Constituents of the Roots and Leaves of Ekebergia capensis and Their Potential Antiplasmodial and Cytotoxic Activities

 and
and
Abstract
:
1. Introduction
2. Results and Discussion
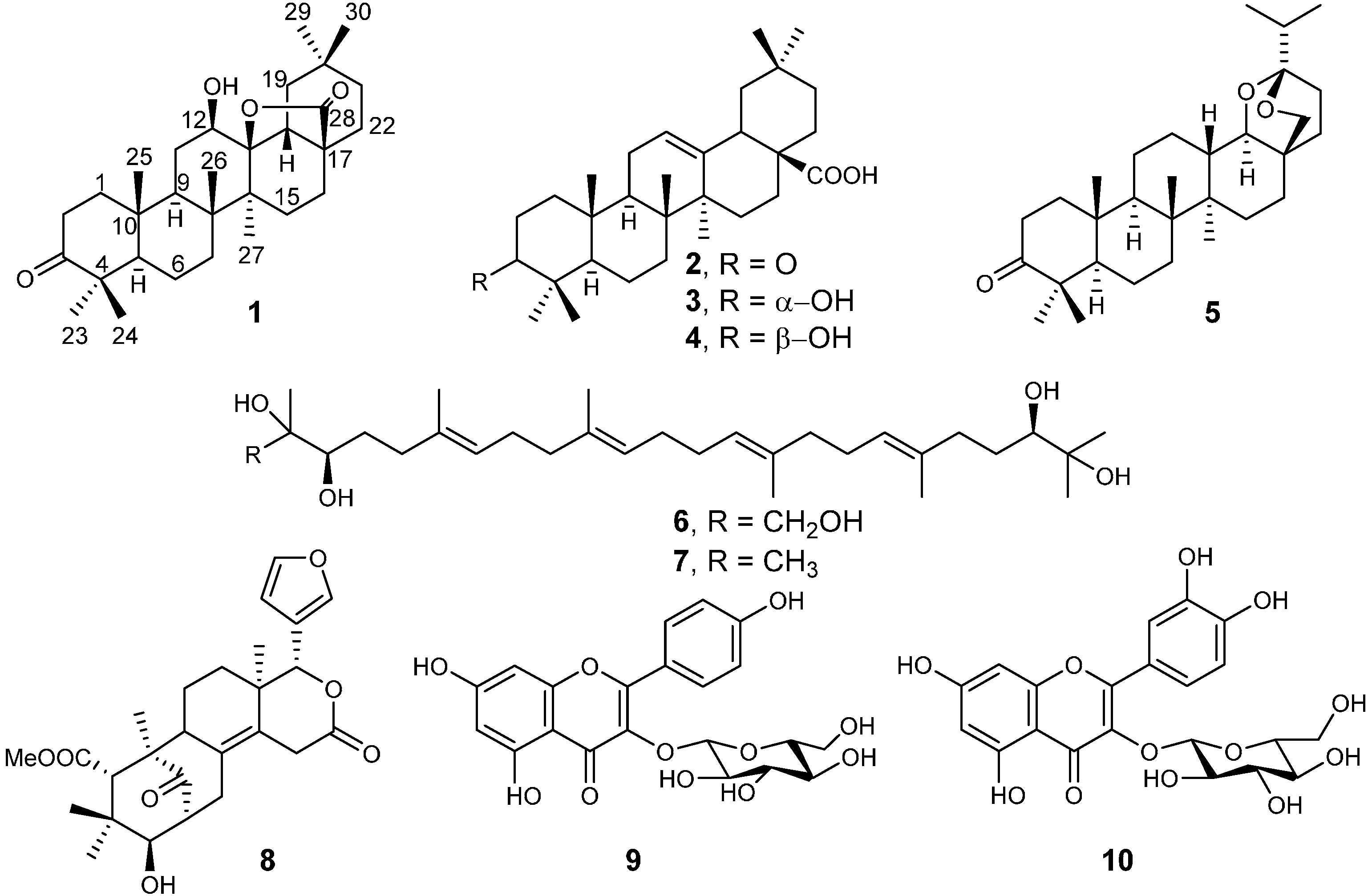
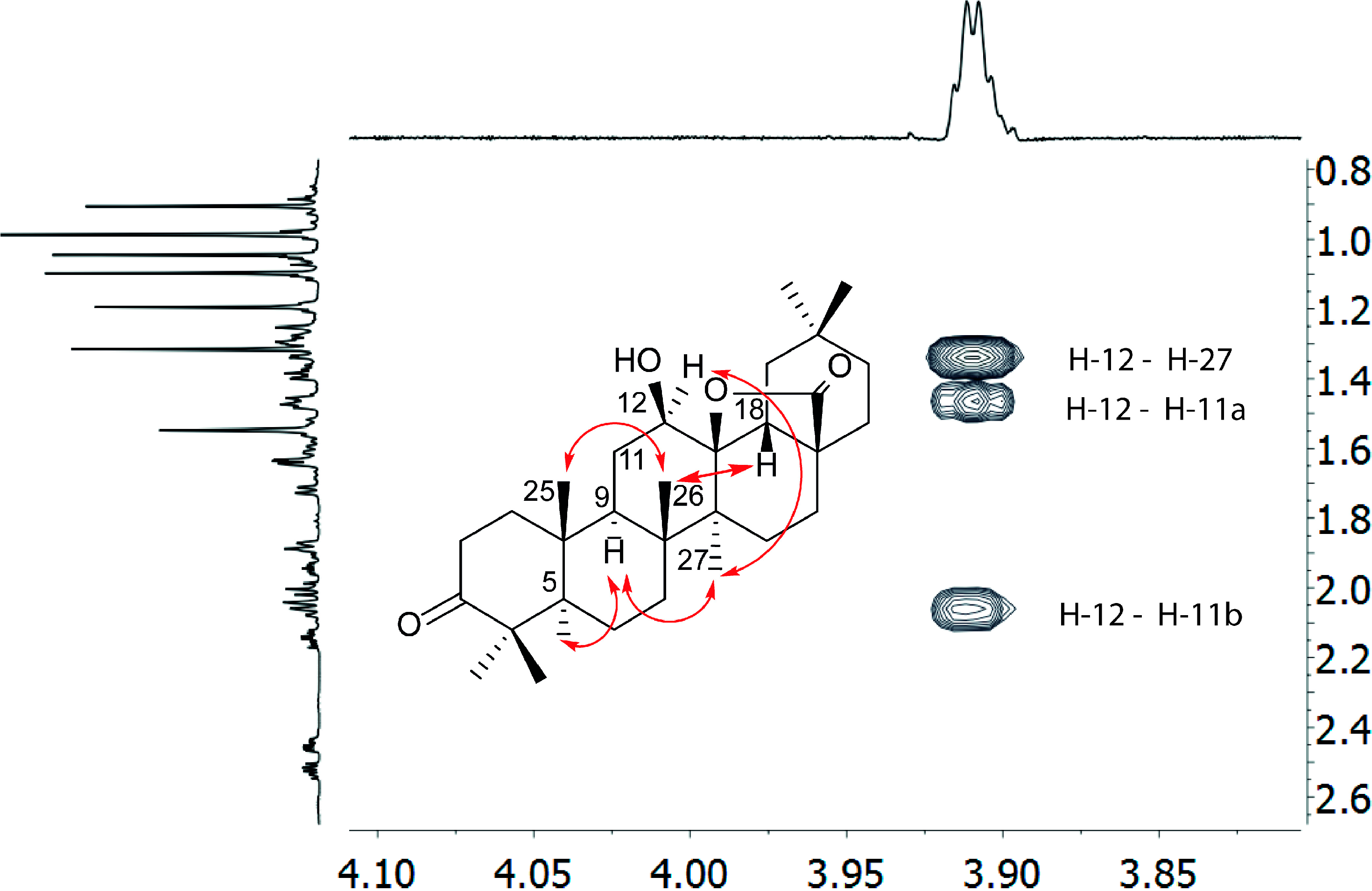
2.1. Isolation and Spectroscopic Identification


{kind=link}
{kind=link}
{kind=link}
| δH (I, multiplicity, J in Hz) | δC | HMBC (2J, 3J) | |
|---|---|---|---|
| 1 | 1.47 (1H, ddd, 7.6, 9.8, 12.5) | 39.8 | C2, C3, C5, C10, C25 |
| 1.94 (1H, ddd, 4.4, 7.6, 12.5) | |||
| 2 | 2.53 (1H, ddd, 7.6, 9.8, 15.7) | 34.1 | C1, C3, C4, C10 |
| 2.45 (1H, ddd, 4.4, 7.6, 15.7) | |||
| 3 | - | 217.7 | - |
| 4 | - | 47.5 | - |
| 5 | 1.39 (1H, dd, 2.7, 12.0) | 55.0 | C4, C6, C7, C9, C10, C23, C25 |
| 6 | 1.55 (1H, m) | 19.2 | C7, C8, C10, C26 |
| 1.47 (1H, m) | C7, C8, C10, C25, C26 | ||
| 7 | 1.61 (1H, m) | 33.5 | C5, C6, C8, C26 |
| 1.30 (1H, m) | C5, C8, C9, C26 | ||
| 8 | - | 42.3 | - |
| 9 | 1.72 (1H, dd, 2.3, 13.1) | 44.0 | C1, C5, C8, C10, C11, C25, C26 |
| 10 | - | 36.3 | - |
| 11 | 2.06 (1H, m) | 29.3 | C8, C9, C10, C12, C13 |
| 1.46 (1H, m) | |||
| 12 | 3.91 (1H, dd, 3.3, 3.3) | 76.2 | C9, C13, C14 |
| 13 | - | 90.6 | - |
| 14 | - | 42.3 | - |
| 15 | 1.88 (1H, ddd, 2.6, 2.6, 12.4) | 28.2 | C14, C16, C17, C18, C27 |
| 1.20 (1H, m) | |||
| 16 | 2.14 (1H, ddd, 5.9, 13.3, 13.3) | 21.3 | C17, C18, C22, C28 |
| 1.29 (1H, m) | - | ||
| 17 | - | 44.9 | - |
| 18 | 2.05 (1H, dd, 3.5, 13.4) | 51.3 | C12, C13, C14, C16, C19, C20 |
| 19 | 2.01 (1H, dd, 13.1, 13.4) | 39.7 | C17, C18, C20, C21, C29, C30 |
| 1.87 (1H, dd, 3.5, 13.1) | |||
| 20 | - | 31.8 | - |
| 21 | 1.38 (1H, m) | 34.3 | C20, C22, C30 |
| 1.27 (1H, m) | |||
| 22 | 1.64 (2H, m) | 27.6 | C16, C17, C18, C20, C28, C29 |
| 23 | 1.10 (3H, s) | 26.7 | C3, C4, C5, C24 |
| 24 | 1.05 (3H, s) | 21.2 | C3, C4, C5, C23 |
| 25 | 0.99 (3H, s) | 16.4 | C1, C5, C9, C10 |
| 26 | 1.20 (3H, s) | 18.4 | C7, C9, C13, C14, C27 |
| 27 | 1.32 (3H, s) | 18.6 | C8, C13, C14, C15 |
| 28 | - | 179.9 | - |
| 29 | 0.99 (3H, s) | 33.4 | C19, C20, C21,C30 |
| 30 | 0.91 (3H, s) | 24.0 | C19, C20, C21,C29 |

2.2. Antimalarial Activity and Cytotoxicity
| IC50 a | ||||||
|---|---|---|---|---|---|---|
| D6 b | W2 b | Vero | 4T1 c | HEp2 | MDA-MB-231 | |
| roots | 18.2 ± 0.1 | 34 ± 0.8 | 2.8 ± 0.1 | 9.3 ± 0.1 | 61 ± 1.4 | n.d. |
| leaves | 44.9 ± 0.8 | 45.3 ± 0.5 | 97.8 ± 0.8 | 82.1 ± 5.7 | 71.6 ± 1.8 | n.d. |
| 2 | 38.8 ± 0.5 | 76.7 ± 4.0 | 35.8 ± 1.3 | 13.3 ± 0.2 | 1.4 ± 0.1 | >212 |
| 3 | 205.0 ± 3.0 | 179.4 ± 6.0 | 58.0 ± 5.2 | 30.3 ± 2.6 | 29.8 ± 0.3 | 36.54 ± 0.02 |
| 4 | 49.6 ± 2.3 | 82.7 ± 2.0 | 112.0 ± 5.1 | 117.6 ± 2.6 | 134.9 ± 0.7 | 39.82 ± 0.03 |
| 5 | 182.2 ± 6.0 | >219 | >219 | 163.2 ± 4.3 | >219 | n.d. |
| 6 | 27.1 ± 0.4 | 66.9 ± 0.6 | 35.7 ± 2.1 | 30.2 ± 1.3 | 38.4 ± 0.8 | 36.69 ± 0.04 |
| 7 | 56.1 ± 0.4 | 64.3 ± 1.0 | 24.7 ± 1.8 | 22.5 ± 3.2 | 35.5 ± 3.1 | > 209 |
| 8 | 84.7 ± 0.8 | 150.2 ± 3.0 | >213 | >213 | >213 | n.d. |
| 9 | 97.1 ± 1.0 | 105.8 ± 0.5 | >223 | >223 | >223 | >223 |
| 10 | 42.9 ± 0.3 | 105.8 ± 1.0 | >216 | >216 | >216 | >216 |
| Compound | IC50 (μg/mL) | |||
|---|---|---|---|---|
| 0:1 a | 1:3 a | 1:1 a | 3:1 a | |
| 3 | 22.5 b | 9.8 c | 5.2 c | 5.8 c |
| 4 | 40.3 b | 3.3 c | 4.7 c | 3.6 c |
| 5 | >100 b | <4 c | <1.4 c | <1.3 c |
| 6 | 13.6 b | 2.2 c | 2.2 c | 2.3 c |
| 7 | 11 b | 2 c | 1.9 c | 2.2 c |
3. Experimental Section
3.1. General Information
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Cytotoxicity Assays
3.5. In Vitro Antiplasmodial Assay
4. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gachathi, M. Kikuyu Botanics Dictionary: A Guide to Plant Names Uses and Culture Values, 2nd ed.; Tropical Botany Press: Nairobi, Kenya, 2007; p. 116. [Google Scholar]
- Beentje, H.J. Kenya Trees, Shrubs, and Lianas; National Museums of Kenya: Nairobi, Kenya, 1994; p. 404. [Google Scholar]
- Sewram, V.; Raynor, M.W.; Mulholland, D.A.; Raidoo, D.M. The uterotonic activity of compounds isolated from the supercritical fluid extract of Ekebergia capensis. J. Pharm. Biomed. Anal. 2000, 24, 133–145. [Google Scholar] [CrossRef]
- Okello, S.V.; Nyunja, R.O.; Netondo, G.W.; Onyango, J.C. Ethnobotanical study of medicinal plants used by Sabaots of Mt. Elgon Kenya. Afr. J. Tradit. Complement. Altern. Med. 2010, 7, 1–10. [Google Scholar] [CrossRef]
- Kamadyaapa, D.R.; Gondwe, M.M.; Moodley, K.; Ojewole, J.A.O.; Musabayane, C.T. Cardiovascular effects of Ekebergia capensis Sparrm (Meliaceae) ethanolic leaf extract in experimental animal paradigms. Cardiovasc. J. Afr. 2009, 20, 162–167. [Google Scholar]
- Lall, N.; Meyer, J.J.M. In vitro inhibition of drug-resistant and drug-sensitive strains of Mycobacterium tuberculosis by ethnobotanically selected South African plants. J. Ethnopharmacol. 1999, 66, 347–354. [Google Scholar] [CrossRef]
- Mulaudzi, R.B.; Ndhlala, A.R.; Kulkarni, M.G.; Finnie, J.F.; Staden, J.V. Anti-inflammatory and mutagenic evaluation of medicinal plants used by Venda people against venereal and related diseases. J. Ethnopharmacol. 2013, 146, 173–179. [Google Scholar] [CrossRef]
- Muregi, F.W.; Chhabra, S.C.; Njagi, E.N.M.; Lang’at-Thoruwa, C.C.; Njue, W.M.; Orago, A.S.; Omar, S.A.; Ndiege, I.O. Anti-plasmodial activity of some Kenyan medicinal plant extracts singly and in combination with chloroquine. Phytother. Res. 2004, 18, 379–384. [Google Scholar] [CrossRef]
- Murata, T.; Miyase, T.; Muregi, F.W.; Naoshima-Ishibashi, Y.; Umehara, K.; Warashina, T.; Kanou, S.; Mkoji, G.M.; Terada, M.; Ishih, A. Antiplasmodial triterpenoids from Ekebergia capensis. J. Nat. Prod. 2008, 71, 167–174. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Moriyasu, M.; Ichimaru, M.; Tachibana, Y.; Kato, A.; Mathenge, S.G.; Nganga, J.N.; Juma, F.D. Acyclic triterpenoids from Ekebergia capensis. Phytochemistry 1996, 42, 803–807. [Google Scholar] [CrossRef]
- Kim, K.H.; Choi, S.U.; Lee, K.R. Cytotoxic triterpenoids from Berberis koreana. Planta Med. 2012, 78, 86–89. [Google Scholar] [CrossRef]
- Kim, H.Y.; Moon, B.H.; Lee, H.J.; Choi, D.H. Flavonol glycosides from the leaves of Eucommia ulmoides O. with glycation inhibitory activity. J. Ethnopharmacol. 2004, 93, 227–230. [Google Scholar] [CrossRef]
- Castellanos, L.; de Correa, R.S.; Martínez, E.; Calderon, J.S. Oleanane triterpenoids from Cedrela montana (Meliaceae). Z. Naturforsch. C 2002, 57, 575–578. [Google Scholar]
- Konoike, T.; Takahashi, K.; Araki, Y.; Horibe, I. Practical partial synthesis of myriceric acid A, and endothelin receptor antagonist, from oleanolic acid. J. Org. Chem. 1997, 62, 960–966. [Google Scholar] [CrossRef]
- Salvador, J.A.R.; Moreira, V.M.; Pinto, R.M.A.; Leal, A.S.; Paixao, J.A. Efficient oxidation of oleanolic acid derivatives using magnesium bis(monoperoxyphtalate) hexahydrate (MMPP): A convenient two-step procedure towards 12-oxo-28-carboxylic acid derivatives. Beilstein J. Org. Chem. 2012, 8, 164–169. [Google Scholar] [CrossRef]
- Leal, A.S.; Wang, R.; Salvador, J.A.R.; Jing, I. Synthesis of novel heterocyclic oleanolic acid derivatives with improved antiproliferative activity in solid tumor cells. Org. Biomol. Chem. 2013, 11, 1726–1738. [Google Scholar] [CrossRef]
- Hu, J.; Wu, G.; Xu, Y.; Xiao, G.; Lei, P. 12α-Hydroxy-3,27-dioxooleanano-28,13-lactone. Acta Crystallogr. Sect. E 2012, 68, o1586. [Google Scholar]
- Ikuta, A.; Tomiyasu, H.; Morita, Y.; Yoshimura, K. Ursane- and oleanane-type triterpenes from Ternstroemia gymnanthera callus tissues. J. Nat. Prod. 2003, 66, 1051–1054. [Google Scholar] [CrossRef]
- Ngouamegne, E.T.; Fongang, R.S.; Ngouela, S.; Boyom, F.F.; Rohmer, M.; Tsamo, E.; Gut, J.; Rosenthal, P.J. Endodesmiadiol, a friedelane triterpenoid, and other antiplasmodial compounds from Endodesmia calophylloides. Chem. Pharm. Bull. 2008, 56, 374–377. [Google Scholar] [CrossRef]
- Sairafianpour, M.; Bahreininejad, B.; Witt, M.; Ziegler, H.L.; Jaroszewski, J.W.; Staerk, D. Terpenoids of Salvia hydrangea: Two new, rearranged 20-norabietanes and the effect of oleanolic acid on erythrocyte membrane. Planta Med. 2003, 69, 846–850. [Google Scholar] [CrossRef]
- Wokaun, A.; Ernst, R.R. Selective detection of multiple quantum transitions in NMR by two-dimensional spectroscopy. Chem. Phys. Lett. 1977, 52, 407–412. [Google Scholar] [CrossRef]
- Kumar, A.; Ernst, R.R.; Wüthrich, K. A Two-dimensional nuclear Overhauser enhancement (2D NOE) Experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem. Biophys. Res. Commun. 1980, 95, 1–6. [Google Scholar] [CrossRef]
- Perpickdumont, M.; Reynolds, W.F.; Enriquez, R.G. C-13-H-1 shift correlation with full H-1-H-1 decoupling. Magn. Reson. Chem. 1988, 26, 358–361. [Google Scholar] [CrossRef]
- Hurd, R.E.; John, B.K. Gradient-enhanced proton-detected heteronuclear multiple-quantum coherence spectroscopy. J. Magn. Reson. 1991, 91, 648–653. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Prayong, P.; Barusrux, S.; Weerapreeyakul, N. Cytotoxic activity screening of some indigenous Thai plants. Fitoterapia 2008, 79, 598–601. [Google Scholar] [CrossRef]
- Negera, A.; Induli, M.; Fitzpatrick, P.; Alao, J.P.; Sunnerhagen, P.; Landberg, G.; Yenesew, A.; Erdélyi, M. Cytotoxic quinones from the roots of Aloe dawei. Molecules 2014, 19, 3264–3273. [Google Scholar] [CrossRef]
- Gupta, S.; Thapar, M.M.; Wernsdorfer, W.H.; Björkman, A. In vitro interactions of artemisinin with atovaquone, quinine, and mefloquine against Plasmodium falciparum. Antimicrob. Agents Chemother. 2002, 46, 1510–1515. [Google Scholar] [CrossRef]
- Trager, W.; Jensen, J.B. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [Google Scholar]
- Desjardins, R.E.; Canfield, C.J.; Haynes, J.D.; Chulay, J.D. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 1979, 16, 710–718. [Google Scholar] [CrossRef]
- Batista, R.; Júnior, A.J.S.; Oliveira, A.B. Plant-derived antimalarial agents: New leads and efficient phytomedicines. Part II. Non-alkaloidal natural products. Molecules 2009, 14, 3037–3072. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 2–10 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Irungu, B.N.; Orwa, J.A.; Gruhonjic, A.; Fitzpatrick, P.A.; Landberg, G.; Kimani, F.; Midiwo, J.; Erdélyi, M.; Yenesew, A. Constituents of the Roots and Leaves of Ekebergia capensis and Their Potential Antiplasmodial and Cytotoxic Activities. Molecules 2014, 19, 14235-14246. https://doi.org/10.3390/molecules190914235
Irungu BN, Orwa JA, Gruhonjic A, Fitzpatrick PA, Landberg G, Kimani F, Midiwo J, Erdélyi M, Yenesew A. Constituents of the Roots and Leaves of Ekebergia capensis and Their Potential Antiplasmodial and Cytotoxic Activities. Molecules. 2014; 19(9):14235-14246. https://doi.org/10.3390/molecules190914235
Chicago/Turabian StyleIrungu, Beatrice N., Jennifer A. Orwa, Amra Gruhonjic, Paul A. Fitzpatrick, Göran Landberg, Francis Kimani, Jacob Midiwo, Máté Erdélyi, and Abiy Yenesew. 2014. "Constituents of the Roots and Leaves of Ekebergia capensis and Their Potential Antiplasmodial and Cytotoxic Activities" Molecules 19, no. 9: 14235-14246. https://doi.org/10.3390/molecules190914235