Synthesis and Biological Evaluation of New Pyridone-Annelated Isoindigos as Anti-Proliferative Agents

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
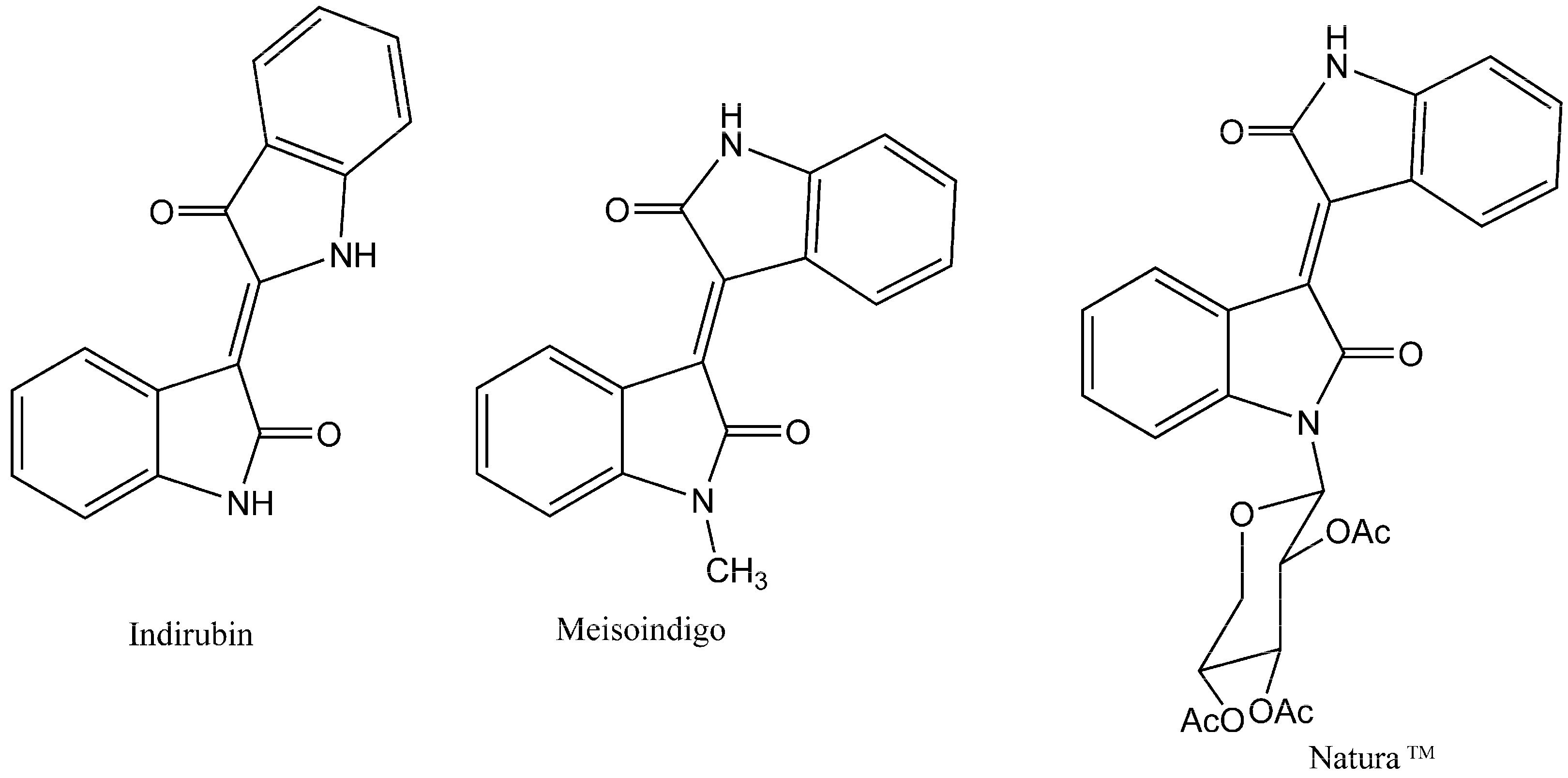
:1. Introduction


2. Results and Discussion
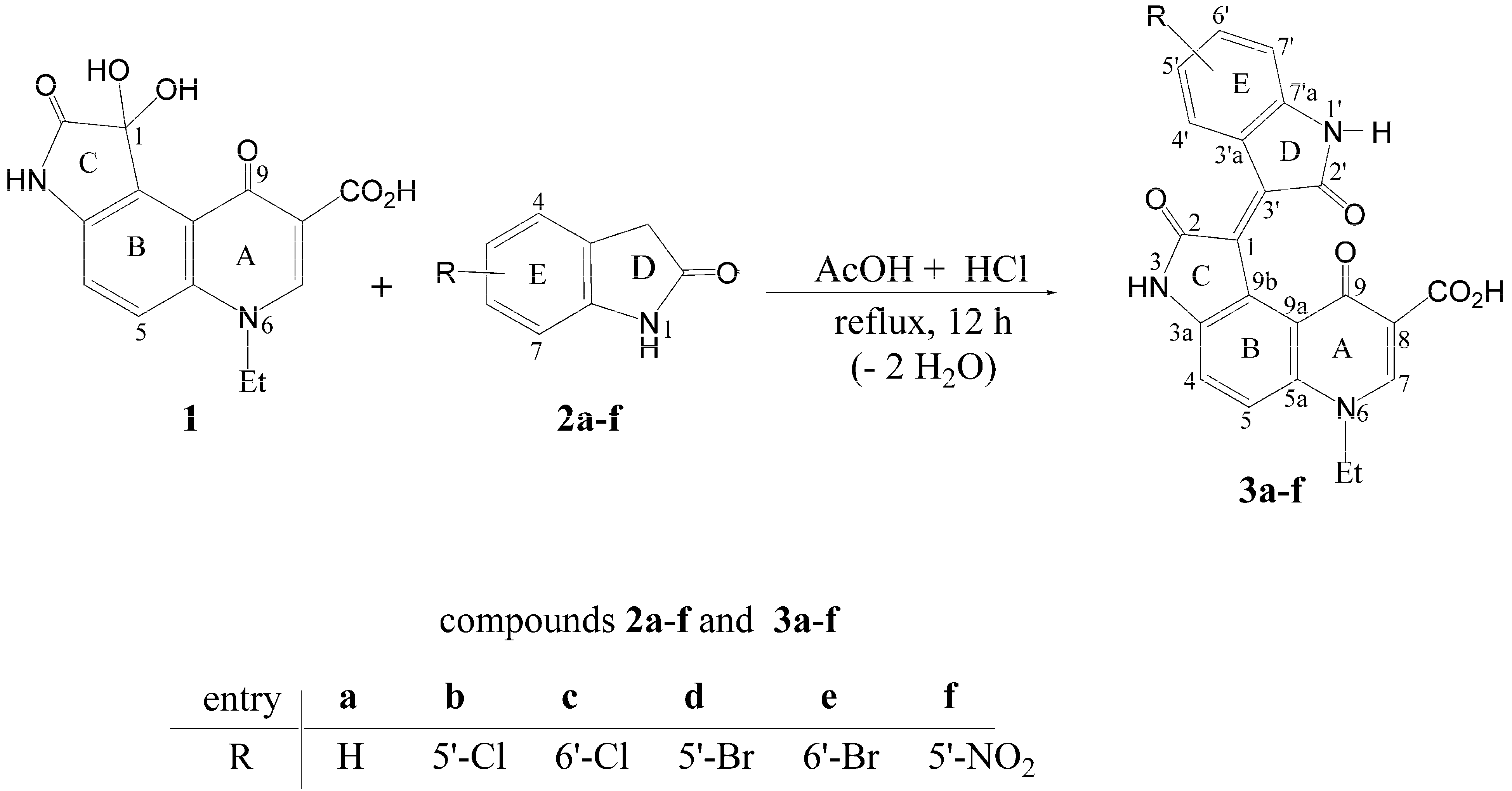
2.1. Chemistry
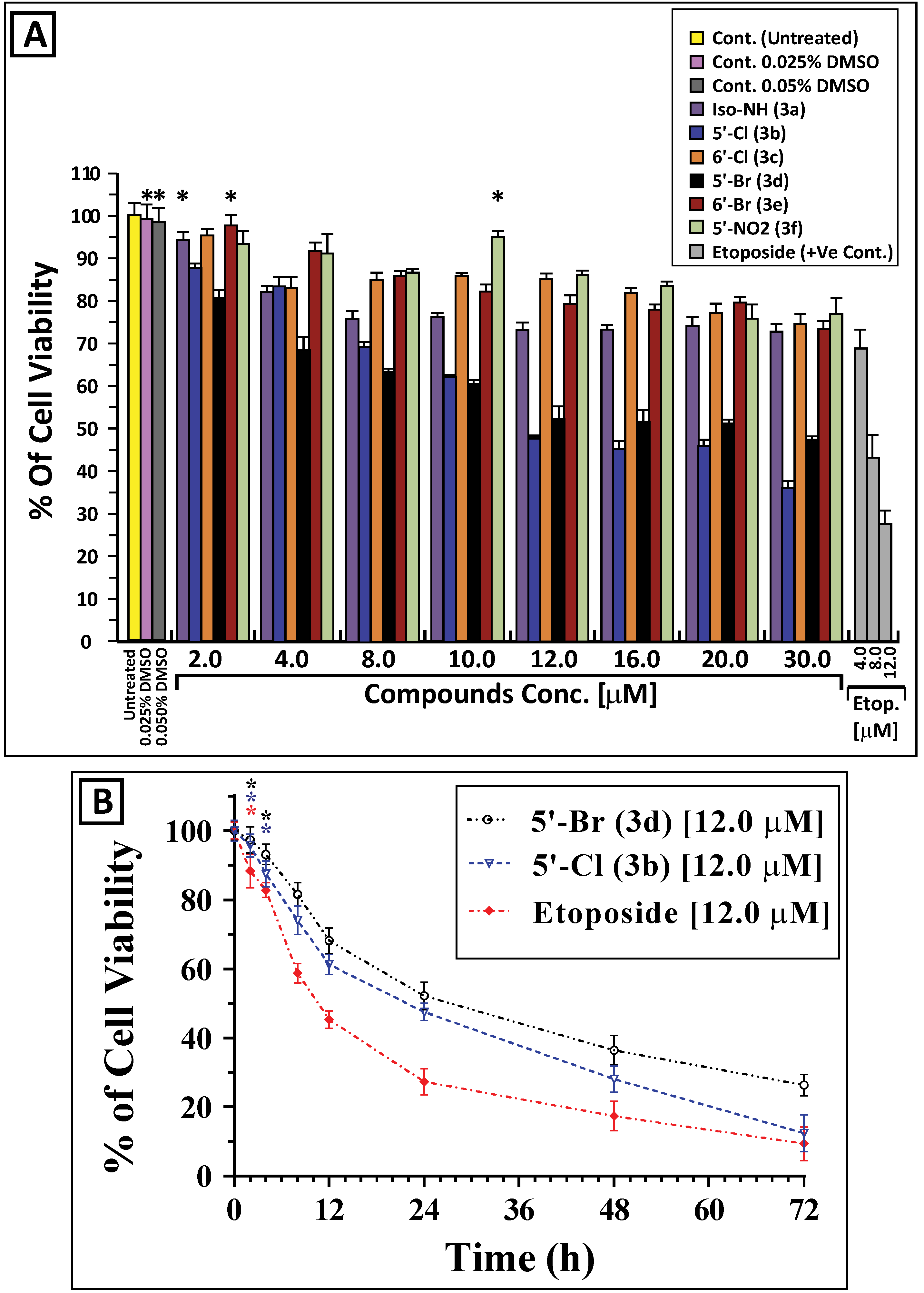
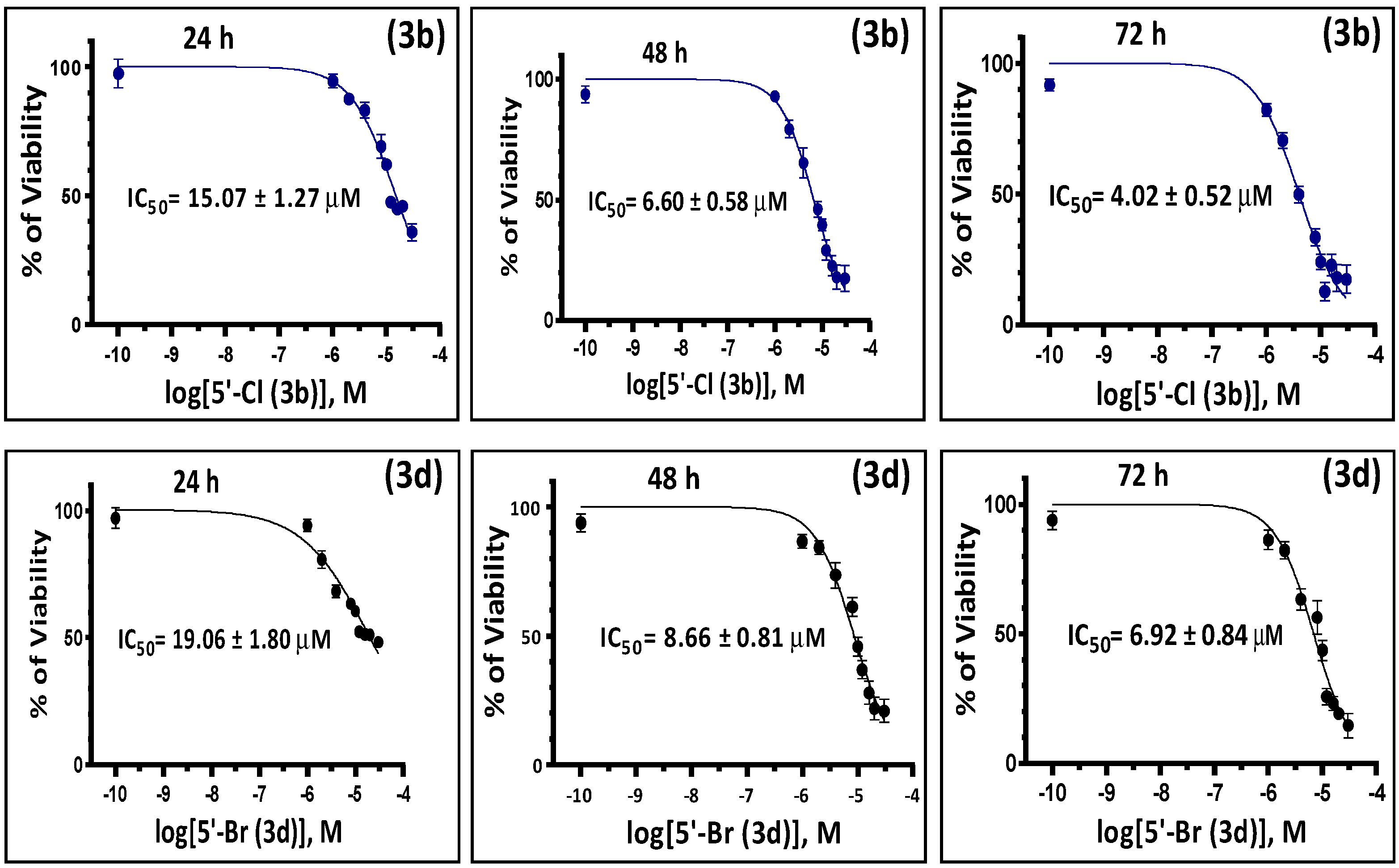
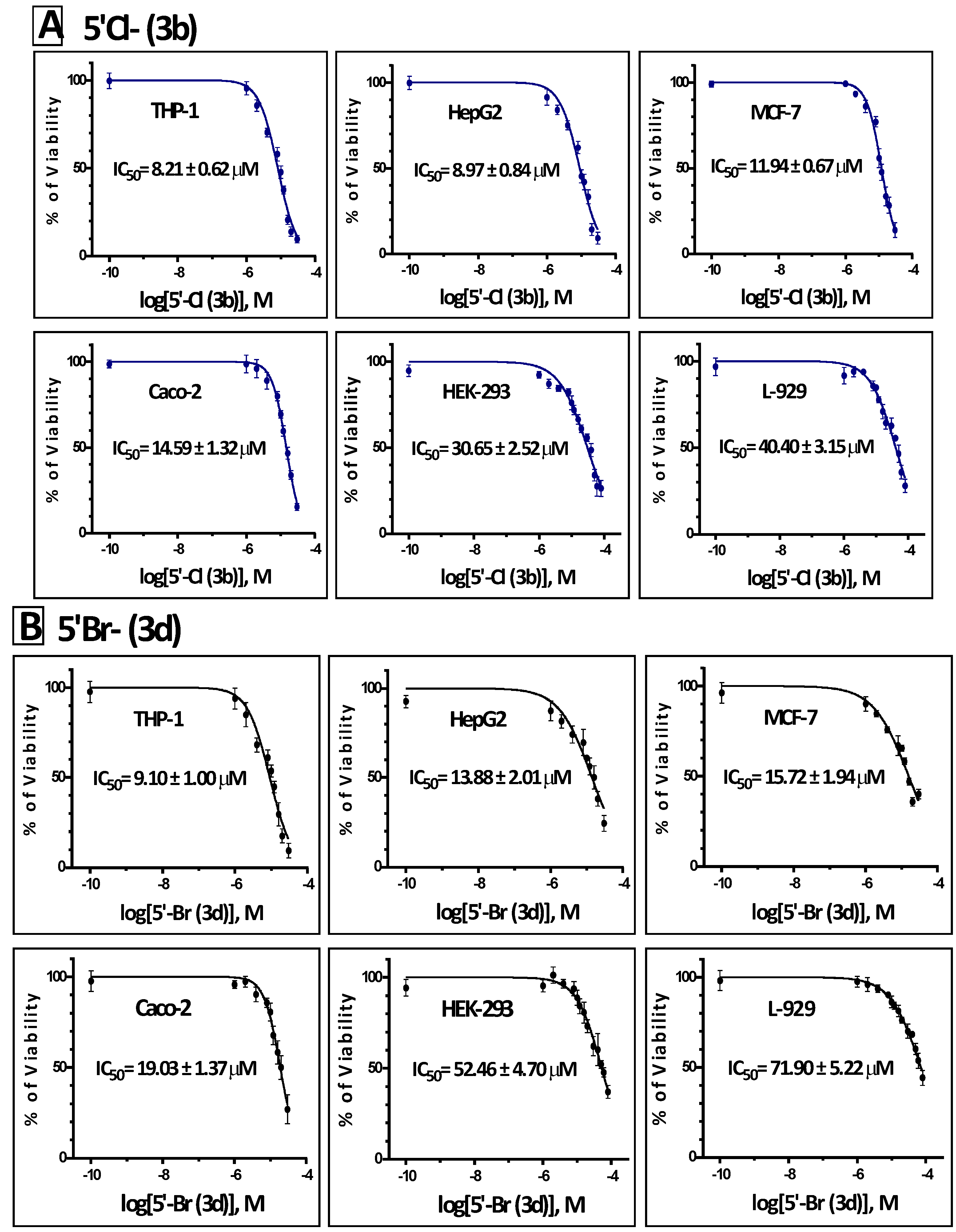
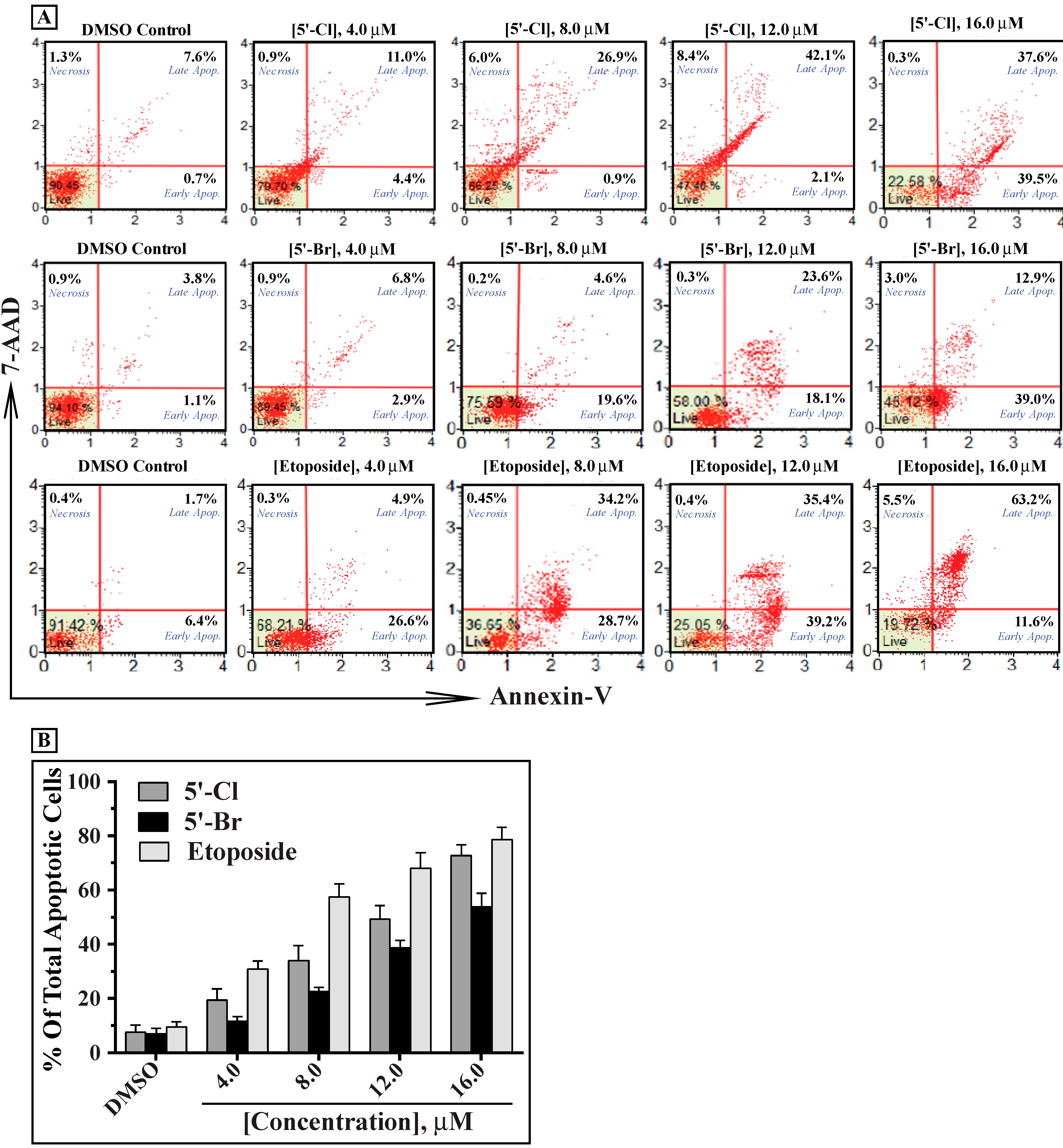
2.2. In Vitro Antiproliferative Activity of the Synthesized Isoindigoid Derivatives 3a–f




3. Experimental Section
3.1. Chemicals
3.2. Instrumentation
3.3. Synthesis of 6-Ethyl-2,9-dioxo-2,3,6,9-tetrahydro-1H-pyrrolo[3,2-f]quinoline-8-carboxylic Acid (1)
3.4. General Procedure for the Synthesis of Pyridone-Annelated Isoindigos 3a–f
3.5. Cell Culture Conditions
3.6. Cell Viability (Antiproliferative) Assay
3.7. Analysis of Apoptosis
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001, 293, 876–880. [Google Scholar] [CrossRef]
- Shah, N.P.; Nicoll, J.M.; Nagar, B.; Gorre, M.E.; Paquette, R.L.; Kuriyan, J.; Sawyers, C.L. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor Imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2002, 2, 117–125. [Google Scholar] [CrossRef]
- Deng, B. Direct colorimetric method for determination of indigo and indirubin in Qingdai. Zhongcaoyao 1986, 17, 163–164. [Google Scholar]
- Hoessel, R.; Leclerc, S., Endicott; Nobel, M.E.; Lawrie, A.; Tunnah, P.; Leost, M.; Damiens, E.; Marie, D.; Marko, D.; et al. Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat. Cell. Biol. 1999, 1, 60–67. [Google Scholar] [CrossRef]
- Tang, W.; Eisenbrand, G. Qingdai. In Chinese Drugs of Plant Origin: Chemistry, Pharmacology and Use in Traditional and Modern Medicine; Springer: Heidelberg, Germany, 1992; pp. 805–812. [Google Scholar]
- Xiao, Z.; Hao, Y.; Liu, B.; Qian, L. Indirubin and meisoindigo in the treatment of chronic myelogenous leukemia in China. Leukemia Lymphoma 2002, 43, 1763–1768. [Google Scholar] [CrossRef]
- Marko, D.; Schatzle, S.; Friedel, A.; Genzlinger, A.; Zankl, H.; Meijer, L.; Eisenbrand, G. Inhibition of cyclin-dependent kinase 1 (CDK1) by indirubin derivatives in human tumour cells. Brit. J. Cancer 2001, 84, 283–289. [Google Scholar] [CrossRef]
- Leclerc, S.; Garnier, M.; Hoessel, R.; Marko, D.; Bibb, J.A.; Snyder, G.L.; Greengard, P.; Biernat, J.; Wu, Y.Z.; Mandelkow, E.M.; et al. Indirubins inhibit glycogen synthase kinase-3 beta and CDK5/P25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s disease—A property common to most cycline-dependent kinase inhibitors. J. Biol. Chem. 2001, 276, 251–260. [Google Scholar] [CrossRef]
- Liu, X.M.; Wang, L.G.; Li, H.Y.; Ji, X.J. Induction of differentiation and down-regulation of c-myb gene expression in ML-1 human myeloblastic leukemia cells by the clinically effective anti-leukemia agent meisoindigo. Biochem. Pharmacol. 1996, 51, 1545–1551. [Google Scholar] [CrossRef]
- Moon, M.J.; Lee, S.K.; Lee, J.W.; Song, W.K.; Kim, S.W.; Kim, J.I.; Cho, C.; Choi, S.J.; Kim, Y.C. Synthesis and structure-activity relationships of novel indirubin derivatives as potent anti-proliferative agents with CDK2 inhibitory activities. Bioorg. Med. Chem. 2006, 14, 237–246. [Google Scholar] [CrossRef]
- Zuo, M.; Li, Y.; Wang, H.; Zhou, J.; Li, H.; Liu, H.; Xin, H.; Zhang, S.; Chen, X. The antitumor activity of meisoindigo against human colorectal cancer HT-29 cells in vitro and in vivo. J. Chemotherapy 2008, 20, 728–733. [Google Scholar] [CrossRef]
- Cooperative Group of Phase III Clinical Trial on Meisoindigo. Phase III clinical trial on meisoindigo in the treatment of chronic myelogenous leukemia. Chin. J. Hematol. 1997, 18, 69–72. [Google Scholar]
- Xiao, Z.; Wang, Y.; Lu, L.; Li, Z.; Peng, Z.; Han, Z.; Hao, Y. Anti-angiogenesis effects of meisoindigoon chronic myelogenous leukemia in vitro. Leukocyte Res. 2006, 30, 54–59. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.; Chen, R. Derivatives of Isoindigo, Indigo and Indirubin and Methods of Treating Cancer. U.S. Patent 6,933,315 B2, 20 May 2003. [Google Scholar]
- Wang, L.; Liu, X.; Chen, R. Derivatives of Isoindigo, Indigo and Indirubin and Use in Treating Cancer. CA 2469649 A1, 26 June 2003. [Google Scholar]
- Sassatelli, M.; Bouchikhi, F.; Messaoudi, S.; Anizon, F.; Debiton, E.; Barthomeuf, C.; Prudhomme, M.; Moreau, P. Synthesis and antiproliferative activities of diversely substituted glycosyl-isoindigo derivatives. Eur. J. Med. Chem. 2006, 41, 88–100. [Google Scholar] [CrossRef] [Green Version]
- Al-As’ad, R.M.; El-abadelah, M.M.; Sabri, S.S.; Zahra, J.A.; Voelter, W. Synthesis of model 6-ethyl-1,2,9-trioxopyrrolo[3,2-f]quinoline-8-carboxylic acid. Z. Naturforsch. 2013, 68b, 700–708. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Ji, X.J.; Liu, X.M.; Li, K.; Chen, R.H.; Wang, L.G. Pharmacological studies of meisoindigo: Absorption and mechanism of action. Biomed. Environ. Sci. 1991, 4, 332–337. [Google Scholar]
- Van Maanen, J.M.; Retèl, J.; de Vries, J.; Pinedo, H.M. Mechanism of action of antitumor drug etoposide: A review. J. Natl. Cancer Inst. 1988, 80, 1526–1533. [Google Scholar] [CrossRef]
- Vine, K.L.; Locke, J.M.; Ranson, M.; Benkendorff, K.; Pyne, S.G.; Bremner, J.B. In vitro cytotoxicity evaluation of some substituted isatin derivatives. Bioorg. Med. Chem. 2007, 15, 931–938. [Google Scholar] [CrossRef]
- Wee, X.K.; Yang, T.; Go, M.L. Exploring the anticancer activity of functionalized isoindigos: Synthesis, drug-like potential, mode of action and effect on tumor-induced xenografts. ChemMedChem 2012, 7, 777–791. [Google Scholar] [CrossRef]
- Damiens, E.; Baratte, B.; Marie, D.; Eisenbrand, G.; Meijer, L. Anti-mitotic properties of indirubin-3'-monoxime, a CDK/GSK-3 inhibitor: Induction of endoreplication following prophase arrest. Oncogene 2001, 20, 3786–3797. [Google Scholar] [CrossRef]
- Xu, J.; Dai, X.; Liu, H.; Guo, W.; Gao, J.; Wang, C.; Li, W.; Yao, Q. A novel 7-azaisoindigo derivative-induced cancer cell apoptosis and mitochondrial dysfunction mediated by oxidative stress. J. Appl. Toxicol. 2011, 31, 164–172. [Google Scholar]
- Bouchikhi, F.; Anizon, F.; Moreau, P. Synthesis and antiproliferative activities of isoindigo and azaisoindigo derivatives. Eur. J. Med. Chem. 2008, 43, 755–762. [Google Scholar] [CrossRef]
- Elgazwy, A.S.H.; Atta-Allah, S.R. Synthesis and reactivity of isoindigo: A revisit. Afinidad 2008, 65, 148–155. [Google Scholar]
- Wee, X.K.; Yeo, W.K.; Zhang, B.; Tan, V.B.C.; Lim, K.M.; Tay, T.E.; Go, M.-L. Synthesis and evaluation of functionalized isoindigos as antiproliferative agents. Bioorg. Med. Chem. 2009, 17, 7562–7571. [Google Scholar] [CrossRef]
- Papageorgiou, C.; Borer, X. Acid-catalyzed rearrangements for a diastereo selective entry into a new fused hexacyclicheterocycle: (5RS,7aRS,12RS,14aRS,)-4,5,7a,11,12,14,14a-octahydro-5,12-dimethyl-diindolo[1,7-bc: 1',7'-gh][2,6]naphthyridine. Helv. Chim. Acta 1988, 71, 1079–1083. [Google Scholar] [CrossRef]
- Bogdanov, A.V.; Mironov, V.F.; Musin, L.I.; Buzykin, B.I.; Konovalov, A.I. Isatin derivatives in the reaction with phosphorous hexaethyltriamide. A new approach to the synthesis of isoindigo derivatives. Russ. J. Gen. Chem. 2008, 78, 1977–1979. [Google Scholar] [CrossRef]
- Lathourakis, G.E.; Litinas, K.E. Synthesis and study of 3-(triphenylphosphoranylidene)-2,3-dihydro-1H-indol-2-one. J. Chem. Soc. Perk. Trans. 1 1996, 5, 491–493. [Google Scholar] [CrossRef]
- Minami, T; Matsumoto, M; Agawa, T. New olefin synthesis from carbonyl compounds and diethyl sodiophosphonate. J. Chem. Soc. Chem. Commun. 1976, 24, 1053–1054. [Google Scholar] [CrossRef]
- Minami, T; Matsuzaki, N; Ohshiro, Y; Agawa, T. New olefin and oxiran syntheses from carbonyl compounds, and diethyl sodiophosphonate anions and 1-aminophosphonate amino-anions. J. Chem. Soc. Perkin Trans. 1 1980, 1980, 1731–1738. [Google Scholar]
- Sandmeyer, T. Isonitroso acetanilides and their condensation to form isatin derivatives. Helv. Chim. Acta 1919, 2, 234–242. [Google Scholar] [CrossRef]
- Marvel, C.S.; Heirs, G.S. Isatin. In Organic Syntheses Collection 2; Blatt, A.H., Ed.; John Wiley and Sons Inc.: New York, NY, USA, 1941; Volume 1, pp. 327–330. [Google Scholar]
- Van Heerde, W.L.; de Groot, P.G.; Reutelingsperger, C.P. The complexity of the phospholipid binding protein Annexin V. Thromb. Haemostasis 1995, 73, 172–179. [Google Scholar]
- Schmid, I.; Wanda, J.K.; Christel, H.U.; Jonathan, B.; Janis, V.G. Dead cell discrimination with 7-amino-actinomycin D in combination with dual color immunofluorescence in single laser flow cytometry. Cytometry 1992, 13, 204–208. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 3b and 3dare available from M.M.E.-A.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Saleh, A.M.; Al-As'ad, R.M.; El-Abadelah, M.M.; Sabri, S.S.; Zahra, J.A.; Alaskar, A.S.; Aljada, A. Synthesis and Biological Evaluation of New Pyridone-Annelated Isoindigos as Anti-Proliferative Agents. Molecules 2014, 19, 13076-13092. https://doi.org/10.3390/molecules190913076
Saleh AM, Al-As'ad RM, El-Abadelah MM, Sabri SS, Zahra JA, Alaskar AS, Aljada A. Synthesis and Biological Evaluation of New Pyridone-Annelated Isoindigos as Anti-Proliferative Agents. Molecules. 2014; 19(9):13076-13092. https://doi.org/10.3390/molecules190913076
Chicago/Turabian StyleSaleh, Ayman M., Randa M. Al-As'ad, Mustafa M. El-Abadelah, Salim S. Sabri, Jalal A. Zahra, Ahmed S. Alaskar, and Ahmad Aljada. 2014. "Synthesis and Biological Evaluation of New Pyridone-Annelated Isoindigos as Anti-Proliferative Agents" Molecules 19, no. 9: 13076-13092. https://doi.org/10.3390/molecules190913076