel α-Aminophosphonates Based on a Mannich-Type Reaction. Eur. J. Org. Chem. 2005, 2, 387–394. [Google Scholar] [CrossRef]
- Schrader, T.; Steglich, W. Phosphorus analogs of amino acids. IV. Syntheses of unusual 1-aminophosphonic acids via Diels-Alder reactions of diethyl (N-acyliminomethyl)phosphonates. Synthesis 1990, 1153–1156. [Google Scholar]
- Mikołajczyk, M.; Costisella, B.; Grejszczak, S.; Zatorski, A. Synthesis of O,S-thioacetals of formylphosphonates. Tetrahedron Lett. 1976, 17, 477–480. [Google Scholar]
- Kim, T.H.; Uh, D.Y. New synthetic method of O,S-thioacetals of formylphosphonates. Tetrahedron Lett. 1985, 26, 3479–3482. [Google Scholar] [CrossRef]
- Costisella, B.; Keitel, I. Synthese und Reaktionen von 1-Acetoxy-1-methylthio- methanphosphorylverbindungen. Phosphorus Sulfur Silicon Relat. Elem. 1988, 40, 161–165. [Google Scholar] [CrossRef]
- Mavrin, V.Y.; Moskva, V.V. Reaction of trihetero-substituted carbonium salts with diethyl phosphite. J. Gen. Chem. USSR 1988, 58, 1930–1931. [Google Scholar]
- Mavrin, V.Y.; Moskva, V.V. Condensation of diethyl phosphite with mixed derivatives of orthocarboxylic acid. J. Gen. Chem. USSR 1988, 58, 1490. [Google Scholar]
- Gross, H.; Seibt, H. Synthese von α-chlor-α-methoxy-methanphosphonsäureestern und α-chlor-α-methylthio- methanphosphonsäureestern. J. Prakt. Chem. 1970, 312, 475–482. [Google Scholar] [CrossRef]
- Petrov, K.A.; Chauzov, V.A.; Agafonov, S.V.; Pazhitkova, I.V. Methoxymethanediphosphonic acid esters. J. Gen. Chem. USSR 1980, 50, 1525–1526. [Google Scholar]
- Galli, R.; Scaglioni, L.; Palla, O.; Gozzo, F. Synthesis of 2-alkylthyo(or trifluoromethylthio)-2-halogenothenyl deryvatives by of Wittig (under phase transfer conditions)or Wittig-Horner reactions. Applications in the field of pyretroids. Applications in the field of pyretroids. Tetrahedron 1984, 40, 1523–1532. [Google Scholar]
- Kim, T.H.; Uh, D.Y. Synthesis of S,S-thioacetals of formylphosphonate from chloro(arylthio)methanephosphonate. Synth. Commun. 1988, 18, 1611–1614. [Google Scholar] [CrossRef]
- Otten, P.A.; Davies, H.M.; van der Gen, A. A Horner-Wittig Synthesis of 1-Chlorovinyl Sulfoxides. Tetrahedron Lett. 1995, 36, 781–784. [Google Scholar] [CrossRef]
- Otten, P.A.; Davies, H.M.; van der Gen, A. A Horner-Wittig Synthesis of 1-Chlorovinyl Sulfoxides. Phosphorus Sulfur Silicon Relat. Elem. 1996, 109, 449–452. [Google Scholar]
- Dinizo, S.E.; Freerksen, R.W.; Rabst, W.E.; Watt, D.S. Synthesis of α-Alkoxyacrylonitriles Using Substituted Diethyl Cyanomethylphoaphonates. J. Org. Chem. 1976, 41, 2846–2849. [Google Scholar] [CrossRef]
- Dinizo, S.E.; Freerksen, R.W.; Rabst, W.E.; Watt, D.S. A One-Carbon Homologation of Carbonyl Compounds to Carboxylyc Acids, Esters and Amides. J. Am. Chem. Soc. 1977, 99, 182–186. [Google Scholar] [CrossRef]
- Diamond, P.M.; Dinizo, S.E.; Freerksen, R.W.; Curtis, R.; Haltiwanger, R.C.; Watt, D.S. Ferric Chloride-catalysed Conversion of α-t-butoxy- or α-acetoxy-acrylonitryles into Imides. J. Chem. Soc. Chem. Commun. 1977, 298–299. [Google Scholar] [CrossRef]
- Koidan, G.N.; Marchenko, A.P.; Oleinik, V.A.; Pinchuk, A.M. 2-Methyl-2-tribromophosphazopropane. J. Gen. Chem. USSR 1988, 58, 1304–1309. [Google Scholar]
- Savignac, P.; Coutrot, P. Preparation of 1,1-dibromoalkanes by Halogen Exchange. Synthesis 1976, 197–199. [Google Scholar] [CrossRef]
- Majewski, P.; Koszuk, J.F. Synthesis of Dichloromethylphosphonates. Phosphorus Sulfur Silicon Relat. Elem. 2009, 184, 956–962. [Google Scholar] [CrossRef]
- Bulpin, A.; Masson, S.; Sene, A. Reaction of Phosphonodithioformates with Nucleophilic Reagents; Potential synthetic Uses. Phosphorus Sulfur Silicon Relat. Elem. 1990, 49/50, 135–138. [Google Scholar]
- Costisella, B.; Oczegowski, S.; Gross, H. α-Substituierte phosphonate. 70 Untersuchungen über Methylthiomethanbisphosphorylderivate. Phosphorus Sulfur Silicon Relat. Elem. 1994, 86, 169–175. [Google Scholar]
- Binder, J.; Zbiral, E. A new procedure for homologation of carbonyl compounds to α-hydroxy-carboxylic esters by means of diethyl-[trimethylsilylethoxymethyl]phosphonate. Tetrahedron Lett. 1986, 27, 5829–5832. [Google Scholar] [CrossRef]
- Dufrechon, S.; Combert, J.-C.; Malhiac, C.; Collignon, N. Efficient synthesis of α-enamino phosphonates in the series of piperidine and morpholine. Phosphorus Sulfur Silicon Relat. Elem. 1997, 127, 1–14. [Google Scholar] [CrossRef]
- Mikołajczyk, M. α-Heterosubstituted phosphonates and phosphineoxides. Pure Appl. Chem. 1987, 59, 983–988. [Google Scholar]
- Gallagher, M.J.; Honegger, H. Dialkoxymethylation of phosphorus with trialkyl orthoformates: Reactions of phosphonic and phosphinic acids via their trivalent tautomers. Tetrahedron Lett. 1977, 18, 2987–2990. [Google Scholar] [CrossRef]
- Coudray, L.; Montchamp, J.-L. Temporary Protection of H-Phosphinic Acids as a Synthetic Strategy. Eur. J. Org. Chem. 2009, 27, 4646–4654. [Google Scholar] [CrossRef]
- Dingwall, Y.G.; Ehrenfreund, I.; Hall, R.G. Diethoxymethylphosphonites and phosphinates. Intermediates for the synthesis of α, β and γ-aminoalkylphosphinous acids. Tetrahedron 1989, 45, 3787–37808. [Google Scholar]
- Hamilton, R.; McKervey, M.A.; Rafferty, M.D.; Walker, B.J. The Reaction of Dimethyl Dioxirane with Diazomethylphosphonates; the First Synthesis of a Formylphosphonate Hydrate. J. Chem. Soc. Chem. Commun. 1994, 37–38. [Google Scholar] [CrossRef]
- Cairns, J.; Dunne, C.; Franczyk, T.S.; Hamilton, R.; Hardacre, C.; Stern, M.K.; Treacy, A.; Walker, B.J. The Synthesis and Chemistry of Formylphosphonate. Phosphorus Sulfur Silicon Relat. Elem. 1999, 144, 385–388. [Google Scholar] [CrossRef]
- Samanta, S.; Perera, S.; Zhao, C.-G. Organocatalytic Enantioselective Synthesis of Both Diastereomers of α-Hydroxyphosphinates. J. Org. Chem. 2010, 75, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Dodda, R.; Zhao, C.-G. Silver(I) triflate-catalyzed direct synthesis of N-PMP protected α-aminopropargylphosphonates from terminal alkynes. Org. Lett. 2007, 9, 165–167. [Google Scholar]
- Hirai, T.; Han, L.-B. Palladium-Catalyzed Ibsertion of Isocyanides into P(O)-H Bonds: Selective Formation of phosphinoyl Imines and Bisphosphinoylaminomethanes. J. Am. Chem. Soc. 2006, 128, 4722–4723. [Google Scholar] [CrossRef]
- Piotrowska, D.G. N-Substituted C-diethoxyphosphorylated nitrones as useful synthons for the synthesis of α-aminophosphonates. Tetrahedron Lett. 2006, 47, 5363–5366. [Google Scholar] [CrossRef]
- Piotrowska, D.G.; Balzarini, J.; Glowacka, I.E. Design, synthesis antiviral and cytostatic evaluation of novel isoxalidine nucleotide analogues with a 1,2,3-triazole linker. Eur. J. Med. Chem. 2012, 47, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Seufert, D.; Marmor, R.S.; Hilbert, P. Some reactions of dimethilphosphono-substituted diazoalkanes. (MeO)2P(O)CR transfer to olefins and 1,3-dipolar additions of (MeO)2P(O)CR'. J. Org. Chem. 1971, 36, 1379–1386. [Google Scholar]
- Maehr, H.; Uskokovic, M.R.; Schaffer, C.P. Concise synthesis of dimethyl(2-Oxopropyl)phosphonate and homologation of aldehydes and to alkynes in a tandem process. Synth. Commun. 2009, 39, 299–310. [Google Scholar] [CrossRef]
- Kosobokov, M.D.; Titnyuk, I.D.; Beletskaya, I.P. An expedient synthesis of diethyl diazomethylphosphonate. Mendeleev Commun. 2011, 21, 142–143. [Google Scholar] [CrossRef]
- Neidlein, R.; Keller, H. Syntheses of 1-Benzyloxyaminoalkylphosphonates. Heterocycles 1993, 36, 1925–1932. [Google Scholar] [CrossRef]
- Carter, W.A. Treatment of Human Viral Infection by Doublestranged RNA Combined with Viral Infections. Eur. Patent 286224, 12 October 1988. [Google Scholar]
- Franczyk, T.S., II. Method for Preparing Formylphosphonic Acid. U.S. Patent 7294733, 13 November 2007. [Google Scholar]
- Franczyk, T.S., II. Method of Making Phosphorus-containing Compounds and Products Thereof. U.S. Patent 6864218, 8 March 2005. [Google Scholar]
- Oediger, H.; Lieb, F.; Disselnkotter, H. Phosphonoformaldehyde,a process for its preparation and its use as an intermediate product for the preparation of medicaments. U.S. Patent 4,348,332, 7 September 1982. [Google Scholar]
- Livantsov, M.V.; Boiko, V.I.; Proskurina, M.V.; Lutsenko, I.F. Phosphorylation of orthoformates. J. Gen. Chem. USSR 1982, 52, 811. [Google Scholar]
- Razumov, A.I.; Moskva, V.V. Reaction of dialkylphosphorous acids with orthoformic esters. Z. Obshch. Khim. 1964, 34, 3125–3126. [Google Scholar]
- Razumov, A.I.; Moskva, V.V. Reaction of orthoformates with hydrogen phosphited and hydrogen phosphinites. J. Gen. Chem. USSR 1965, 35, 1599. [Google Scholar]
- Beznosko, B.K.; Usanova, V.M.; Zhuravleva, L.V.; Kharitonov, A.V.; Bondarenko, N.A.; Yarkevich, A.N.; Antoshin, A.E.; Tsvetkov, E.N. Antiinflammatory and analgesic activity of quaternary phosphine oxides. Pharm. Chem. J. 1990, 24, 244–247. [Google Scholar] [CrossRef]
- Gross, H.; Costisella, B. Reaktion von phosphoriger Säure bzw. deren Anhydrid mit Orthoameisensäureestern. J. Prakt. Chem. 1974, 311, 550–556. [Google Scholar]
- Gross, H.; Costisella, B. O,O-and S,S-Acetals of formylphosphoniumsalts. J. Prakt. Chem. 1978, 320, 128–132. [Google Scholar] [CrossRef]
- Gross, H.; Freiberg, J.; Costisella, B. Zur existanz von halogendialkoxyalkanen, eine einfache synthese von dialkoxymethanphosphonaten. Chem. Ber. 1968, 101, 1250–1256. [Google Scholar] [CrossRef]
- Malenko, D.M.; Gololobov, Y.G. New method for synthesis of phosphorylated formals. J. Gen. Chem. USSR 1981, 51, 1214–1215. [Google Scholar]
- Groß, H.; Costisella, B. Zur Umsetzung von Dimethylformamid-dimethyl-acetal mit Phosphortrichlorid. Z. Chem. 1970, 10, 404–405. [Google Scholar]
- Costisella, B.; Gross, H. Zur Reaktion von Orthoameisensäureestern mit Phosphor-III-Verbindungen. J. Prakt. Chem. 1969, 311, 571–576. [Google Scholar] [CrossRef]
- Krokhina, S.S.; Pyrkin, R.I.; Levin, Y.A.; Ivanov, B.E. Effect of triethyl othoformate on some trivalent phosphorus derivatives. Russ. Chem. Bull. 1968, 17, 1349. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Petrosyan, V.S. Synthesis and reactivity of substituted α-carbonylphosphonites and their derivatives. Heteroat. Chem. 2012, 23, 352–372. [Google Scholar] [CrossRef]
- Dietsche, W. Darstellung von C-phosphorylierten Formaldehydacetalen. Liebigs Ann. Chem. 1968, 712, 21–27. [Google Scholar] [CrossRef]
- Moskva, V.V.; Maikova, A.I.; Razumov, A.I. Reaction of orthocarboxylates and acetals with trivalent phosphorus acid chlorides. J. Gen. Chem. USSR 1969, 39, 563–566. [Google Scholar]
- Costisella, B.; Gross, H. Notiz zur Synthese unsymmetrisch substituirten derivate der diätoxymethanphosphonsäure. J. Prakt. Chem. 1977, 319, 343–346. [Google Scholar] [CrossRef]
- Krutskaya, L.V.; Safiulina, O.Z.; Voronina, S.G.; Bryukhovetskaya, L.V.; Tsivunin, V.S. On the reaction of 2-chloro-1,3,2-dioxaphospholanes with 2-ethoxy-1,3-dioxolanes. J. Gen. Chem. USSR 1989, 59, 2408–2411. [Google Scholar]
- Tsivunin, V.S.; Krutskii, L.N.; Ernazarov, M.; Kamai, G.K. Reaction of the diethylamido chloride of ethylphosphinous acid and ethyldichlorophosphine with orthoformic esters. J. Gen. Chem. USSR 1970, 40, 2551–2553. [Google Scholar]
- Costisella, B.; Gross, H. Zur reaktion von P(III) amiden mit orthoameisensäureesternchloriden. Phosphorus Sulfur Silicon Relat. Elem. 1980, 8, 99–103. [Google Scholar] [CrossRef]
- Młоtkowska, B.; Costisella, B.; Gross, H. Eine einfache Methode zur Synthese cyclischer Acetale des Formylphosphonsäreesters. J. Prakt. Chem. 1974, 316, 913–916. [Google Scholar]
- Bennett, S.N.L.; Hall, R.G. New syntheses of arylphosphinic acids from the reaction of ethyl diethoxymethylphosphinate with aryl bromides and phenols. J. Chem. Soc. Perkin Trans. I. 1995, 1145–1151. [Google Scholar] [CrossRef]
- Costisella, B.; Gross, H. Zur reaktivität von Acetalen des Formylphosphonsäureesters. J. Prakt. Chem. 1971, 313, 265–276. [Google Scholar] [CrossRef]
- Niyazymbetov, M.S.; Costisella, B.; Kaitel, I.; Shvatrz, K.K. Cathode-catalyzed transesterification of alkylphosphonates. Russ. Chem. Bull. 1991, 40, 182–186. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Graczyk, P.P.; Wiechorek, M.W.; Bujacz, G. A solution and solid state conformation of 2-diphenylphosphinoyl-1,3-dioxanes. The nature of O-C-P anomeric interactions. Tetrahedron 1992, 48, 4209–4230. [Google Scholar]
- Gross, H.; Böck, C.; Costisella, B.; Gloede, J. Entalkylierung von Phosphonoesthers mit labilen funktionellen Gruppen mittels Trimethylsilyl-Bromid. J. Prakt. Chem. 1978, 320, 344–350. [Google Scholar] [CrossRef]
- Rol’nik, L.Z.; Pastushenko, E.V.; Livantsov, M.V.; Proskurnina, M.V.; Zlotskii, S.S.; Rakhmankulov, D.L. Free-radical reactions of dialkyl formylphosphonate phosphonacetals. J. Gen. Chem. USSR 1983, 53, 1104–1109. [Google Scholar]
- Razumov, A.I.; Gurevich, P.A.; Liorber, B.G.; Borisova, T.B. A study in the series of phosphinic and phosphinous acid derivatives. J. Gen. Chem. USSR 1969, 39, 369–372. [Google Scholar]
- Nguyen-Ba, P.; Lee, N.; Mitchell, H.; Chan, L.; Quimpere, M. Design and synthesis of a novel class nucleotide analogs with anti-HCMV activity. Bioorg. Med. Chem. Lett. 1998, 8, 3555–3560. [Google Scholar]
- Kruse, C.G.; Broekhof, N.L.Y.M.; Wijsman, A.; van der Gen, A. Synthetic application of 2-chloro-1,3-dithiane preparation of ketene dithioacetals. Tetrahedron Lett. 1977, 18, 885–888. [Google Scholar] [CrossRef]
- Ishikawa, K.; Akiba, K.; Inamoto, N. Synthesis of 1, 4-benzodithiofulvenes via Wittig reaction. Tetrahedron Lett. 1976, 17, 3695–3698. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Graczyk, P.P.; Wieczorek, M.W. Conformational preference in 1,3-dithianes containing 2-phosphoryl, -(thiophosphoryl), and (selenophosphoryl)groups. Chemical and crystallographic implications of the nature of the anomeric effect. J. Org. Chem. 1994, 59, 1672–1679. [Google Scholar]
- Mikołajczyk, M.; Łuczak, T.; Graczyk, P.P.; Wieczorek, M.W.; Błaszczyk, Y.; Bujacz, G.D.; Majzner, W.R. Solid state conformation of the anomeric effect in 2-phosphoryl, 2- thiophosphoryl and 2-selenophosphoryl-substituted 1,3-dithiolans. J. Organomet. Chem. 1997, 536–537, 355–360. [Google Scholar]
- Aggarwal, V.K.; Barnell, J.K.; Worrall, J.M.; Alexander, R. Highly diastereoselective epoxidation of ketene dithioacetal dioxides: A new approach to asymmetric synthesis of α-amino amides. J. Org. Chem. 1998, 63, 7128–7129. [Google Scholar] [CrossRef] [PubMed]
- Mikołajczyk, M.; Grejszczak, S.; Chefchyńska, A.; Zatorski, A. Addition of elemental sulfur to phosphonate carbanions and its application for synthesis of α-phosphoryl organosulfur compounds, synthesis of oaromatic ketones. J. Org. Chem. 1979, 44, 2967–2972. [Google Scholar]
- Mikołajczyk, M.; Bałczewski, P.; Grejszczak, S. Sulphenylation of phosphonates. A facile synthesis of α-phosphoryl sulfides and S,S-acetals of oxomethanephosphonates. Synthesis 1980, 127–129. [Google Scholar]
- Grayson, J.I.; Warren, S. Acyl Anion Equivalents: Synthesis of Ketones and Enones from α-Phenylthioalkylphosphine Oxides. J. Chem. Soc. Perkin Trans. I 1977, 2263–2272. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Bałczewski, P. Synthesis and reactivity of diethyl(methylthio)(trimethylsilyl)methylphosphonate. Synthesis 1989, 101–106. [Google Scholar]
- Makomo, H.; Masson, S.; Saquet, M. Reduction of phosphonodithioformates: Syntheses of α-phosphoryl thiols and hemidithioacetals. Tetrahedron Lett. 1994, 50, 10277–10288. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Mikina, M.; Graczyk, P.P.; Bałczewski, P. Synthesis of dithio- and diselenoacetals of formylphosphonates. Synthesis 1996, 1232–1238. [Google Scholar]
- Yuaristi, E.; Cordillo, B.; Valle, L. Relative reactivity of 2-diphenylphosphinoil- and 2-diphenylthiophosphinoil-2-[1,3]-dithianyllithium as reagents Wittig-Horner. Tetrahedron 1986, 42, 1963–1970. [Google Scholar] [CrossRef]
- Yuaristi, E.; Valle, L.; Valenzuela, B.A.; Aguilar, M.A. S-C-P anomeric interactions. 4. Conformational analys of 2-(diphenylphosphinoil)-1,3-dithyane. J. Am. Chem. Soc. 1996, 108, 2000–2005. [Google Scholar]
- Iorga, B.; Mouriès, V.; Savigniac, P. Carbanionic displacement reactions of phosphorus. Synthesis and reactivity of 5,5-dimethyl-2-oxo-2-(1,1-dithian-2-yl)-1, 3, 2-dioxaphosphorinane. Bull. Chem. Soc. Fr. 1997, 134, 891–985. [Google Scholar]
- El-Wareth, A.; Sarhan, A.O.; Murakami, M.; Izumi, T. Synthesis of functionalized compounds related to π-extended tetrathiafulvenes with quinonoidal bearing a ferrocene moiety. Monatsh. Chem. 2002, 133, 1055–1066. [Google Scholar]
- Costisella, B.; Gross, H. 1-Trimethylammonium-1-diethylphosphono-1-cyanomethylid, ein stabilеs N-Ylid. J. Prakt. Chem. 1987, 324, 545–549. [Google Scholar] [CrossRef]
- Costisella, B. 1H, 13C, 31P, 15N, 17O-NMR investigations of α-substituted aminophosphonates. Phosphorus Sulfur Silicon Relat. Elem. 1990, 51/52, 226. [Google Scholar]
- Prishchenko, A.A.; Livantsov, M.V.; Petrosyan, V.S. Reaction of some CH acids with dimethylformamide dimethylacetal. J. Gen. Chem. USSR 1993, 63, 1326–1327. [Google Scholar]
- Gross, H.; Costisella, B.; Gnauk, T.; Brenneke, L. Derivate der Aminomethan-bis-phosphonsäure. J. Prakt. Chem. 1976, 318, 116–126. [Google Scholar] [CrossRef]
- Nesterov, L.V.; Krepysheva, N.E.; Aleksandrova, N.A. Reaction of dimethylformamide dimethylacetal with silyl phosphites. J. Gen. Chem. USSR 1989, 59, 641. [Google Scholar]
- Prishchenko, A.A.; Livantsov, M.V.; Boganova, N.V.; Zhutskii, P.V.; Lutsenko, I.F. Synthesis of tetraalkyl dimethylaminomethylenediphosphorus-containing acids. J. Gen. Chem. USSR 1989, 59, 2132–2133. [Google Scholar]
- Prishchenko, A.A.; Livantsov, M.V.; Petrosyan, V.S. New types of aminomethyl organophosphorus compounds. J. Gen. Chem. USSR 1994, 63, 1181–1193. [Google Scholar]
- Degenhardt, C.R. Use of tetraethyl dimethylaminomethylene diphosphonate in the synthesis of benzothiophene-2-acetic acid and other carboxylic acids. Synth. Commun. 1982, 12, 415–421. [Google Scholar] [CrossRef]
- Gross, H.; Costisella, B. Bis(diethoxyphosphinyl)(trimethylammonio)-methylide, a Stable N-Ylide. Angew. Chem. Int. Ed. Engl. 1968, 7, 463. [Google Scholar] [CrossRef]
- Gross, H.; Costisella, B.; Bürger, W. Synthese von Alkylphosphonbetainen und stabilen N-Ylyden aus α-Trimethylammoniummethylphosphonaten. J. Prakt. Chem. 1969, 311, 563–570. [Google Scholar] [CrossRef]
- Monenschein, H.; Dräger, G.; Alexander, Jung; Kirschning, A. Asymmetric Nucleophilic Acylation of Aldehydes via 1, 1-Heterodisubstituted Alkenes. Chem. A Eur. J. 1999, 5, 2270–2280. [Google Scholar]
- Kirschning, A.; Kujat, C.; Luiken, S.; Schaumann, E. Small and Versatile—Formyl Anion and Dianion Equivalents. Eur. J. Org. Chem. 2007, 2387–2400. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Midura, W.; Grejszczak, S. Synthesis of mono- and 1, 4-dicarbonyl compounds based on the oxygenation of phosphonate carbanions. Synthesis of dihydrojasmone, allethone and methylenomycin B. Tetrahedron Lett. 1984, 25, 2489–2492. [Google Scholar]
- Morita, T.; Okamoto, Y.; Sackurai, H. Dealkylation reaction of acetals, phosphonate, and phosphate esters with chlorotrimethylsilane/metal halide reagent in acetonitrile, and its application to the synthesis of phosphonic acids and vinyl phosphonates. Bull. Chem. Soc. Jpn. 1981, 54, 267–273. [Google Scholar] [CrossRef]
- Morita, T.; Okamoto, Y.; Sackurai, H. A convenient dealkylation of dialkyl phosphonates by chlorotrimethylsilane in the presence of sodium iodide. Tetrahedron Lett. 1978, 28, 2523–2526. [Google Scholar] [CrossRef]
- Gross, H.; Keitel, I. Heterosubstituierte olefine durch horner-reaktion mit α-substituierten Phosphonaten. Z. Chem. 1982, 22, 117–126. [Google Scholar]
- Van Schaik, T.A.M.; Henzen, A.V.; van der Gen, A. A Horner-Wittig solution to the synthesis of ketene, O, O-acetals. Tetrahedron Lett. 1983, 24, 1303–1306. [Google Scholar]
- Streitwieser, A., Jr.; Juaristi, E. Carbon acidity. Equilibrium ion pair acidies of some phosphorus-substituted carbon acids. J. Org. Chem. 1982, 47, 768–770. [Google Scholar]
- Afarinkia, K.; Faller, A.; Twist, A.J. A new synthesis of α-ketophosphonates. Synthesis 2003, 357–360. [Google Scholar] [CrossRef]
- Coutrot, P.; Grison, C.; Lecouvey, M. Preparation of the phosphonic acid analogue of 3-deoxy-d-manno-2-octulosonic acid (KDO). Tetrahedron Lett. 1996, 37, 1595–1598. [Google Scholar] [CrossRef]
- Horner, L.; Hoffmann, H.; Wippel, H.S. Phosphinoxide als Olefinierungsreagenzien. Chem. Ber. 1958, 91, 61–63. [Google Scholar] [CrossRef]
- Horner, L.; Hoffmann, H.; Wippel, H.S.; Klahre, G. Phosphinoxide als olefinierungsreagenzien. Chem. Ber. 1959, 92, 2499–2505. [Google Scholar] [CrossRef]
- Wittig, G.; Schöllkopf, U. Über Triphenyl-phosphin-methylene als olefinbildende Reagenzien (I Mitteilung). Chem. Ber. 1954, 87, 1318–1330. [Google Scholar] [CrossRef]
- Wardsworth, W.S.; Emmons, W.D. The Utility of Phosphonate Carbanions in Olefins Synthesis. J. Am. Chem. Soc. 1961, 83, 1733–1738. [Google Scholar] [CrossRef]
- Martin, S.F. Synthesis of Aldehydes, Ketones, and Carboxylic Acids from Lower Carbonyl Compounds by C-C Coupling Reactions. Synthesis 1979, 633–665. [Google Scholar] [CrossRef]
- Korotchenko, V.N.; Nenaidenko, V.G.; Balenkova, E.S.; Shastin, A.V. Olefination of carbonyl compounds. The newest and classical methods. Russ. Chem. Rev. 2004, 73, 957–989. [Google Scholar]
- Costisella, B.; Keitel, I.; Gross, H. α-Phosphononoenamine und Acyl phosphonate durch Horner-Olefinierung. Tetrahedron 1981, 37, 1227–1232. [Google Scholar] [CrossRef]
- Gross, H.; Costisella, B.; Schick, H. γ-Butyrolactone aus metallierten Enamine. Tetrahedron 1984, 40, 733–736. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Greene, L.M.; Bergin, O.; Fichet, J.-B.; McCabe, T.; Lloyd, D.G.; Zisterer, D.M.; Meegan, M.J. Synthesis, evaluation and structural studies of antiproliferative tubulin-targeting azetidin-2-ones. Bioorg. Med. Chem. 2011, 19, 2306–2325. [Google Scholar]
- Mikołajczyk, M.; Grzejszczak, S.; Zatorski, A.; Młotkowska, B. A new general synthesis of ketene thioacetals. Tetrahedron Lett. 1976, 31, 2731–2734. [Google Scholar]
- Mikołajczyk, M.; Bałczewski, P. Diverse reactivity of α-carbanions derived from α-phosphoryl dithioacetals and α-phosphoryl sulphides towards α, β-unsaturated carbonyl compounds. A general synthesis of conjugated ketene dithioacetals. Tetrahedron 1992, 8, 8697–8710. [Google Scholar]
- Ceruti, M.; Degani, I.; Fochi, R. A new synthetic application of 1, 2-benzodithiolium cations: synthesis of aldehydes by 1-carbon homologation of carbonyl compounds. Synthesis 1987, 79–83. [Google Scholar]
- Hanessian, S.; Maji, D.K.; Govindan, S.; Matera, R.; Tintelnot-Blomley, M. Substrate-Controlled and Organocatalytic Asymmetric Synthesis of Carbocyclic Amino Acid Dipeptide Mimetics. J. Org. Chem. 2010, 75, 2861–2876. [Google Scholar] [CrossRef] [PubMed]
- Hanessian, S.; Desilets, D.; Rancourt, G.; Fortin, R. The total stereocontrolled synthesis of a chemical precursor to (+)-thienamycin. A formal synthesis of the antibiotic. Can. J. Chem. 1982, 60, 2292–2294. [Google Scholar]
- Moore, A.J.; Bryce, M.R. Generation and trapping of phosphorus stabilized 4,5-ethylenedithio-1,3-dithiol-2-ide carbanions: Synthesis of ethylenedithio-1,3-dithiafulvalenes. Synthesis 1991, 26–28. [Google Scholar] [CrossRef]
- Misaki, Y.; Nishikawa, H.; Kawakami, K.; Uehara, T.; Yamabe, T. Bis(2-methylidene-1,3-dithiolo[4,5-d])tetrathiafulvalene (BDT-TTF): A tetrathiafulvalene condensed with 1,3-dithiol-2-ylidene moieties. Tetrahedron Lett. 1992, 33, 4321–4324. [Google Scholar] [CrossRef]
- Heynderick, A.; Kaou, A.M.; Moustrou, C.; Samat, A.; Guglielmetti, R. Synthesis and photochromic behaviour of new Dipyrrolylperfluorocyclopentenes. New J. Chem. 2003, 27, 1425–1432. [Google Scholar] [CrossRef]
- Seebach, D.; Bürstinghaus, R. S-methyl thiocarboxylates from aldehydes and ketones through ketene thioacetals. Reductive nucleophile thiocarboxylation. Synthesis 1975, 461–462. [Google Scholar]
- Theil, F.; Costisella, B.; Mahrwald, R.; Gross, H.; Schick, H.; Schwarz, S. Prostaglandins and Prostaglandin Intermediates. XVII. Synthesis of the Main Metabolite of the PGF2α Analogue Cloprostenol. J. Prakt. Chem. 1986, 325, 435–440. [Google Scholar]
- Theil, F.; Costisella, B.; Groß, H.; Schick, H.; Schwarz, S. A Three-step Procedure for the Conversion of γ-Lactones into δ-Lactones. J. Chem. Soc. Perkin Trans. I 1987, 2469–2472. [Google Scholar] [CrossRef]
- Zimmerman, H.E.; Baker, M.R.; Bottner, R.C.; Morrissey, M.M.; Murphy, S. Photochemistry of some allenic counterparts of cyclohexenones and 2, 5-cyclohexadienones. J. Am. Chem. Soc. 1993, 115, 459–466. [Google Scholar] [CrossRef]
- Monenschein, H.; Brünjes, M.; Kirschning, A. Lithiated dimethoxymethyl diphenyl phosphine oxide. A versatile formiate carbanion equivalent. Synlett 2002, 525–527. [Google Scholar]
- Okada, H.; Mori, T.; Saikawa, Y.; Nakata, M. Formation of α-hydroxyketones via irregular Wittig reaction. Tetrahedron Lett. 2009, 50, 1276–1278. [Google Scholar] [CrossRef]
- Kirschning, A.; Dräger, G.; Yung, A. A new asymmetric formylation of aldehydes. Angew. Chem. Int. Ed. Engl. 1997, 36, 253–255. [Google Scholar] [CrossRef]
- Becker, H.; Sharpless, B. A new ligand class for the asymmetric dihydroxylation of olefins. Angew. Chem. Int. Ed. Engl. 1996, 35, 448–451. [Google Scholar] [CrossRef]
- Wittenberg, R.; Beier, C.; Dräger, G.; Jas, G.; Jasper, C.; Monenschein, H.; Kirschning, A. Towards the total synthesis of tonantzitlolone––Preparation of key fragments and the complete carbon backbone. Tetrahedron Lett. 2004, 45, 4457–4460. [Google Scholar] [CrossRef]
- Morgalyuk, V.P.; Strelkova, T.V.; Nifant’ev, E.E. Synthesis of polyfunctionalized methylphosphine oxides. Russ. Chem. Bull. 2012, 61, 380–385. [Google Scholar] [CrossRef]
- Schrader, T.; Kober, R.; Steglich, W. Synthese von 1-aminophosphonsäure-derivaten über acyliminophosphonsäure-ester. Synthesis 1986, 372–375. [Google Scholar] [CrossRef]
- Costisella, B.; Keitel, I.; Ozegowski, S. α-Substituierte Phosphonate. 69. Diastereoselektivität bei der Knüpfung der Phosphor-kohlenstoffbindung. Phosphorus Sulfur Silicon Relat. Elem. 1993, 84, 115–120. [Google Scholar]
- Marson, C.M. Reactions of carbonyl compounds with (monohalo) methyleniminium salts (Vilsmeier reagents). Tetrahedron 1992, 48, 3659–3726. [Google Scholar] [CrossRef]
- Winter, A.; Risch, N. The vinylogous mannich reaction: An efficient access to substituted nicotinonitriles. Synlett 2003, 13, 1959–1964. [Google Scholar]
- Morgalyuk, V.P.; Petrovskii, P.V.; Lysenko, K.A.; Nifant’ev, E.E. Synthesis of polyfunctionalyzed methylphosphine oxides. Russ. Chem. Bull. 2009, 58, 248–250. [Google Scholar] [CrossRef]
- Morgalyuk, V.P.; Strelkova, T.V. Stepwise scheme for the reaction of chlorodiphenylphosphine with N,N-dialkylformamides in the presence of NaI. Russ. J. Gen. Chem. 2011, 81, 2096–2101. [Google Scholar] [CrossRef]
- Morgalyuk, V.P.; Strelkova, T.V.; Nifant’ev, E.E. Reactivity of Chloro(dialkylamino)(diphenylphosphinoyl)methanes. Bull. Chem. Soc. Jpn. 2012, 85, 93–100. [Google Scholar] [CrossRef]
- Schrader, T.; Steglich, W. Phosphoranaloge von Aminosäuren IV. Synthesen ungevönlicher 1-Aminophosphonsäuren über Diels-Alder-Reaktionen von (N-Acyliminomethyl)phosphonsäurendiethylestern. Synthesis 1990, 1153–1156. [Google Scholar]
- Boukallaba, K.; Elachqar, A.; El Hallaoui, A.; Alami, A.; El Hajji, S.; Labriti, B.; Atmani, A. Synthesis of α-Heterocyclic α-Aminophosphonates, Part II: Morpholine, Piperidine, Pyrrolidine, Tetrahydrofurylmethylamine, N-Benzyl-N-Methylamine, and Aniline Derivatives. Phosphorus Sulfur Silicon Relat. Elem. 2007, 182, 1045–1052. [Google Scholar] [CrossRef]
- Elachqar, A.; El Hallaouiq, A.; Roumestant, M.L.; Viallefont, P. Synthesis of Heterocyclic α-Aminophosphonic Acids. Synth. Commun. 1994, 24, 1279–1286. [Google Scholar] [CrossRef]
- Achamlale, S.; Mabrouk, H.; Elachqar, A.; El Hallaoui, A.; El Hajji, S.; Alami, A.; Bellan, J.; Mazières, M.R.; Wolf, J.G.; Pierrot, M. Synthesis and Thermal Isomerization of Carboxylic and Phosphonic α-Aminoesters Substituted With a Triazole Ring. Phosphorus Sulfur Silicon Relat. Elem. 2007, 182, 357–367. [Google Scholar] [CrossRef]
- Gallagher, M.J.; Honegger, H. Organophosphorus intermediates. VI. The acid-catalysed reaction of trialkyl orthoformates with phosphinic acid. Aust. J. Chem. 1980, 33, 287–294. [Google Scholar]
- Fougère, C.; Guénin, E.; Hardouin, J.; Lecouve, M. Rapid and Efficient Synthesis of Unsymmetrical Phosphinic Acids R'P(O)OHR''. Eur. J. Org. Chem. 2009, 6048–6054. [Google Scholar]
- Baylis, E.K. 1,1-Diethoxyethylphosphinates and Phosphinites. Intermediates for the Synthesis Functional Phosphorus Acids. Tetrahedron Lett. 1995, 36, 9385–9388. [Google Scholar]
- Hall, R.G.; Riebli, P. Preparation of New Phosphine Oxides: Synthesis of an Analogue of Muscarinic Antagonists. Synlett 1999, 1633–1635. [Google Scholar] [CrossRef]
- Buckler, S.A.; Epstein, M. The preparation and reactions of primary phosphine oxides. Tetrahedron 1962, 3, 1221–1230. [Google Scholar] [CrossRef]
- Zhang, D.; Yuan, C. A concise and first synthesis of α-aminophosphinates with two stereogenic atoms leading to optically pure α-amino-H-phosphinic Acids. Chem. A Eur. J. 2008, 14, 6049–6052. [Google Scholar] [CrossRef]
- Alstermark, C.; Amin, K.; Dinn, S.R.; Elebring, T.; Fjellström, O.; Fitzpatrick, K.; Geiss, W.B.; Gottfries, J.; Guzzo, P.R.; Harding, J.P.; et al. Synthesis and pharmacological evaluation of novel γ-aminobutyric acid type B (GABAB) receptor agonists as gastroesophageal reflux inhibitors. J. Med. Chem. 2008, 51, 4315–4320. [Google Scholar]
- Froestl, W.; Mickel, S.J.; von Sprecher, G.; Diel, P.J.; Hall, R.G.; Maier, L.; Strub, D.; Melillo, V.; Baumann, P.A.; Bernasconi, R.; et al. Phosphinic acid analogues of GABA. 2. Selective, orally active GABAB antagonists. J. Med. Chem. 1995, 38, 3313–3331. [Google Scholar]
- Seebach, D. Methoden und möglichkeiten der nucleophilen acylierung. Angew. Chem. Int. Ed. Engl. 1969, 81, 690–700. [Google Scholar] [CrossRef]
- Seebach, D. Methods of reactivity umpolung. Angew. Chem. Int. Ed. Engl. 1979, 18, 39–336. [Google Scholar]
- Dodda, R.; Zhao, C.-G. Organocatalytic highly enantioselective synthesis of secondary α-hydroxyphosphonates. Org. Lett. 2006, 8, 4911–4914. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Morgalyuk, V.P. Chemistry of Phosphorylated Formaldehyde Derivatives. Part I. Molecules 2014, 19, 12949-13009. https://doi.org/10.3390/molecules190912949
AMA Style
Morgalyuk VP. Chemistry of Phosphorylated Formaldehyde Derivatives. Part I. Molecules. 2014; 19(9):12949-13009. https://doi.org/10.3390/molecules190912949
Chicago/Turabian StyleMorgalyuk, Vasily P. 2014. "Chemistry of Phosphorylated Formaldehyde Derivatives. Part I" Molecules 19, no. 9: 12949-13009. https://doi.org/10.3390/molecules190912949