Graphene-Based Nanomaterials as Heterogeneous Acid Catalysts: A Comprehensive Perspective

Abstract
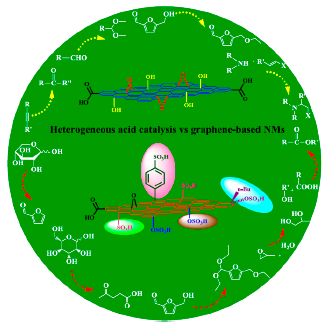
:
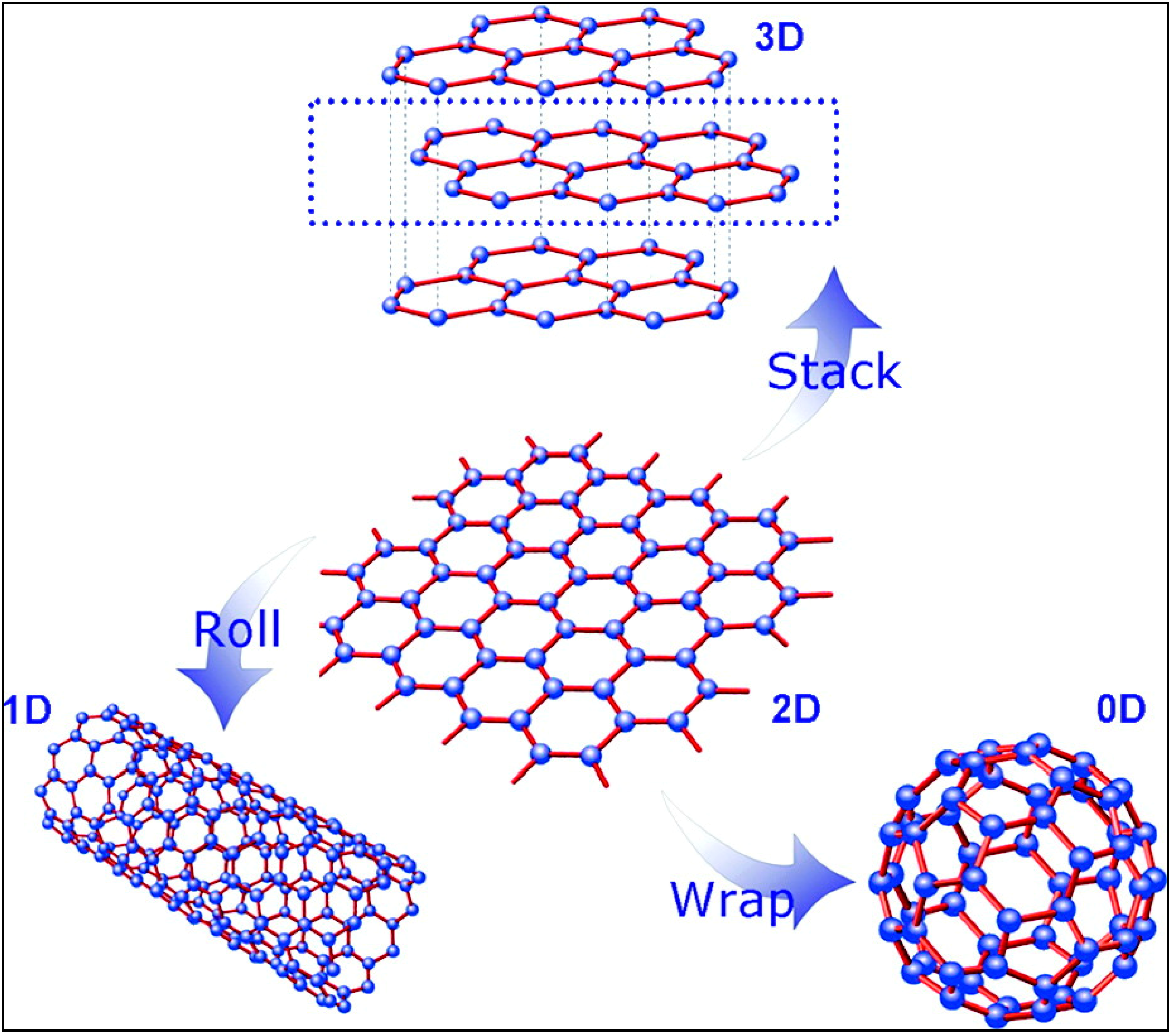
1. Introduction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
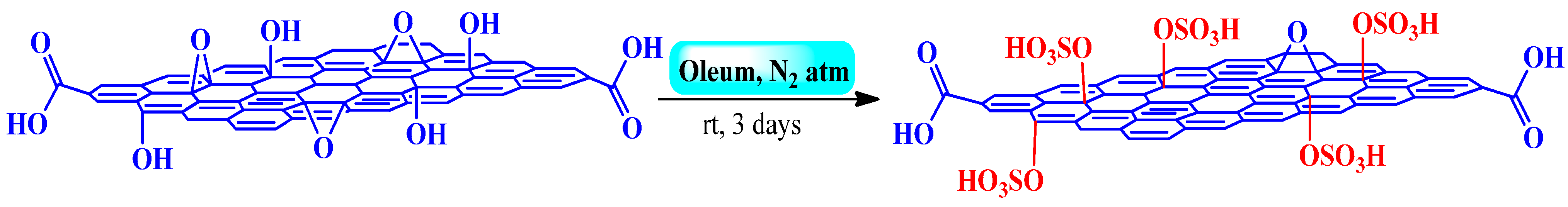{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | Sulfuric Acid | Amberlyst™-15 | G-NMs |
|---|---|---|---|
| Physical state | Liquid | Solid | Solid |
| Acid activity | High | High | High |
| Surface Area | - | Relatively low | High |
| Operating conditions | Typically harsh | Relatively Mild | Variable a |
| Diffusion/Leaching | None | Possible | Negligible |
| Recoverability | Difficult | Easy b | Easy b |
| Reusability/cycles | Difficult | Easy/Less cycles c | Easy/More cycles c |
| Water tolerance/stability | High | Relatively low | High |
| Cost-effectiveness | Cost-effective | Relatively expensive | Variable a |
| Mass transfer | - | Relatively low | High |
| Reaction selectivity | High | Variable a | Variable a |
| Eco-friendly use | No | Yes | Yes |
| Industrial use | Frequent | Selected d | Awaiting |
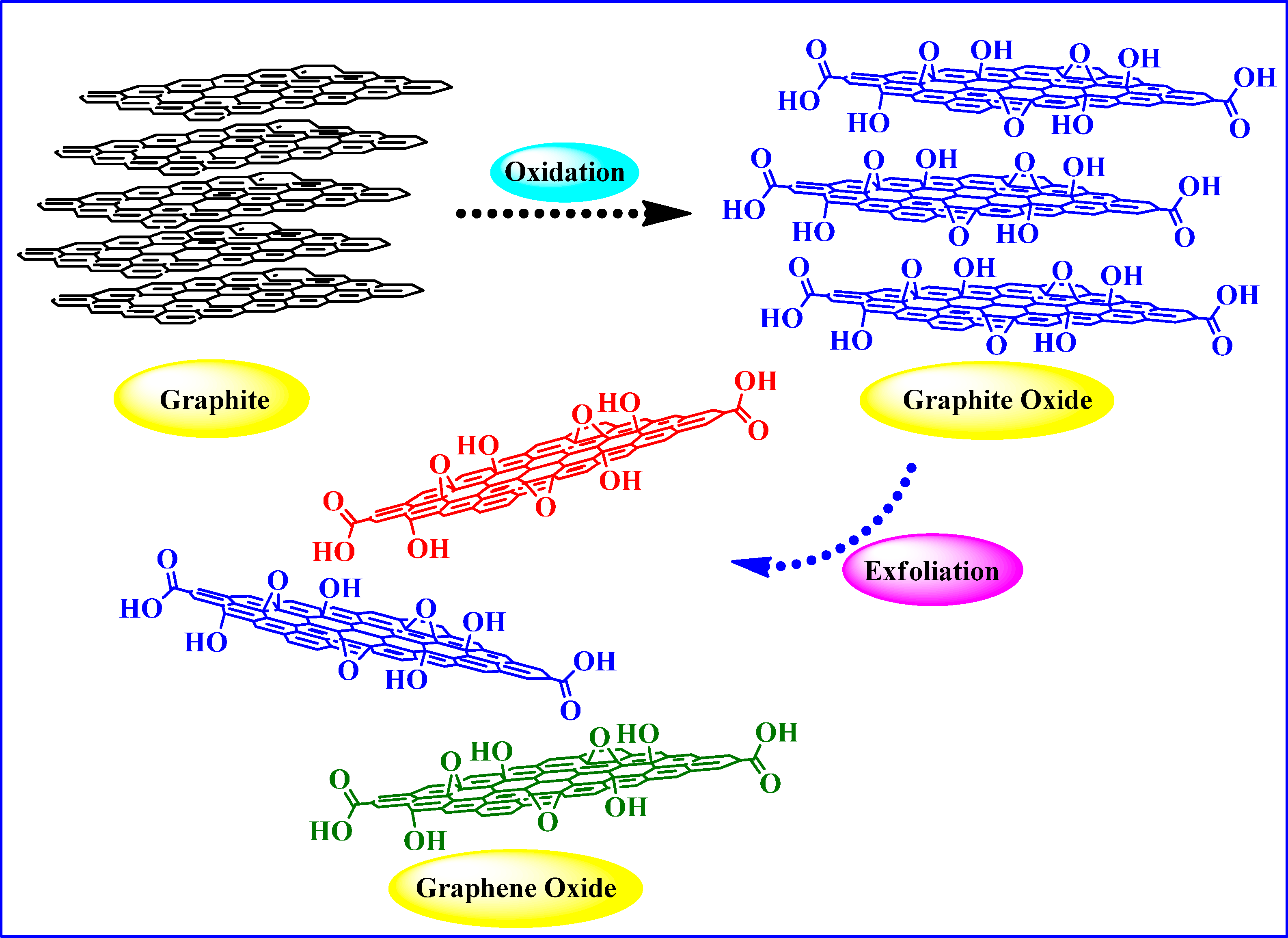
2. Graphite Oxide and Graphene Oxide (GO)
2.1. Synthesis and Structure

2.2. Acidic Properties of Graphite Oxide and GO



3. G-NMs vs. Sulfonation and Sulfation

3.1. Reduction of GO
3.1.1. Hydrazine (N2H4)
3.1.2. Sodium Borohydride (NaBH4)
3.1.3. Lithium Aluminum Hydride (LiAlH4)
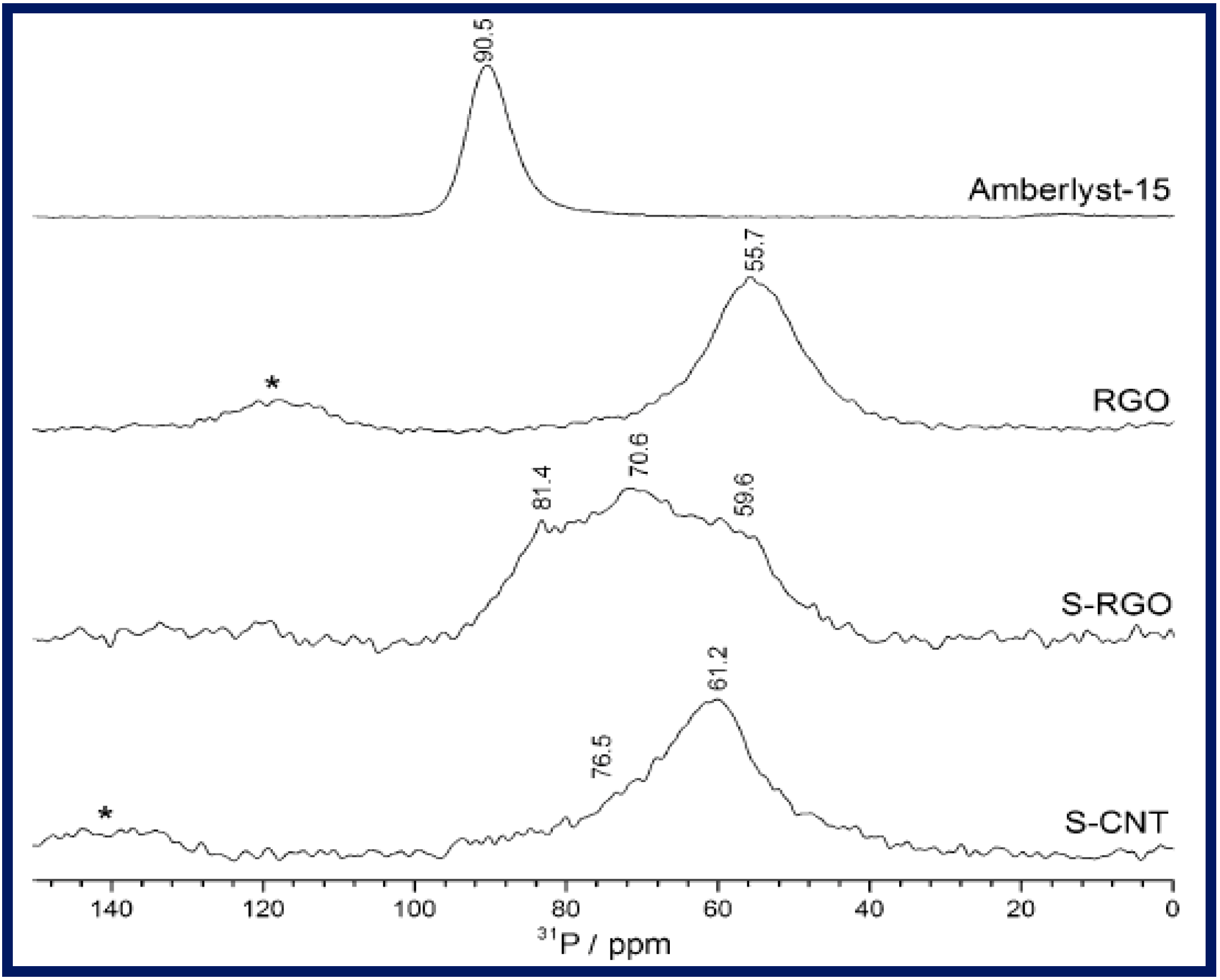
3.2. Sulfonation of rGO and GO
| Sulfonating Agents | Reducing Agents a | Reaction Conditions b | Acid Density c | Applications d | Ref. |
|---|---|---|---|---|---|
| 4-Benzenediazonium sulfonate | N2H4·H2O | EtOH/H2O, H3PO2, 3–5 °C, 1.5 h | 1.55 mmol H+ g−1 | Hydrolysis of ethyl acetate | [82] |
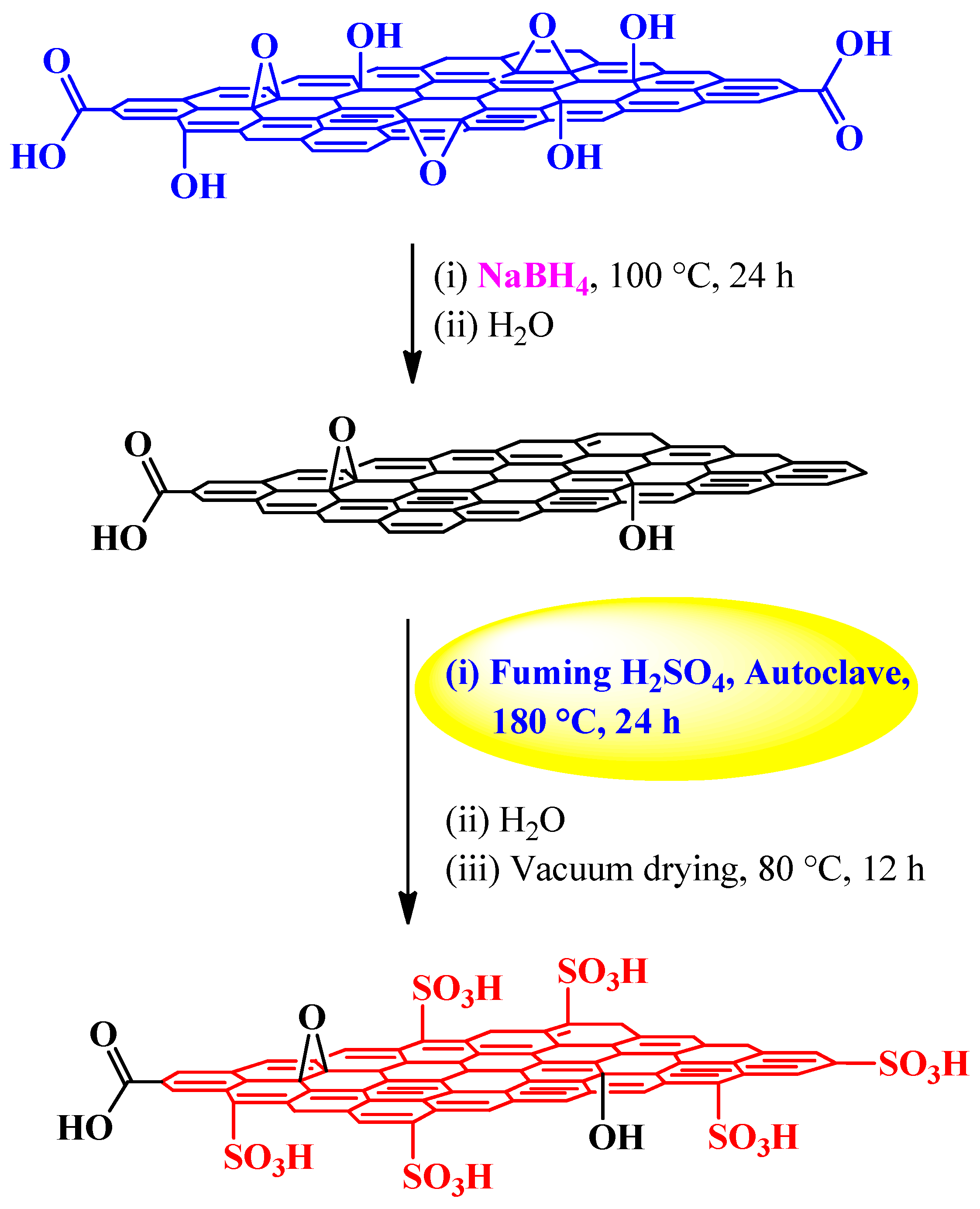
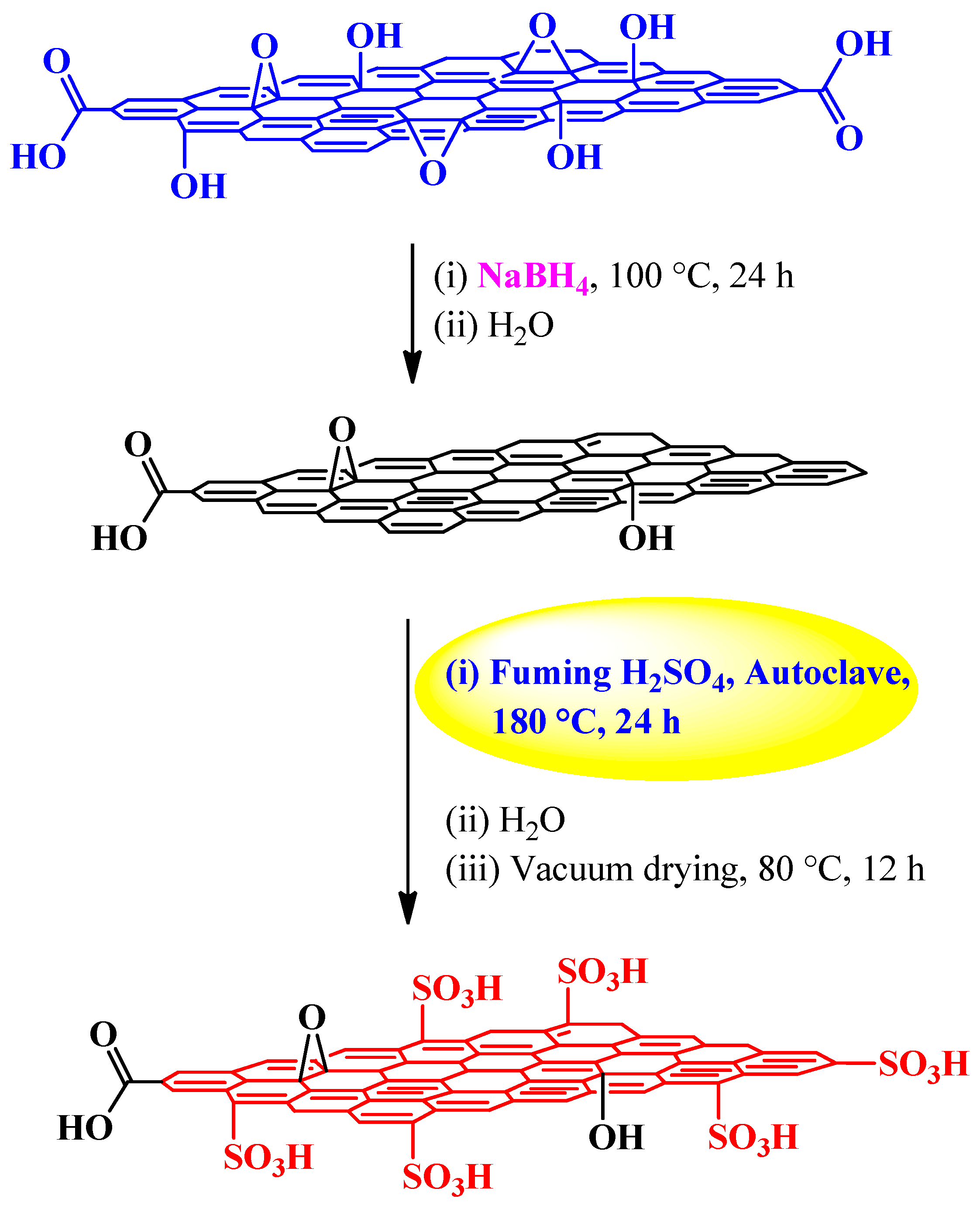
| Fuming sulfuric acid | NaBH4 | Autoclave, 180 °C, 24 h | 1.2 mmol H+ g−1 | (1) Esterification reactions (2) Peckmann reaction (3) Hydration reaction | [83] |
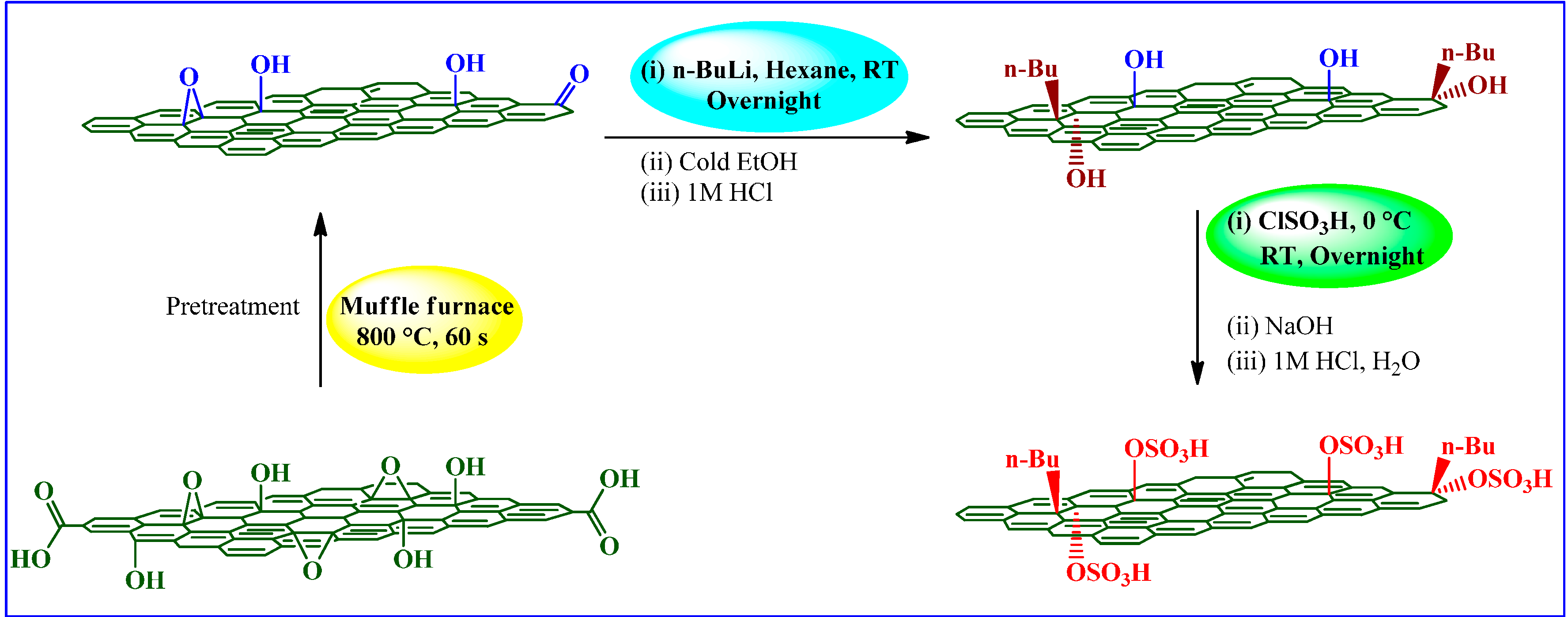
| Chlorosulfonic acid | n-butyl lithium | ClSO3H, 0 °C, RT e, overnight, NaOH, HCl, H2O | - | Ester exchange reactions | [84] |
| 4-Benzenediazonium sulfonate | NaBH4 | 0 °C, 2 h, Hydrazine, 100 °C, 24 h | - | Synthesis of xanthenes and benzoxanthenes | [85] |
| Chlorosulfonic acid | - | CHCl3, 70 °C, 4 h | 1.2 mmol H+ g−1 | Chemical conversion of biomass derived carbohydrates | [86] |
| 4-Benzenediazonium sulfonate | (i) NaBH4 (ii) N2H4 | RT e, overnight | 0.5–1.7 mmol H+ g−1 | Dehydration of xylose to furfural | [87] |
| Ammonium sulfate | - | 235 °C, 30 min, argon atmosphere | EW f = 725 ± 5 g/mol | As support for metal nanocatalysts | [88] |
| Sulfuric acid | Microwave irradiation, 190 °C | 160 °C, 5 h nitrogen atmosphere | 1.9 mmol H+ g−1 | Production of biofuels | [89] |
3.2.1. Diazonium Salt of Sulfanilic Acid; 4-Benzenediazonium Sulfonate

3.2.2. Fuming Sulfuric Acid
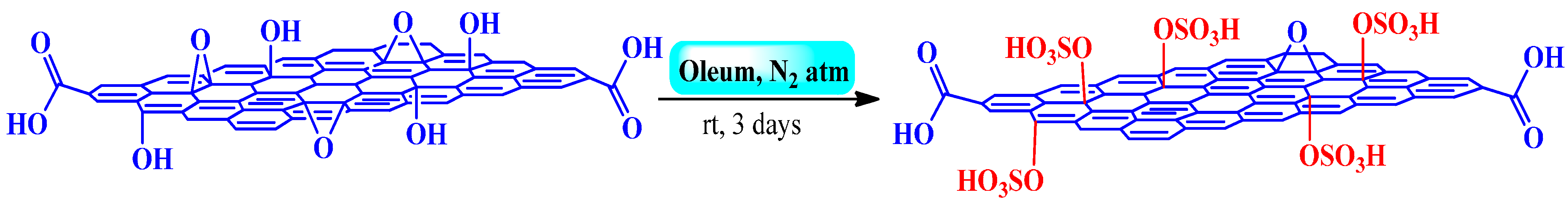
3.2.3. Chlorosulfonic Acid

3.2.4. Sulfuric Acid

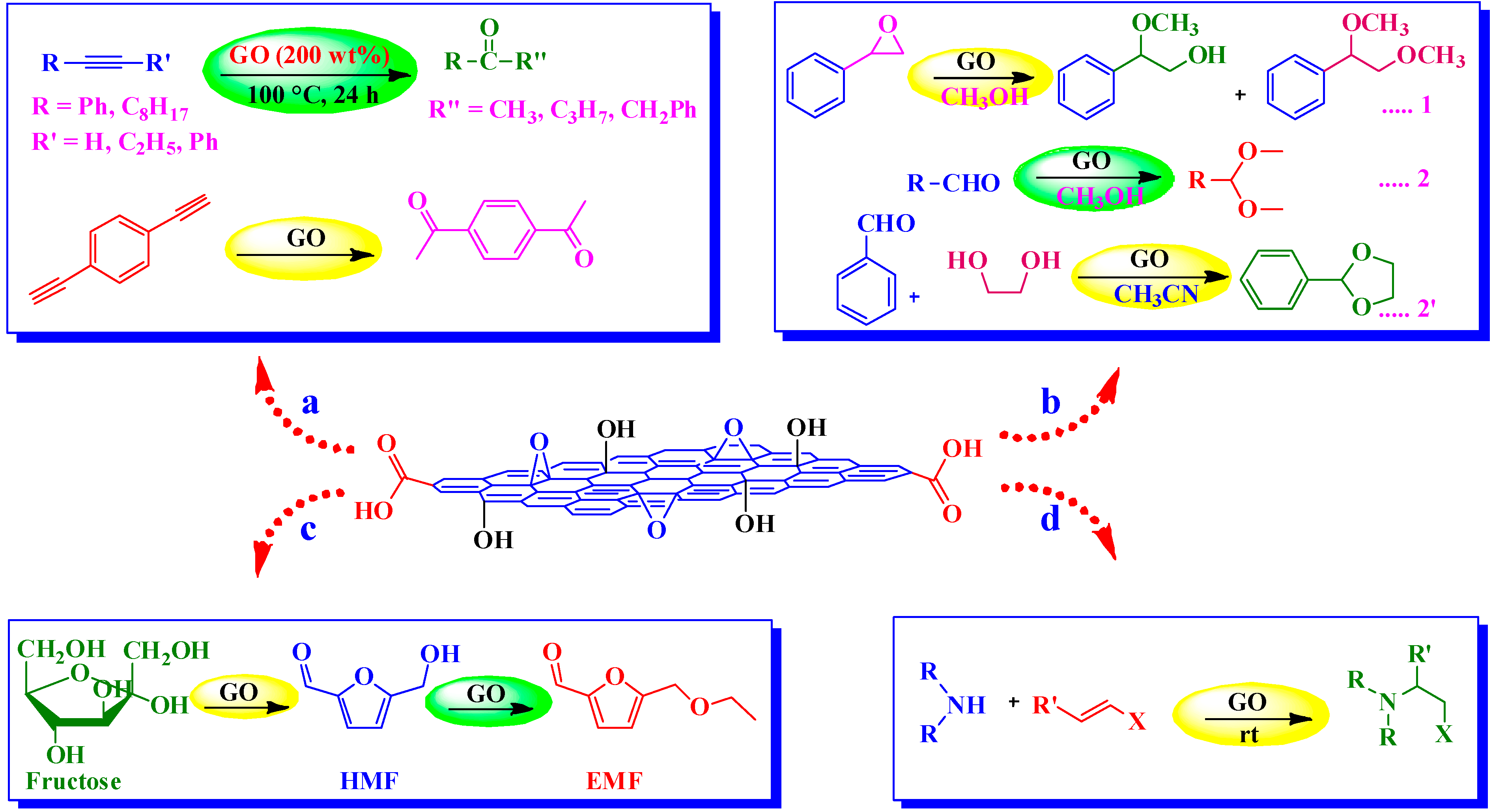
4. Applications of G-NMs in Heterogeneous Acid Catalysis
4.1. Graphite Oxide as Acid Catalyst
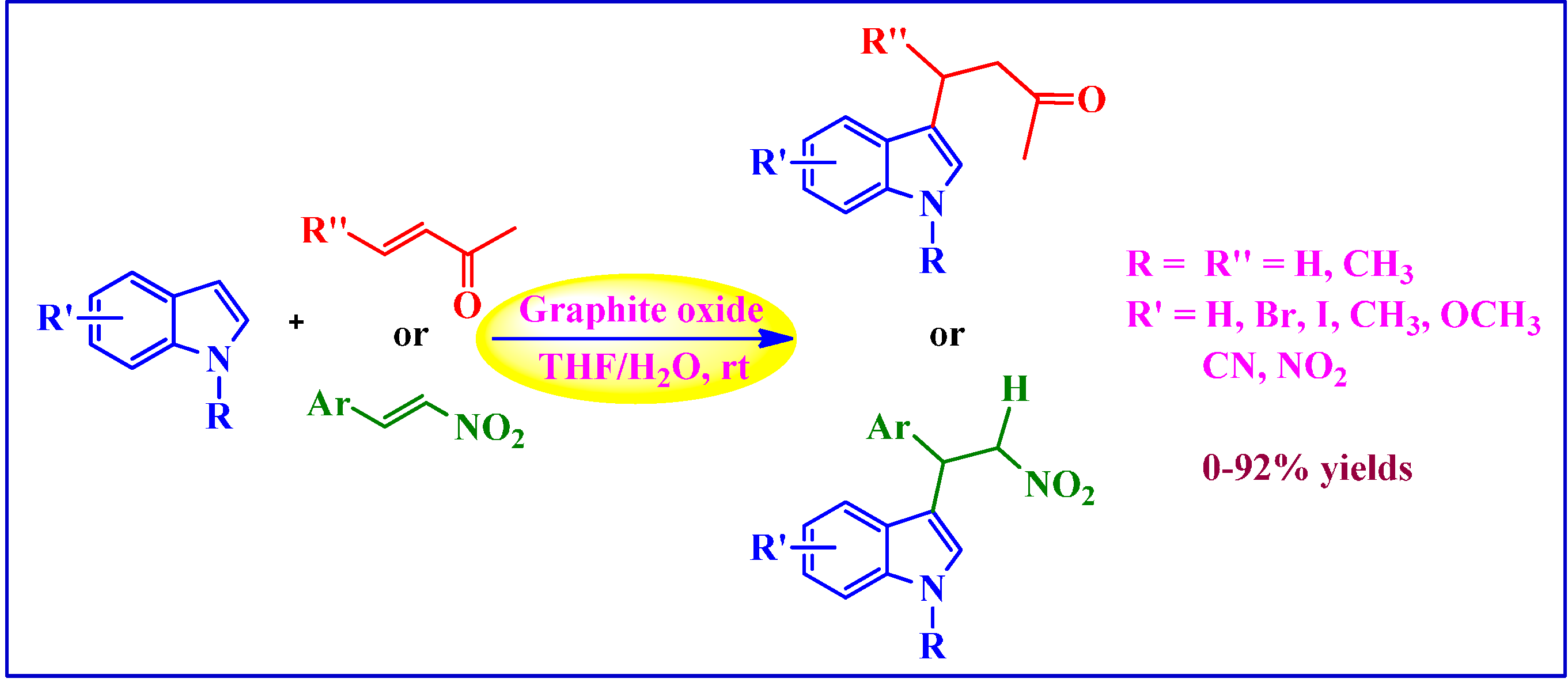
4.1.1. Michael-Type Friedel-Crafts Addition

4.1.2. Polymerization
4.1.3. Synthesis of Dipyrromethanes
4.2. Graphene Oxide (GO) as Acid Catalyst
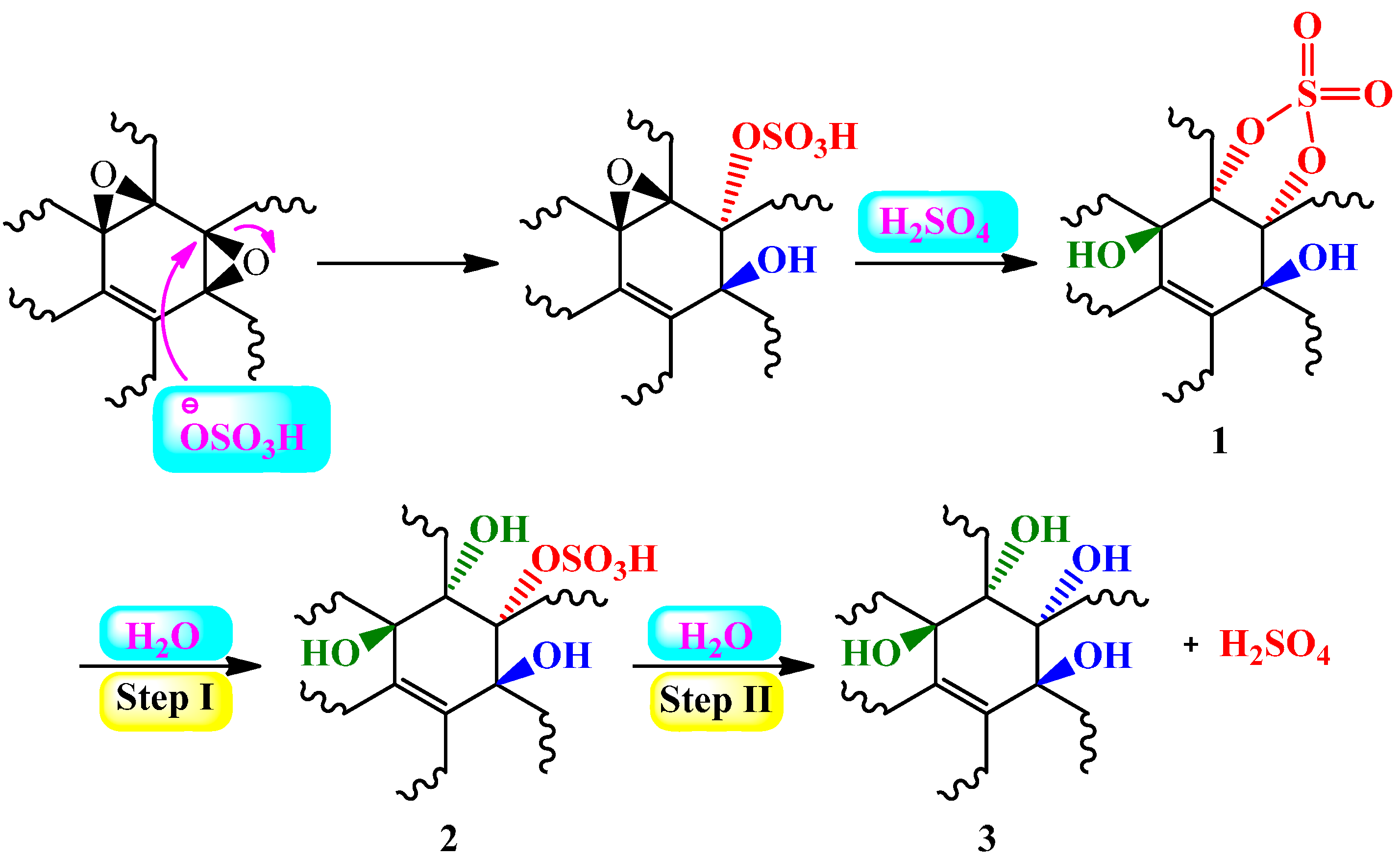
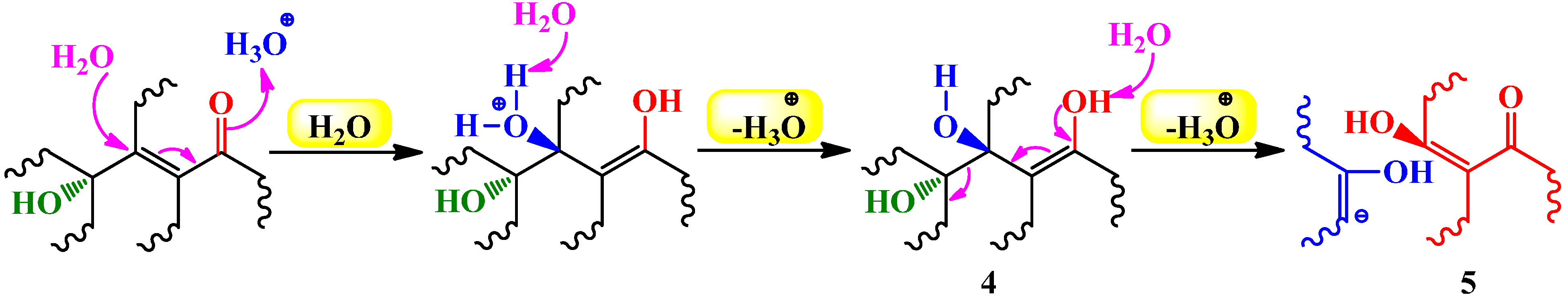
4.2.1. Hydration of Alkynes

4.2.2. Aza-Michael Addition
4.2.3. Ring Opening of Epoxides and Acetalization of Aldehydes
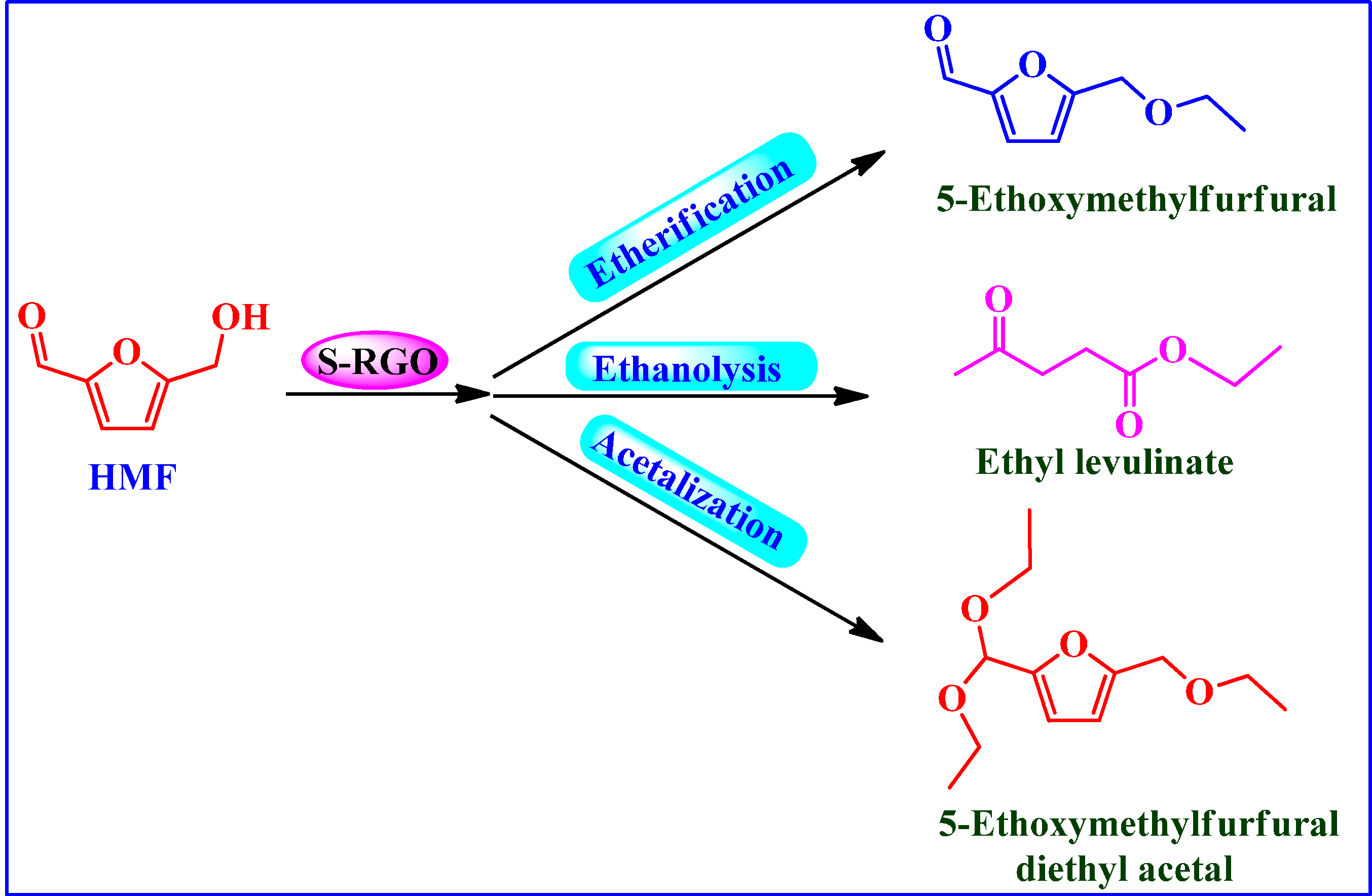
4.2.4. Conversion of Carbohydrates into 5-Ethoxymethylfurfural (EMF)
4.3. Sulfonated/Sulfated Graphene and Graphene Oxide (GO) as Acid Catalysts
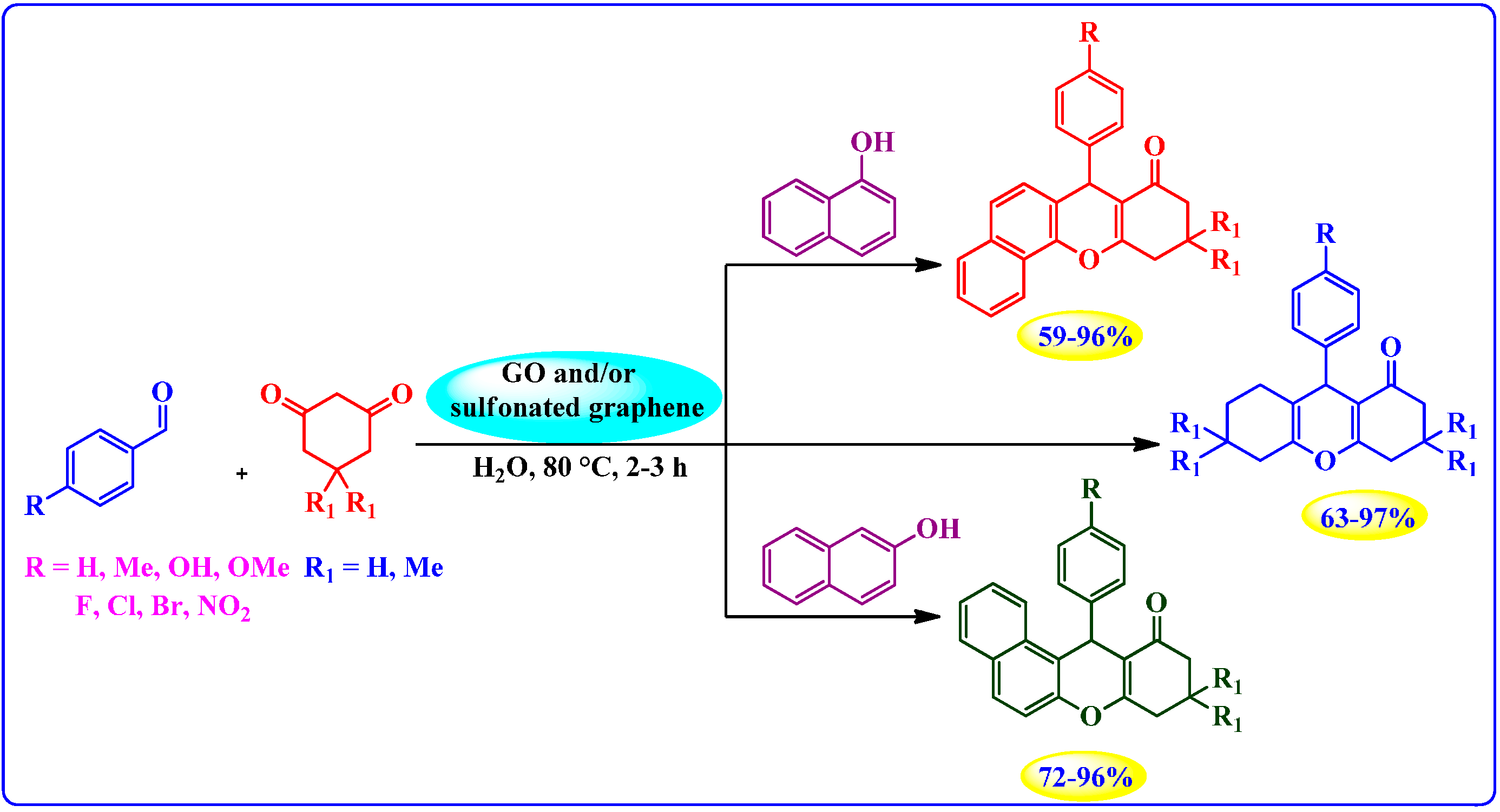
4.3.1. Sulfonated Graphene and GO as Acid Catalysts Prepared by Diazonium Salt
Hydrolysis of Ethyl Acetate
Synthesis of Xanthenes and Benzoxanthenes
Dehydration of Xylose


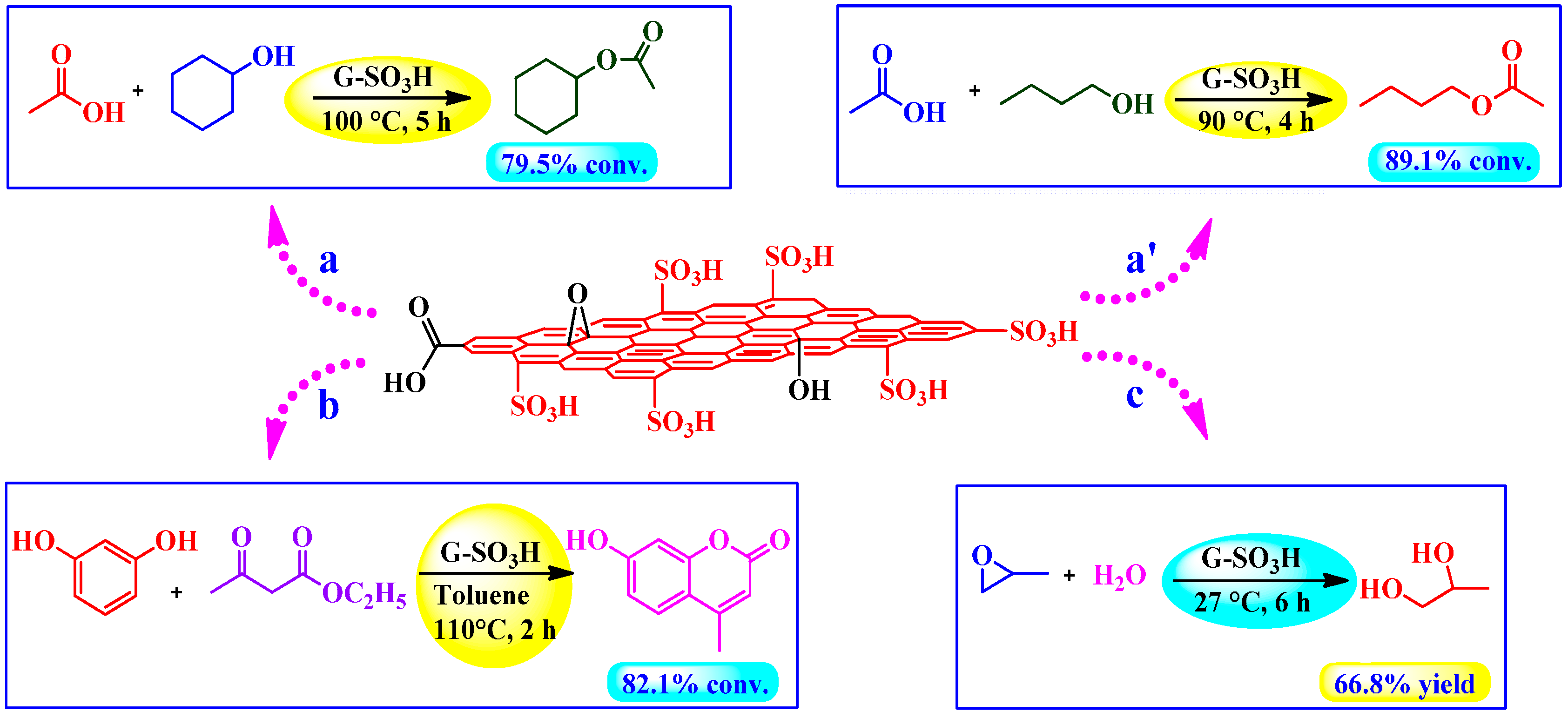
4.3.2. Sulfonated Graphene as Acid Catalyst Prepared by Fuming Sulfuric Acid or Oleum
Esterification, Peckmann, and Hydration Reactions

4.3.3. Sulfated Graphene as Acid Catalyst Prepared by ClSO3H
Ester-Exchange Reactions
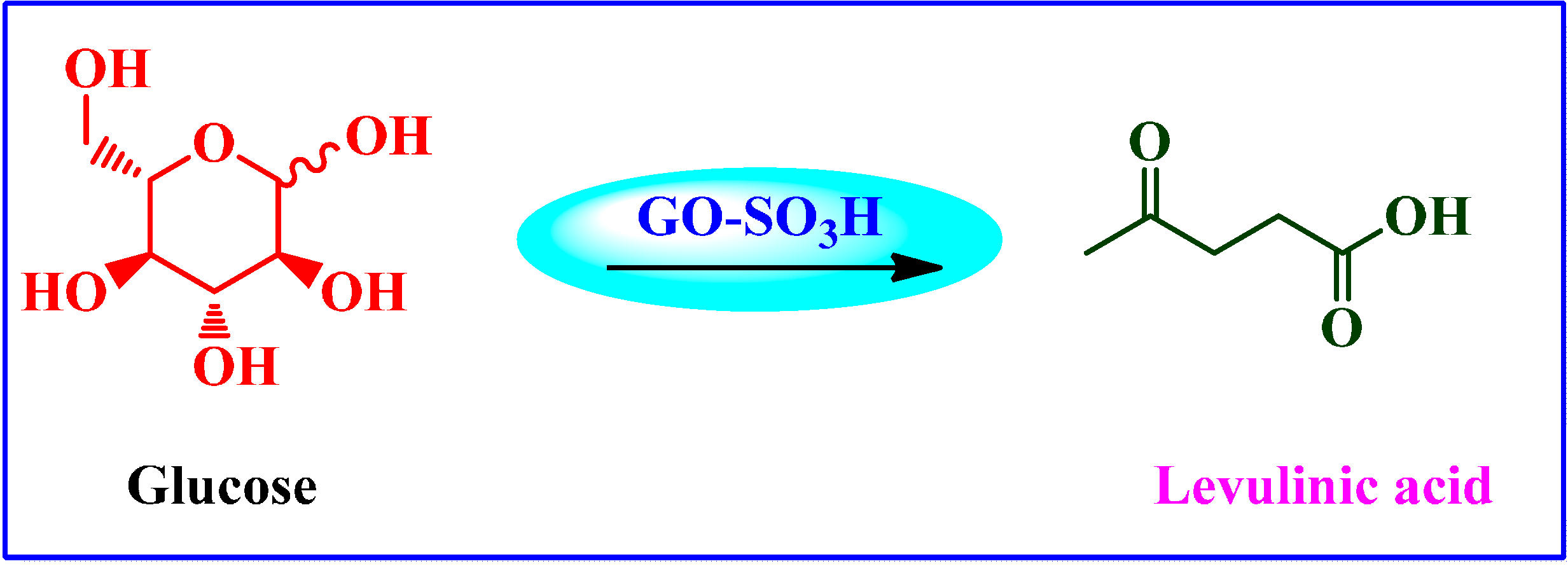
Chemical Conversion of Carbohydrates to Levulinic Acid (LA)

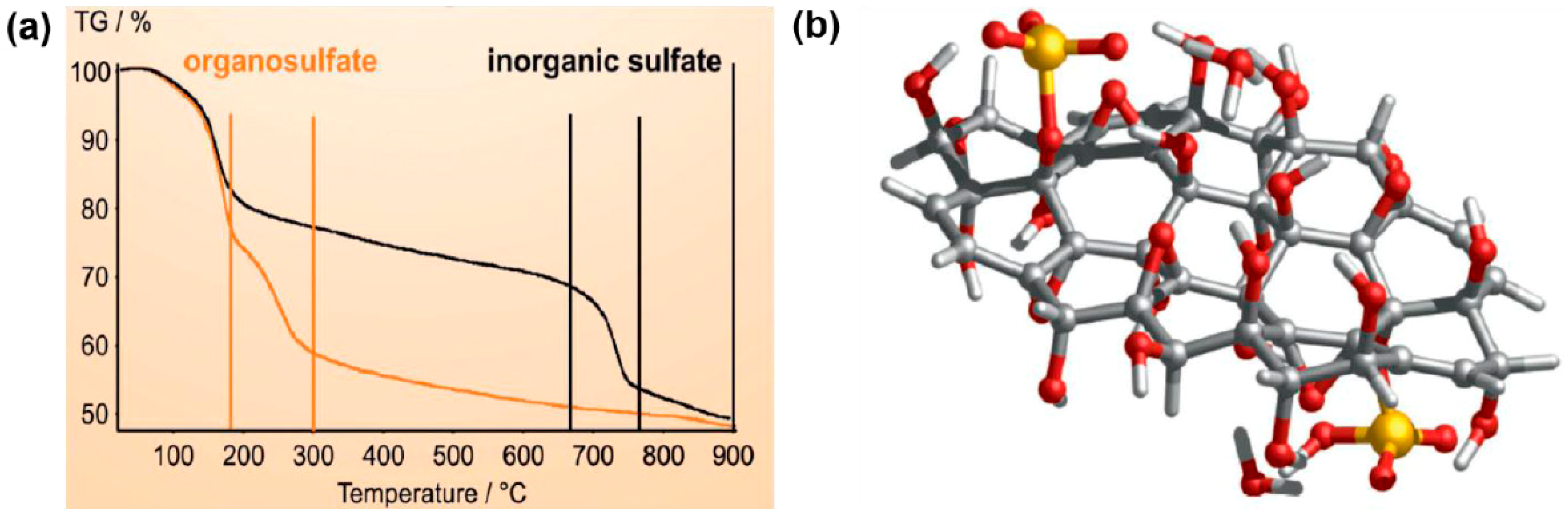
4.3.4. Sulfated rGO as Acid Catalyst Prepared by H2SO4
Conversion of HMF into Biofuels

5. Conclusions: Final Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mizuno, N.; Misono, M. Heterogeneous catalysis. Chem. Rev. 1998, 98, 199–217. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Sarkar, T.; Khasnobis, S. Amberlyst-15 in organic synthesis. ARKIVOC 2012, 570–609. [Google Scholar] [CrossRef]
- Okuhara, T. Water-tolerant solid acid catalysts. Chem. Rev. 2002, 102, 3641–3666. [Google Scholar] [CrossRef] [PubMed]
- Harmer, M.A.; Sun, Q. Solid acid catalysis using ion-exchange resins. Appl. Catal. A: Gen. 2001, 221, 45–62. [Google Scholar]
- Harmer, M.A.; Farneth, W.E.; Sun, Q. High surface area nafion resin/silica nanocomposites: A new class of solid acid catalyst. J. Am. Chem. Soc. 1996, 118, 7708–7715. [Google Scholar] [CrossRef]
- Shylesh, S.; Thiel, W.R. Bifunctional acid–base cooperativity in heterogeneous catalytic reactions: Advances in silica supported organic functional groups. ChemCatChem 2011, 3, 278–287. [Google Scholar] [CrossRef]
- Harmer, M.A. Industrial processes using solid acid catalysts. In Handbook of Green Chemistry and Technology; Clark, J.H., Macquarris, D.J., Eds.; Blackwell Publishers: London, UK, 2002; pp. 86–117. [Google Scholar]
- Chauhan, S.M.S.; Garg, B.; Bisht, T. Syntheses of calix[4]pyrroles by amberlyst-15 catalyzed cyclocondensations of pyrrole with selected ketones. Molecules 2007, 12, 2458–2466. [Google Scholar] [CrossRef]
- Suwannakarn, K.; Lotero, E.; Goodwin, J.G., Jr.; Lu, C. Stability of sulfated zirconia and the nature of the catalytically active species in the transesterification of triglycerides. J. Catal. 2008, 255, 279–286. [Google Scholar] [CrossRef]
- Onda, A.; Ochi, T.; Yanagisawa, K. Selective hydrolysis of cellulose into glucose over solid acid catalysts. Green Chem. 2008, 10, 1033–1037. [Google Scholar] [CrossRef]
- Suwannakarn, K.; Lotero, E.; Goodwin, J.G., Jr. A comparative study of gas phase esterification on solid acid catalysts. Catal. Lett. 2007, 114, 3–4. [Google Scholar] [CrossRef]
- Wang, X.; Liu, R.; Waje, M.M.; Chen, Z.; Yan, Y.; Bozhilov, K.N.; Feng, P. Sulfonated ordered mesoporous carbon as a stable and highly active protonic acid catalysts. Chem. Mater. 2007, 19, 2395–2397. [Google Scholar] [CrossRef]
- Nakajima, K.; Hara, M. Amorphous carbon with SO3H groups as a solid Brønsted acid catalyst. ACS Catal. 2012, 2, 1296–1304. [Google Scholar]
- Peng, F.; Zhang, L.; Wang, H.; Lv, P.; Yu, H. Sulfonated carbon nanotubes as a strong protonic acid catalyst. Carbon 2005, 43, 2405–2408. [Google Scholar] [CrossRef]
- Yu, H.; Jin, Y.; Li, Z.; Peng, F.; Wang, H. Synthesis and characterization of sulfonated single-walled carbon nanotubes and their performance as solid acid catalyst. J. Solid State Chem. 2008, 181, 432–438. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Guest Editorial. Nomenclature of sp2 carbon nanoforms. Carbon 2012, 50, 741–747.
- Wan, X.; Huang, Y.; Chen, Y. Focusing on energy and optoelectronic applications: A journey for graphene and graphene oxide at large scale. Acc. Chem. Res. 2012, 45, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Compton, O.C.; Nguyen, S.T. Graphene oxide, highly reduced graphene oxide, and graphene: Versatile building blocks for carbon-based materials. Small 2010, 6, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Chang, D.W.; Baek, J.-B.; Lu, W. Carbon nanomaterials for advanced energy conversion and storage. Small 2012, 8, 1130–1166. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Qi, X.; Boey, F.; Zhang, H. Graphene-based composites. Chem. Soc. Rev. 2012, 41, 666–686. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, B.; Liu, G.; Zhuang, X.; Kang, E.-T. Graphene and its derivatives: Switching ON and OFF. Chem. Soc. Rev. 2012, 41, 4688–4707. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.C.; Deokar, A.R.; Liao, J.H.; Shih, P.Y.; Ling, Y.-C. Graphene-based photothermal agent for rapid and effective killing of bacteria. ACS Nano 2013, 7, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.X.; Ng, S.R.; Khoo, S.Y.; Zheng, X.; Chen, P.; Li, C.M. RGD-peptide functionalized graphene biomimetic live-cell sensor for real-time detection of nitric oxide molecules. ACS Nano 2012, 6, 6944–6951. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wen, T.; Xue, C.; Han, Q.; Wang, Y.; Hong, J.; Zhou, X.; Jiang, H. RGO LBL modified biomimetic electrochemical sensor for detection of Sildenafil in herbal sexual health products. Biosens. Bioelectron. 2013, 42, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, G.; Chang, C.C.; Ling, Y.-C. Facile synthesis of smart magnetic graphene for safe drinking water: Heavy metal removal and disinfection control. ACS Sustain. Chem. Eng. 2013, 1, 462–471. [Google Scholar] [CrossRef]
- Liu, J.Y.; Chang, H.Y.; Truong, Q.D.; Ling, Y.-C. Synthesis of nitrogen-doped graphene by pyrolysis of ionic-liquid-functionalized graphene. J. Mater. Chem. C 2013, 1, 1713–1716. [Google Scholar] [CrossRef]
- Ganesh, G.; Ling, Y.-C. Magnetic and fluorescent graphene for dual modal imaging and single light induced photothermal and photodynamic therapy of cancer cells. Biomaterials 2014, 35, 4499–4507. [Google Scholar] [CrossRef] [PubMed]
- Garg, B.; Ling, Y.-C. Versatilities of graphene-based catalysts in organic transformations. Green Mater. 2013, 1, 47–61. [Google Scholar] [CrossRef]
- Dreyer, D.R.; Jia, H.-P.; Bielawski, C.W. Graphene oxide: A convenient carbocatalyst for facilitating oxidation and hydration reactions. Angew. Chem. Int. Ed. 2010, 49, 6813–6816. [Google Scholar]
- Su, C.; Loh, K.P. Carbocatalysts: Graphene oxide and its derivatives. Acc. Chem. Res. 2013, 46, 2275–2285. [Google Scholar] [CrossRef] [PubMed]
- Machado, B.F.; Serp, P. Graphene-based materials for catalysis. Catal. Sci. Technol. 2012, 2, 54–75. [Google Scholar] [CrossRef]
- Dreyer, D.R.; Todd, A.D.; Bielawski, C.W. Harnessing the chemistry of graphene oxide. Chem. Soc. Rev. 2014, 43, 5288–5301. [Google Scholar] [CrossRef] [PubMed]
- Navalon, S.; Dhakshinamoorthy, A.; Alvaro, M.; Garcia, H. Carbocatalysis by graphene-based materials. Chem. Rev. 2014, 114, 6179–6212. [Google Scholar] [CrossRef] [PubMed]
- Brodie, B.C. On the atomic weight of graphite. Philos. Trans. R. Soc. Lond. 1859, 149, 249–259. [Google Scholar] [CrossRef]
- Staudenmaier, L. Verfahren zür darstellung der graphitsäure. Ber. Dtsch. Chem. Ges. 1898, 31, 1481–1487. [Google Scholar] [CrossRef]
- Staudenmaier, L. Verfahren zür darstellung der graphitsäure. Ber. Dtsch. Chem. Ges. 1899, 32, 1394–1399. [Google Scholar] [CrossRef]
- Hummers, W.S., Jr.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339–1339. [Google Scholar] [CrossRef]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved Synthesis of Graphene Oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, U.; Holst, R. Über die säurenatur und die methylierung von graphitoxyd. Ber. Dtsch. Chem. Ges. A/B 1939, 72, 754–771. [Google Scholar]
- Ruess, G. Uber das graphitoxyhydroxyd (graphitoxyd). Monatshefte Chem. 1947, 76, 381–417. [Google Scholar] [CrossRef]
- Clauss, A.; Plass, R.; Boehm, H.-P.; Hofmann, U. Untersuchungen zür Struktur des Graphitoxyds. Z. Anorg. Allg. Chem. 1957, 291, 205–220. [Google Scholar] [CrossRef]
- Scholz, W.; Boehm, H.P. Untersuchungen am Graphitoxid. VI. Betrachtungen zür Struktur des Graphitoxids. Z. Anorg. Allg. Chem. 1969, 369, 327–340. [Google Scholar]
- Nakajima, T.; Mabuchi, A.; Hagiwara, R. A new structure model of graphite oxide. Carbon 1988, 26, 357–361. [Google Scholar] [CrossRef]
- Nakajima, T.; Matsuo, Y. Formation process and structure of graphite oxide. Carbon 1994, 32, 469–475. [Google Scholar] [CrossRef]
- He, H.; Riedl, T.; Lerf, A.; Klinowski, J. Solid-state NMR studies of the structure of graphite oxide. J. Phys. Chem. 1996, 100, 19954–19958. [Google Scholar] [CrossRef]
- Lerf, A.; He, H.; Riedl, T.; Forster, M.; Klinowski, J. 13C and 1H MAS NMR studies of graphite oxide and its chemically modified derivatives. Solid State Ion. 1997, 101–103 Part 2, 857–862. [Google Scholar]
- Lerf, A.; He, H.; Forster, M.; Klinowski, J. Structure of graphite oxide revisited. J. Phys. Chem. B 1998, 102, 4477–4482. [Google Scholar] [CrossRef]
- Szabó, T.; Berkesi, O.; Forgó, P.; Josepovits, K.; Sanakis, Y.; Petridis, D.; Dékány, I. Evolution of surface functional groups in a series of progressively oxidized graphite oxides. Chem. Mater. 2006, 18, 2740–2749. [Google Scholar] [CrossRef]
- Gao, W.; Alemany, L.B.; Ci, L.J.; Ajayan, P.M. New insights into the structure and reduction of graphite oxide. Nat. Chem. 2009, 1, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Erickson, K.; Erni, R.; Lee, Z.; Alem, N.; Gannett, W.; Zettl, A. Determination of the local chemical structure of graphene oxide and reduced graphene oxide. Adv. Mater. 2010, 22, 4467–4472. [Google Scholar] [CrossRef] [PubMed]
- Szabó, T.; Tombácz, E.; Illés, E.; Dékány, I. Enhanced acidity and pH-dependent surface charge characterization of successively oxidized graphite oxides. Carbon 2006, 44, 537–538. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Alvaro, M.; Concepción, P.; Fornés, V.; Garcia, H. Graphene oxide as an acid catalyst for the room temperature ring opening of epoxides. Chem. Commun. 2012, 48, 5443–5445. [Google Scholar] [CrossRef]
- Wang, H.; Deng, T.; Wang, Y.; Cui, X.; Qi, Y.; Mu, X.; Hou, X.; Zhu, Y. Graphene oxide as a facile acid catalyst for the one-pot conversion of carbohydrates into 5-ethoxymethylfurfural. Green Chem. 2013, 15, 2379–2383. [Google Scholar] [CrossRef]
- Dimiev, A.; Kosynkin, D.V.; Alemany, L.B.; Chaguine, P.; Tour, J.M. Pristine graphite oxide. J. Am. Chem. Soc. 2012, 134, 2815–2822. [Google Scholar] [CrossRef] [PubMed]
- Dimiev, A.; Alemany, L.B.; Tour, J.M. Graphene oxide. Origin of acidity, its instability in water, and a new dynamic structural model. ACS Nano 2013, 7, 576–588. [Google Scholar]
- Eigler, S.; Dotzer, C.; Hof, F.; Bauer, W.; Hirsch, A. Sulfur species in graphene oxide. Chem. Eur. J. 2013, 19, 9490–9496. [Google Scholar] [CrossRef] [PubMed]
- Toda, M.; Takagaki, A.; Okamura, M.; Kondo, J.N.; Hayashi, S.; Domen, K.; Hara, M. Green chemistry: Biodiesel made with sugar catalyst. Nature 2005, 438, 178–178. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-Y.; Hunag, M.; Ma, H.-L.; Zhang, Z.-Q.; Gao, J.-M.; Zhu, Y.-L.; Han, X.-J.; Guo, X.-Y. Preparation of a carbon-based solid acid catalyst by sulfonating activated carbon in a chemical reduction process. Molecules 2010, 15, 7188–7196. [Google Scholar] [CrossRef] [PubMed]
- McAllister, M.J.; Li, J.-L.; Adamson, D.H.; Schniepp, H.C.; Abdala, A.A.; Liu, J.; Herrera-Alonso, M.; Milius, D.L.; Car, R.; Prud’homme, R.K.; et al. Single sheet functionalized graphene by oxidation and thermal expansion of graphite. Chem. Mater. 2007, 19, 4396–4404. [Google Scholar] [CrossRef]
- Schniepp, H.C.; Li, J.-L.; McAllister, M.J.; Sai, H.; Herrera-Alonso, M.; Adamson, D.H.; Prud’homme, R.K.; Car, R.; Saville, D.A.; Aksay, I.A. Functionalized single graphene sheets derived from splitting graphite oxide. J. Phys. Chem. B 2006, 110, 8535–8539. [Google Scholar] [CrossRef] [PubMed]
- Kaniyoor, A.; Baby, T.T.; Ramaprabhu, S. Graphene synthesis via hydrogen induced low temperature exfoliation of graphite oxide. J. Mater. Chem. 2010, 20, 8467–8469. [Google Scholar] [CrossRef]
- Zhou, M.; Wang, Y.; Zhai, Y.; Zhai, J.; Ren, W.; Wang, F.; Dong, S. Controlled synthesis of large-area and patterned electrochemically reduced graphene oxide films. Chem. Eur. J. 2009, 15, 6116–6120. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, D.R.; Park, S.; Bielawski, C.W.; Ruoff, R.S. The chemistry of graphene oxide. Chem. Soc. Rev. 2010, 39, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Amarnath, C.A.; Hong, C.E.; Kim, N.H.; Ku, B.-C.; Kuila, T.; Lee, J.H. A; Hong, C.E.; Kim, N.H; Ku, B.-C.; Kuila, T.; Lee, J.H. Efficient synthesis of graphene sheets using pyrrole as a reducing agent. H. Efficient synthesis of graphene sheets using pyrrole as a reducing agent. Carbon 2011, 49, 3497–3502. [Google Scholar]
- Liu, J.; Tang, J.; Gooding, J.J. Strategies for chemical modification of graphene and applications of chemically modified graphene. J. Mater. Chem. 2012, 22, 12435–12452. [Google Scholar] [CrossRef]
- Pei, S.; Cheng, H.-M. The reduction of graphene oxide. Carbon 2012, 50, 3210–3228. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Ma, G.; Wang, Z.; Liu, K.; Liu, H. Ethylene glycol reduced graphene oxide/polypyrrole composite for supercapacitor. Electrochim. Acta 2013, 88, 519–525. [Google Scholar] [CrossRef]
- Chua, C.K.; Pumera, M. Chemical reduction of graphene oxide: A synthetic chemistry viewpoint. Chem. Soc. Rev. 2014, 43, 291–312. [Google Scholar] [CrossRef] [PubMed]
- Kudin, K.N.; Ozbas, B.; Schniepp, H.C.; Prud’homme, R.K.; Aksay, I.A.; Car, R. Raman spectra of graphite oxide and functionalized graphene sheets. Nano Lett. 2008, 8, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Chakraborty, B.; Sood, A.K. Raman spectroscopy of graphene on different substrates and influence of defects. Bull. Mater. Sci. 2008, 31, 579–584. [Google Scholar] [CrossRef]
- Gupta, A.; Chen, G.; Joshi, P.; Tadigadapa, S.; Eklund, P.C. Raman scattering from high-frequency phonons in supported n-graphene layer films. Nano Lett. 2006, 6, 2667–2673. [Google Scholar] [CrossRef] [PubMed]
- Graf, D.; Molitor, F.; Ensslin, K.; Stampfer, C.; Jungen, A.; Hierold, C.; Wirtz, L. Spatially resolved Raman spectroscopy of single- and few-layer graphene. Nano Lett. 2007, 7, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Stankovich, S.; Dikin, D.A.; Piner, R.D.; Kohlhaas, K.A.; Kleinhammes, A.; Jia, Y.; Wu, Y.; Nguyen, S.T.; Ruoff, R.S. Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon 2007, 45, 1558–1565. [Google Scholar] [CrossRef]
- Muszynski, R.; Seger, B.; Kamat, P.V. Decorating graphene sheets with gold nanoparticles. J. Phys. Chem. C 2008, 112, 5263–5266. [Google Scholar] [CrossRef]
- Shin, H.-J.; Kim, K.K.; Benayad, A.; Yoon, S.-M.; Park, H.K.; Jung, I.-S.; Jin, M.H.; Jeong, H.-K.; Kim, J.M.; Choi, J.-Y.; et al. Efficient reduction of graphite oxide by sodium borohydride and its effect on electrical conductance. Adv. Funct. Mater. 2009, 19, 1987–1992. [Google Scholar] [CrossRef]
- Ambrosi, A.; Chua, C.K.; Bonanni, A.; Pumera, M. Lithium aluminium hydride as reducing agent for chemically reduced graphene oxides. Chem. Mater. 2012, 24, 2292–2298. [Google Scholar] [CrossRef]
- Stankovich, S.; Piner, R.D.; Chen, X.; Wu, N.; Nguyen, S.T.; Ruoff, R.S. Stable aqueous dispersions of graphitic nanoplatelets via the reduction of exfoliated graphite oxide in the presence of poly(sodium 4-styrenesulfonate). J. Mater. Chem. 2006, 16, 155–158. [Google Scholar] [CrossRef]
- Bai, H.; Xu, Y.; Zhao, L.; Li, C.; Shi, G. Non-covalent functionalization of graphene sheets by sulfonated polyaniline. Chem. Commun. 2009, 1667–1669. [Google Scholar] [CrossRef]
- Hao, R.; Qian, W.; Zhang, L.; Hou, Y. Aqueous dispersions of TCNQ-anion-stabilized graphene sheets. Chem. Commun. 2008, 6576–6578. [Google Scholar] [CrossRef]
- Xu, Y.; Bai, H.; Lu, G.; Li, C.; Shi, G. Flexible graphene films via the filtration of water-soluble noncovalent functionalized graphene sheets. J. Am. Chem. Soc. 2008, 130, 5856–5857. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Zhang, G.; Chen, H.; Wang, S.; Zhang, G.; Zhang, F.; Fan, X. Sulfonated graphene as water-tolerant solid acid catalyst. Chem. Sci. 2011, 2, 484–487. [Google Scholar] [CrossRef]
- Liu, F.; Sun, J.; Zhu, L.; Meng, X.; Qi, C.; Xiao, F.-S. Sulfated graphene as an efficient solid catalyst for acid-catalyzed liquid reactions. J. Mater. Chem. 2012, 22, 5495–5502. [Google Scholar] [CrossRef]
- Wang, L.; Wang, D.; Zhang, S.; Tian, H. Synthesis and characterization of sulfonated graphene as a highly active solid acid catalyst for the ester-exchange reaction. Catal. Sci. Technol. 2013, 3, 1194–1197. [Google Scholar] [CrossRef]
- Shaabani, A.; Mahyari, M.; Hajishaabanha, F. The synthesis of xanthenes and benzoxanthenes on graphene oxide and sulfated graphene nanosheets in water. Res. Chem. Intermed. 2013. [Google Scholar] [CrossRef]
- Upare, P.P.; Toon, J.-W.; Kim, M.Y.; Kang, H.-Y.; Hwang, D.W.; Hwang, Y.K.; Kung, H.H.; Chang, J.-S. Chemical conversion of biomass-derived hexose sugars to levulinic acid over sulfonic acid-functionalized graphene oxide catalysts. Green Chem. 2013, 15, 2935–2943. [Google Scholar] [CrossRef]
- Lam, E.; Chong, J.H.; Majid, E.; Liu, Y.; Hrapovic, S.; Leung, A.C.W.; Luong, J.H.T. Carbocatalytic dehydration of xylose to furfural in water. Carbon 2012, 50, 1033–1043. [Google Scholar] [CrossRef]
- He, D.; Kou, Z.; Xiong, Y.; Cheng, K.; Chen, X.; Pan, M.; Mu, S. Simultaneous sulfonation and reduction of graphene oxide as highly efficient supports for metal nanocatalysts. Carbon 2014, 66, 312–319. [Google Scholar] [CrossRef]
- Antunes, M.M.; Russo, P.A.; Wiper, P.V.; Veiga, J.M.; Pillinger, M.; Mafra, L.; Evtuguin, D.V.; Pinna, N.; Valente, A.A. Sulfonated graphene oxide as effective catalyst for conversion of 5-(hydroxymethyl)-2-furfural into biofuels. ChemSusChem 2014, 7, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xue, Y.; Dai, L. Sulfated graphene oxide as a hole-extraction layer in high performance polymer solar cells. Phys. Chem. Lett. 2012, 3, 1928–1933. [Google Scholar] [CrossRef]
- Garg, B.; Bisht, T.; Chauhan, S.M.S. Synthesis and anion binding properties of novel 3,12- and 3,7-bis(4'-nitrophenyl)-azo-calix[4]pyrrole receptors. New J. Chem. 2010, 34, 1251–1254. [Google Scholar] [CrossRef]
- Chauhan, S.M.S.; Garg, B.; Bisht, T. Synthesis and anion binding of 2-arylazo-meso-octamethylcalix[4]pyrroles. Supramol. Chem. 2009, 21, 394–400. [Google Scholar] [CrossRef]
- Chauhan, S.M.S.; Bisht, T.; Garg, B. 1-arylazo-5,5-dimethyl Dipyrromethanes: Versatile chromogenic probes for anions. Sens. Actuators B: Chem. 2009, 141, 116–123. [Google Scholar]
- Garg, B.; Bisht, T.; Chauhan, S.M.S. 2,2'-Diaminoazo-benzene, a potential scaffold for the synthesis of bis-ureas and thioureas: Solution phase anion sensing and binding studies. Sens. Actuators B: Chem. 2012, 168, 318–328. [Google Scholar]
- Lomeda, J.R.; Doyle, C.D.; Kosynkin, D.V.; Hwang, W.-F.; Tour, J.M. Diazonium functionalization of surfactant-wrapped chemically converted graphene sheets. J. Am. Chem. Soc. 2008, 130, 16201–16206. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Samulski, E.T. Synthesis of water soluble graphene. Nano Lett. 2008, 8, 1679–1682. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Jiang, L.; He, Y.; Li, J.; Dong, H.; Wang, X.; Hu, W. Sulfonated graphene for persistent aromatic pollutant management. Adv. Mater. 2011, 23, 3959–3963. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Low, W.P.; Lee, H.K. Evaluation of sulfonated graphene sheets as sorbent for micro-solid-phase extraction combined with gas chromatography-mass spectrometry. J. Chromatogr. A 2012, 1233, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Coşkun, E.; Zaragoza-Contreras, E.A.; Salavagione, H.J. Synthesis of sulfonated graphene/polyaniline composites with improved electroactivity. Carbon 2012, 50, 2235–2243. [Google Scholar] [CrossRef]
- Ankamwar, B.; Surti, F. Water soluble graphene synthesis. Chem. Sci. Trans. 2012, 1, 500–507. [Google Scholar] [CrossRef]
- Geim, A.K. Graphene: Status and prospects. Science 2009, 324, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Garg, B.; Ling, Y.-C. Highly efficient synthesis of N-confused meso-tetraspirocyclohexyl calix[4]pyrrole using Brønsted acidic ionic liquids as catalysts. Tetrahedron Lett. 2012, 53, 5674–5677. [Google Scholar] [CrossRef]
- Garg, B.; Lei, S.-L.; Liu, S.-C.; Bisht, T.; Liu, J.-Y.; Ling, Y.-C. Rapid identification of trimethyl and triethyl amines using sulfonic acidic ionic liquids: A TOF-SIMS study of fragmentation reactions. Anal. Chim. Acta 2012, 757, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Garg, B.; Ling, Y.-C. One-pot green synthesis of azides from alcohols using Brønsted acidic ionic liquid [HMIM][BF4] as solvent and catalyst. J. Chin. Chem. Soc. 2014, 61, 737–742. [Google Scholar] [CrossRef]
- Pei, S.; Zhao, J.; Du, J.; Ren, W.; Cheng, H. Direct reduction of graphene oxide films into highly conductive and flexible graphene films by hydrohalic acids. Carbon 2010, 48, 4466–4474. [Google Scholar] [CrossRef]
- Behabtu, N.; Lomeda, J.R.; Green, M.J.; Higginbotham, A.L.; Sinitskii, A.; Kosynkin, D.V.; Tsentalovich, D.; Parra-Vasquez, A.N.G.; Schimidt, J.; Kesselman, E.; et al. Spontaneous high-concentration dispersions and liquid crystals of graphene. Nat. Nanotechnol. 2010, 5, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Liu, S.; Qin, X.; Wang, L.; Tian, J.; Luo, Y.; Asiri, A.M.; Al-You, A.O.; Sun, X. High-yield, large-scale production of few-layer graphene flakes within seconds: Using chlorosulfonic acid and H2O2 as exfoliating agents. J. Mater. Chem. 2012, 22, 8775–8777. [Google Scholar] [CrossRef]
- Mutlay, I.; Tudoran, L.B. Chlorosulfonic acid-based room temperature chemical expansion route for the bulk production of graphite nanoplatelets. Fuller. Nanotub. Carbon Nanostruct. 2013, 21, 149–157. [Google Scholar] [CrossRef]
- Dixon, P. Formation of sulphamic acid during the decomposition of ammonium sulphate. Nature 1944, 154, 706–706. [Google Scholar] [CrossRef]
- Kiyoura, R.; Urano, K. Mechanism, kinetics, and equilibrium of thermal decomposition of ammonium sulfate. Ind. Eng. Chem. Process Des. Dev. 1970, 9, 489–494. [Google Scholar] [CrossRef]
- Li, J.-H.; Zhang, G.-E. Investigation of the kinetics and mechanism of decomposition of ammonium hydrogen sulfate. Acta Phys. Chim. Sin. 1992, 8, 123–127. [Google Scholar] [CrossRef]
- Li, J.; Zhang, G.; Wang, J. The identification of the mechanism function and the kinetic investigation of the energy-storing reaction of ammonium hydrogen sulfate. Thermochem. Acta 1992, 207, 219–225. [Google Scholar] [CrossRef]
- Halstead, W.D. Thermal decomposition of ammonium sulphate. J. Appl. Chem. 1970, 20, 129–132. [Google Scholar] [CrossRef]
- Xu, Z.; Qi, Z.; Kaufman, A. Superior catalysts for proton exchange membrane fuel cells sulfonation of carbon-supported catalysts using sulfate salts. Electrochem. Solid-State Lett. 2005, 8, A313–A315. [Google Scholar]
- Kumar, A.V.; Rao, K.R. Recyclable graphite oxide catalyzed Friedel-Crafts addition of indoles to α,β-unsaturated ketones. Tetrahedron Lett. 2011, 52, 5188–5191. [Google Scholar] [CrossRef]
- Dreyer, D.R.; Jarvis, K.A.; Ferreirab, P.J.; Bielawski, C.W. Graphite oxide as a carbocatalyst for the preparation of fullerene-reinforced polyester and polyamide nanocomposites. Polym. Chem. 2012, 3, 757–766. [Google Scholar] [CrossRef]
- Chauhan, S.M.S.; Mishra, S. Use of graphite oxide and graphene oxide as catalysts in the synthesis of dipyrromethane and calix[4]pyrrole. Molecules 2011, 16, 7256–7266. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Mungse, H.P.; Kumar, N.; Choudhary, S.; Jain, S.L.; Sain, B.; Khatri, O.P. Graphene oxide: An efficient and reusable carbocatalyst for aza-Michael addition of amines to activated alkenes. Chem. Commun. 2011, 47, 12673–12675. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Alvaro, M.; Puche, M.; Fornes, V.; Garcia, H. Graphene oxide as catalyst for the acetalization of aldehydes at room temperature. ChemCatChem 2012, 4, 2026–2030. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Garg, B.; Bisht, T.; Ling, Y.-C. Graphene-Based Nanomaterials as Heterogeneous Acid Catalysts: A Comprehensive Perspective. Molecules 2014, 19, 14582-14614. https://doi.org/10.3390/molecules190914582
Garg B, Bisht T, Ling Y-C. Graphene-Based Nanomaterials as Heterogeneous Acid Catalysts: A Comprehensive Perspective. Molecules. 2014; 19(9):14582-14614. https://doi.org/10.3390/molecules190914582
Chicago/Turabian StyleGarg, Bhaskar, Tanuja Bisht, and Yong-Chien Ling. 2014. "Graphene-Based Nanomaterials as Heterogeneous Acid Catalysts: A Comprehensive Perspective" Molecules 19, no. 9: 14582-14614. https://doi.org/10.3390/molecules190914582