Inhibition of Glutamine Synthetase: A Potential Drug Target in Mycobacterium tuberculosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
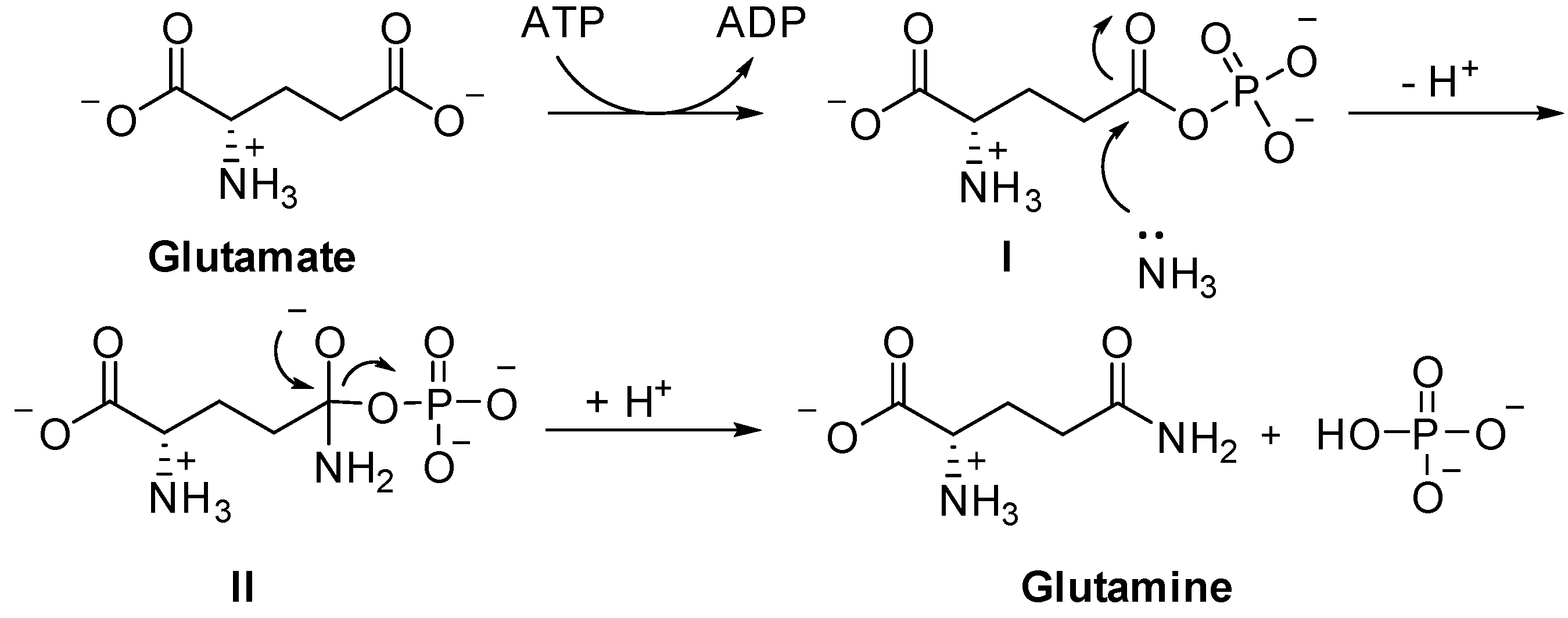
:1. Introduction


2. Inhibitors of GS
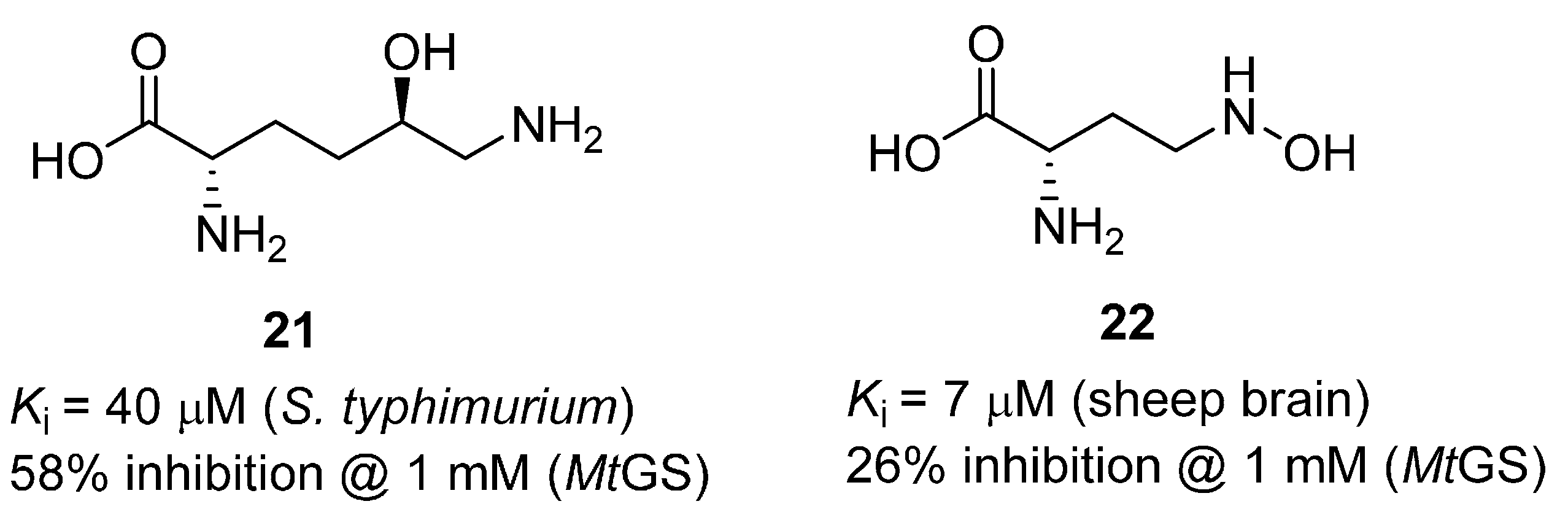
2.1. Amino Acid Analogues






2.2. Heterocycles
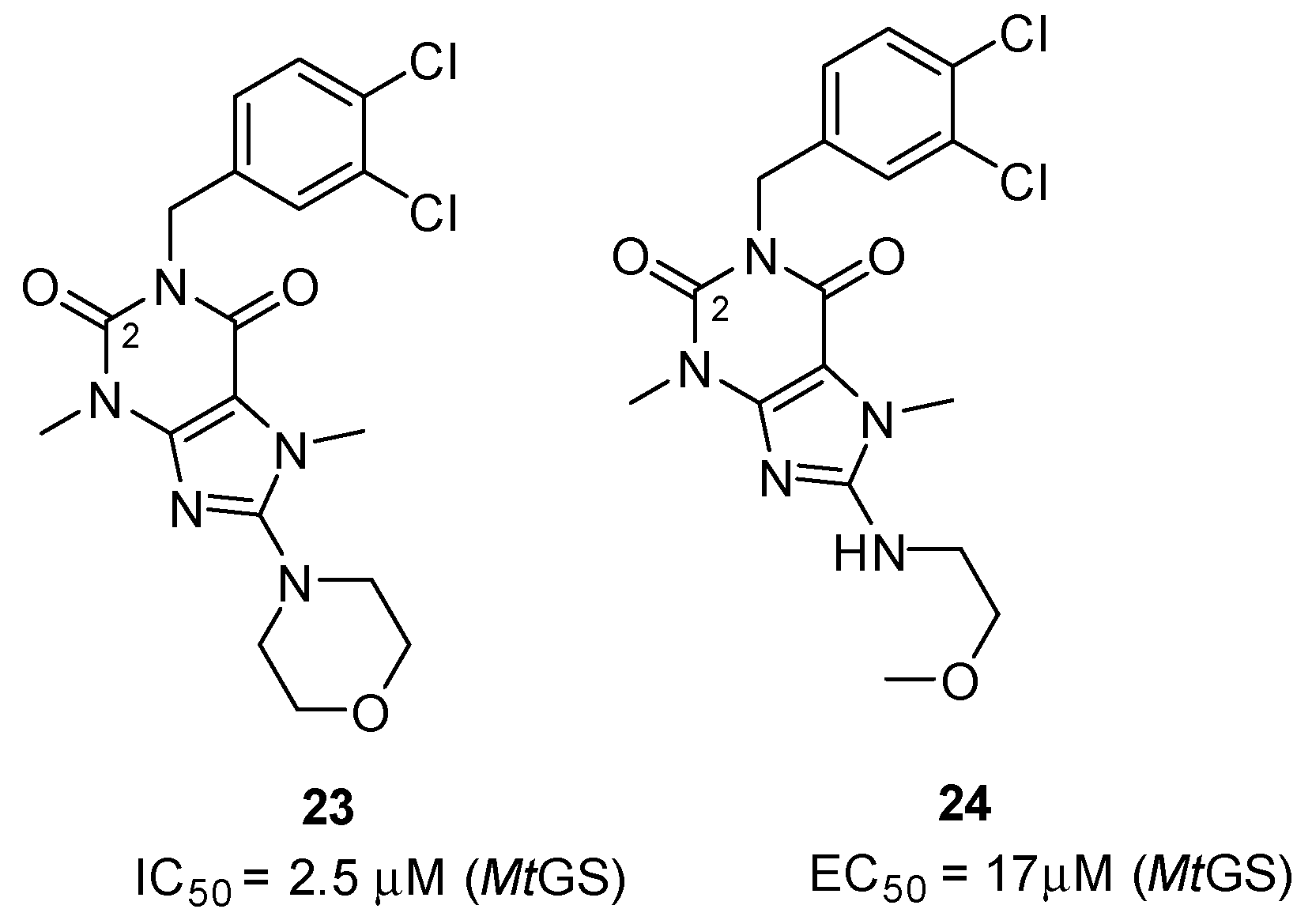
2.2.1. Purine Analogues as Novel ATP-Competitive Inhibitors


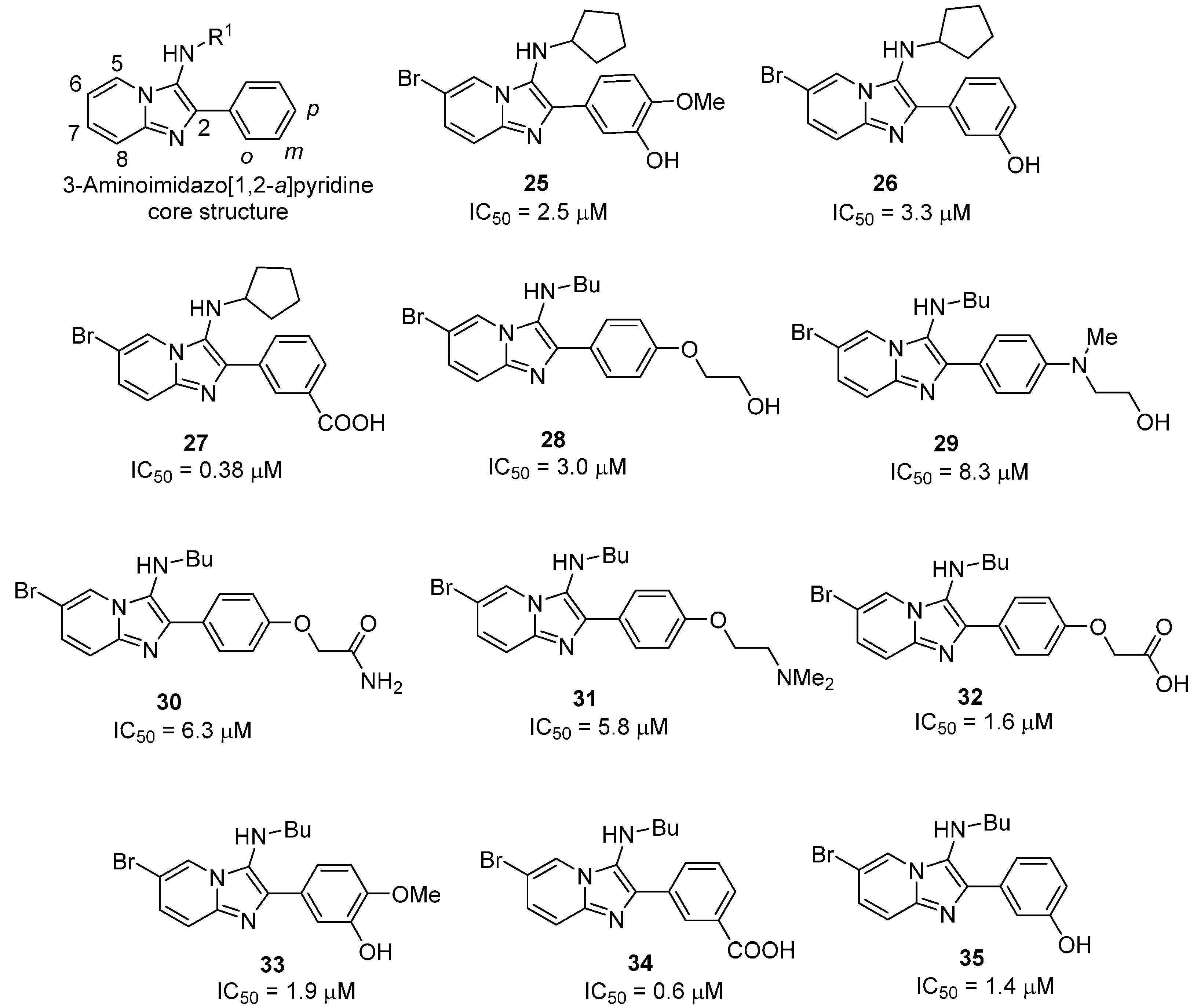
2.2.2. 3-Aminoimidazo[1,2-a]pyridines

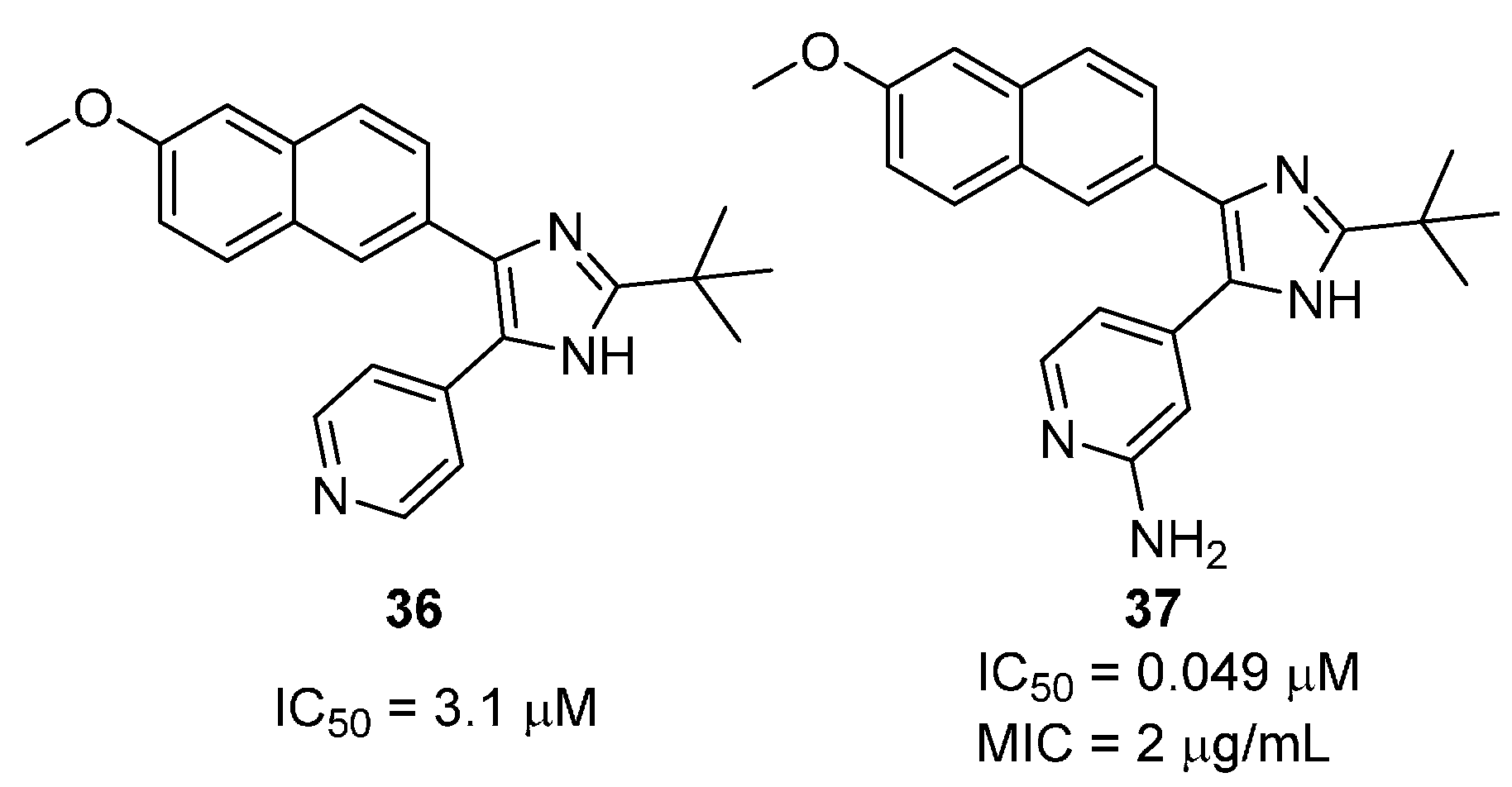
2.2.3. 2,4,5-Trisubtituted Imidazoles

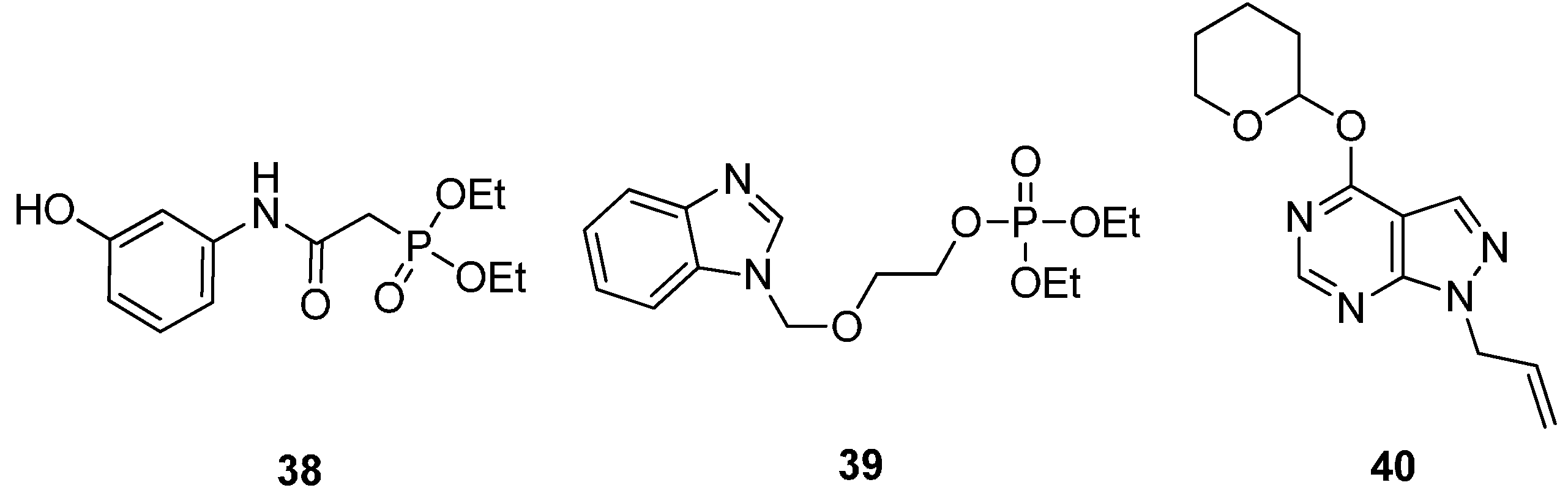
2.2.4. Potential ATP-Competitive Inhibitors

3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization: Global Tuberculosis Report 2013. Available online: http://apps.who.int/iris/bitstream/10665/91355/1/9789241564656_eng.pdf (accessed on 22 August 2014).
- Shenoi, S.; Heysell, S.; Moll, A.; Friedland, G. Multidrug-resistant and extensively drug-resistant tuberculosis: Consequences for the global HIV community. Curr. Opin. Infect. Dis. 2009, 22, 11–17. [Google Scholar] [CrossRef]
- Dheda, K.; Gumbo, T.; Gandhi, N.R.; Murray, M.; Theron, G.; Udwadia, Z.; Migliori, G.B.; Warren, R. Global control of tuberculosis: from extensively drug-resistant to untreatable tuberculosis. Lancet Respir. Med. 2014, 2, 321–338. [Google Scholar] [CrossRef]
- Rivers, E.C.; Mancera, R.L. New anti-tuberculosis drugs in clinical trials with novel mechanisms of action. Drug Discov. Today 2008, 13, 1090–1098. [Google Scholar] [CrossRef]
- Harth, G.; Clemens, D.L.; Horwitz, M.A. Glutamine synthetase of Mycobacterium tuberculosis—Extracellular release and characterization of its enzymatic activity. Proc. Natl. Acad. Sci. USA 1994, 91, 9342–9346. [Google Scholar] [CrossRef]
- Berlicki, L.; Kafarski, P. Computer-aided analysis of the interactions of glutamine synthetase with its inhibitors. Bioorg. Med. Chem. 2006, 14, 4578–4585. [Google Scholar] [CrossRef]
- Harth, G.; Horwitz, M.A. Inhibition of Mycobacterium tuberculosis glutamine synthetase as a novel antibiotic strategy against tuberculosis: Demonstration of efficacy in vivo. Infect. Immun. 2003, 71, 456–464. [Google Scholar] [CrossRef]
- Tullius, M.V.; Harth, G.; Horwitz, M.A. High extracellular levels of Mycobacterium tuberculosis glutamine synthetase and superoxide dismutase in actively growing cultures are due to high expression and extracellular stability rather than to a protein-specific export mechanism. Infect. Immun. 2001, 69, 6348–6363. [Google Scholar] [CrossRef]
- Harth, G.; Horwitz, M.A. An inhibitor of exported Mycobacterium tuberculosis glutamine synthetase selectively blocks the growth of pathogenic mycobacteria in axenic culture and in human monocytes: Extracellular proteins as potential novel drug targets. J. Exp. Med. 1999, 189, 1425–1435. [Google Scholar] [CrossRef]
- Almassy, R.J.; Janson, C.A.; Hamlin, R.; Xuong, N.H.; Eisenberg, D. Novel subunit subunit interactions in the structure of glutamine synthetase. Nature 1986, 323, 304–309. [Google Scholar] [CrossRef]
- Harth, G.; Maslesa-Galic, S.; Tullius, M.V.; Horwitz, M.A. All four Mycobacterium tuberculosis glnA genes encode glutamine synthetase activities but only GlnA1 is abundantly expressed and essential for bacterial homeostasis. Mol. Microbiol. 2005, 58, 1157–1172. [Google Scholar] [CrossRef]
- Jiang, P.; Peliska, J.A.; Ninfa, A.J. The regulation of Escherichia coli glutamine synthetase revisited: role of 2-ketoglutarate in the regulation of glutamine synthetase adenylylation state. Biochemistry 1998, 37, 12802–12810. [Google Scholar] [CrossRef]
- Krajewski, W.W.; Jones, T.A.; Mowbray, S.L. Structure of Mycobacterium tuberculosis glutamine synthetase in complex with a transition-state mimic provides functional insights. Proc. Natl. Acad. Sci. USA 2005, 102, 10499–10504. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, version 1.2r3pre; Schrödinger, LLC. Available online: http://www.pymol.org (accessed on 22 August 2014).
- Gill, H.S.; Pfluegl, G.M.U.; Eisenberg, D. Multicopy crystallographic refinement of a relaxed glutamine synthetase from Mycobacterium tuberculosis highlights flexible loops in the enzymatic mechanism and its regulation. Biochemistry 2002, 41, 9863–9872. [Google Scholar] [CrossRef]
- Krishnaswamy, P.R.; Pamiljans, V.; Meister, A. Studies on the mechanism of glutamine synthesis: Evidence for the formation of enzyme-bound activated glutamic acid. J. Biol. Chem. 1962, 237, 2932–2940. [Google Scholar]
- Midelfort, C.F.; Rose, I.A. A stereochemical method for detection of ATP terminal phosphate transfer in enzymatic reactions. Glutamine synthetase. J. Biol. Chem. 1976, 251, 5881–5887. [Google Scholar]
- Meek, T.D.; Villafranca, J.J. Kinetic mechanism of Escherichia coli glutamine synthetase. Biochemistry 1980, 19, 5513–5519. [Google Scholar] [CrossRef]
- Berlicki, L. Inhibitors of glutamine synthetase and their potential application in medicine. Mini Rev. Med. Chem. 2008, 8, 869–878. [Google Scholar] [CrossRef]
- Nilsson, M.T.; Krajewski, W.W.; Yellagunda, S.; Prabhumurthy, S.; Chamarahally, G.N.; Siddamadappa, C.; Srinivasa, B.R.; Yahiaoui, S.; Larhed, M.; Karlen, A.; et al. Structural basis for the inhibition of Mycobacterium tuberculosis glutamine synthetase by novel ATP-competitive inhibitors. J. Mol. Biol. 2009, 393, 504–513. [Google Scholar] [CrossRef]
- Bentley, H.R.; Mcdermott, E.E.; Pace, J.; Whitehead, J.K.; Moran, T. Action of nitrogen trichloride (Agene) on proteins—Isolation of crystalline toxic factor. Nature 1949, 164, 438–439. [Google Scholar] [CrossRef]
- Gershoff, S.N.; Elvehjem, C.A. The relative effect of methionine sulfoximine on different animal species. J. Nutr. 1951, 45, 451–458. [Google Scholar]
- Logusch, E.W.; Walker, D.M.; McDonald, J.F.; Franz, J.E. Substrate variability as a factor in enzyme inhibitor design: Inhibition of ovine brain glutamine synthetase by α- and γ-substituted phosphinothricins. Biochemistry 1989, 28, 3043–3051. [Google Scholar] [CrossRef]
- Jeitner, T.M.; Cooper, A.J. Inhibition of human glutamine synthetase by l-methionine-S,R-sulfoximine-relevance to the treatment of neurological diseases. Metab. Brain Dis. 2013. [Google Scholar] [CrossRef]
- Leason, M.; Cunliffe, D.; Parkin, D.; Lea, P.J.; Miflin, B.J. Inhibition of pea leaf glutamine synthetase by methionine sulphoximine, phosphinothricin and other glutamate analogues. Phytochemistry 1982, 21, 855–857. [Google Scholar] [CrossRef]
- Logusch, E.W.; Walker, D.M.; McDonald, J.F.; Franz, J.E.; Villafranca, J.J.; DiIanni, C.L.; Colanduoni, J.A.; Li, B.; Schineller, J.B. Inhibition of Escherichia coli glutamine synthetase by α- and γ-substituted phosphinothricins. Biochemistry 1990, 29, 366–372. [Google Scholar] [CrossRef]
- Lamichhane, G.; Freundlich, J.S.; Ekins, S.; Wickramaratne, N.; Nolan, S.T.; Bishai, W.R. Essential metabolites of Mycobacterium tuberculosis and their mimics. mBio 2011, 2. [Google Scholar] [CrossRef]
- Odell, L.R.; Nilsson, M.T.; Gising, J.; Lagerlund, O.; Muthas, D.; Nordqvist, A.; Karlen, A.; Larhed, M. Functionalized 3-amino-imidazo[1,2-a]pyridines: A novel class of drug-like Mycobacterium tuberculosis glutamine synthetase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 4790–4793. [Google Scholar] [CrossRef]
- Carroll, P.; Waddell, S.J.; Butcher, P.D.; Parish, T. Methionine sulfoximine resistance in Mycobacterium tuberculosis is due to a single nucleotide deletion resulting in increased expression of the major glutamine synthetase, GlnA1. Microb. Drug Resist. 2011, 17, 351–355. [Google Scholar] [CrossRef]
- Ronzio, R.A.; Rowe, W.B.; Meister, A. Studies on the mechanism of inhibition of glutamine synthetase by methionine sulfoximine. Biochemistry 1969, 8, 1066–1075. [Google Scholar] [CrossRef]
- Rowe, W.B.; Meister, A. Studies on the inhibition of glutamine synthetase by methionine sulfone. Biochemistry 1973, 12, 1578–1582. [Google Scholar] [CrossRef]
- Griffith, O.W.; Meister, A. Differential inhibition of glutamine and gamma-glutamylcysteine synthetases by α-alkyl analogs of methionine sulfoximine that induce convulsions. J. Biol. Chem. 1978, 253, 2333–2338. [Google Scholar]
- Griffith, O.W.; Anderson, M.E.; Meister, A. Inhibition of glutathione biosynthesis by prothionine sulfoximine (S-N-propyl homocysteine sulfoximine), a selective inhibitor of γ-glutamylcysteine synthetase. J. Biol. Chem. 1979, 254, 1205–1210. [Google Scholar]
- Lagerlund, O.; Odell, L.R.; Mowbray, S.L.; Nilsson, M.T.; Krajewski, W.W.; Nordqvist, A.; Karlen, A.; Larhed, M. Microwave-enhanced α-arylation of a protected glycine in water: Evaluation of 3-phenylglycine derivatives as inhibitors of the tuberculosis enzyme, glutamine synthetase. Comb. Chem. High Throughput Screen. 2007, 10, 783–789. [Google Scholar] [CrossRef]
- Bayer, E.; Zahner, H.; Konig, W.A.; Jessipow, S.; Gugel, K.H.; Hagele, K.; Hagenmai, H. Stoffwechselprodukte von Mikroorganismen. 98. Mitteilung. Phosphinothricin und Phosphinothricyl-Alanyl-Alanin. Helv. Chim. Acta 1972, 55, 224–239. [Google Scholar] [CrossRef]
- Ogawa, Y.; Tsuruoka, T.; Inouye, S.; Niida, T. Studies on a New Antibiotic SF-1293 Part 2 Chemical Structure of Antibiotic SF-1293. Sci. Rep. Meiji Seika Kaisha 1973, 13, 42–48. [Google Scholar]
- Logusch, E.W.; Walker, D.M.; Mcdonald, J.F.; Leo, G.C.; Franz, J.E. Synthesis of α-alkyl-substituted and γ-alkyl-substituted phosphinothricins—Potent new inhibitors of glutamine synthetase. J. Org. Chem. 1988, 53, 4069–4074. [Google Scholar] [CrossRef]
- Johnson, C.R.; Boettcher, B.R.; Cherpeck, R.E.; Dolson, M.G. Design and synthesis of potent inhibitors of glutamine synthetase. 1. Cyclic analogs of phosphinothricin. Bioorg. Chem. 1990, 18, 154–159. [Google Scholar] [CrossRef]
- Berlicki, L.; Obojska, A.; Forlani, G.; Kafarski, P. Design, synthesis, and activity of analogues of phosphinothricin as inhibitors of glutamine synthetase. J. Med. Chem. 2005, 48, 6340–6349. [Google Scholar] [CrossRef]
- Maier, L.; Lea, P.J. Organic phosphorus compounds 76. Synthesis and properties of phosphinothricin derivatives. Phosphorus Sulfur Silicon Relat. Elem. 1983, 17, 1–19. [Google Scholar] [CrossRef]
- Maier, L.; Rist, G.; Lea, P.J. Synthesis and properties of phosphinothricin derivatives. Phosphorus Sulfur Silicon Relat. Elem. 1983, 18, 349–352. [Google Scholar] [CrossRef]
- Farrington, G.K.; Kumar, A.; Wedler, F.C. Design and synthesis of phosphonate inhibitors of glutamine synthetase. J. Med. Chem. 1987, 30, 2062–2067. [Google Scholar] [CrossRef]
- Walker, D.M.; McDonald, J.F.; Logusch, E.W. Synthesis of d,l-y-hydroxyphosphinothricin, a potent new inhibitor of glutamine synthetase. J. Chem. Soc. Chem. Commun. 1987, 22, 1710–1711. [Google Scholar]
- Gill, H.S.; Eisenberg, D. The crystal structure of phosphinothricin in the active site of glutamine synthetase illuminates the mechanism of enzymatic inhibition. Biochemistry 2001, 40, 1903–1912. [Google Scholar] [CrossRef]
- Obojska, A.; Berlicki, L.; Kafarski, P.; Lejczak, B.; Chicca, M.; Forlani, G. Herbicidal pyridyl derivatives of aminomethylene-bisphosphonic acid inhibit plant glutamine synthetase. J. Agric. Food Chem. 2004, 52, 3337–3344. [Google Scholar] [CrossRef]
- Berlicki, L.; Kafarski, P. The use of molecular modelling for comparison of three possible modes of action of herbicidally active derivatives of aminomethylenebisphosphonic acid. Pestic. Biochem. Phys. 2002, 73, 94–103. [Google Scholar] [CrossRef]
- Nordqvist, A.; Nilsson, M.T.; Rottger, S.; Odell, L.R.; Krajewski, W.W.; Andersson, C.E.; Larhed, M.; Mowbray, S.L.; Karlen, A. Evaluation of the amino acid binding site of Mycobacterium tuberculosis glutamine synthetase for drug discovery. Bioorg. Med. Chem. 2008, 16, 5501–5513. [Google Scholar] [CrossRef]
- Correia, C.; Carvalho, M.A.; Proenca, M.F. Synthesis and in vitro activity of 6-amino-2,9-diarylpurines for Mycobacterium tuberculosis. Tetrahedron 2009, 65, 6903–6911. [Google Scholar] [CrossRef]
- Nordqvist, A.; Nilsson, M.T.; Lagerlunda, O.; Muthas, D.; Gising, J.; Yahiaoui, S.; Odell, L.R.; Srinivasa, B.R.; Larhed, M.; Mowbray, S.L.; et al. Synthesis, biological evaluation and X-ray crystallographic studies of imidazo[1,2-a]pyridine-based Mycobacterium tuberculosis glutamine synthetase inhibitors. MedChemComm 2012, 3, 620–626. [Google Scholar] [CrossRef]
- Gising, J.; Nilsson, M.T.; Odell, L.R.; Yahiaoui, S.; Lindh, M.; Iyer, H.; Sinha, A.M.; Srinivasa, B.R.; Larhed, M.; Mowbray, S.L.; et al. Trisubstituted imidazoles as Mycobacterium tuberculosis glutamine synthetase inhibitors. J. Med. Chem. 2012, 55, 2894–2898. [Google Scholar] [CrossRef]
- Mutorwa, M.; Salisu, S.; Blatch, G.L.; Kenyon, C.; Kaye, P.T. 3-Substituted anilines as scaffolds for the construction of glutamine synthetase and DXP-reductoisomerase inhibitors. Synth. Commun. 2009, 39, 2723–2736. [Google Scholar] [CrossRef]
- Gxoyiya, B.S.B.; Kaye, P.T.; Kenyon, C. Benzimidazole-derived ATP analogues as potential glutamine synthetase inhibitors. Synth. Commun. 2010, 40, 2578–2587. [Google Scholar] [CrossRef]
- Sarkar, D.; Joshi, S.P.; Singh, U.; Shurpali, K.D.; Kulkarni, R.R. Antituberculosis Composition of Byttneria Species. US 20130040007 A1, 14 February 2013. [Google Scholar]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mowbray, S.L.; Kathiravan, M.K.; Pandey, A.A.; Odell, L.R. Inhibition of Glutamine Synthetase: A Potential Drug Target in Mycobacterium tuberculosis. Molecules 2014, 19, 13161-13176. https://doi.org/10.3390/molecules190913161
Mowbray SL, Kathiravan MK, Pandey AA, Odell LR. Inhibition of Glutamine Synthetase: A Potential Drug Target in Mycobacterium tuberculosis. Molecules. 2014; 19(9):13161-13176. https://doi.org/10.3390/molecules190913161
Chicago/Turabian StyleMowbray, Sherry L., Muthu K. Kathiravan, Abhishek A. Pandey, and Luke R. Odell. 2014. "Inhibition of Glutamine Synthetase: A Potential Drug Target in Mycobacterium tuberculosis" Molecules 19, no. 9: 13161-13176. https://doi.org/10.3390/molecules190913161