Carbonylation of Ethene Catalysed by Pd(II)-Phosphine Complexes

{kind=link}
{kind=link}
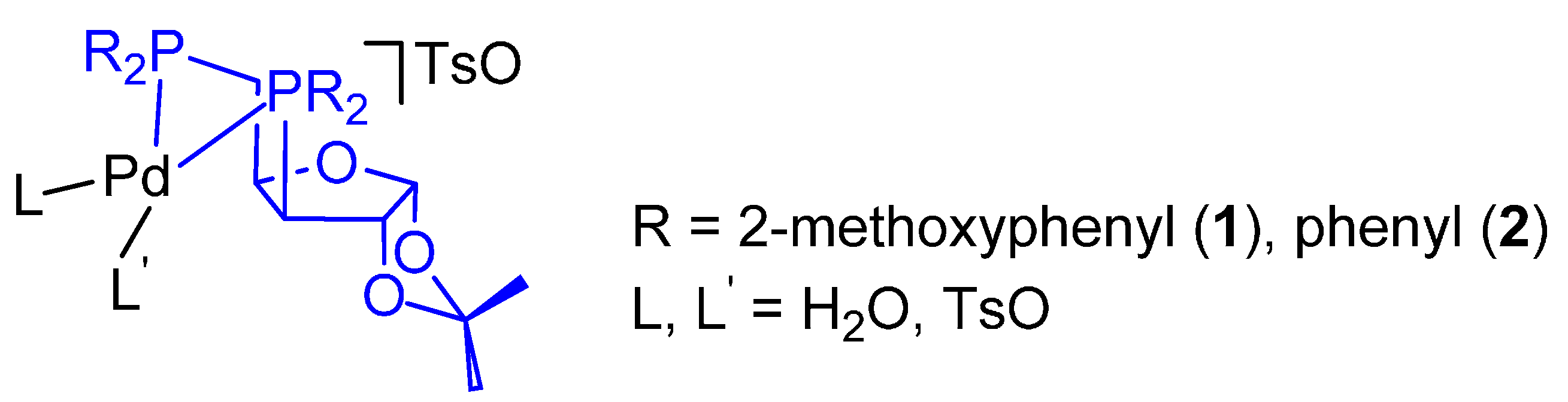{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
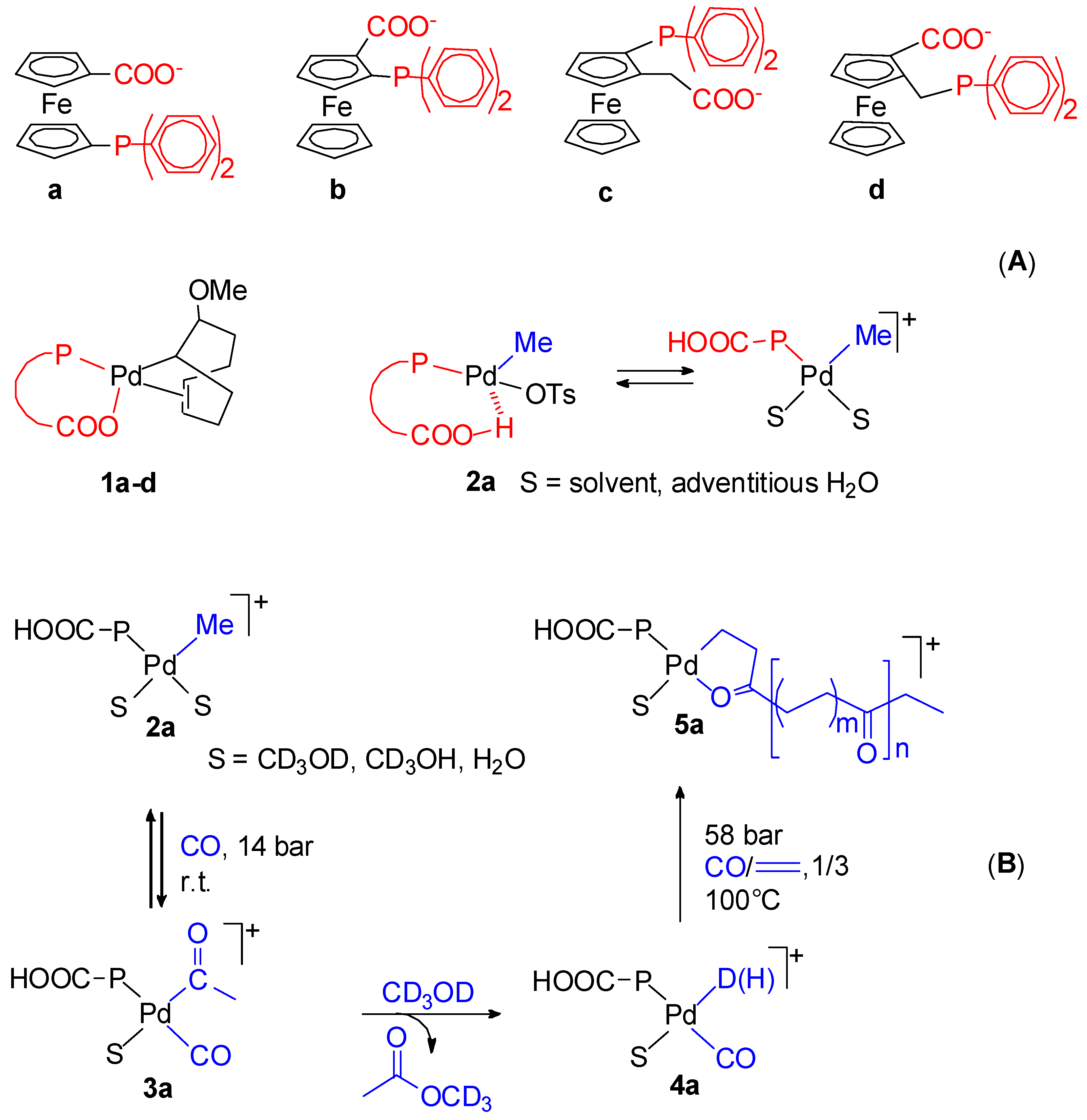{kind=link}
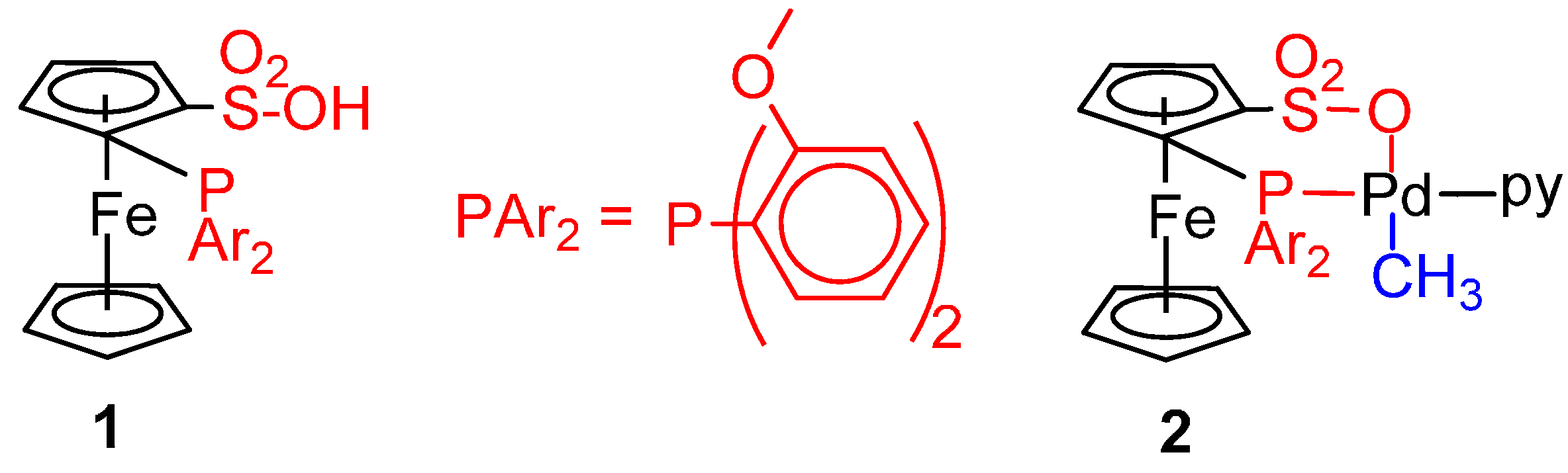{kind=link}
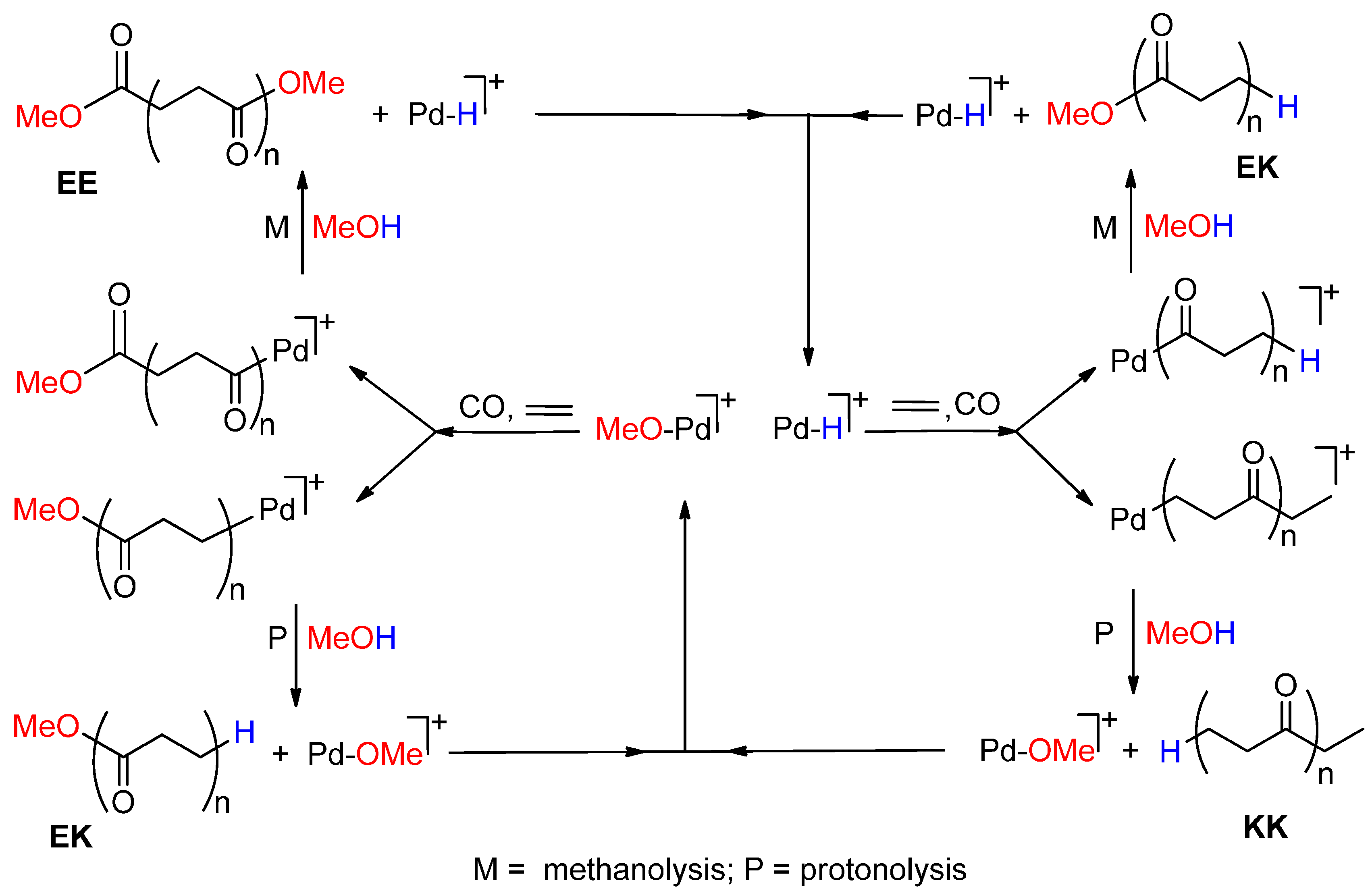{kind=link}
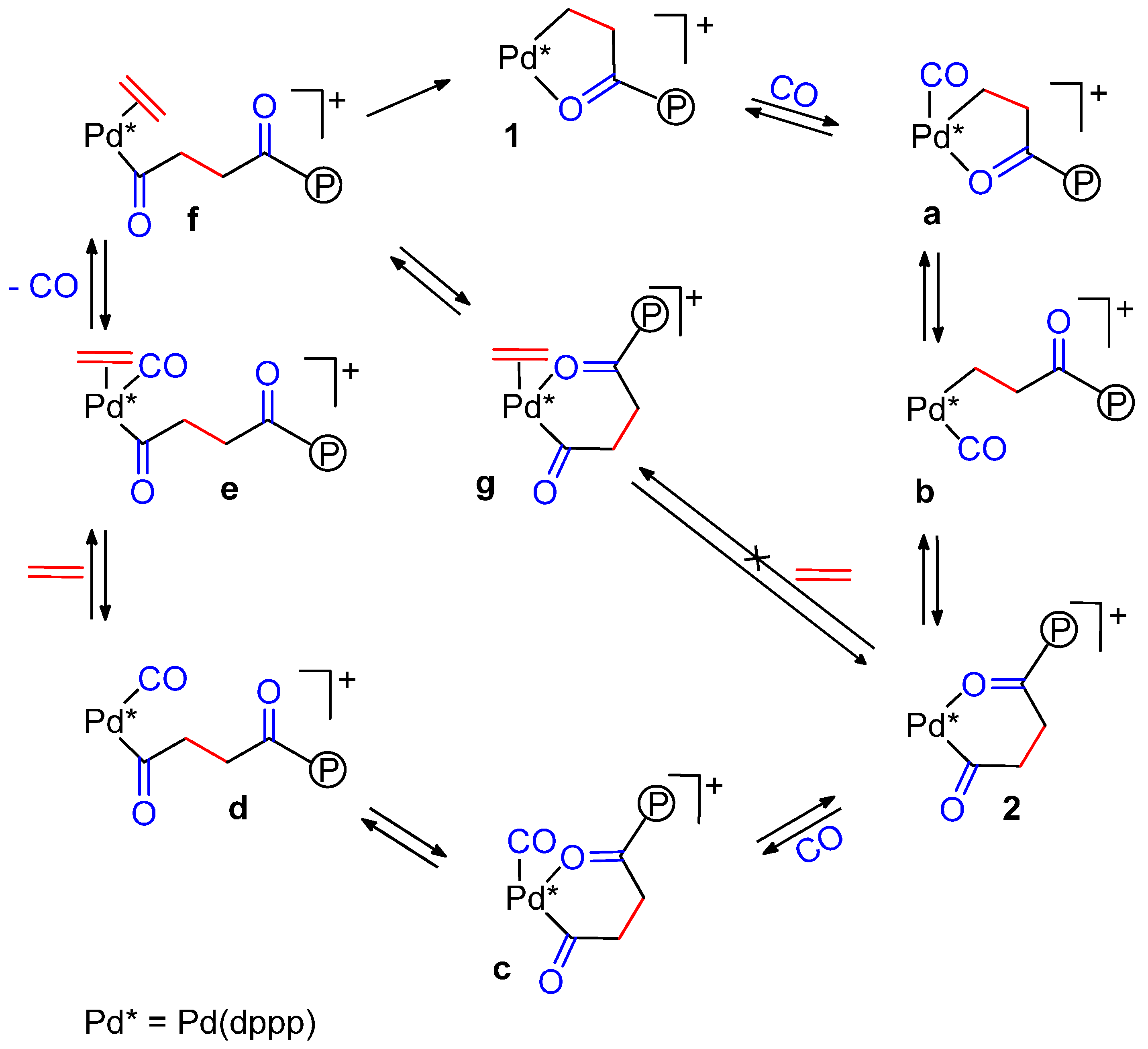{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Basic Concepts of CO-Ethene Copolymerization in MeOH
2.1. General Aspects
- (i)
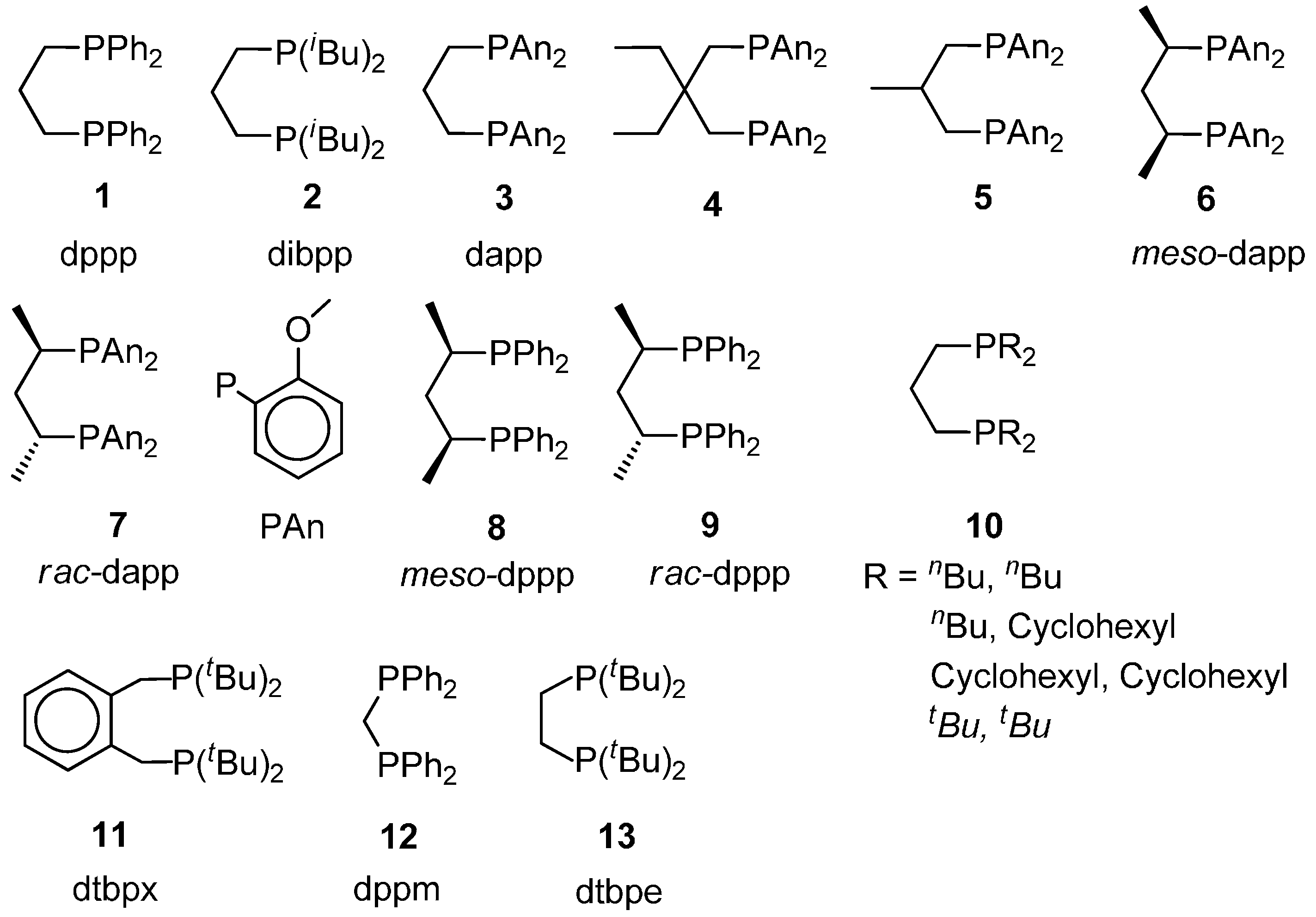
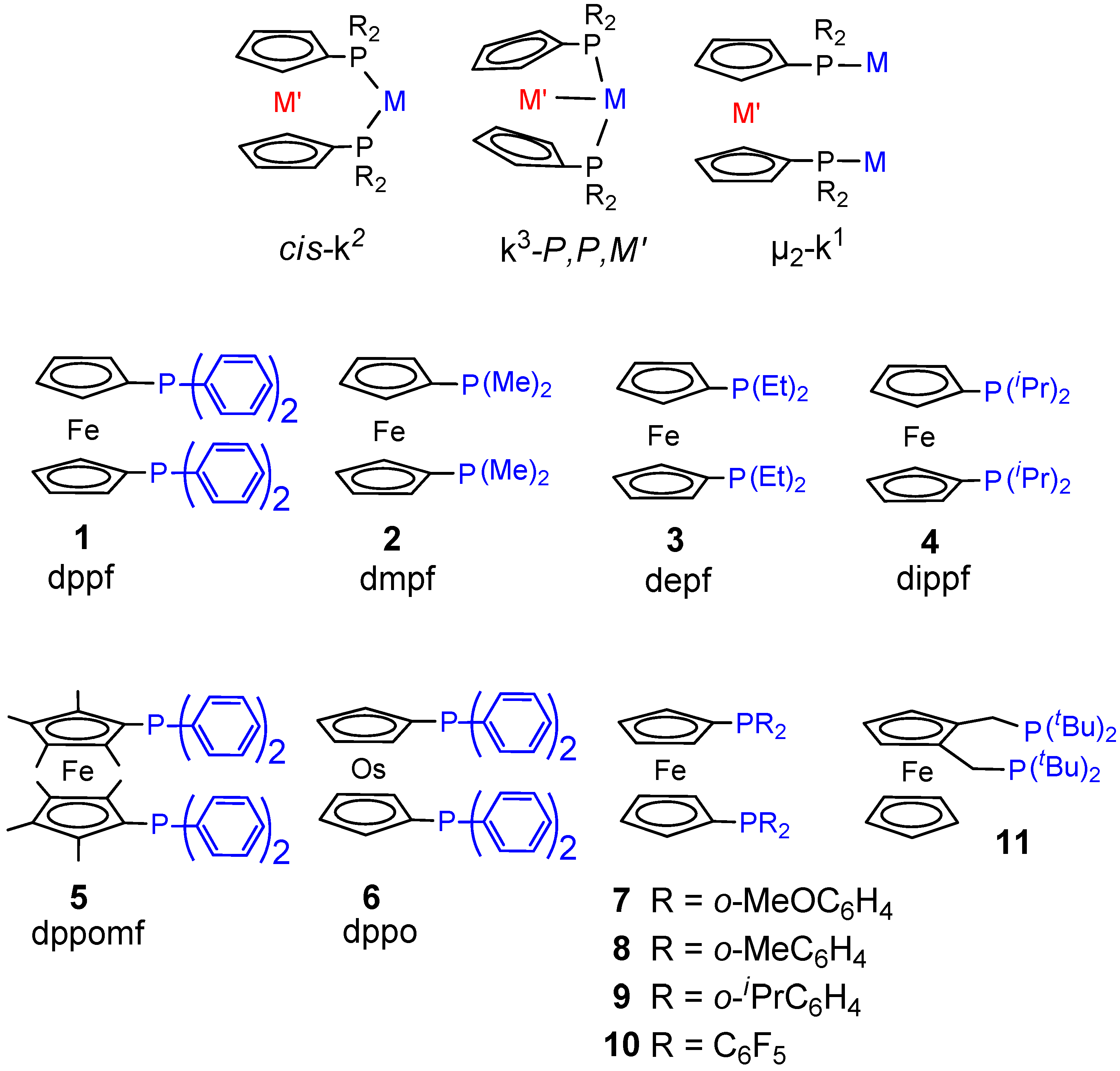
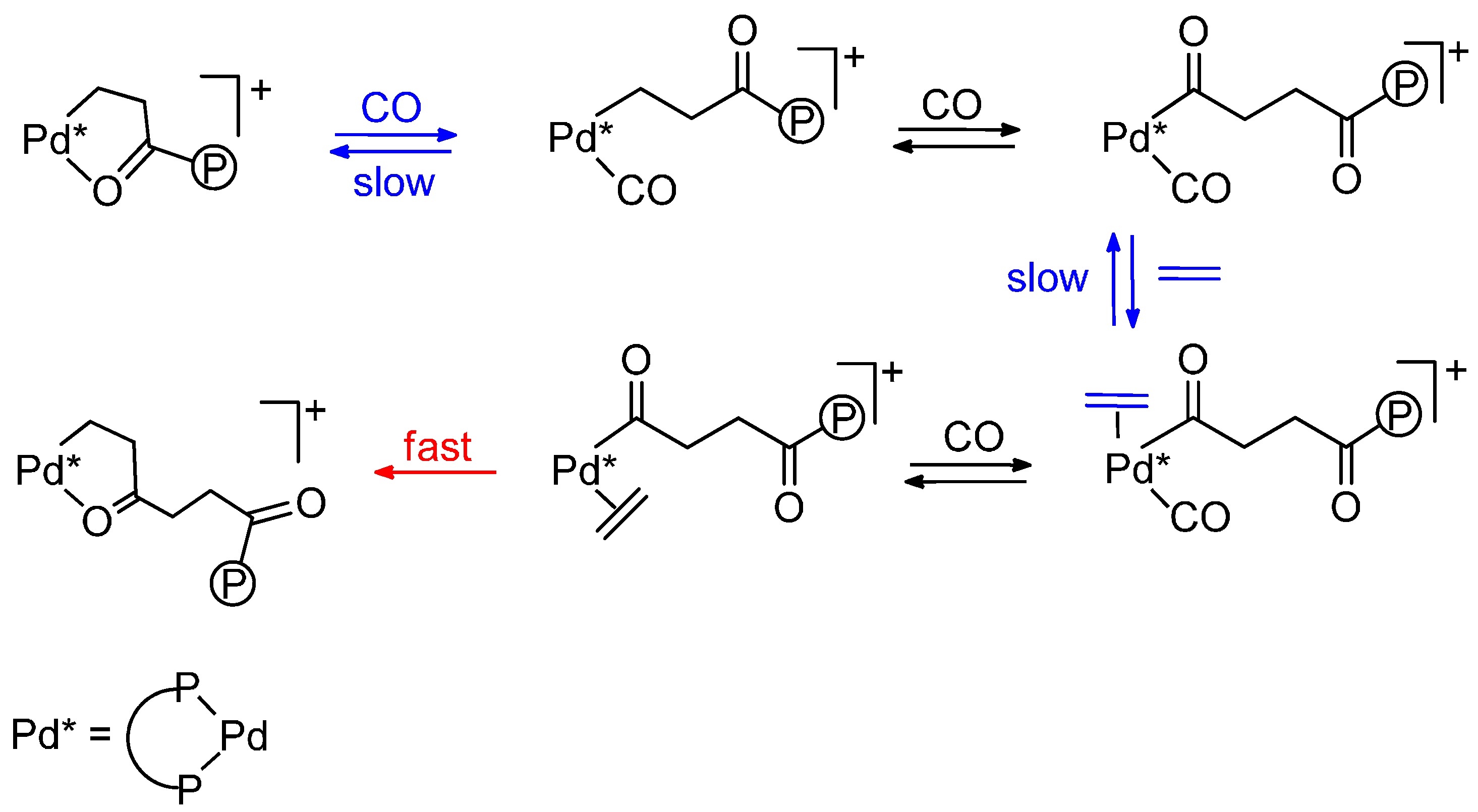
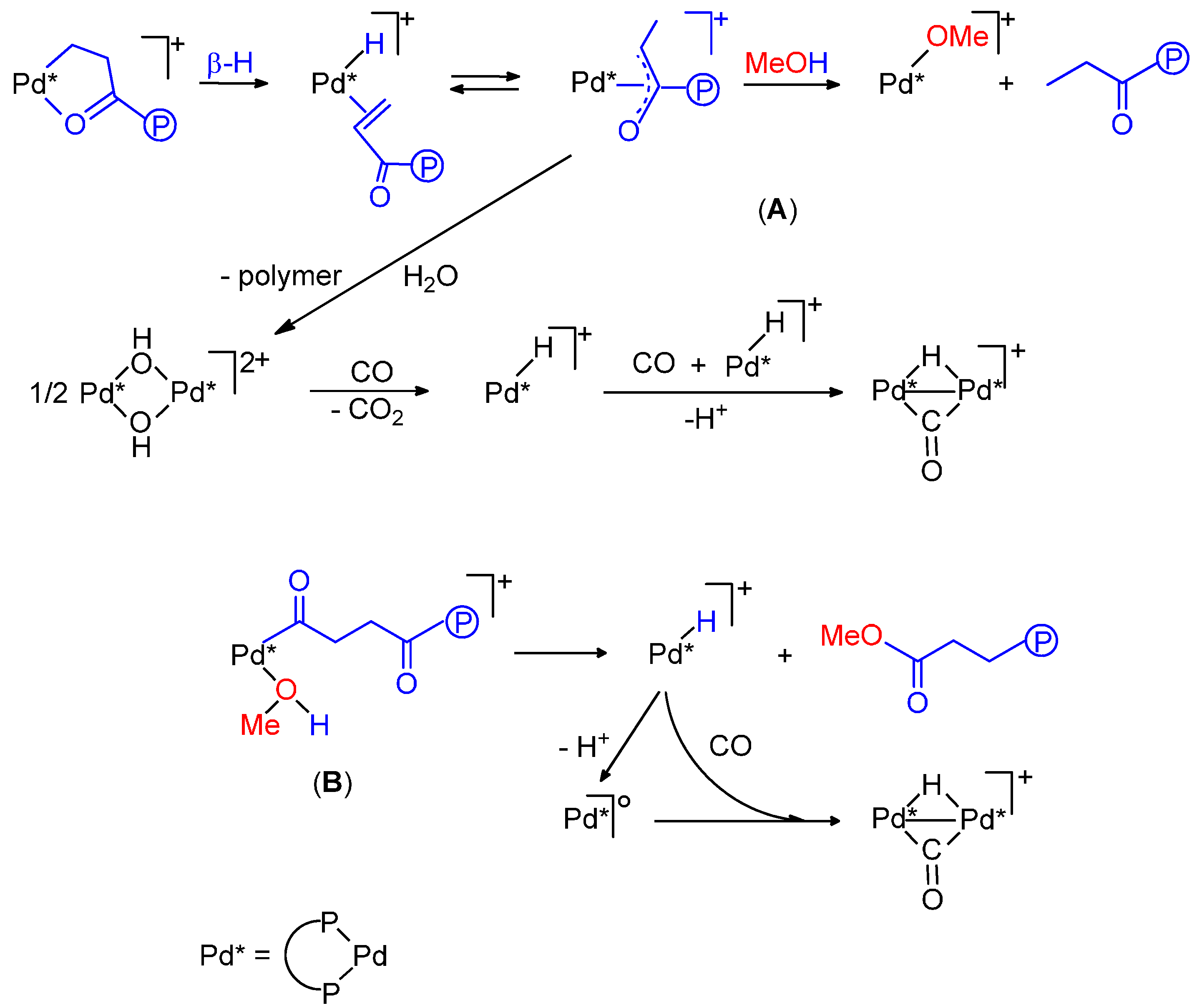
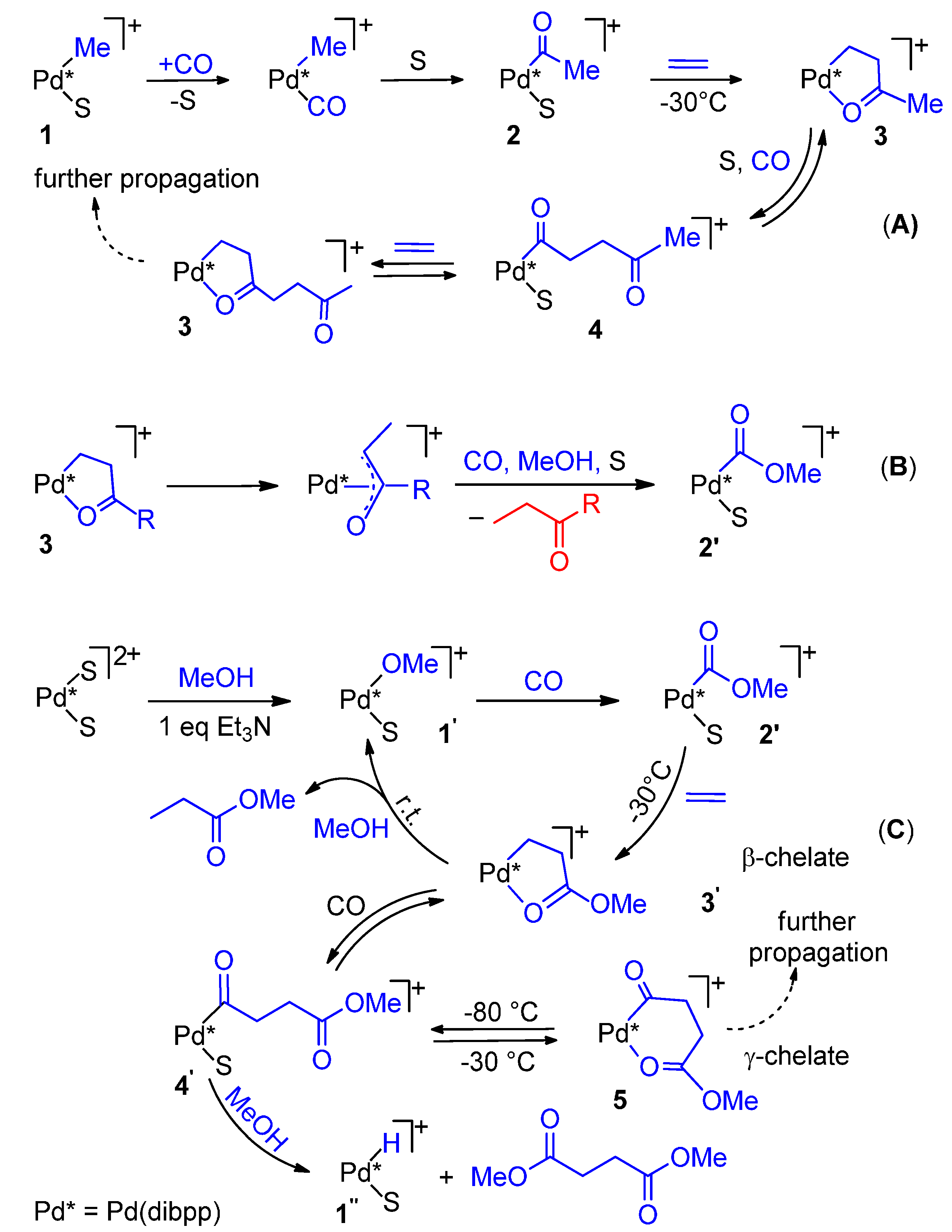
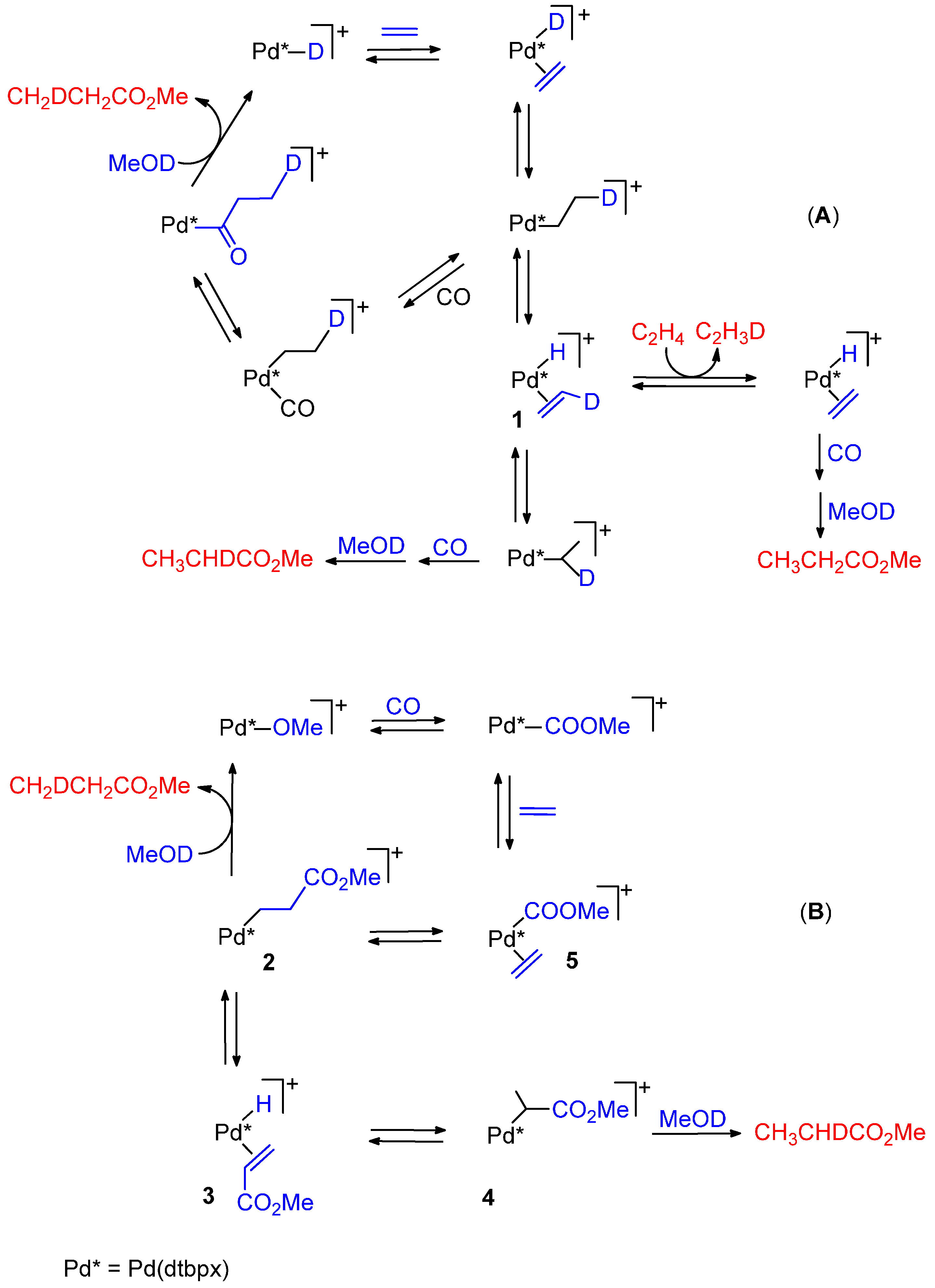
- Monophosphine catalysts lead to MP and eventually to low molecular weight co-oligomers, whereas diphosphine ones give PKs. Diphosphines, having the two P atoms linked by a C3 chain, give PKs with higher productivities and molecular weights than the others [8,10]. The striking difference between monophosphine- and diphosphine-catalysts was explained as the result that the diphosphine is always cis-chelated, so that the other two coordination sites of the d8-square planar palladium centre, one occupied by the growing polymer chain and the other by the monomer, are also always cis to each other, which is ideal in favouring the chain growth through migratory insertions of the monomers. Differently, Pd(II) coordinated by a monophosphine can assume both cis and trans geometries, so that chain growth is relatively slow compared to chain termination [8,10].
- (ii)
- (iii)
- (iv)
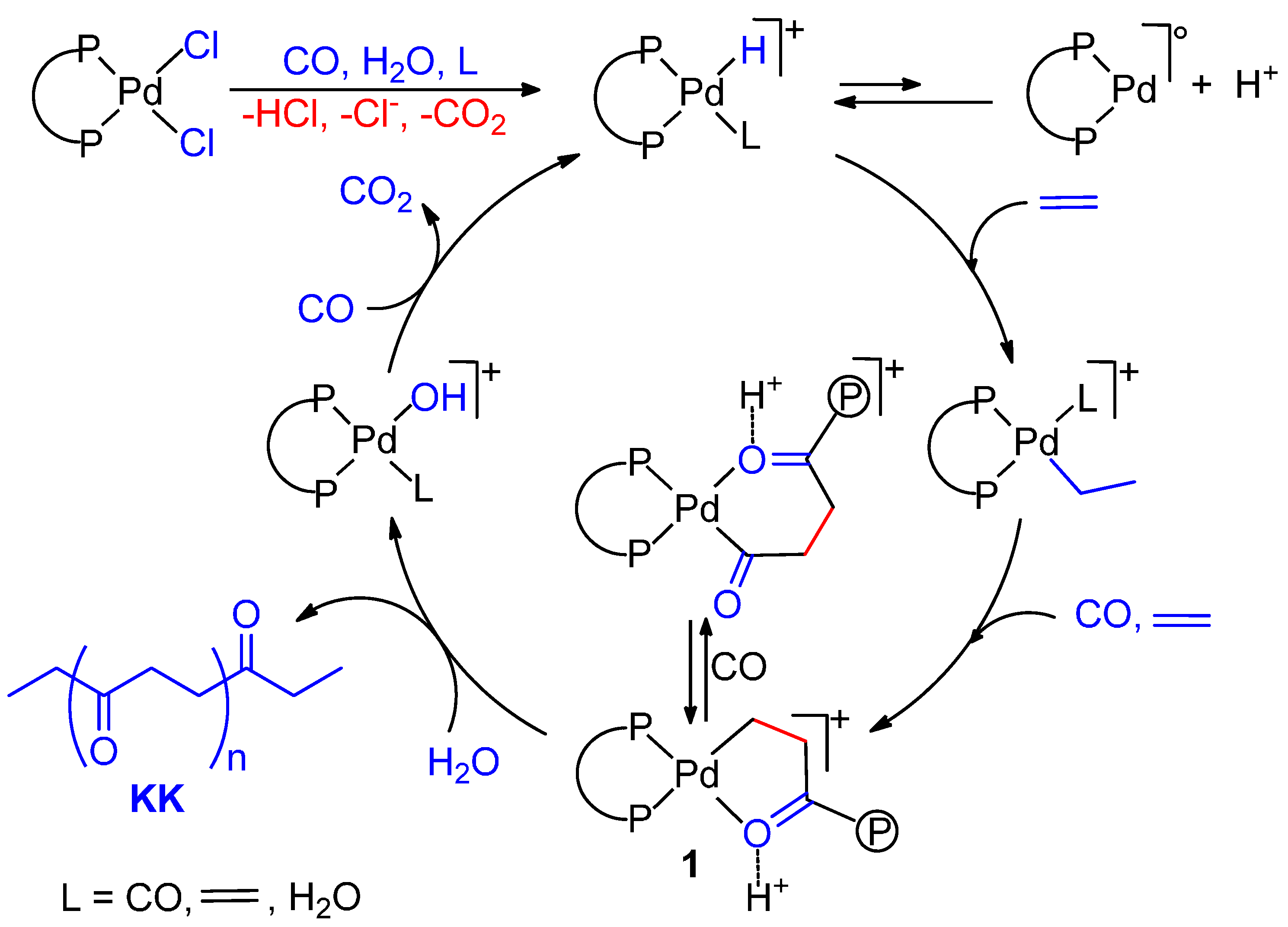
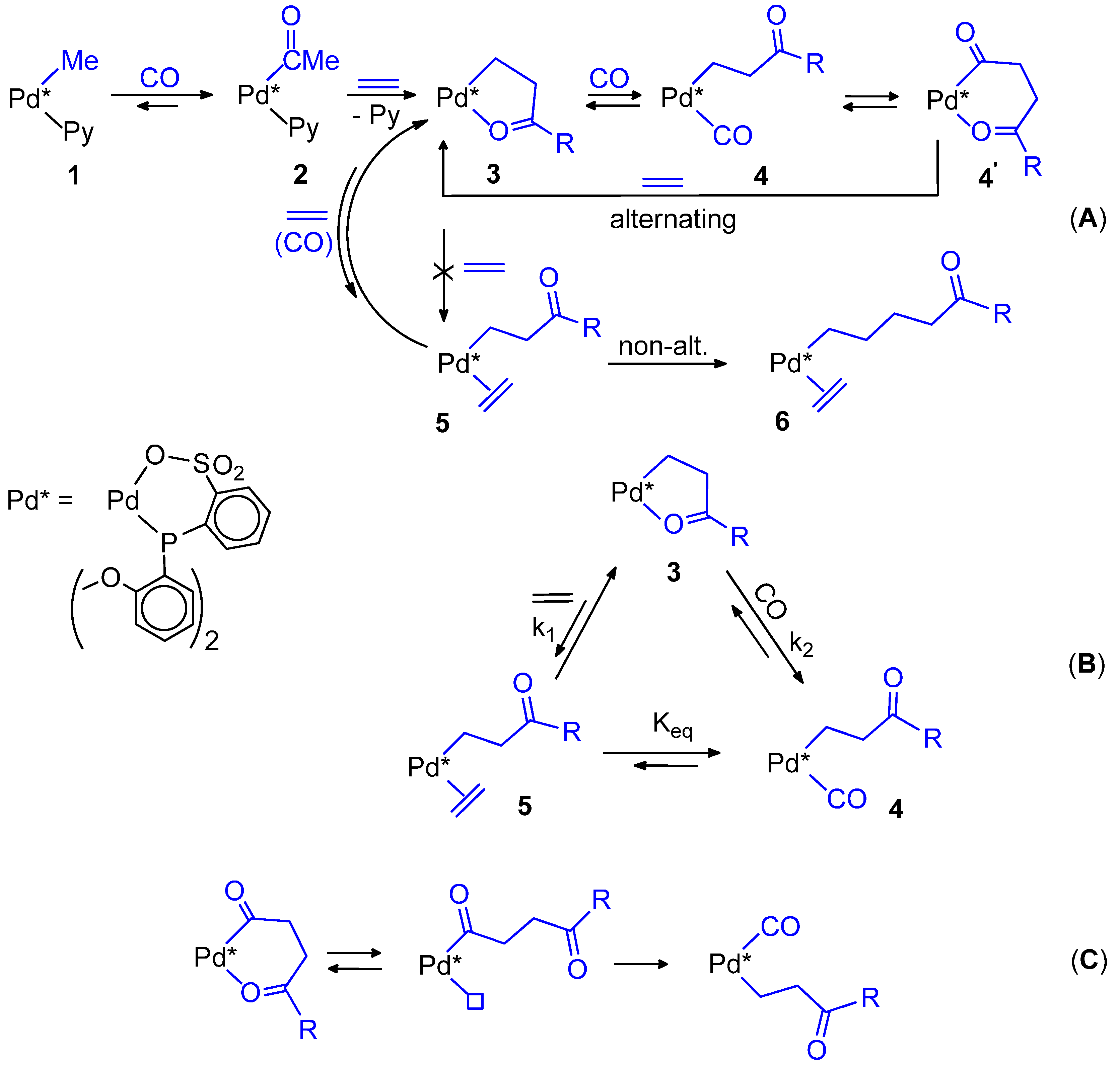
2.2. Alternating Chain Growing

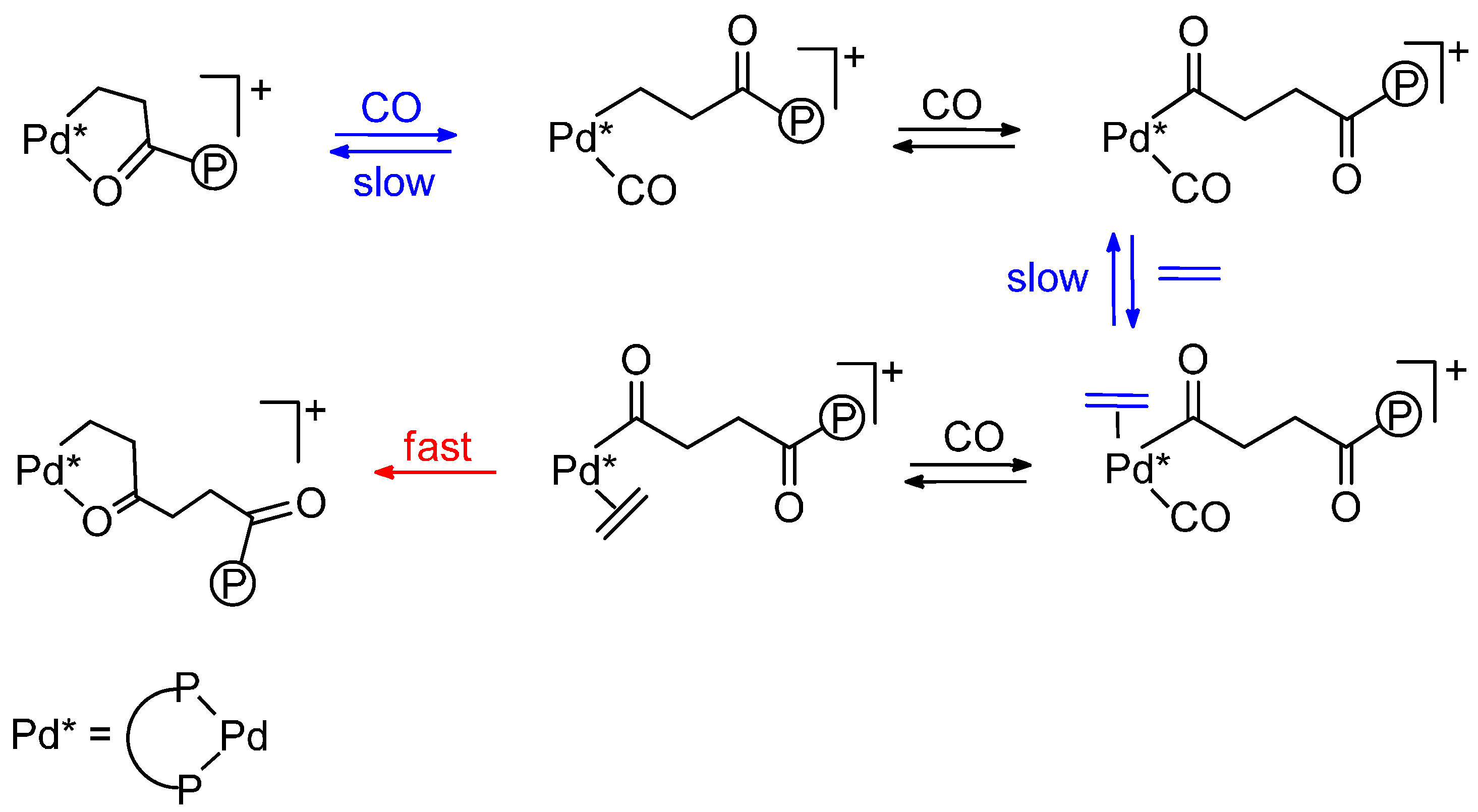
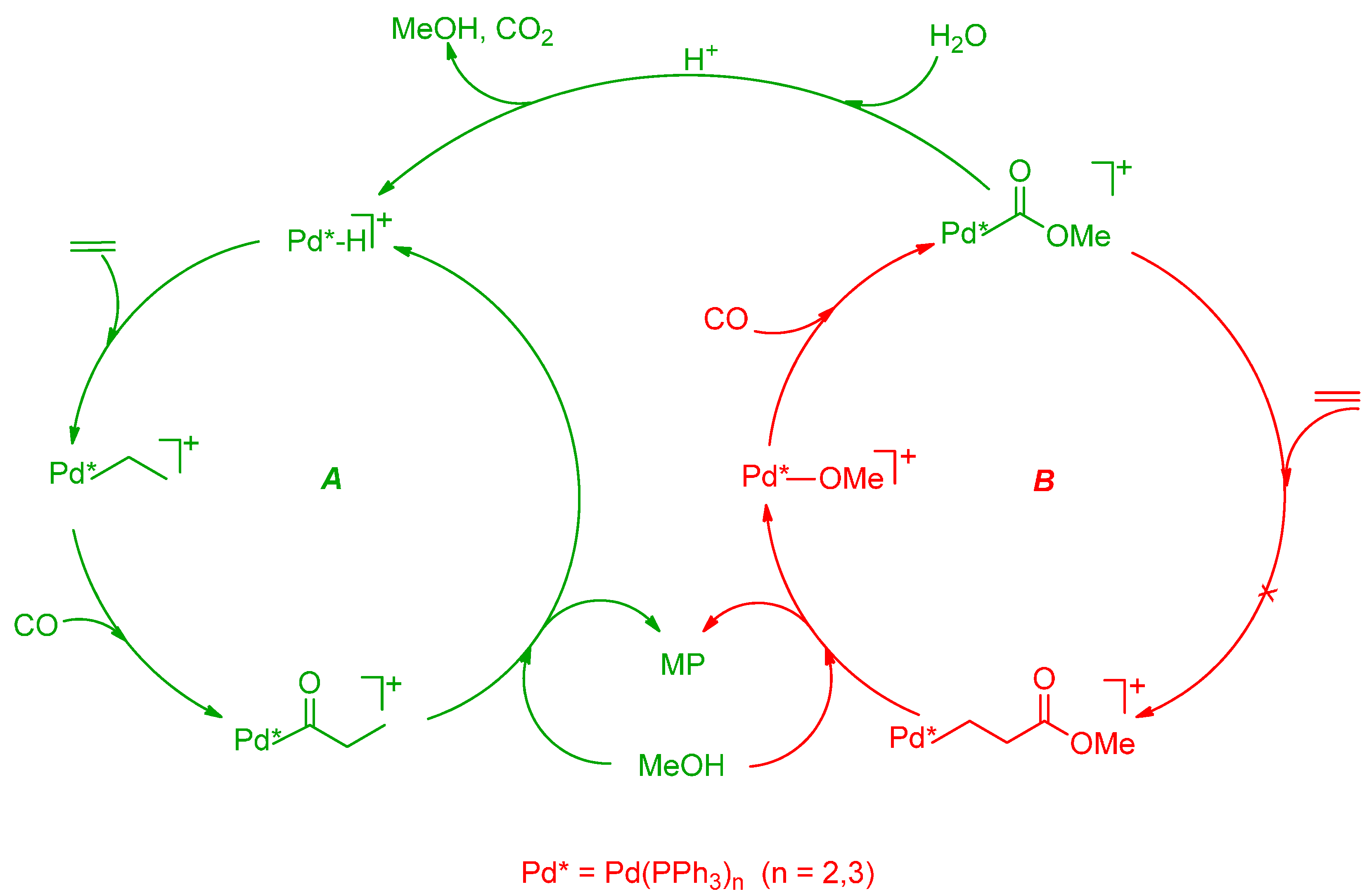
2.3. Termination-Initiation
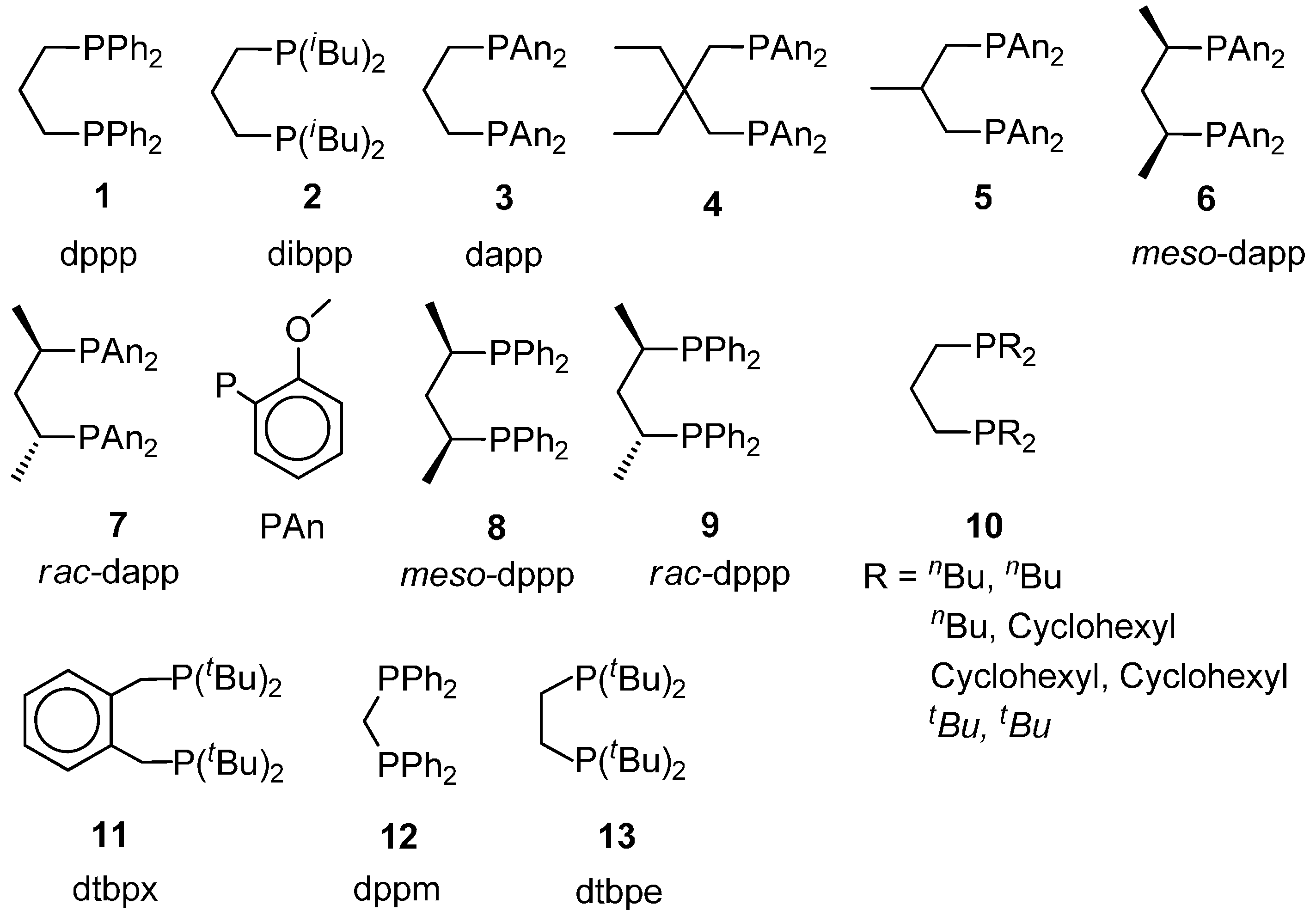
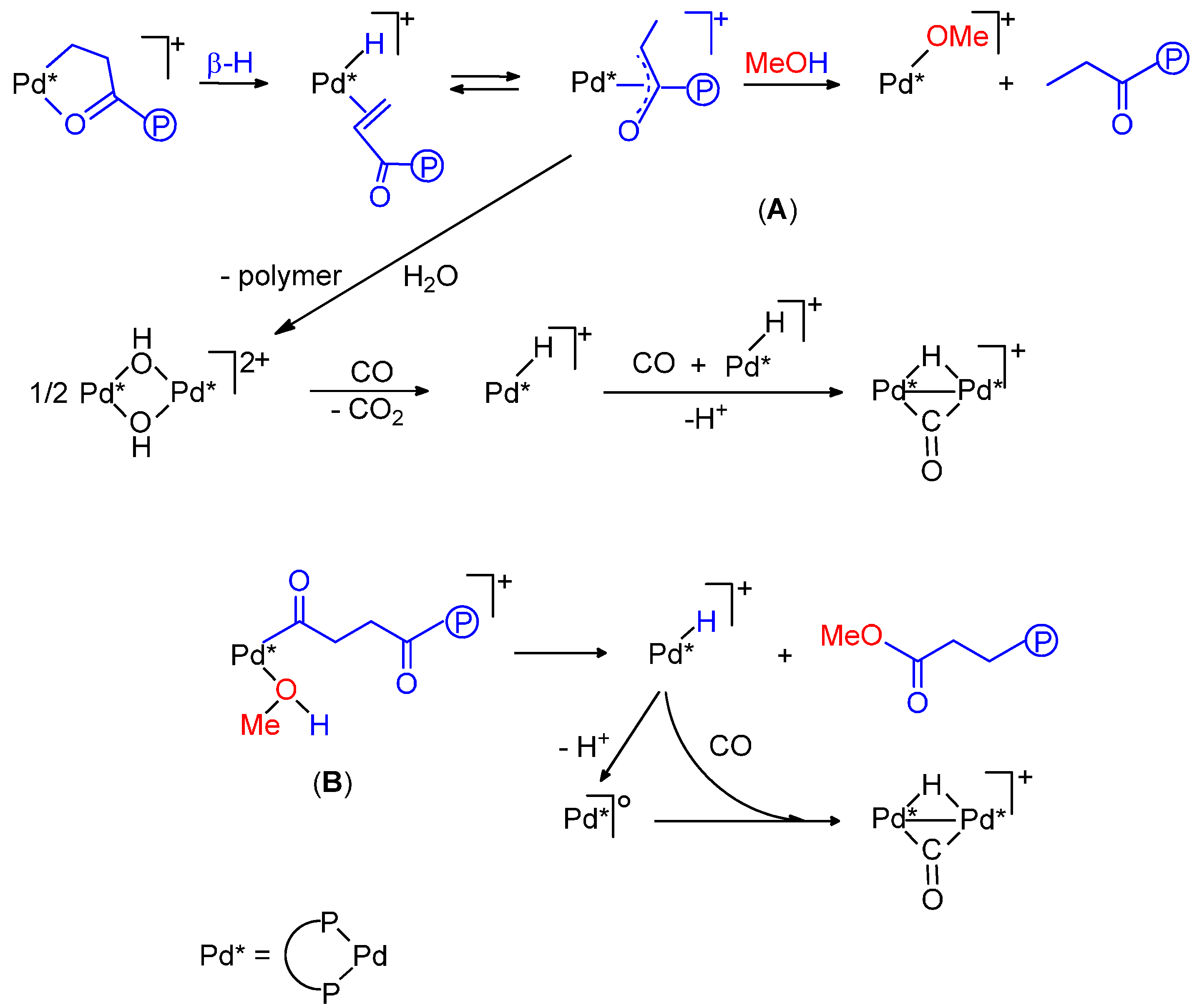

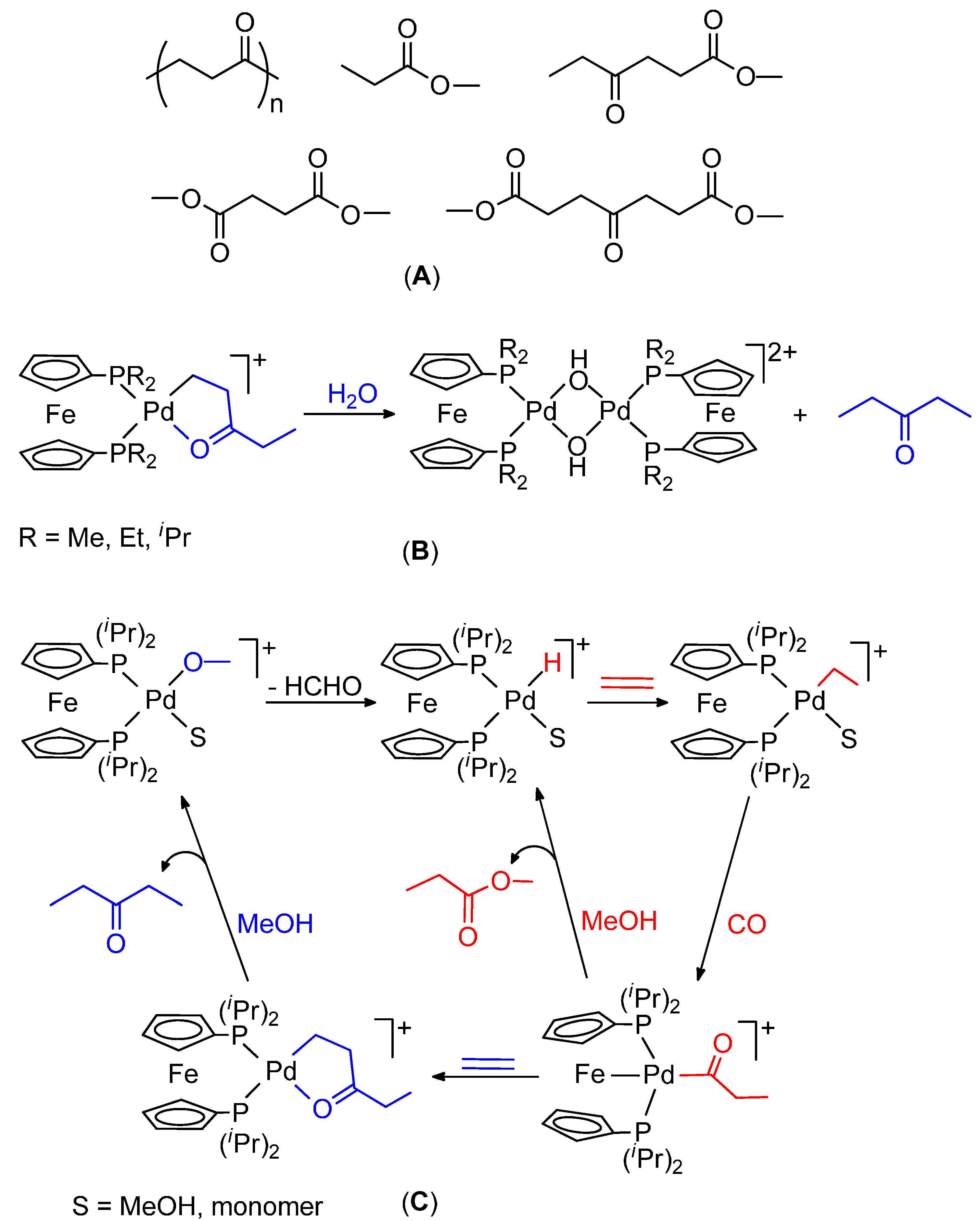
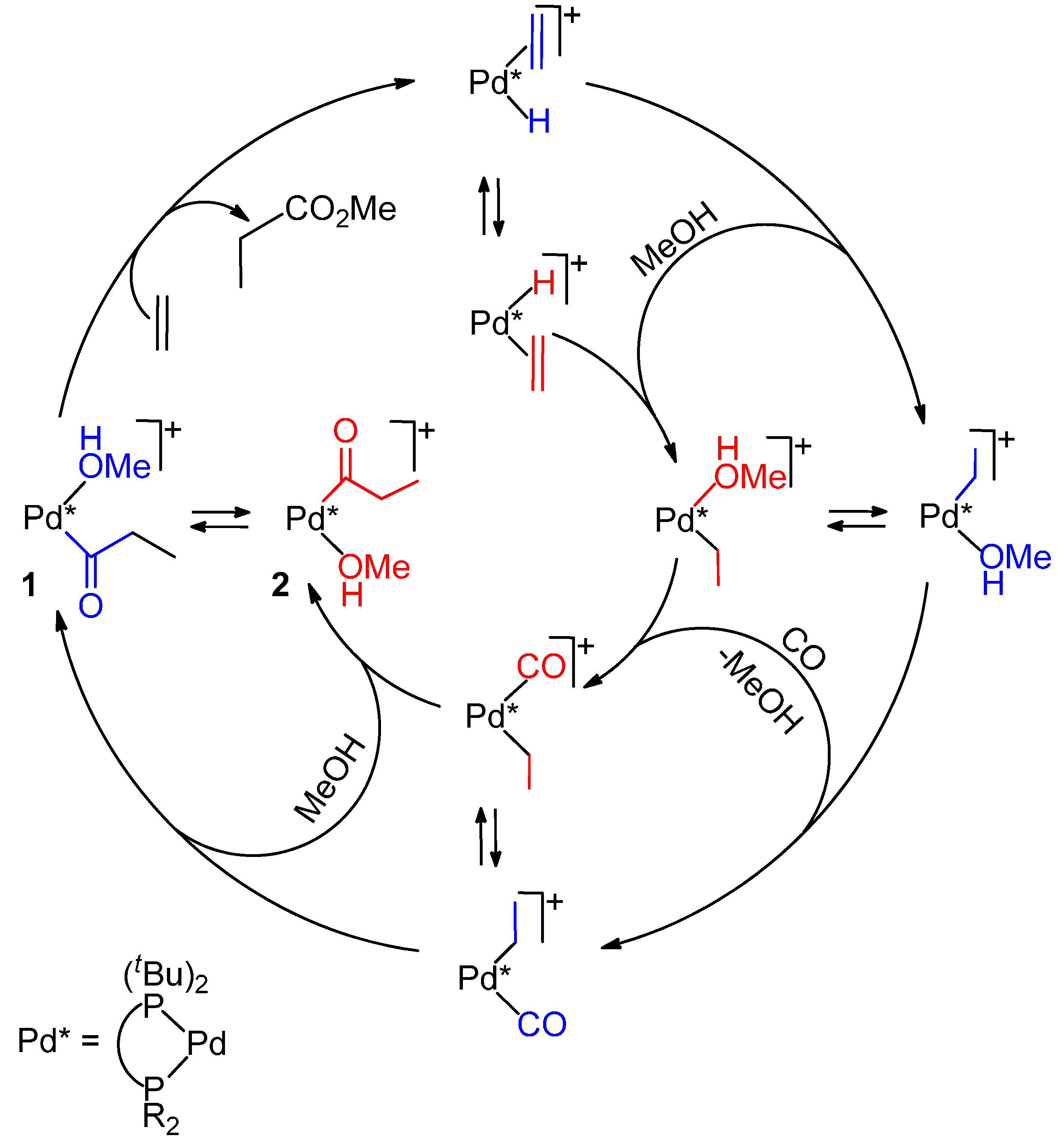
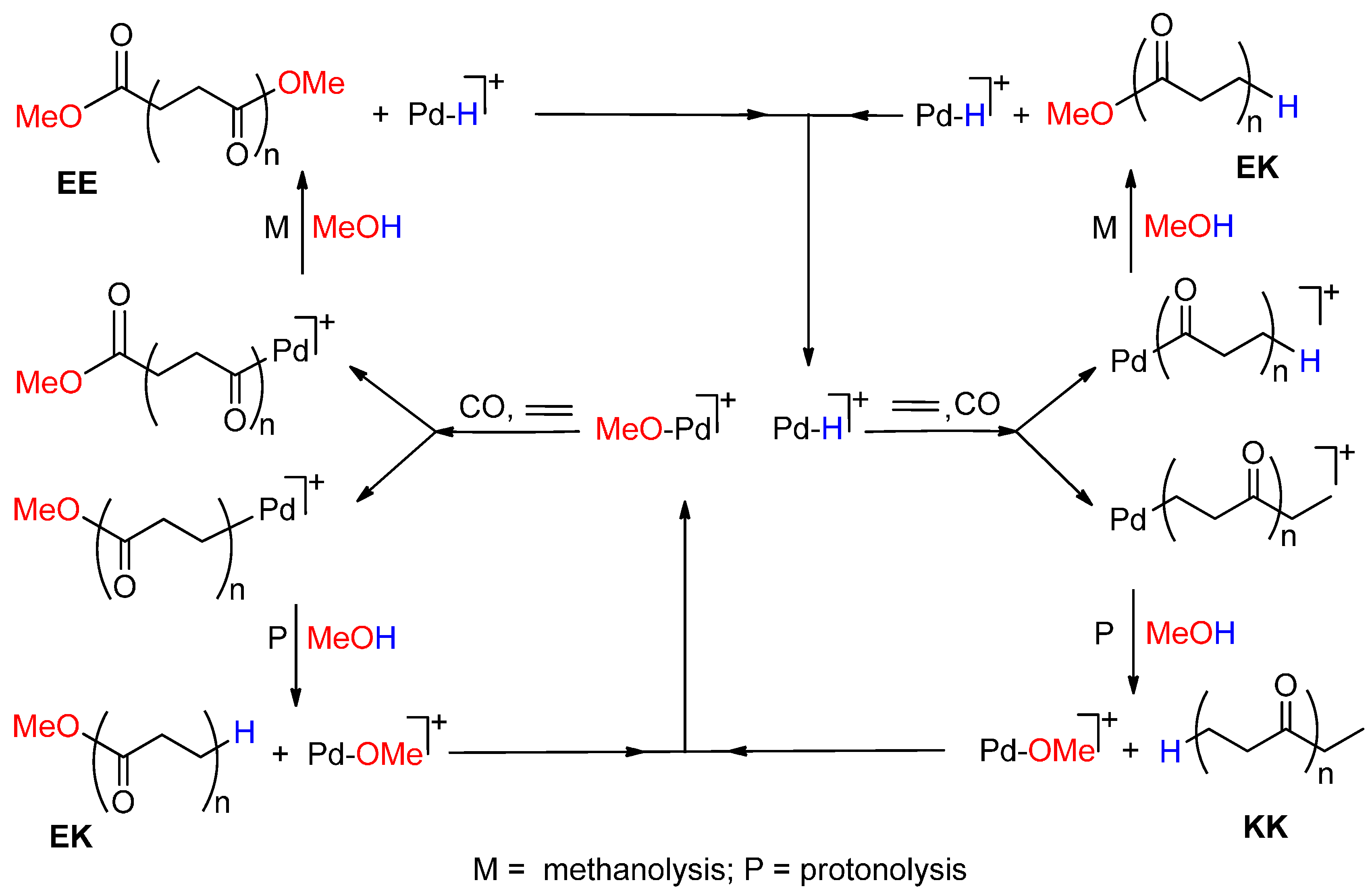
2.4. Shift from the Hydride Mechanism to the Methoxy One and Vice Versa
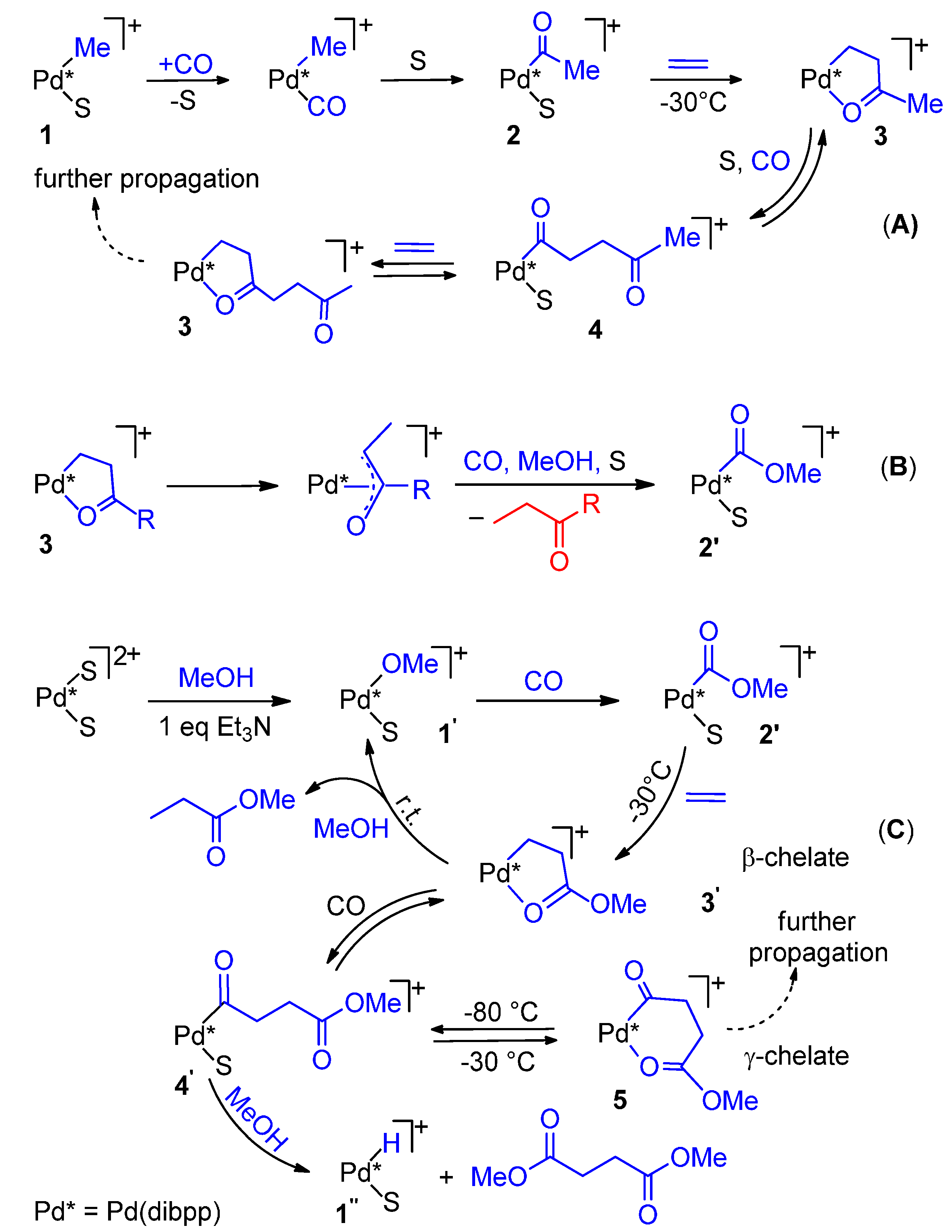
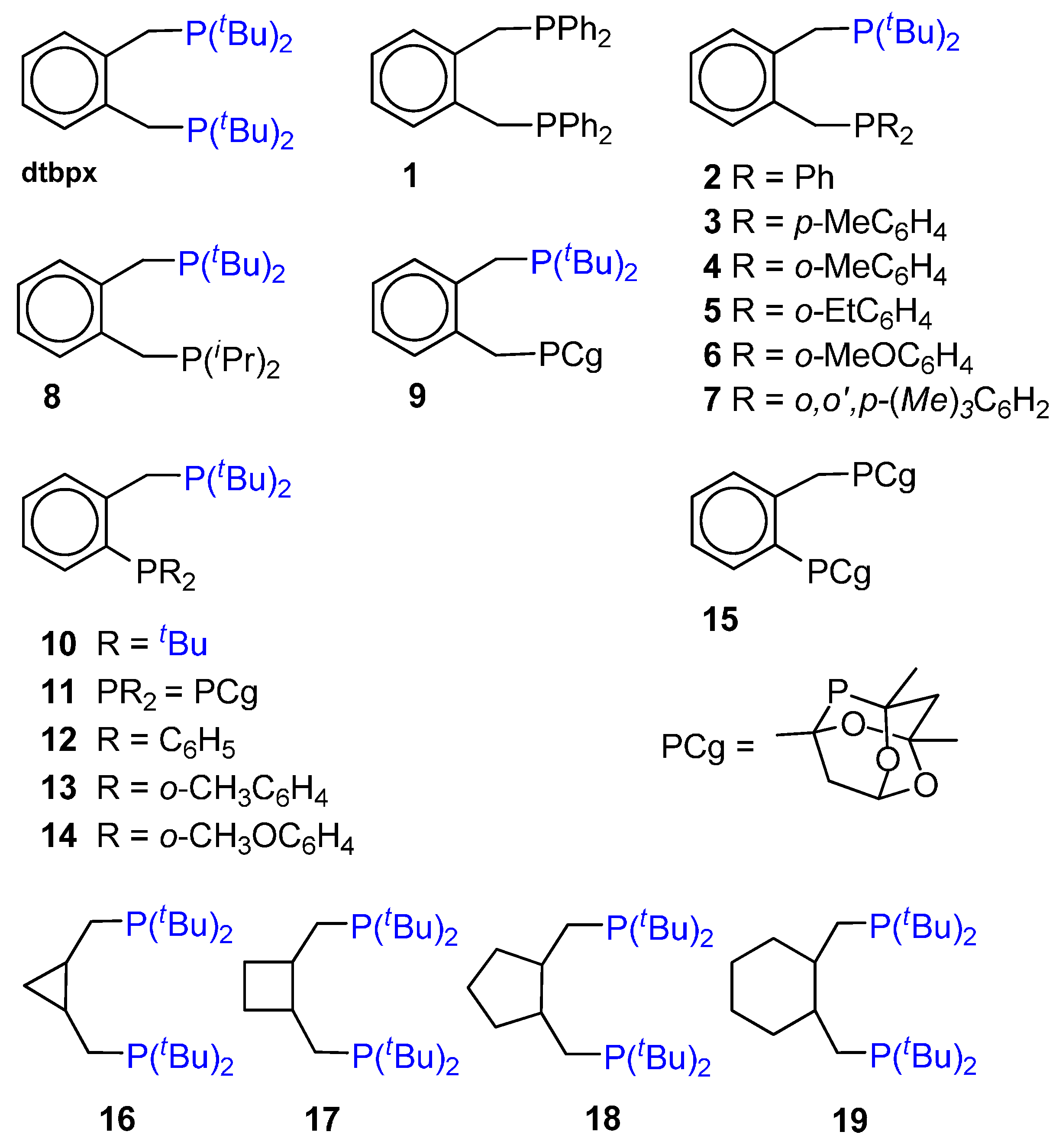
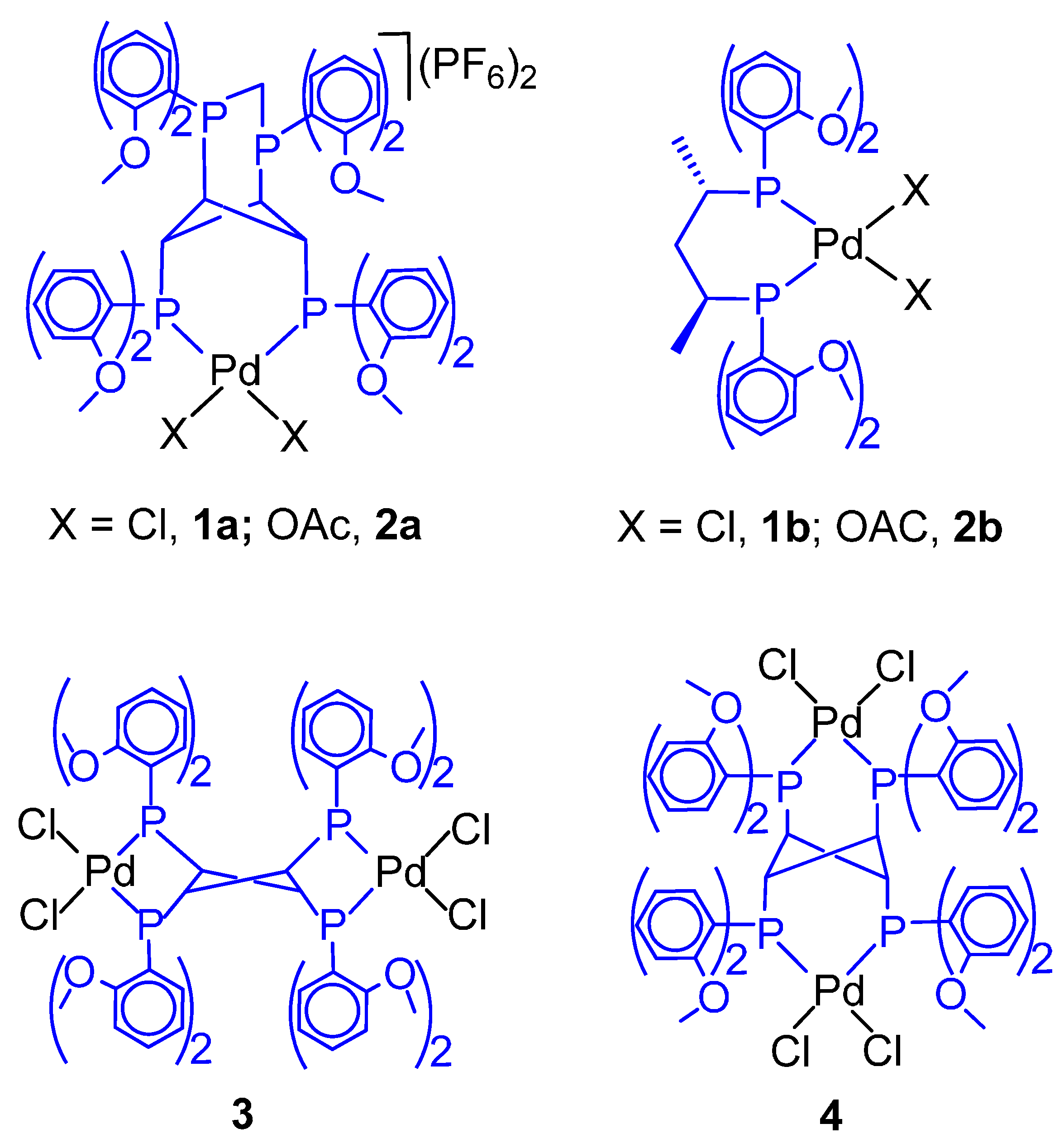
3. Influence of the Ligand on the Product Formation
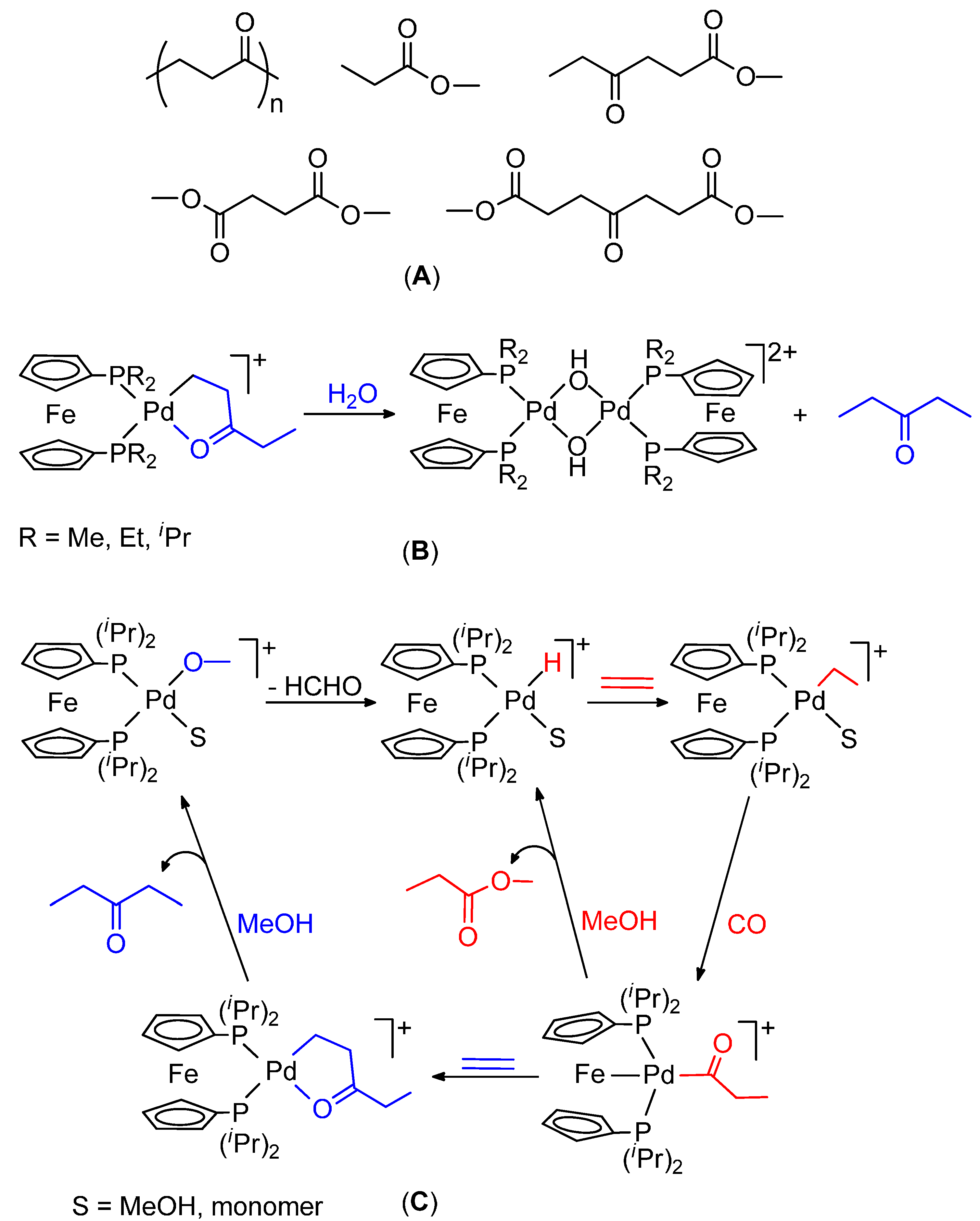
CO-Ethene Chain Propagation
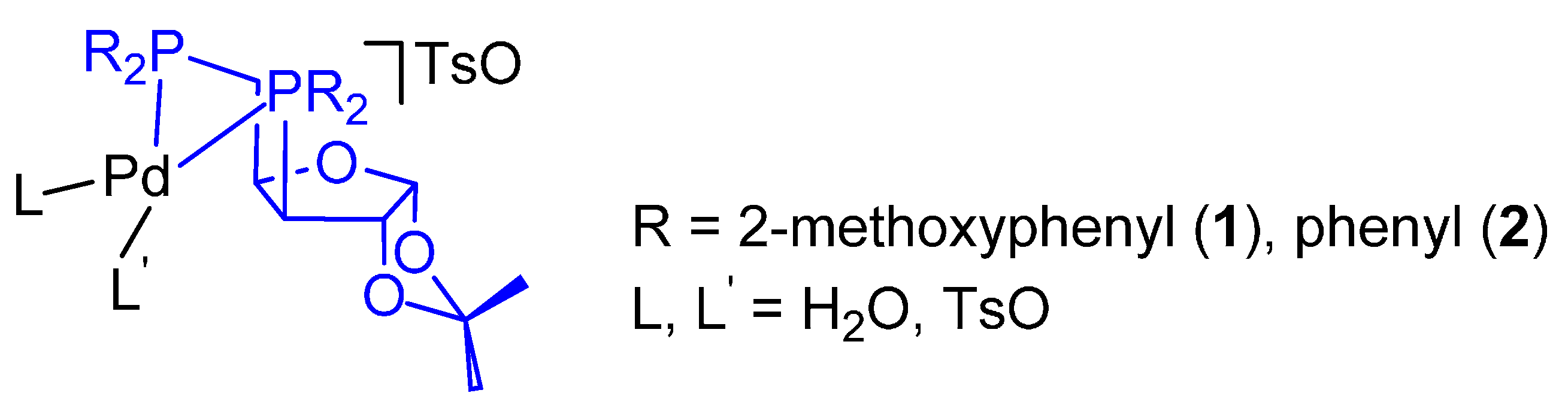
4. Highly Active and Selective Catalysts for the Ethene Hydromethoxycarbonylation
5. Mechanism of the Ethene Hydromethoxycarbonylation
5.1. Diphosphine Catalysts
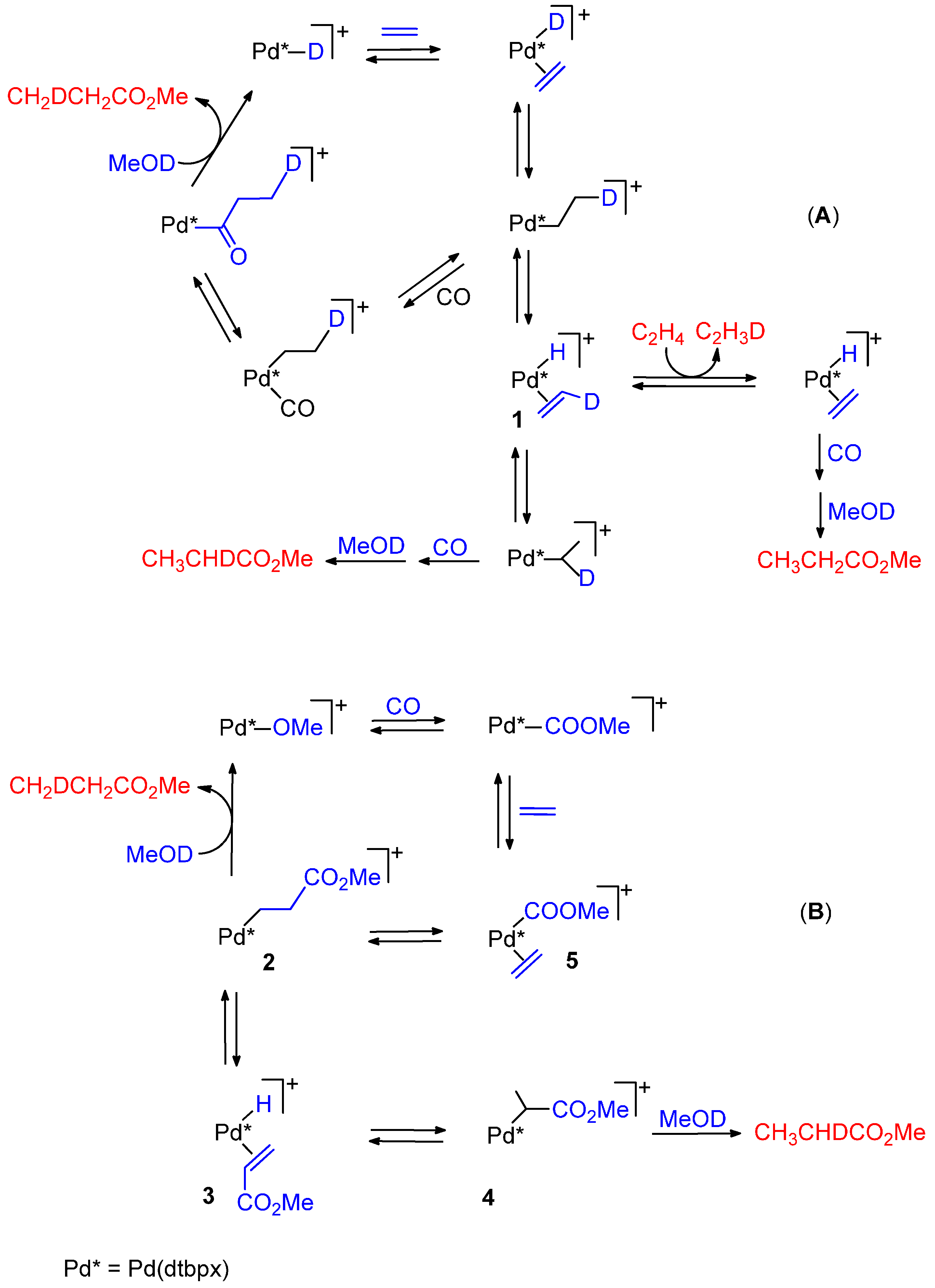
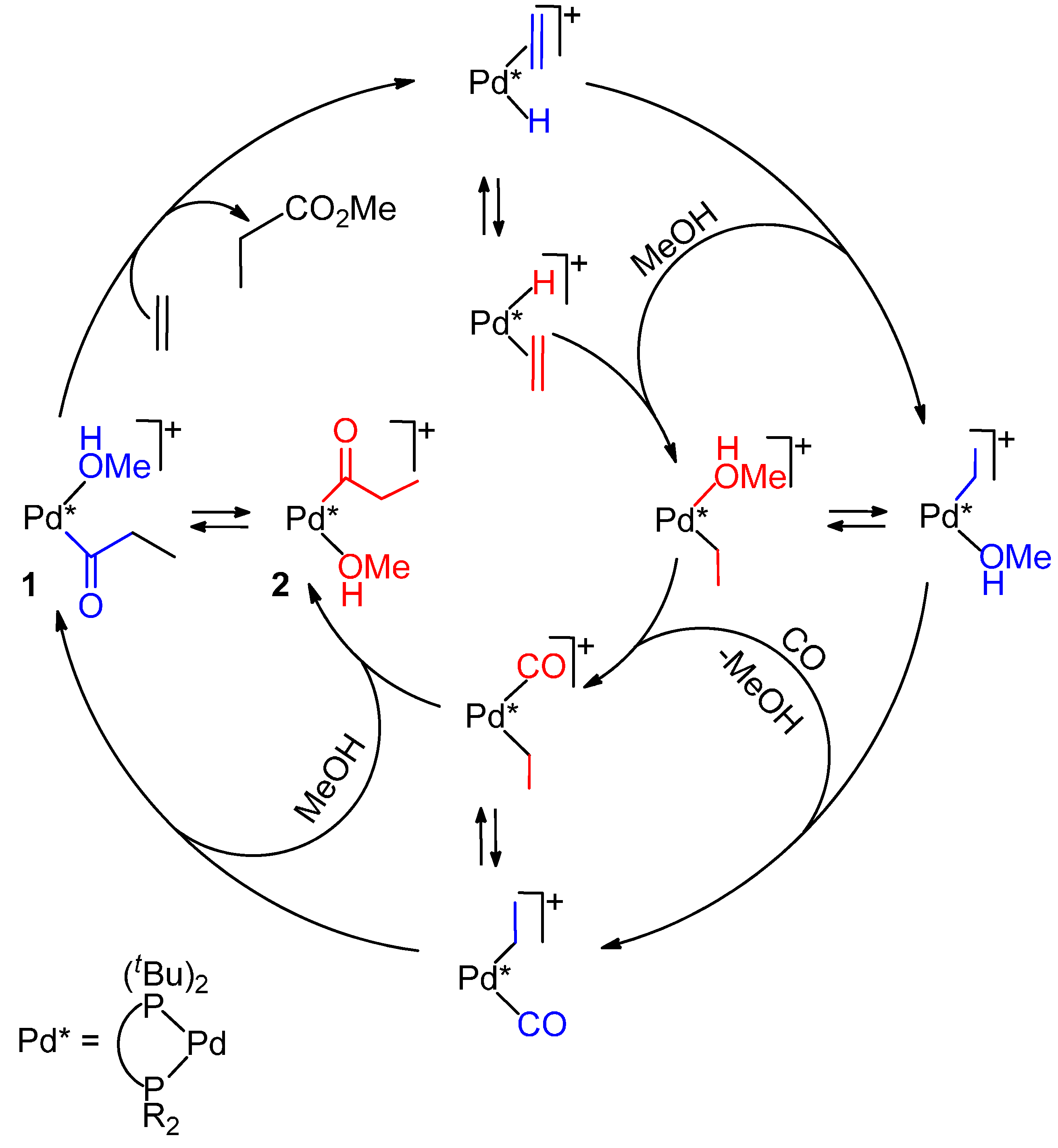
5.2. Monophosphine Catalysts
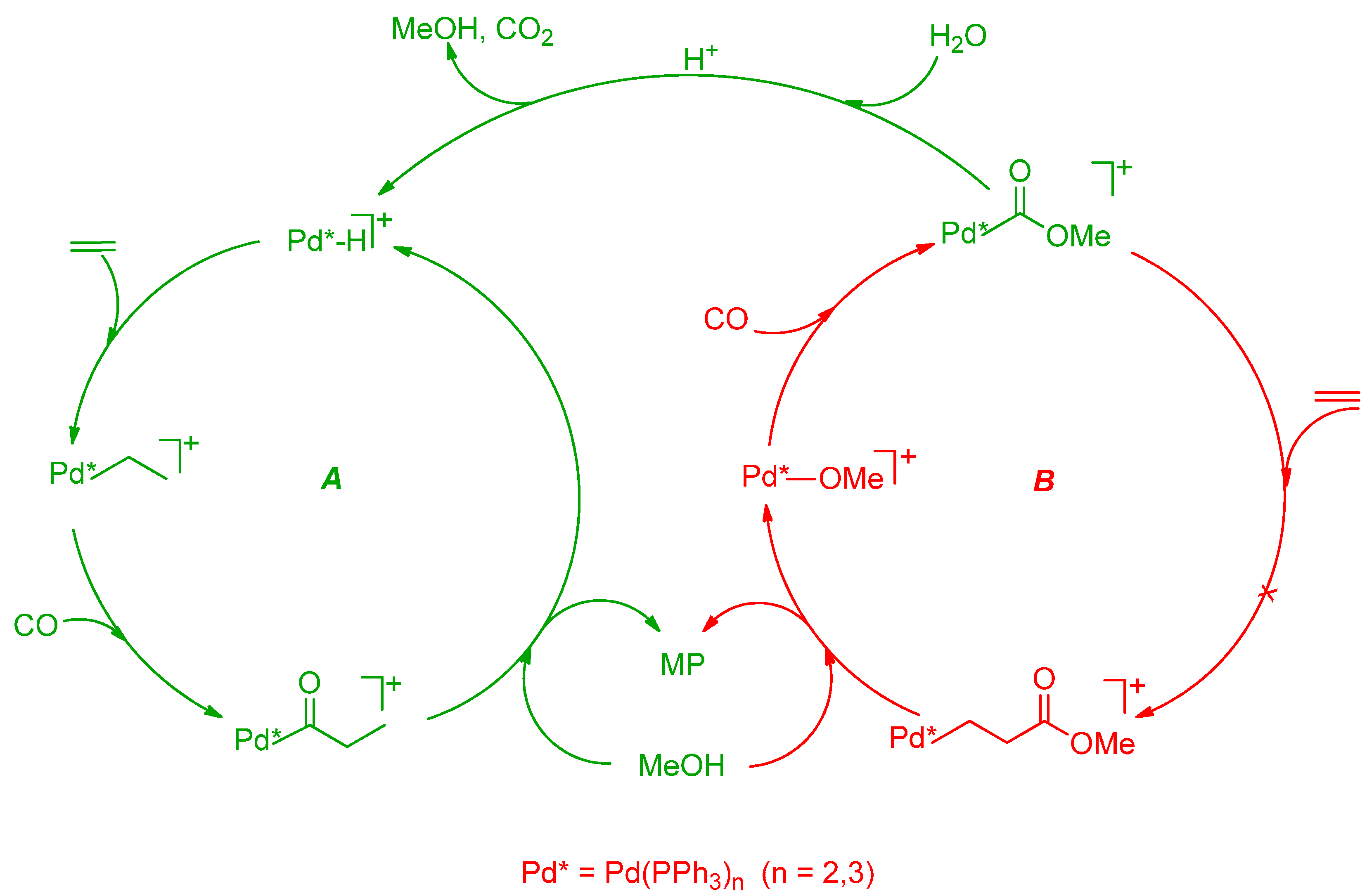

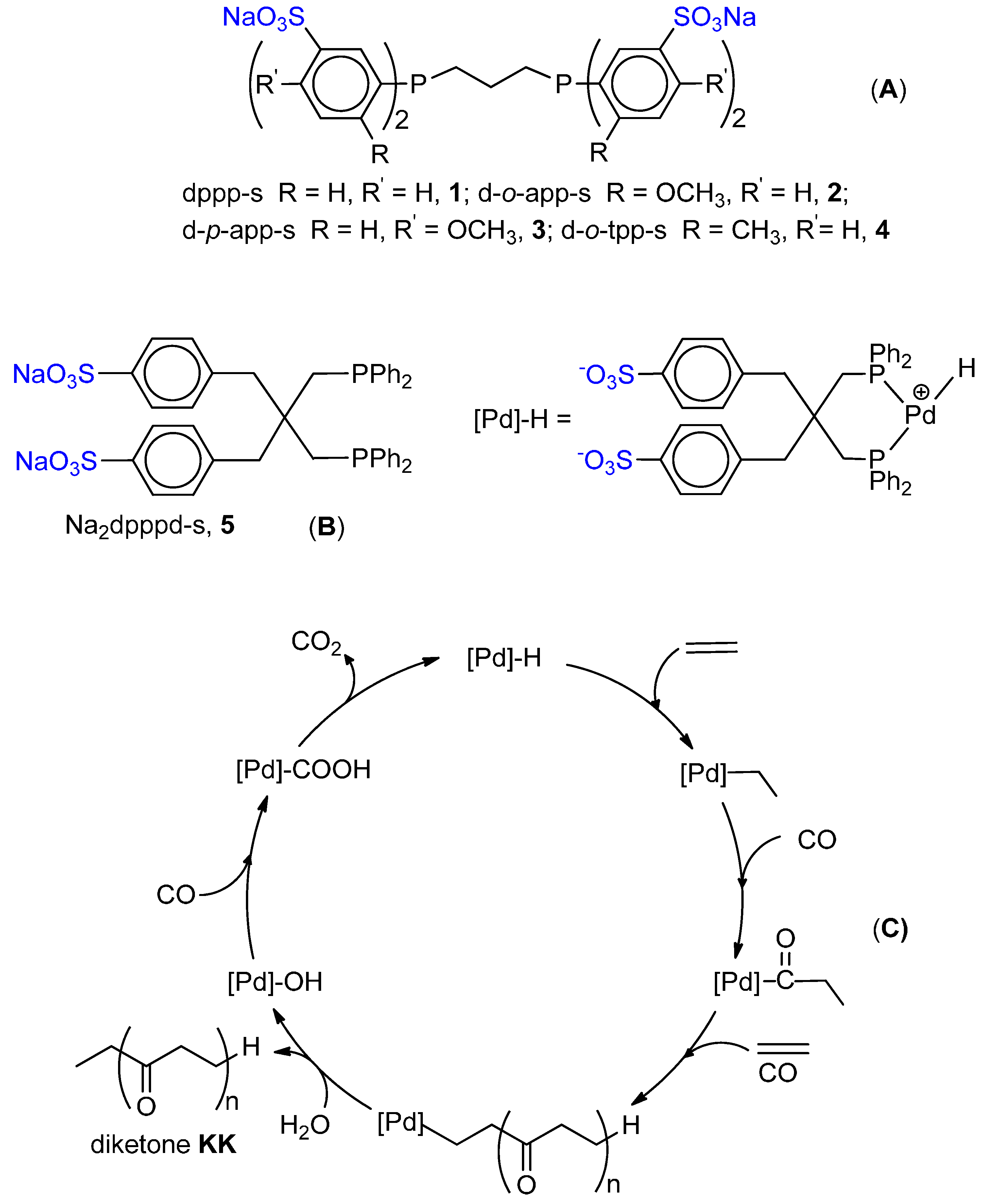
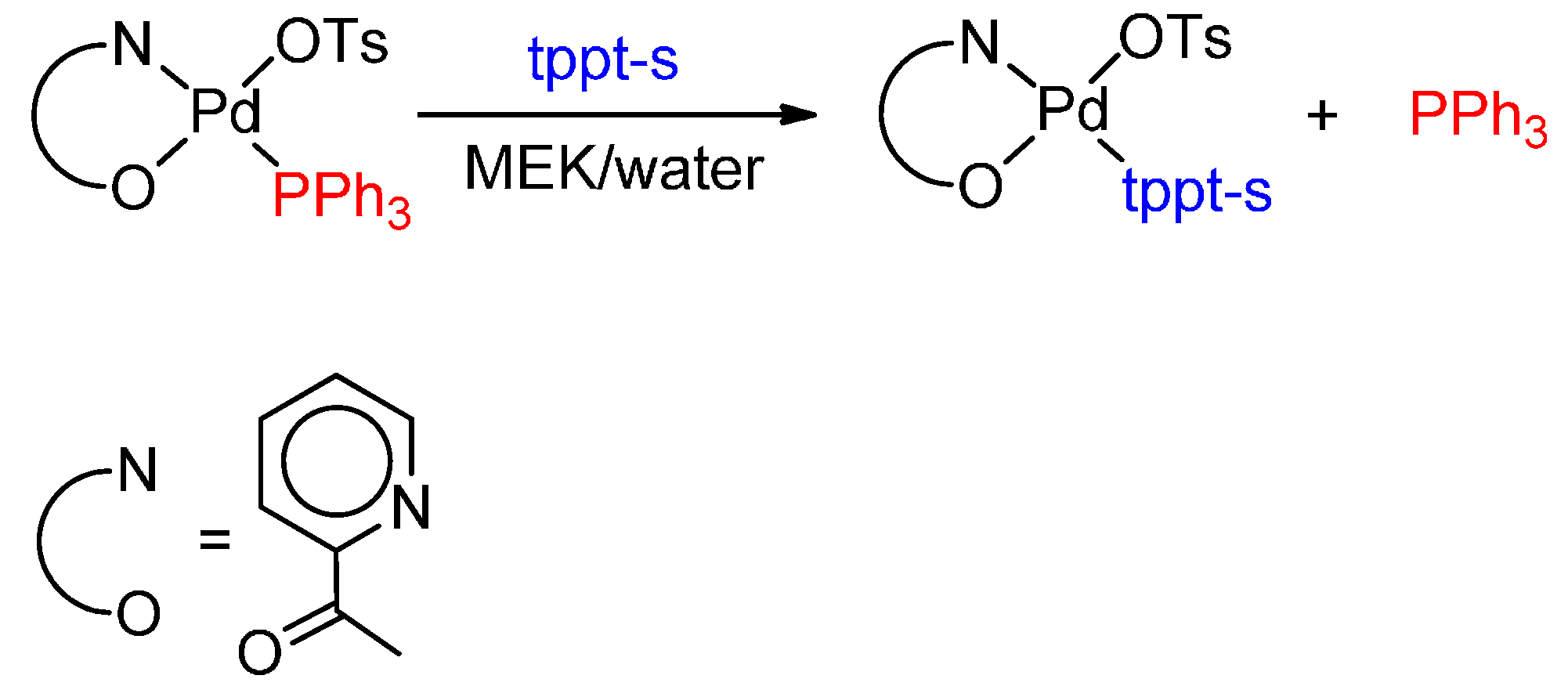
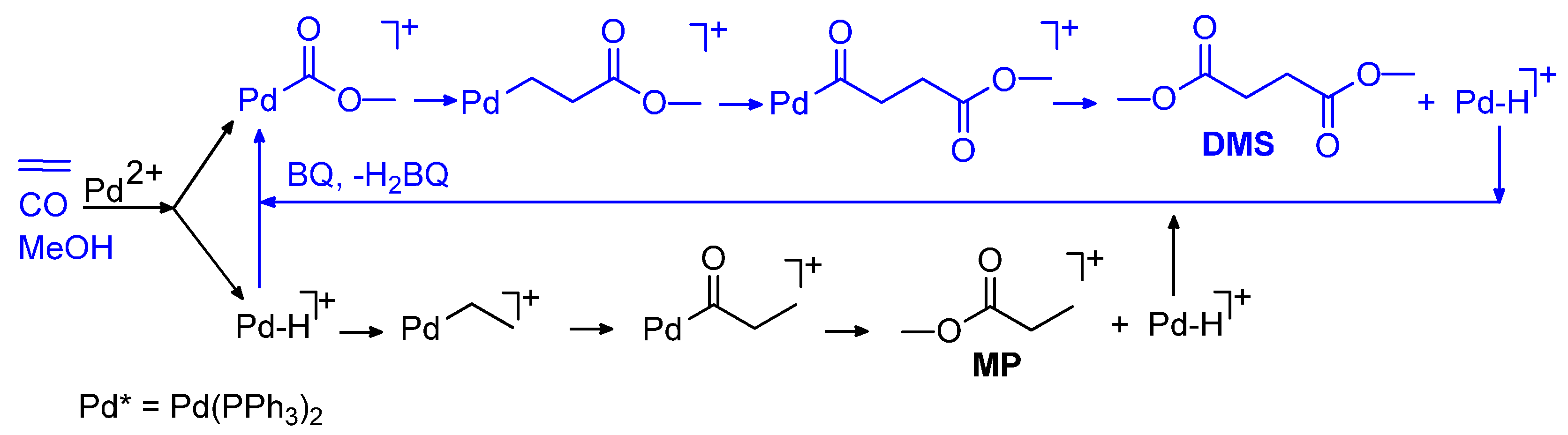
6. Carbonylation in Aqueous Solvents
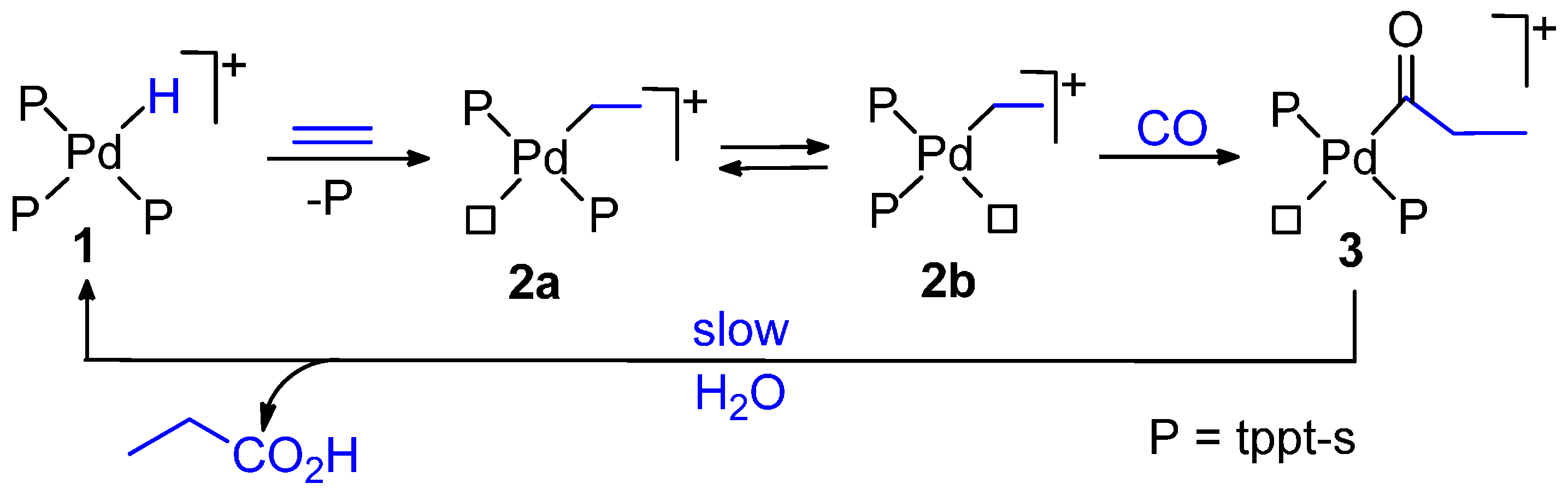
6.1. CO-Ethene Copolymerization with Water-Soluble Catalysts
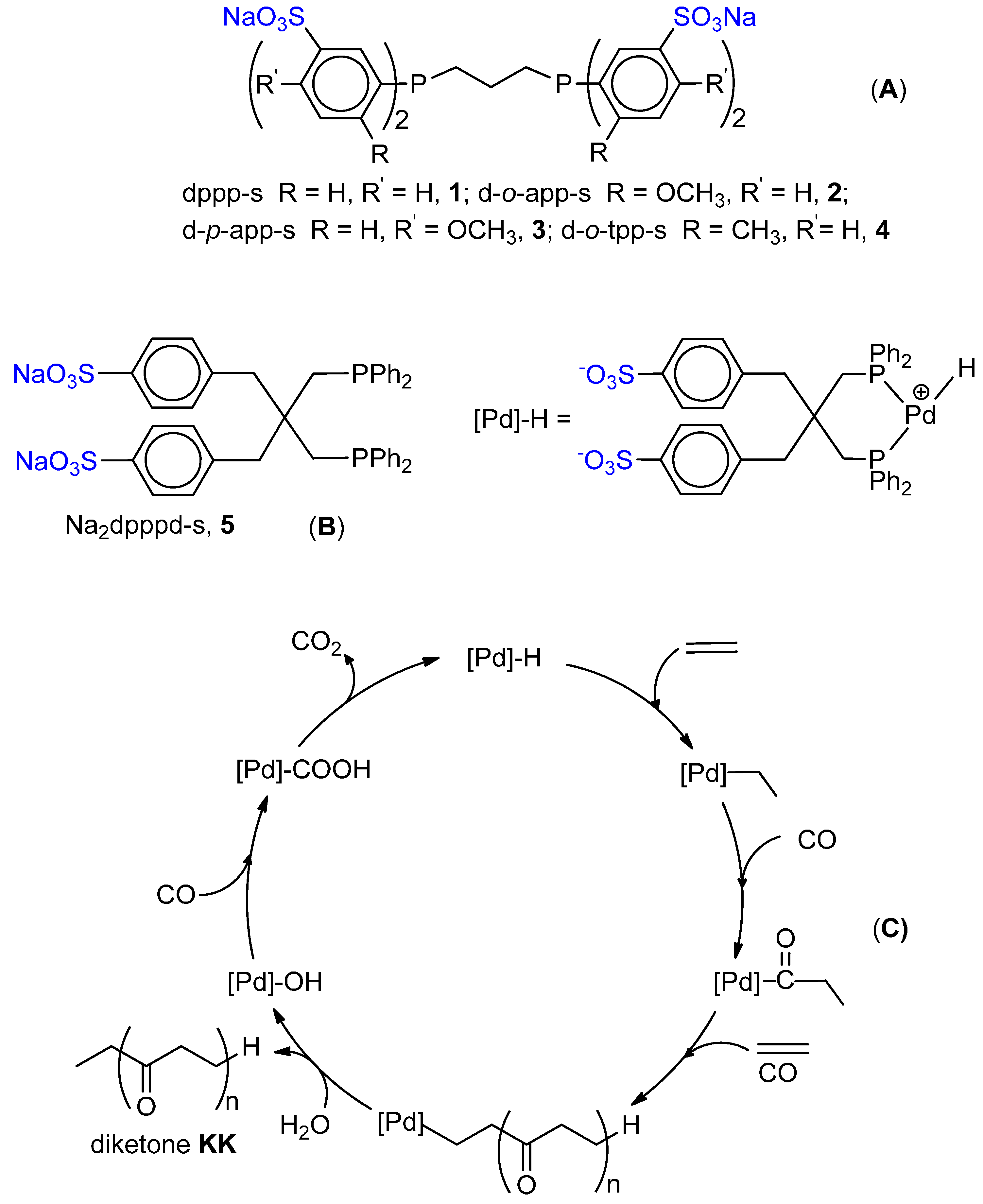
6.2. Monocarbonylation of Olefins
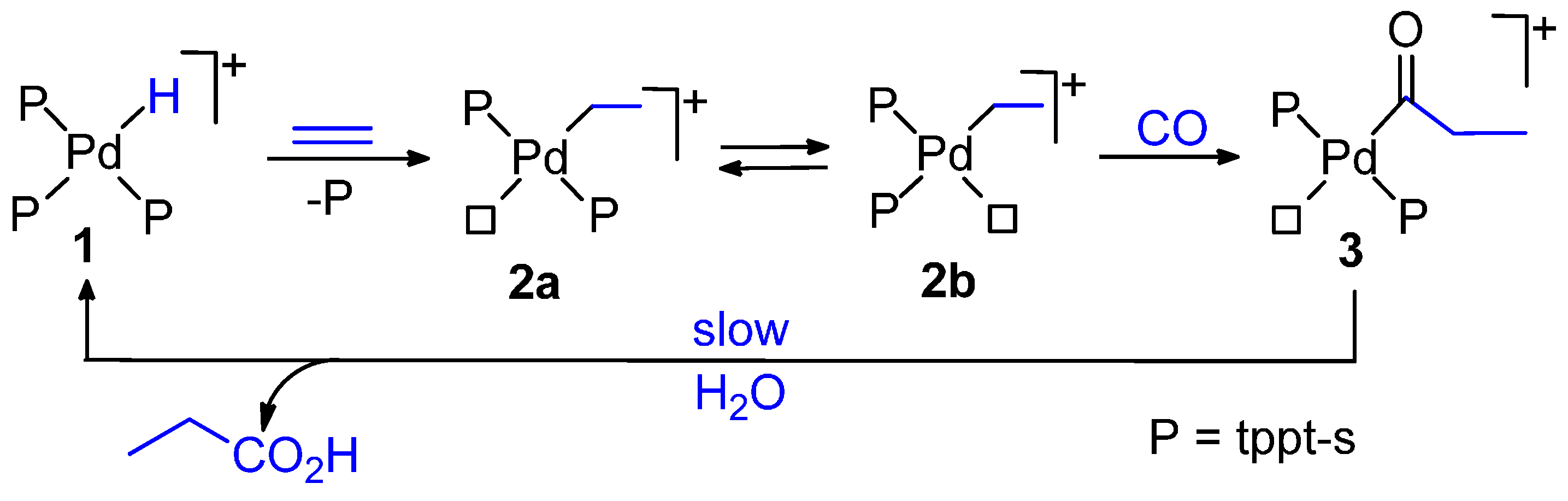

6.3. Copolymerization in Water-Organic Acid
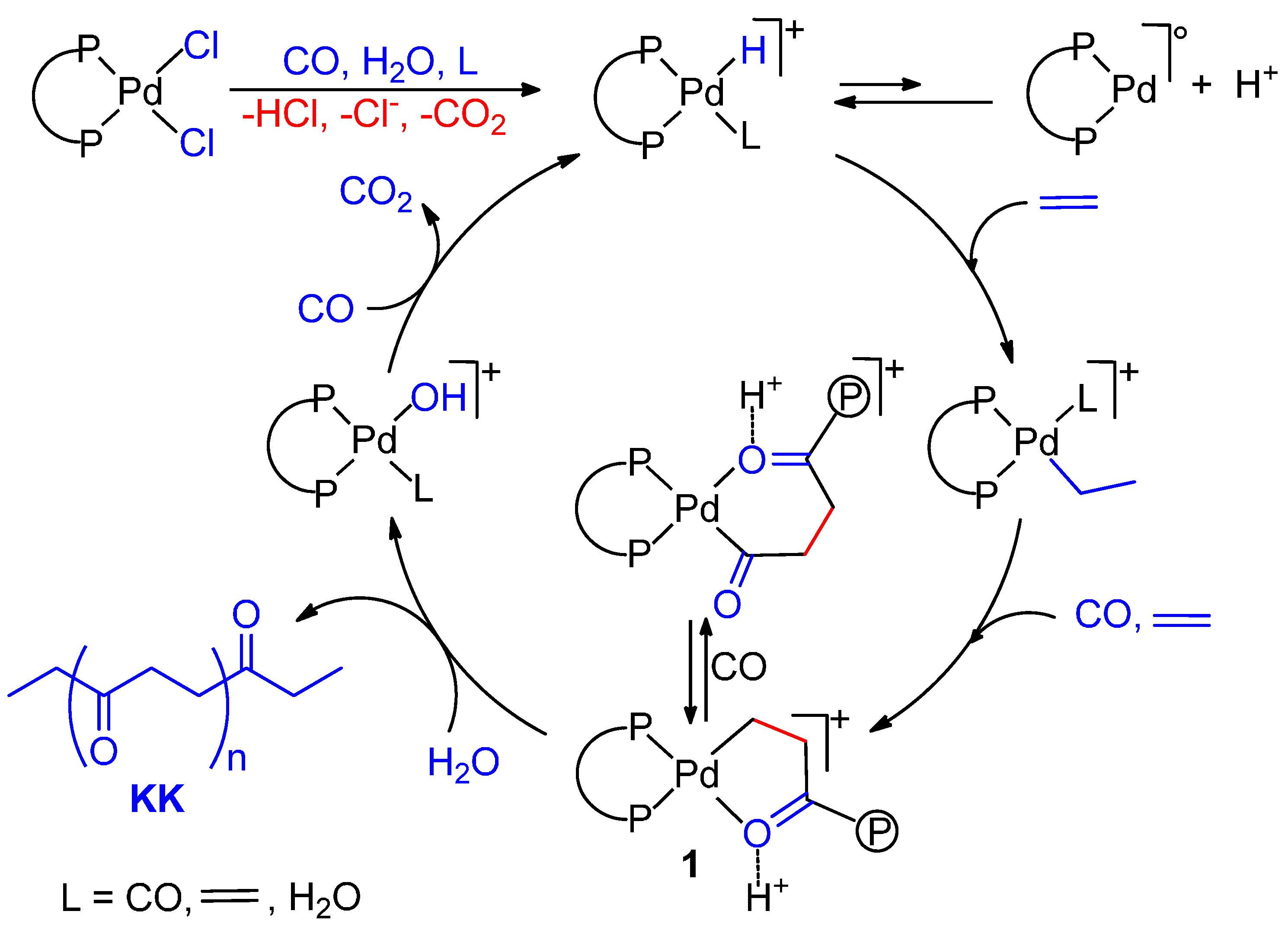
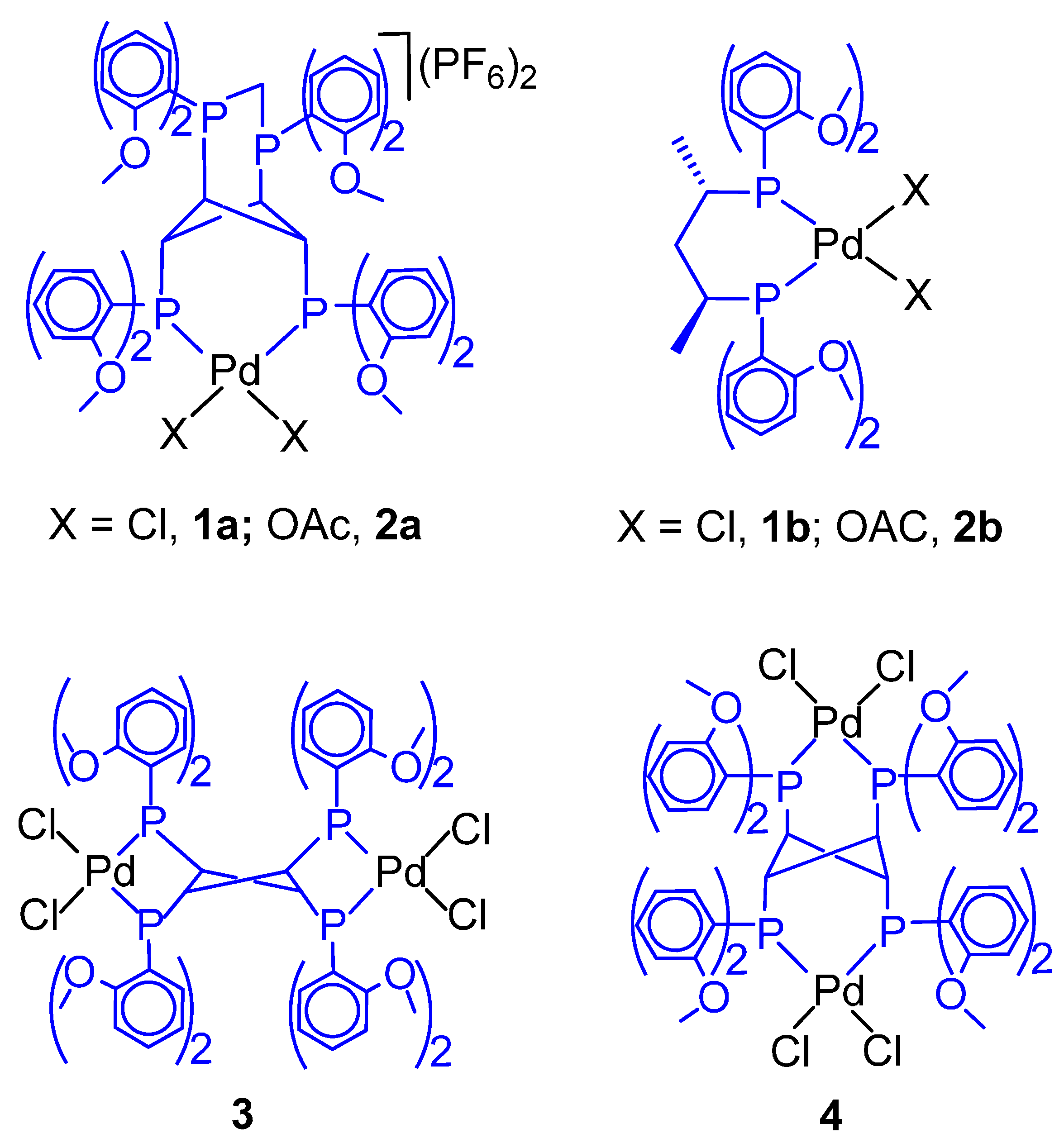
6.4. Biphasic Copolymerization in the Presence of an Emulsifier
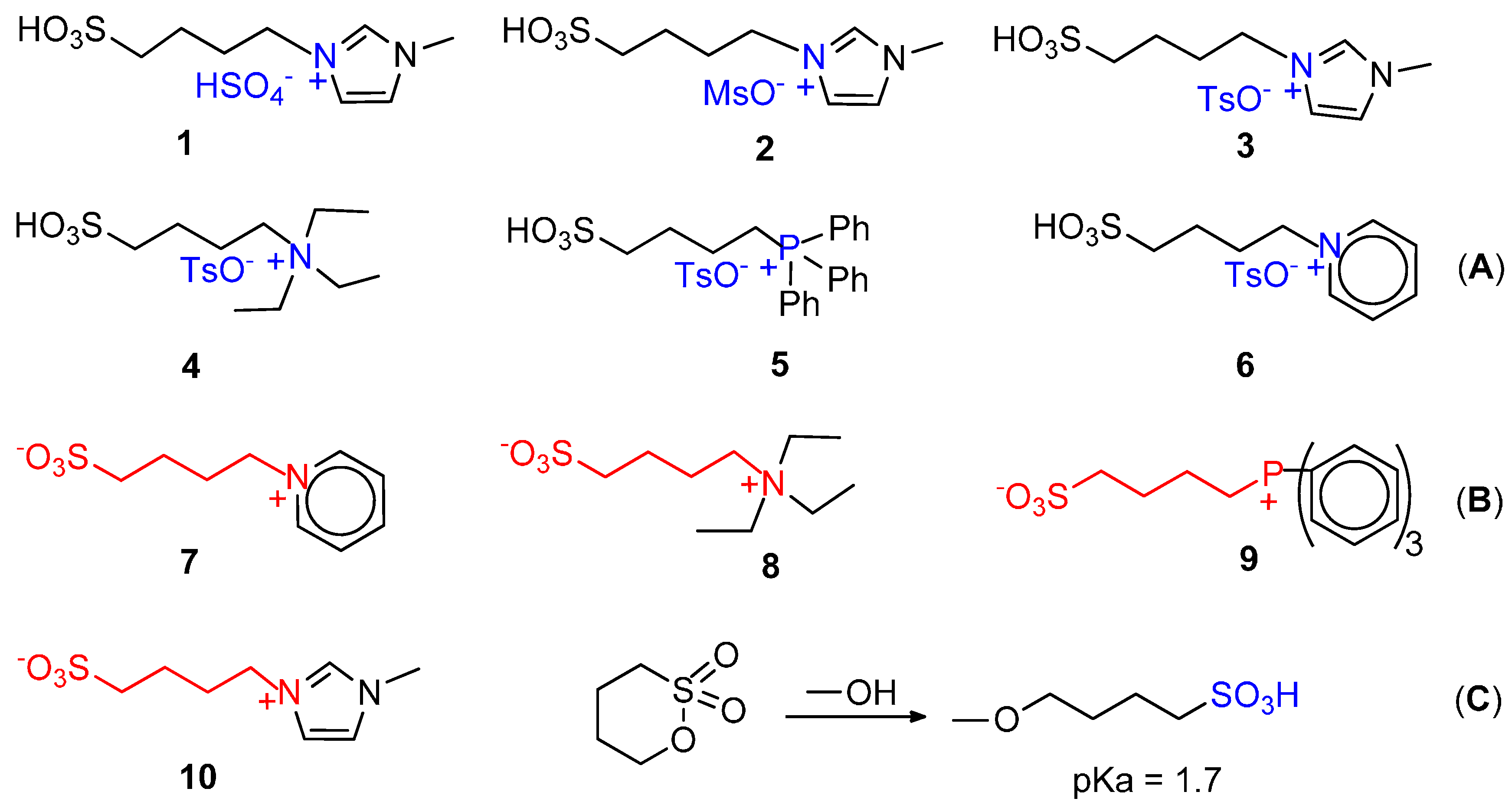
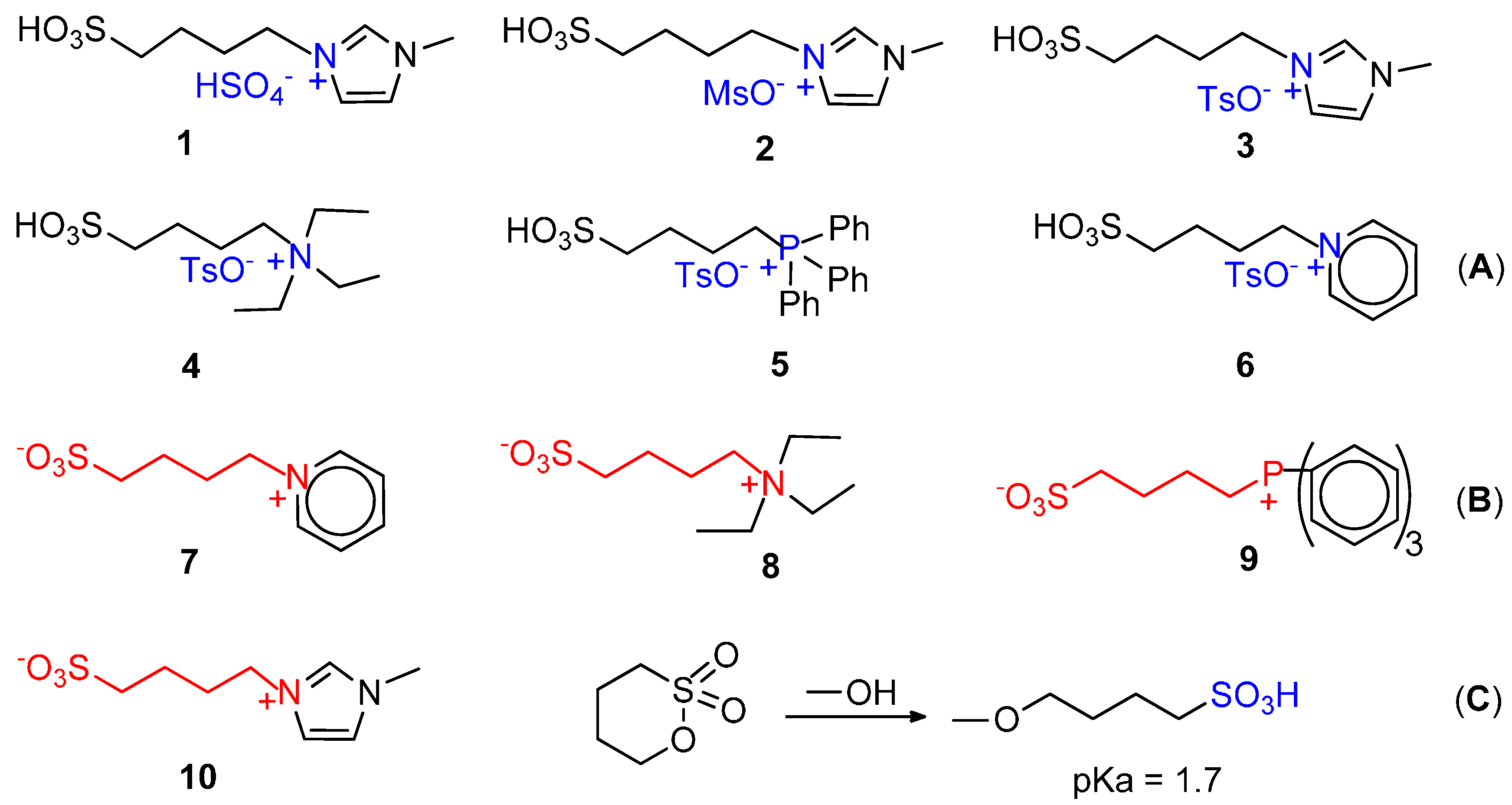
7. Carbonylation in Ionic Liquids
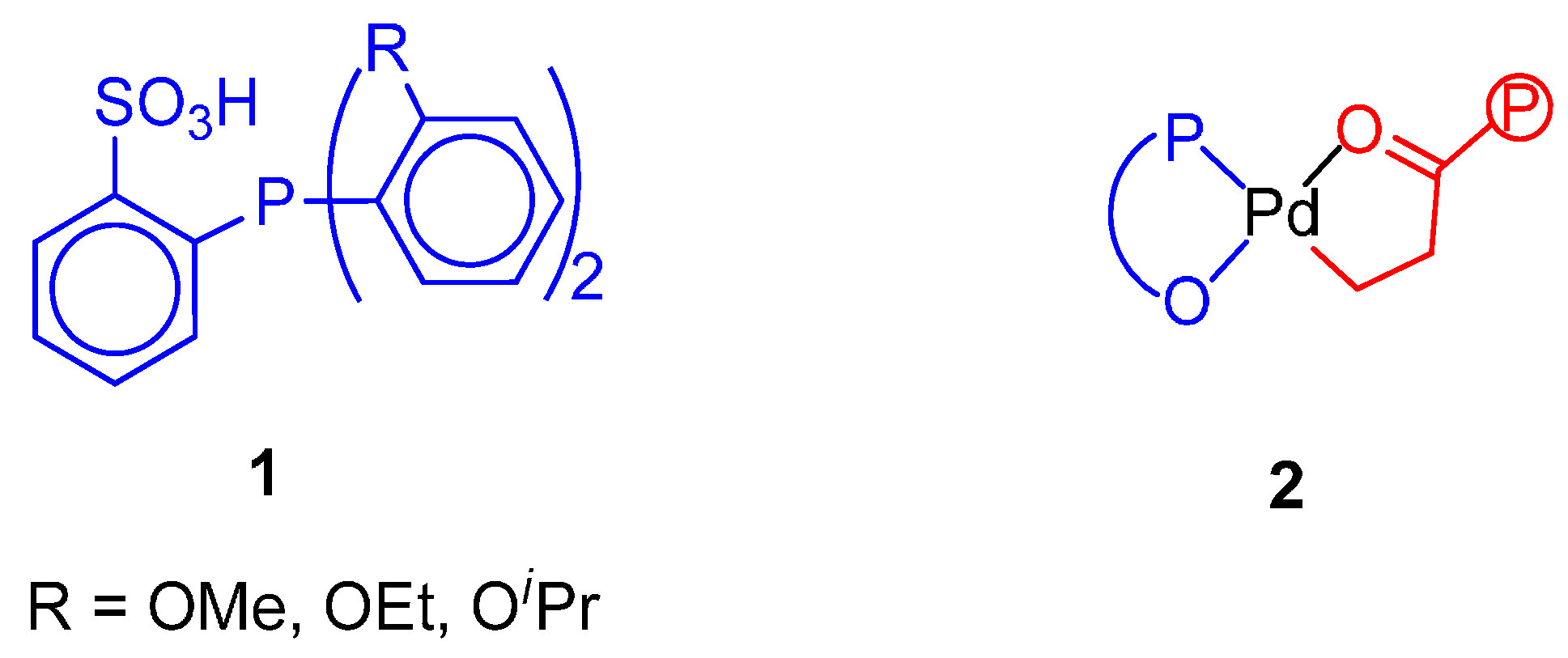
8. Supported Copolymerization
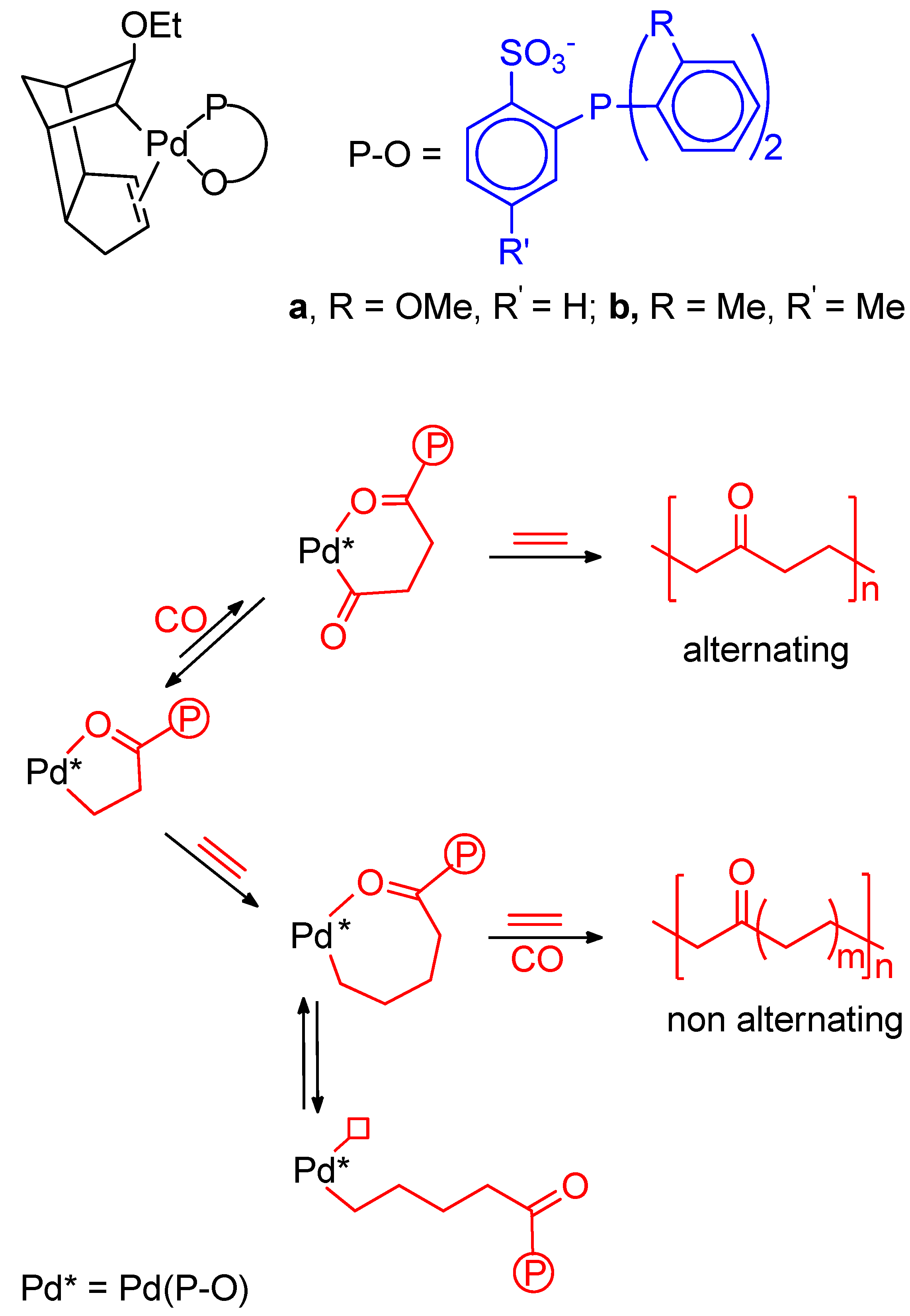
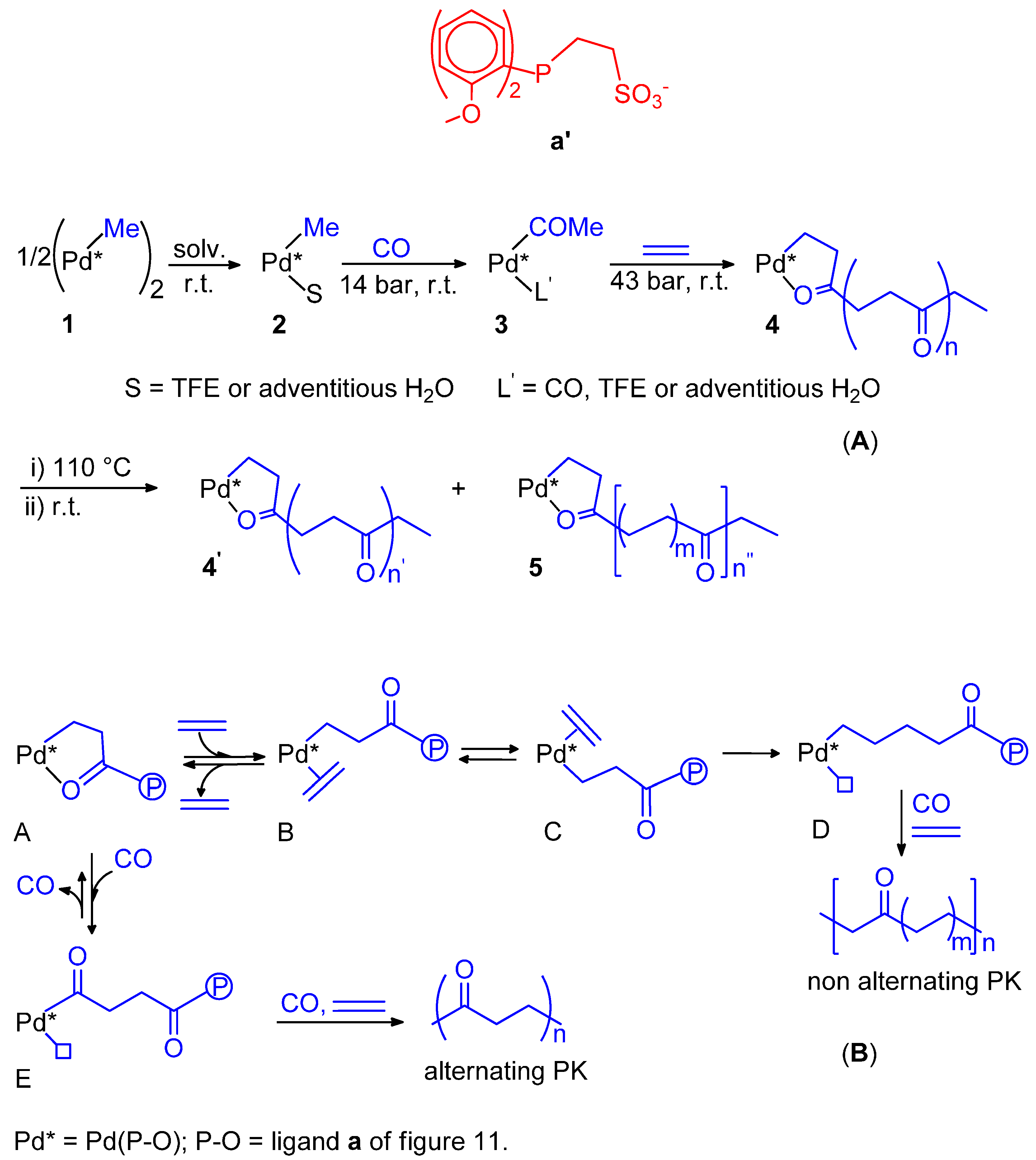
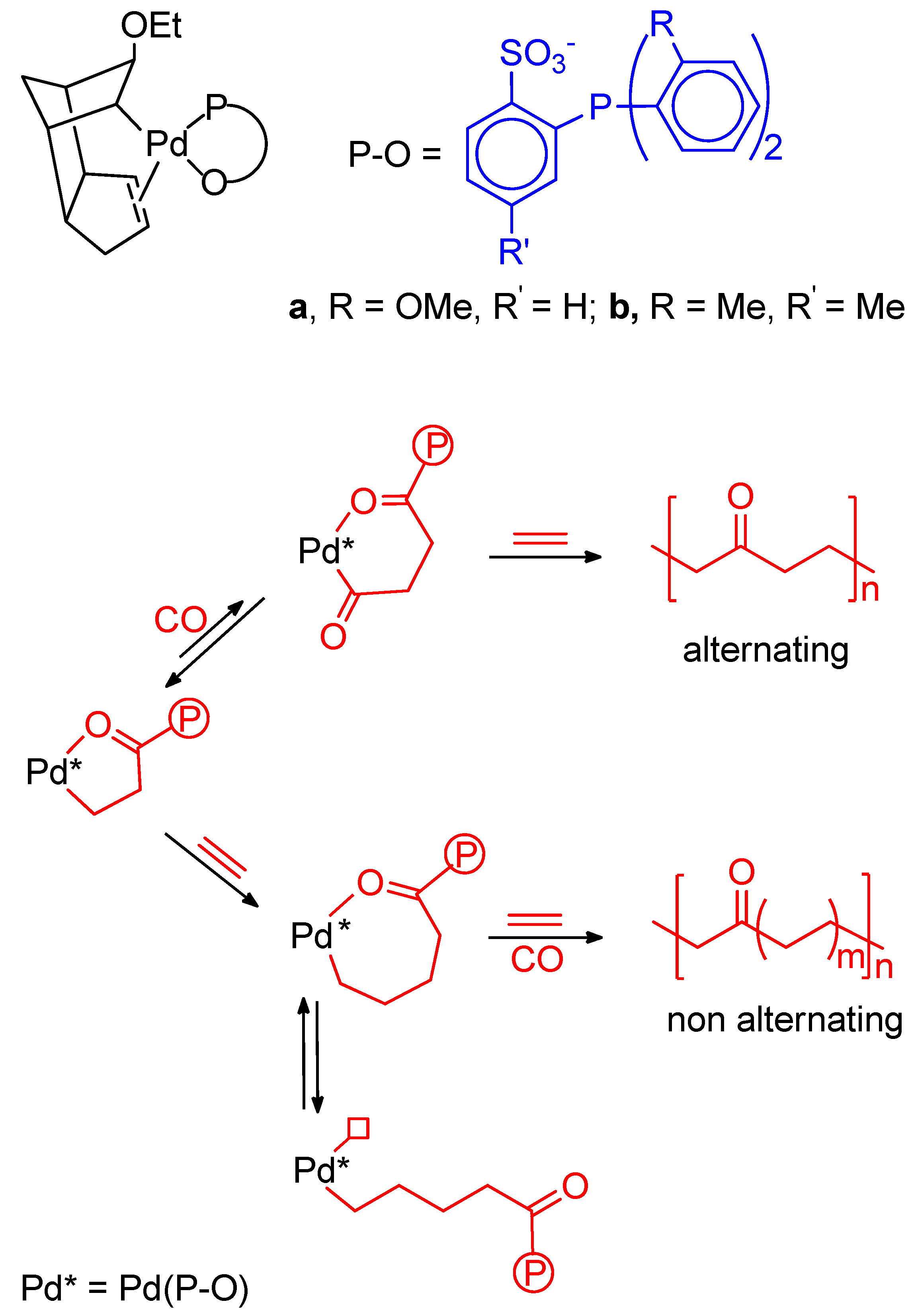
9. Nonalternating CO-Ethene Copolymerization
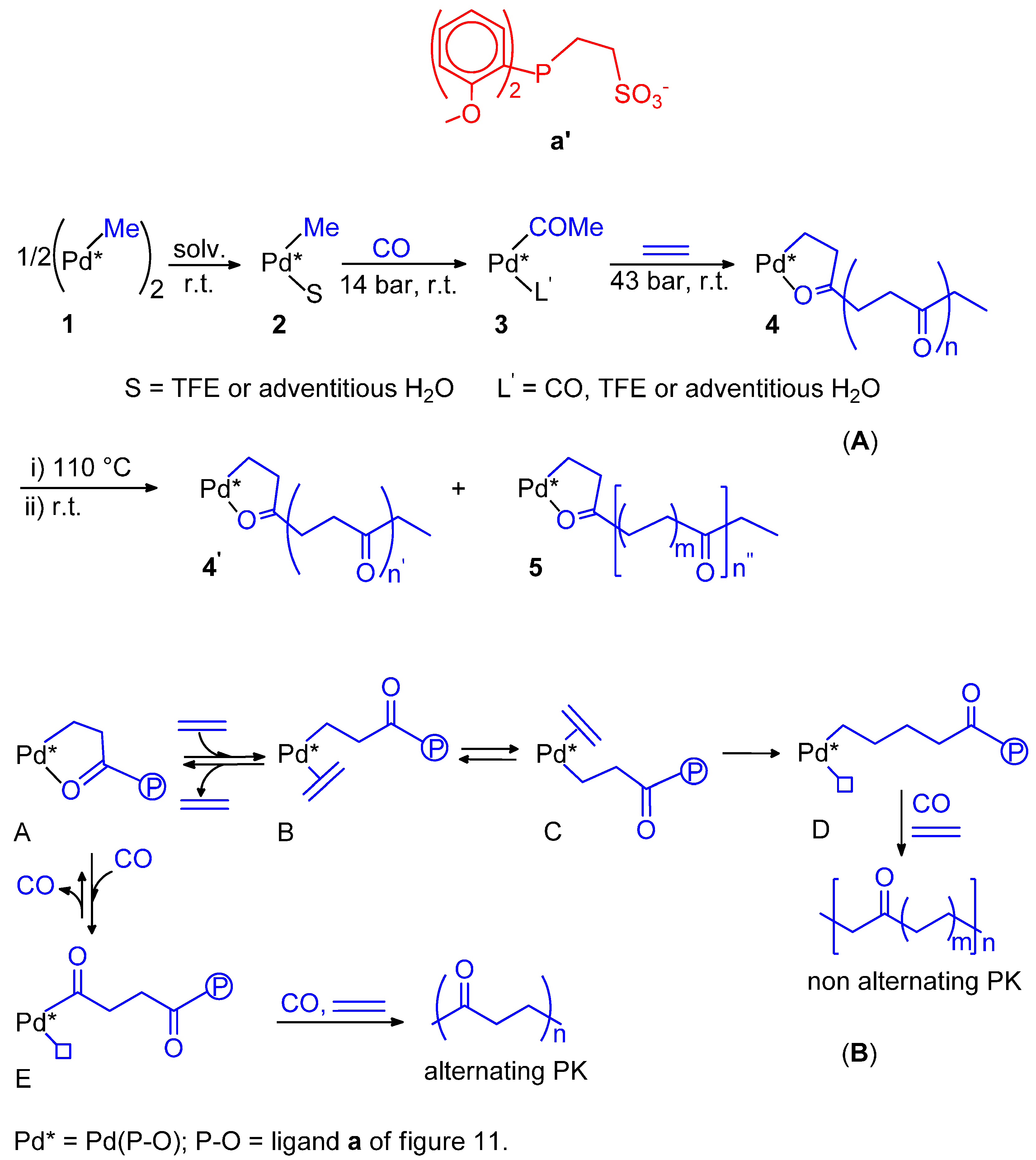
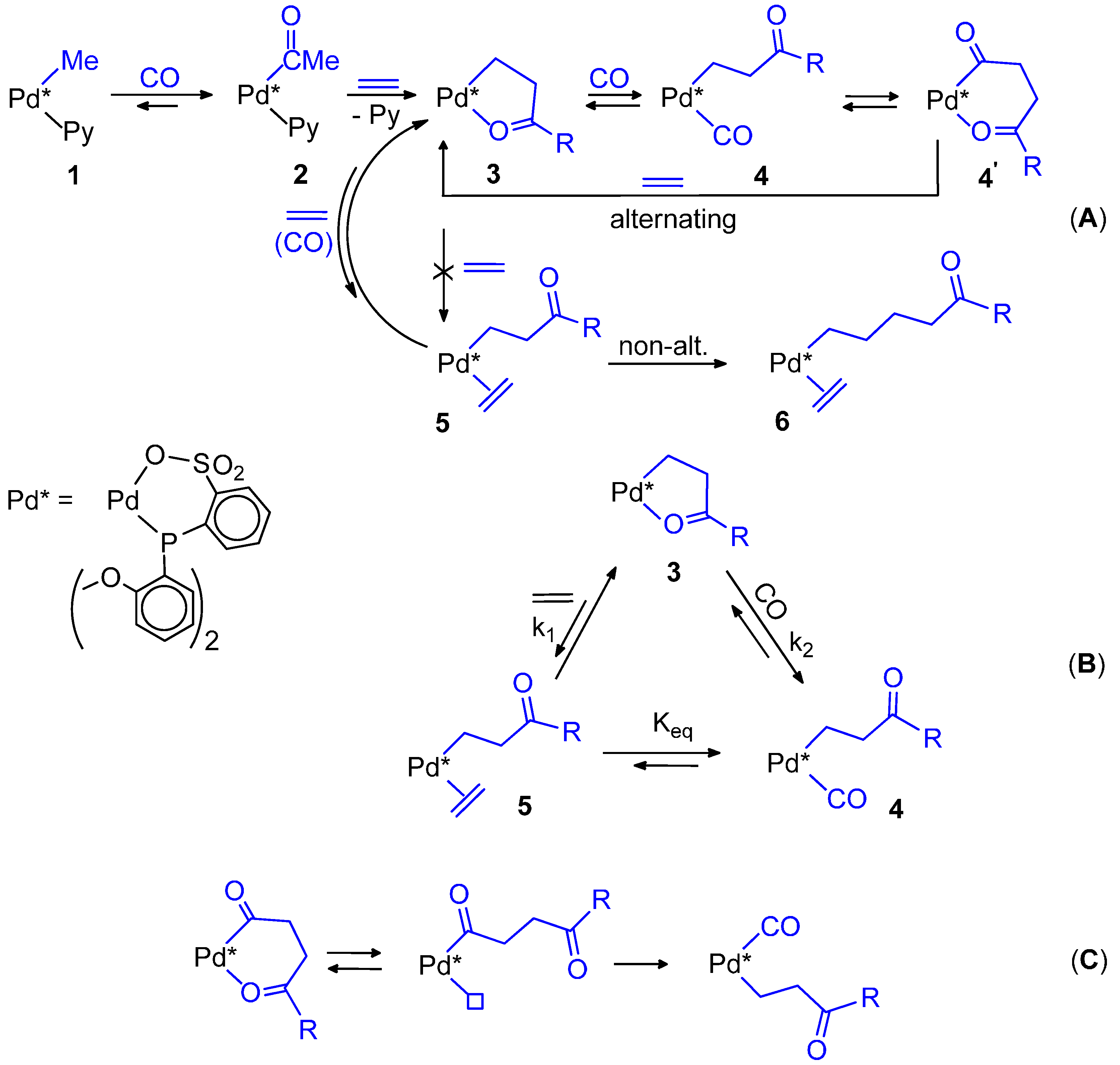
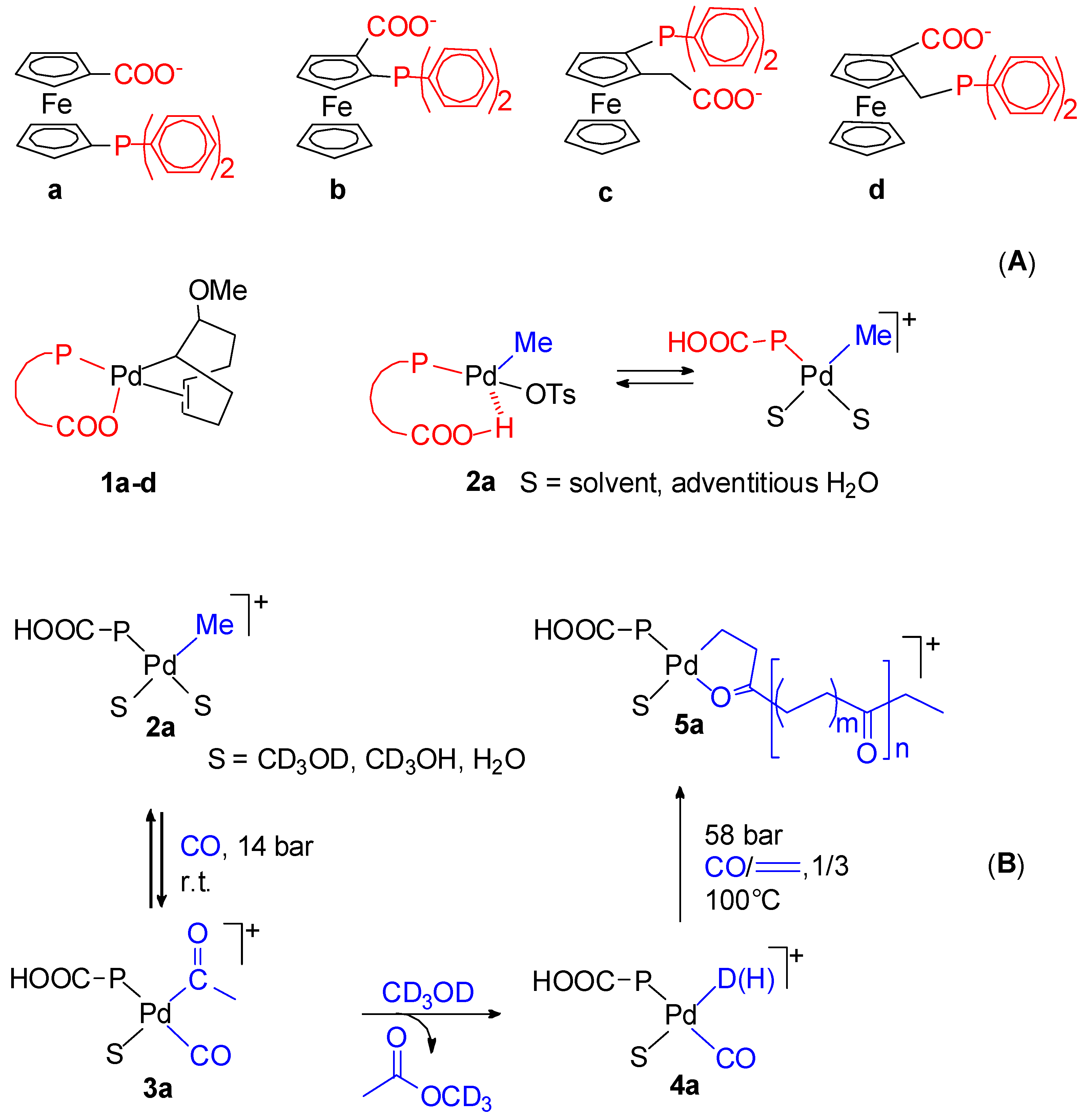
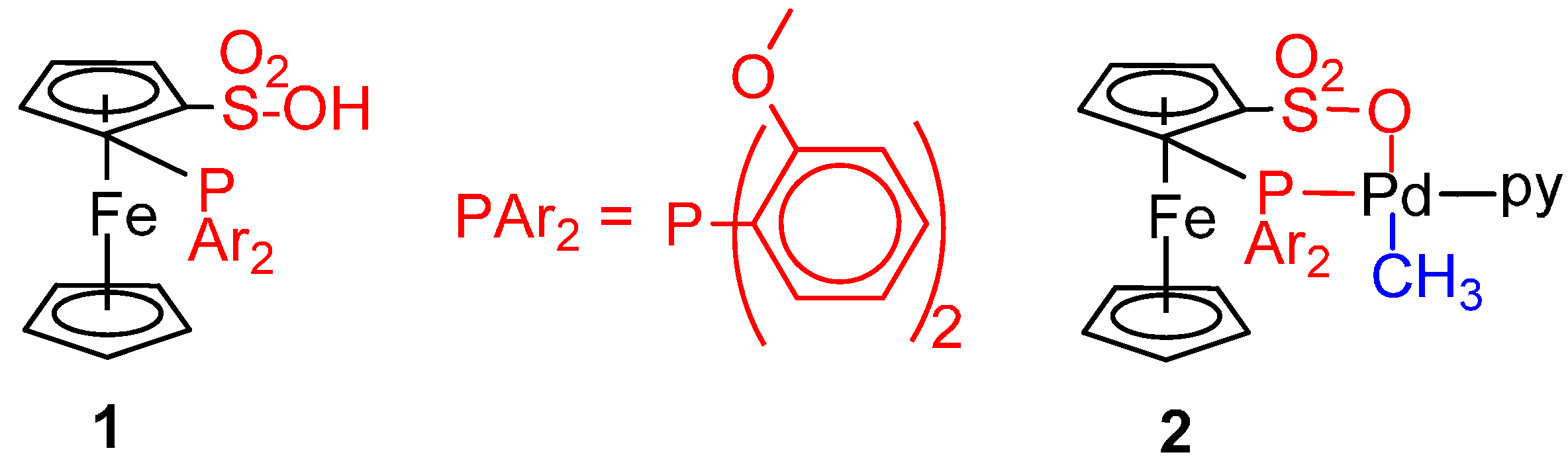
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bittler, K.; von Kutepow, N.; Neubauer, D. Carbonylation of Olefins under Mild Temperature Conditions in the Presence of Palladium Complexes. Angew. Chem. Int. Ed. 1968, 7, 329–335. [Google Scholar] [CrossRef]
- Grushin, V.V. Hydrido Complexes of Palladium. Chem. Rev. 1996, 96, 2011–2033. [Google Scholar] [CrossRef] [PubMed]
- Kiss, G. Palladium-Catalyzed Reppe Carbonylation. Chem. Rev. 2001, 101, 3435–3456. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, I.; Claver, C.; van Leeuwen, P.W.N.M. On the Mechanism of the Hydroxycarbonylation of Styrene with Palladium Systems. Eur. J. Inorg. Chem. 2001, 2719–2738. [Google Scholar]
- Kalck, P.; Urrutigoïty, M.; Dechy-Cabaret, O. Hydroxy- and alkoxycarbonylations of alkenes and alkynes. Top. Organomet. Chem. 2006, 18, 97–123. [Google Scholar]
- Sen, A.; Lai, T.W. Novel Palladium(II)-Catalyzed Copolymerization of Carbon Monoxide with Olefins. J. Am. Chem. Soc. 1982, 104, 3520–3522. [Google Scholar] [CrossRef]
- Lai, T.W.; Sen, A. Palladium(II)-Catalyzed Copolymerization of Carbon Monoxide with Ethylene. Direct Evidence for a Single Mode of Chain Growth. Organometallics 1984, 3, 866–870. [Google Scholar]
- Drent, E.; van Broekhoven, J.A.M.; Doyle, M.J. Efficient palladium catalysts for the copolymerization of carbon monoxide with olefins to produce perfectly alternating polyketones. J. Organomet. Chem. 1991, 417, 235–251. [Google Scholar] [CrossRef]
- Sen, A. Mechanistic Aspects of Metal-Catalyzed Alternating Copolymerization of Olefins with Carbon Monoxide. Acc. Chem. Res. 1993, 26, 303–310. [Google Scholar] [CrossRef]
- Drent, E.; Budzelaar, P.H.M. Palladium-Catalyzed Alternating Copolymerization of Alkenes and Carbon Monoxide. Chem. Rev. 1996, 96, 663–682. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, C.; Meli, A. Alternating copolymerization of carbon monoxide and olefins by single-site metal catalysis. Coord. Chem. Rev. 2002, 225, 35–66. [Google Scholar] [CrossRef]
- Robertson, R.A.M.; Cole-Hamilton, D.J. The production of low molecular weight oxygenates from carbon monoxide and ethene. Coord. Chem. Rev. 2002, 225, 67–90. [Google Scholar] [CrossRef]
- Bianchini, C.; Meli, A.; Oberhauser, W. Catalyst design and mechanistic aspects of the alternating copolymerisation of ethene and carbon monoxide by diphosphine-modified palladium catalysis. Dalton Trans. 2003, 2627–2635. [Google Scholar] [CrossRef]
- Sen, A. Chain Propagation Mechanisms. In Catalytic Synthesis of Alkene-Carbon Monoxide Copolymers and Cooligomers; Sen, A., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003; pp. 237–263. [Google Scholar]
- Belov, G.P.; Novikova, E.V. Polyketones as alternating copolymers of carbon monoxide. Russ. Chem. Rev. 2004, 73, 267–291. [Google Scholar] [CrossRef]
- Cavinato, G.; Toniolo, L.; Vavasori, A. Carbonylation of Ethene in Methanol Catalysed by Cationic Phosphine Complexes of Pd(II): From Polyketones to Monocarbonylated Products. Top. Organomet. Chem. 2006, 18, 125–164. [Google Scholar]
- Garcia Suarez, E.J.; Godard, C.; Ruiz, A.; Claver, C. Alternating and Non-Alternating Pd-Catalysed Co- and Terpolymerisation of Carbon Monoxide and Alkenes. Eur. J. Inorg. Chem. 2007, 2582–2593. [Google Scholar] [CrossRef]
- Pascu, S.I. CO/alkene copolymerization reactions catalyzed by chelating diphosphine, diimine and hemilabile N/O, P/O and P/N late transition metal complexes revisited. Rev. Roum. Chim. 2009, 54, 477–500. [Google Scholar]
- Consiglio, G.; Milani, B. Stereochemical Aspects of Co-oligomerization and Co-polymerization. In Catalytic Synthesis of Alkene-Carbon Monoxide Copolymers and Cooligomers; Sen, A., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003; pp. 189–215. [Google Scholar]
- Nosaki, K. Synthesis of Chiral, Optically Active Copolymers. In Catalytic Synthesis of Alkene-Carbon Monoxide Copolymers and Cooligomers; Sen, A., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003; pp. 217–235. [Google Scholar]
- Margl, P.; Michalak, A.; Ziegler, T. Theoretical Studies on Copolymerization of Polar Monomers. In Catalytic Synthesis of Alkene-Carbon Monoxide Copolymers and Cooligomers; Sen, A., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003; pp. 265–307. [Google Scholar]
- Durand, J.; Milani, B. The role of nitrogen-donor ligands in the palladium-catalyzed polyketones synthesis. Coord. Chem. Rev. 2006, 250, 542–560. [Google Scholar] [CrossRef]
- Carfagna, C.; Gatti, G.; Mosca, L.; Natanti, P.; Paoli, P.; Rossi, P.; Gabriele, B.; Salerno, G. Carbonylation of styrenes catalyzed by bioxazoline Pd(II) complexes: Mechanism of enantioselectivity. Dalton Trans. 2011, 40, 6792–6801. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Ito, S.; Nozaki, K. Coordination-Insertion Copolymerization of Fundamental Polar Monomers. Chem. Rev. 2009, 109, 5215–5244. [Google Scholar] [CrossRef]
- Abu-Surrah, A.S.; Wursche, R.; Rieger, B. Control of Molecular Weight in α-Olefin-Carbon Monoxide Alternating Copolymerization. A Way to High Molecular Weight Propene-Carbon Monoxide Thermoplastic Elastomers. Macromolecules 1996, 29, 4806–4807. [Google Scholar]
- Keim, W.; Maas, H. Copolymerization of ethylene and carbon monoxide by phosphinite-modified palladium catalysts. J. Organomet. Chem. 1996, 514, 271–276. [Google Scholar] [CrossRef]
- Doherty, S.; Eastham, G.R.; Tooze, R.; Scanlan, T.H.; Williams, D.; Elsegood, M.R.J.; Clegg, W. Palladium Complexes of C2-, C3-, and C4-Bridged Bis(phospholyl) Ligands: Remarkably Active Catalysts for the Copolymerization of Ethylene and Carbon Monoxide. Organometallics 1999, 18, 3558–3560. [Google Scholar] [CrossRef]
- Schwarz, J.; Herdtweck, E.; Herrmann, W.A. Highly Efficient Monocationic Palladacycles of Chelating Diphosphines in C2H4/CO Copolymerization. Organometallics 2000, 19, 3154–3160. [Google Scholar] [CrossRef]
- Liptau, P.; Seki, T.; Kehr, G.; Abele, A.; Froehlich, R.; Erker, G.; Grimme, S. Formation of a Chelate Bis(phosphino)[3]ferrocenophane Ligand and Its Use in Palladium-Catalyzed Alternating CO/Ethene Copolymerization. Organometallics 2003, 22, 2226–2232. [Google Scholar] [CrossRef]
- Meier, U.W.; Hollmann, F.; Thewalt, U.; Klinga, M.; Leskelä, M.; Rieger, B. Nonsymmetric Palladium Complexes of Partly Fluorinated Bisphosphine Ligands: Efficient Catalysts for Flexible Propene/CO Copolymer Materials of Ultrahigh Molecular Weight. Organometallics 2003, 22, 3905–3914. [Google Scholar] [CrossRef]
- Meinhard, D.; Hollmann, F.; Huhn, W.; Thewalt, U.; Klinga, M.; Rieger, B. Novel Single-Component Palladium(II) Catalysts for the Alternating CO/Propene Copolymerization Reaction. Organometallics 2004, 23, 5637–5639. [Google Scholar] [CrossRef]
- Sen, A.; Chen, J.T.; Vetter, W.M.; Whittle, R.R. Synthesis, Characterization, and Reactivity of α-Ketoacyl Complexes of Platinum(II) and Palladium(II). Crystal Structures of trans-Pt(PPh3)2(Cl)(COCOPh) and cis-Pt(PPh3)2(COPh)(COOMe). J. Am. Chem. Soc. 1987, 109, 148–156. [Google Scholar]
- Brunbaugh, S.; Sen, A. Mechanism of Acyl Group Isomerization in Palladium(II) Complexes. Development of a Catalytic Process for the Isomerization of Carboxylic Acid Chlorides. Development of a Catalytic Process for the Isomerization of Carboxylic Acid Chlorides. J. Am. Chem. Soc. 1988, 110, 803–816. [Google Scholar]
- Liu, J.; Heaton, B.T.; Iggo, J.A.; Whyman, R. The Complete Delineation of the Initiation, Propagation, and Termination Steps of the Carbomethoxy Cycle for the Carboalkoxylation of Ethene by Pd-Diphosphane Catalysts. Angew. Chem. Int. Ed. 2004, 43, 90–94. [Google Scholar] [CrossRef]
- Liu, J.; Heaton, B.T.; Iggo, J.A.; Whyman, R.; Bickley, J.F.; Steiner, A. The Mechanism of the Hydroalkoxycarbonylation of Ethene and Alkene-CO Copolymerization Catalyzed by PdII-Diphosphine Cations. Chem. Eur. J. 2006, 12, 4417–4430. [Google Scholar] [CrossRef] [PubMed]
- Mul, W.P.; Oosterbeek, H.; Beitel, G.A.; Kramer, G.J.; Drent, E. In Situ Monitoring of a Heterogeneus Palladium-Based Polyketone Catalyst. Angew. Chem. Int. Ed. 2000, 39, 1848–1851. [Google Scholar] [CrossRef]
- Shultz, C.S.; Ledford, J.; DeSimone, J.M.; Brookhart, M. Kinetic Studies of Migratory Insertion Reactions at the (1,3-Bis(diphenylphosphino)propane)Pd(II) Center and Their Relationship to the Alternating Copolymerization of Ethylene and Carbon Monoxide. J. Am. Chem. Soc. 2000, 122, 6351–6356. [Google Scholar] [CrossRef]
- Zuideveld, M.A.; Kamer, P.C.J.; van Leeuwen, P.W.N.M.; Klusener, P.A.A.; Stil, H.A.; Roobeek, C.F. Chain-Transfer Mechanisms of the Alternating Copolymerization of Carbon Monoxide and Ethene Catalyzed by Palladium(II) Complexes: Rearrangement to Highly Reactive Enolates. J. Am. Chem. Soc. 1998, 120, 7977–7978. [Google Scholar] [CrossRef]
- Ledford, J.; Shultz, C.S.; Gates, D.P.; White, P.S.; DeSimone, J.M.; Brookhart, M. Bond Angle Effects on the Migratory Insertion of Ethylene and Carbon Monoxide into Palladium(II)-Methyl Bonds in Complexes Bearing Bidentate Phosphine Ligands. Organometallics 2001, 20, 5266–5276. [Google Scholar] [CrossRef]
- Zuidema, E.; Bo, C.; van Leeuwen, P.W.N.M. Ester versus Polyketone Formation in the Palladium-Diphosphine Catalyzed Carbonylation of Ethene. J. Am. Chem. Soc. 2007, 129, 3989–4000. [Google Scholar] [CrossRef] [PubMed]
- Zudin, V.N.; Chinakov, V.D.; Nekipelov, V.M.; Rogov, V.A.; Likholobov, V.A.; Yermakov, Y.I. Determination of key intermediates for homogeneous water-gas shift reaction and hydrocarbonylation of ethylene to diethyl ketone catalyzed by the ‘Pd(OAc)2PPh3-CF3COOH/H2O’ system. J. Mol. Catal. 1989, 52, 27–48. [Google Scholar] [CrossRef]
- Pugh, R.I.; Drent, E. Methoxycarbonylation versus Hydroacylation of Ethene; Dramatic Influence of the Ligand in Cationic Palladium Catalysis. Adv. Synth. Catal. 2002, 344, 837–840. [Google Scholar] [CrossRef]
- Van Leeuwen, P.W.N.M.; Zuideveld, M.A.; Swennenhuis, B.H.G.; Freixa, Z.; Kamer, P.C.J.; Goubitz, K.; Fraanje, J.; Lutz, M.; Spek, A.L. Alcoholysis of Acylpalladium(II) Complexes Relevant to the Alternating Copolymerization of Ethene and Carbon Monoxide and the Alkoxycarbonylation of Alkenes: The Importance of Cis-Coordinating Phosphines. J. Am. Chem. Soc. 2003, 125, 5523–5539. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Heaton, B.T.; Iggo, J.A.; Whyman, R. Methanolysis of acyl-Pd(II) complexes Relevant to CO/ethene coupling reactions. Chem. Commun. 2004, 1326–1327. [Google Scholar] [CrossRef]
- Bianchini, C.; Meli, A.; Oberhauser, W.; van Leeuwen, P.W.N.M.; Zuideveld, M.A.; Freixa, Z.; Kamer, P.C.J.; Spek, A.L.; Gusev, O.V.; Kal’sin, A.M. Methoxycarbonylation of Ethene by Palladium(II) Complexes with 1,1'-Bis(diphenylphosphino)ferrocene (dppf) and 1,1'-Bis(diphenylphosphino)octamethylferrocene (dppomf). Organometallics 2003, 22, 2409–2421. [Google Scholar] [CrossRef]
- Gusev, O.V.; Kalsin, A.M.; Petrovskii, P.V.; Lyssenko, K.A.; Oprunenko, Y.F.; Bianchini, C.; Meli, A.; Oberhauser, W. Synthesis, Characterization, and Reactivity of 1,1'-Bis(diphenylphosphino)osmocene: Palladium(II) Complexes and Their Use as Catalysts in the Methoxycarbonylation of Olefins. Organometallics 2003, 22, 913–915. [Google Scholar] [CrossRef]
- Bianchini, C.; Meli, A.; Muller, G.; Oberhauser, W.; Passaglia, E. Studies of Ligand and Solvent Effects in the Alternating Copolymerization of Carbon Monoxide and Ethene by Palladium-Diphosphine Catalysis. Organometallics 2002, 21, 4965–4977. [Google Scholar] [CrossRef]
- Vavasori, A.; Toniolo, L. Carbon monoxide-ethylene copolymerization catalyzed by a Pd(AcO)2/dppp/TsOH system: The promoting effect of water and of the acid. J. Mol. Catal. A Chem. 1996, 110, 13–23. [Google Scholar] [CrossRef]
- Vavasori, A.; Cavinato, G.; Toniolo, L. Carbon monoxide–ethylene copolymerization catalyzed by a Pd(OAc)2/dppp/formic acid system [dppp = 1,3-bis(diphenylphosphino)propane]. J. Mol. Catal. A Chem. 2003, 191, 209–215. [Google Scholar] [CrossRef]
- Fabrello, A.; Vavasori, A.; Dall’Acqua, F.; Toniolo, L. Influence of the reaction conditions on the productivity and on the molecular weight of the polyketone obtained by the CO-ethene copolymerisation catalysed by [Pd(TsO)(H2O)(dppp)](TsO) in MeOH. J. Mol. Catal. A Chem. 2007, 276, 211–218. [Google Scholar] [CrossRef]
- Pugh, R.I.; Drent, E. Palladium-Catalyzed Synthesis of Mono-esters, -ketones, and-aldehydes/alcohols. In Catalytic Synthesis of Alkene-Carbon Monoxide Copolymers and Cooligomers; Sen, A., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003; pp. 9–35. [Google Scholar]
- Vavasori, A.; Toniolo, L.; Cavinato, G.; Visentin, A. Highly active [Pd(AcO)2(dppp)] catalyst for the CO-C2H4 copolymerization in H2O-CH3COOH solvent [dppp = 1,3-bis(diphenylphosphino)propane]. J. Mol. Catal. A Chem. 2003, 204–205, 295–303. [Google Scholar]
- Bianchini, C.; Lee, H.M.; Meli, A.; Oberhauser, W.; Peruzzini, M.; Vizza, F. Ligand and Solvent Effects in the Alternating Copolymerization of Carbon Monoxide and Olefins by Palladium-Diphosphine Catalysis. Organometallics 2002, 21, 16–33. [Google Scholar] [CrossRef]
- Bianchini, C.; Meli, A.; Oberhauser, W.; Claver, C.; Garcia Suarez, E.J. Unraveling the o-Methoxy Effect in the CO/Ethene Copolymerization Reaction by Diphosphanepalladium(II). Eur. J. Inorg. Chem. 2007, 2702–2710. [Google Scholar] [CrossRef]
- Bianchini, C.; Lee, H.M.; Meli, A.; Moneti, S.; Vizza, F.; Fontani, M.; Zanello, P. Copolymerization of Carbon Monoxide with Ethene Catalyzed by Palladium(II) Complexes of 1,3-Bis(diphenylphosphino)propane Ligands Bearing Different Substituents on the Carbon Backbone. Macromolecules 1999, 32, 4183–4193. [Google Scholar] [CrossRef]
- Mul, W.P.; van der Made, A.W.; Smaardijk, A.A.; Drent, E. Catalytic Synthesis of Copolymers and Terpolymers. In Catalytic Synthesis of Alkene-Carbon Monoxide Copolymers and Cooligomers; Sen, A., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003; pp. 87–140. [Google Scholar]
- Muñoz-Moreno, B.K.; Claver, C.; Ruiz, A.; Bianchini, C.; Meli, A.; Oberhauser, W. Synthesis of palladium(II) complexes containing a new α-d-xylofuranose-modified diphosphine and their application as catalyst precursors in the co- and terpolymerization of CO-ethene and propene. Dalton Trans. 2008, 2741–2750. [Google Scholar] [CrossRef]
- Xu, F.Y.; Zhao, A.X.; Chien, J.C.W. Regio- and stereo-selective alternating copolymerization of carbon monoxide with propene. Macromol. Chem. Phys. 1993, 194, 2579–2603. [Google Scholar] [CrossRef]
- Batistini, A.; Consiglio, G.; Suter, U.W. Regioselectivity Control in the Palladium-Catalyzed Copolymerization of Propylene with Carbon Monoxide. Angew. Chem. Int. Ed. 1992, 31, 303–305. [Google Scholar] [CrossRef]
- Mul, W.P.; Dirkzwager, H.; Broekhuis, A.A.; Heeres, H.J.; van der Linden, A.J.; Orpen, A.G. Highly active, recyclable catalyst for the manufacture of viscous, low molecular weight, CO-ethene-propene-based polyketone, base component for a new class of resins. Inorg. Chim. Acta 2002, 327, 147–159. [Google Scholar] [CrossRef]
- Clegg, W.; Eastham, G.R.; Elsegood, M.R.J.; Tooze, R.P.; Wang, X.L.; Whiston, K. Highly active and selective catalysts for the production of methyl propanoate via the methoxycarbonylation of ethene. Chem. Commun. 1999, 1877–1878. [Google Scholar] [CrossRef]
- Eastham, G.R.; Heaton, B.T.; Iggo, J.A.; Tooze, R.P.; Whyman, R.; Zacchini, S. Synthesis and spectroscopic characterisation of all the intermediates in the Pd-catalysed methoxycarbonylation of ethene. Chem. Commun. 2000, 609–610. [Google Scholar] [CrossRef]
- Clegg, W.; Eastham, G.R.; Elsegood, M.R.J.; Heaton, B.T.; Iggo, J.A.; Tooze, R.P.; Whyman, R.; Zacchini, S. Characterization and Dynamics of [Pd(L-L)H(solv)]+, [Pd(L-L)(CH2CH3)]+, and [Pd(L-L)(C(O)Et)(THF)]+ (L-L =1,2-(CH2PBut2)2C6H4): Key Intermediates in the Catalytic Methoxycarbonylation of Ethene to Methylpropanoate. Organometallics 2002, 21, 1832–1840. [Google Scholar] [CrossRef]
- Eastham, G.R.; Tooze, R.P.; Kilner, M.; Foster, D.F.; Cole-Hamilton, D.J. Deuterium labelling evidence for a hydride mechanism in the formation of methyl propanoate from carbon monoxide, ethene and methanol catalysed by a palladium complex. J. Chem. Soc. Dalton Trans. 2002, 1613–1617. [Google Scholar] [CrossRef]
- Clegg, W.; Easthman, G.R.; Elsegood, M.R.J.; Heaton, B.T.; Iggo, J.A.; Tooze, R.P.; Whyman, R.; Zacchini, S. Synthesis and reactivity of palladium hydrido-solvento complexes, including a key intermediate in the catalytic methoxycarbonylation of ethene to methyl propanoate. J. Chem. Soc. Dalton Trans. 2002, 3300–3308. [Google Scholar] [CrossRef]
- Puddephatt, R.J. Chemistry of bis(diphenylphosphino)methane. Chem. Soc. Rev. 1983, 12, 99–127. [Google Scholar] [CrossRef]
- Dossett, S.J.; Gillon, A.; Orpen, A.G.; Fleming, J.S.; Pringle, P.G.; Wass, D.F.; Jones, M.D. Steric activation of chelate catalysts: Efficient polyketone catalysts based on four-membered palladium(II) diphosphine chelates. Chem. Commun. 2001, 699–700. [Google Scholar] [CrossRef]
- Cavinato, G.; Vavasori, A.; Amadio, E.; Toniolo, L. CO–ethene copolymerisation catalysed by [PdCl2(PPh3)2]/PPh3/HCl in MeOH. J. Mol. Catal. A Chem. 2007, 278, 251–257. [Google Scholar] [CrossRef]
- Bianchini, C.; Meli, A.; Oberhauser, W.; Parisel, S.; Passaglia, E.; Ciardelli, F.; Gusev, O.V.; Kal’sin, A.M.; Vologdin, N.V. Ethylene Carbonylation in Methanol and in Aqueous Media by Palladium(II) Catalysts Modified with 1,1'-Bis(dialkylphosphino)ferrocenes. Organometallics 2005, 24, 1018–1030. [Google Scholar] [CrossRef]
- Gusev, O.V.; Kalsin, A.M.; Peterleitner, M.G.; Petrovskii, P.V.; Lyssenko, K.A.; Akhmedov, N.G.; Bianchini, C.; Meli, A.; Oberhauser, W. Palladium(II) Complexes with 1,1'-Bis(diphenylphosphino)ferrocenes [Fe(η5-C5R4PPh2)2]n+ (dppf, R = H, n = 0; dppomf, R = Me, n = 0; dppomf+, R = Me, n = 1). Synthesis, Characterization, and Catalytic Activity in Ethene Methoxycarbonylation. Organometallics 2002, 21, 3637–3649. [Google Scholar]
- Kalsin, A.M.; Vologdin, N.V.; Peganova, T.A.; Petrovskii, P.V.; Lyssenko, K.A.; Dolgushin, F.M.; Gusev, O.V. Palladium(II) complexes with o-aryl substituted 1,1'-bis(phosphino)ferrocenes [Fe(η5-C5H4PR2)2Pd(NCMe)n](OTf)2 (R = o-MeOC6H4, o-MeC6H4, o-PriC6H4, C6F5): Synthesis, structure and catalytic properties in methoxycarbonylation of ethylene. J. Organomet. Chem. 2006, 691, 921–927. [Google Scholar] [CrossRef]
- Butler, I.R.; Horton, P.N.; Fortune, K.M.; Morris, K.; Greenwell, C.H.; Eastham, G.R.; Hursthouse, M.B. The first 1,2,3-tris(phosphinomethyl)ferrocene. Inorg. Chem. Commun. 2004, 7, 923–928. [Google Scholar] [CrossRef]
- Butler, I.R.; Baker, P.K.; Eastham, G.R.; Fortune, K.M.; Horton, P.N.; Hursthouse, M.B. Ferrocenylmethyl phosphines ligands in the palladium-catalysed synthesis of methyl propionate. Inorg. Chem. Commun. 2004, 7, 1049–1052. [Google Scholar] [CrossRef]
- Fanjul, T.; Eastham, G.; Fey, N.; Hamilton, A.; Orpen, A.G.; Pringle, P.G.; Waugh, M. Palladium Complexes of the Heterodiphosphine o-C6H4(CH2PtBu2)(CH2PPh2) Are Highly Selective and Robust Catalysts for the Hydromethoxycarbonylation of Ethene. Organometallics 2010, 29, 2292–2305. [Google Scholar] [CrossRef]
- Fanjul, T.; Eastham, G.; Haddow, M.F.; Hamilton, A.; Pringle, P.G.; Orpen, A.G.; Turner, T.P.W.; Waugh, M. Efficient and chemoselective ethene hydromethoxycarbonylation catalysts based on Pd-complexes of heterodiphosphines o-C6H4(CH2PtBu2)(CH2PR2). Catal. Sci. Technol. 2012, 2, 937–950. [Google Scholar] [CrossRef]
- Fanjul, T.; Eastham, G.; Floure, J.; Forrest, S.J.K.; Haddow, M.F.; Hamilton, A.; Pringle, P.G.; Orpen, A.G.; Waugh, M. Interplay of bite angle and cone angle effects. A comparison between o-C6H4(CH2PR2)(PR'2) and o-C6H4(CH2PR'2)(CH2PR'2) as ligands for Pd-catalysed ethene hydromethoxycarbonylation. Dalton Trans. 2013, 42, 100–115. [Google Scholar]
- De la Fuente, V.; Waugh, M.; Eastham, G.R.; Iggo, J.A.; Castillón, S.; Claver, C. Phosphine Ligands in the Palladium-Catalysed Methoxycarbonylation of Ethene: Insights into the Catalytic Cycle through an HP NMR Spectroscopic Study. Chem. Eur. J. 2010, 16, 6919–6932. [Google Scholar] [CrossRef] [PubMed]
- Hidai, M.; Kokura, M.; Uchida, Y. Reactions of palladium(II) compounds with carbon monoxide in alcohol/amine systems: A new route to palladium(0) carbonyl and carboalkoxy-palladium(II) complexes. J. Organomet. Chem. 1973, 52, 431–435. [Google Scholar] [CrossRef]
- Smith, G.D.; Hanson, B.E.; Merola, J.S.; Waller, F.J. Palladium Methoxide and Carbomethoxy Complexes: Synthesis and Molecular Structure of (bipy)Pd(CO2CH3)2. Organometallics 1993, 12, 568–570. [Google Scholar] [CrossRef]
- Bellabarba, R.M.; Tooze, R.P.; Slawin, A.M.Z. Synthesis, X-ray characterisation and reactions of a trigonal planar palladium(0) carbonyl complex, (tbpx)PdCO. Chem. Commun. 2003, 1916–1917. [Google Scholar] [CrossRef]
- Jimenez Rodriguez, C.; Foster, D.F.; Eastham, G.R.; Cole-Hamilton, D.J. Highly selective formation of linear esters from terminal and internal alkenes catalysed by palladium complexes of bis-(di-tert-butylphosphinomethyl)benzene. Chem. Commun. 2004, 1720–1721. [Google Scholar] [CrossRef]
- Vavasori, A.; Cavinato, G.; Toniolo, L. Effect of a hydride source (water, hydrogen, p-toluenesulfonic acid) on the hydroesterification of ethylene to methyl propionate using a Pd(PPh3)2(TsO)2 (TsO = p-toluenesulfonate anion) catalyst precursor. J. Mol. Catal. A Chem. 2001, 176, 11–18. [Google Scholar] [CrossRef]
- Cavinato, G.; Toniolo, L.; Vavasori, A. Characterization and catalytic activity of trans-[Pd(COCH2CH3)(TsO)(PPh3)2], isolated from the hydro-methoxycarbonylation of ethene catalyzed by [Pd(TsO)2(PPh3)2]. J. Mol. Catal. A Chem. 2004, 219, 233–240. [Google Scholar] [CrossRef]
- Cavinato, G.; Toniolo, L.; Vavasori, A.; Benetollo, F. Synthesis, characterization and X-ray structure of trans-[Pd(COOCH3)(H2O)(PPh3)2](TsO), a possible intermediate in the catalytic hydroesterification of olefins (TsO = p-toluenesulfonate). Inorg. Chim. Acta 2003, 343, 183–188. [Google Scholar] [CrossRef]
- Otsuka, S.; Nakamura, A.; Yoshida, T.; Naruto, M.; Ataka, K. Chemistry of Alkoxycarbonyl, Acyl, and Alkyl Compounds of Nickel(II) and Palladium(II). J. Am. Chem. Soc. 1973, 95, 3180–3188. [Google Scholar] [CrossRef]
- Fenton, D.M. Noble Metal Catalysis. II. Hydratocarbonylation Reaction of Olefins with Carbon Monoxide to Give Saturated Acids. J. Org. Chem. 1973, 38, 3192–3198. [Google Scholar]
- Milstein, D. Aspects of Intermediacy of Carbalkoxymetal Complexes in CO Reactions. Acc. Chem. Res. 1988, 21, 428–438. [Google Scholar] [CrossRef]
- Amadio, E.; Cavinato, G.; Dolmella, A.; Ronchin, L.; Toniolo, L.; Vavasori, A. New carboalkoxybis(triphenylphosphine)palladium(II) cationic complexes: Synthesis, characterization, reactivity and role in the catalytic hydrocarboalkoxylation of ethene. X-ray structure of trans-[Pd(COOMe)(TsO)(PPh3)2]·2CHCl3. J. Mol. Catal. A: Chem. 2009, 298, 103–110. [Google Scholar]
- Cavinato, G.; Facchetti, S.; Toniolo, L. Ethene hydromethoxycarbonylation catalyzed by cis-[Pd(SO4)(PPh3)2]/H2SO4/PPh3. J. Mol. Catal. A Chem. 2010, 333, 180–185. [Google Scholar] [CrossRef]
- Amadio, E.; Cavinato, G.; Harter, P.; Toniolo, L. An NMR study on the mechanism of ethane hydromethoxycarbonylation catalyzed by cationic Pd(II)-PPh3 complexes. J. Organomet. Chem. 2013, 745–746, 115–119. [Google Scholar]
- Tooze, R.P.; Whiston, K.; Malyan, A.P.; Taylor, M.J.; Wilson, N.W. Evidence for the hydride mechanism in the methoxycarbonylation of ethene catalysed by palladium-triphenylphosphine complexes. J. Chem. Soc. Dalton Trans. 2000, 3441–3444. [Google Scholar] [CrossRef]
- Bardi, R.; Del Pra, A.; Piazzesi, A.M.; Toniolo, L. Metals in organic syntheses. III. Highly regioselective propene hydrocarboxylation promoted by a PdCl2(PPh3)2-PPh3 catalyst precursor: Trans-Pd(COPr-n)Cl(PPh3)2 as an active catalytic species. Inorg. Chim. Acta 1979, 35, L345–L346. [Google Scholar]
- Bardi, R.; Piazzesi, A.M.; Del Pra, A.; Cavinato, G.; Toniolo, L. Metals in organic syntheses. XIII. The Isolation and Molecular Structure of trans-[PdCl(COC6H13-n)(PPh3)2], an Intermediate in the Hydrocarboalcoxylation of 1-hexene Catalyzed by the Precursor trans-[PdCl2(PPh3)2]. Inorg. Chim. Acta 1985, 102, 99–103. [Google Scholar]
- Cavinato, G.; Toniolo. On the mechanism of the hydrocarbalkoxylation of olefins catalyzed by palladium complexes. J. Organomet. Chem 1990, 398, 187–195. [Google Scholar]
- Knifton, J.F. Linear Carboxylic Acid Esters from α-Olefins. I. Catalysis by Homogeneous Platinum Complexes. J. Org. Chem. 1976, 41, 793–767. [Google Scholar]
- Knifton, J.F. Linear carboxylic acid esters from α-olefins. 2. Catalysis by homogeneous palladium complexes. J. Org. Chem. 1976, 41, 2885–2890. [Google Scholar]
- Benedek, C.; Toros, S.; Heil, B. Comparative investigation of the regioselectivity in styrene and α -methylstyrene hydroalkoxycarbonylation as a function of palladium catalyst structure. J. Organomet. Chem. 1999, 586, 85–93. [Google Scholar] [CrossRef]
- Benedek, C.; Szalontai, G.; Gomory, A.; Toros, S.; Heil, B. A study on the catalytic pathways of the hydroalkoxycarbonylation reaction of styrene. J. Organomet. Chem. 1999, 579, 147–155. [Google Scholar] [CrossRef]
- Seayad, A.; Kelkar, A.A.; Toniolo, L.; Chaudhari, R.V. Hydroesterification of styrene using in situ formed Pd(OTs)2(PPh3)2 complex catalyst. J. Mol. Catal. A Chem. 2000, 151, 47–59. [Google Scholar] [CrossRef]
- Seayad, A.; Jayasree, S.; Damoran, K.; Toniolo, L.; Chaudhari, R.V. On the mechanism of hydroesterification of styrene using an in situ-formed cationic palladium complex. J. Organomet. Chem. 2000, 601, 100–107. [Google Scholar] [CrossRef]
- Benedek, C.; Gomory, A.; Heil, B.; Toros, S. Reversibility of palladium-alkyl intermediate formation in deuterioalkoxycarbonylation of 1-hexene. J. Organomet. Chem. 2001, 622, 112–120. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Coppel, Y.; Urrutigoïty, M.; Kalck, P. [Pd(H)(SnCl3)L2]: The key active species in the catalysed alkoxycarbonylation reaction of terminal alkenes. J. Organomet. Chem. 2005, 690, 2947–2951. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Hebrard, F.; Duran, J.; Polo, A.; Urrutigoïty, M.; Kalck, P. An efficient catalytic system for cyclocarbonylation of terpenes into lactones. Appl. Organometal. Chem. 2005, 19, 30–34. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Laurenczy, G.; Urrutigoïty, M.; Kalck, P. Hydride Route for the Palladium-Catalysed Cyclocarbonylation of Monoterpenes. Eur. J. Inorg. Chem. 2005, 4215–4225. [Google Scholar]
- Cavinato, G.; Facchetti, S.; Toniolo, L. Oxidative carbonylation of ethene catalyzed by Pd(II)-PPh3 complexes in MeOH using benzoquinone as stoichiometric oxidant. J. Mol. Catal. A Chem. 2012, 352, 63–69. [Google Scholar] [CrossRef]
- Bertani, R.; Cavinato, G.; Toniolo, L.; Vasapollo, G. New alkoxycarbonyl complexes of palladium (II) and their role in carbonylation reactions carried out in the presence of an alkanol. J. Mol. Catal. 1993, 84, 165–176. [Google Scholar] [CrossRef]
- Jiang, Z.; Sen, A. Water-Soluble Palladium(II) Compounds as Catalysts for the Alternating Copolymerization of Olefins with Carbon Monoxide in an Aqueous Medium. Macromolecules 1994, 27, 7215–7216. [Google Scholar] [CrossRef]
- Bianchini, C.; Lee, H.M.; Meli, A.; Moneti, S.; Patinec, V.; Petrucci, G.; Vizza, F. Water-Soluble Palladium(II) Catalysts for the Alternating Co- and Terpolymerization of CO and Olefins in Aqueous Phase. Macromolecules 1999, 32, 3859–3866. [Google Scholar] [CrossRef]
- Verspui, G.; Feiken, J.; Papadogianakis, G.; Sheldon, R.A. Catalytic conversions in water Part 11: Highly active water-soluble palladium-catalysts in the hydrocarboxylation of olefins and the alternating copolymerization of CO and olefins in water. J. Mol. Catal. A Chem. 1999, 146, 299–307. [Google Scholar] [CrossRef]
- Verspui, G.; Schanssema, F.; Sheldon, R.A. Catalytic conversions in water Part 14. The alternating copolymerization of ethene and CO catalyzed by water-soluble Pd catalysts. Appl. Catal. A General 2000, 198, 5–11. [Google Scholar]
- Verspui, G.; Schanssema, F.; Sheldon, R.A. A Stable, Conspicuously Active, Water-Soluble Pd Catalyst for the Alternating Copolymerization of Ethene and CO in Water. Angew. Chem. Int. Ed. 2000, 39, 804–806. [Google Scholar] [CrossRef]
- Dunbar, K.R.; Sun, J.-S. Synthesis and structure of the distorted octahedral palladium(II) complex [Pd(tmpp)2][BF4]2[tmpp = tris(2,4,6-trimethoxyphenyl)phosphine]. J. Chem. Soc. Chem. Commun. 1994, 2387–2388. [Google Scholar]
- Papadogianakis, G.; Verspui, G.; Maat, L.; Sheldon, R.A. Catalytic conversions inwater. Part 6. A novel biphasic hydrocarboxylation of olefins catalysed by palladium TPPTS complexes (TPPTS = P(C6H4-m-SO3Na)3). Catal. Lett. 1997, 47, 43–46. [Google Scholar]
- Tilloy, S.; Monflier, E.; Bertoux, F.; Castanet, Y.; Mortreux, A. A new fruitful development in biphasic catalysis: The palladium-catalyzed hydrocarboxylation of alkenes. New J. Chem. 1997, 21, 529–531. [Google Scholar]
- Verspui, G.; Moiseev, I.I.; Sheldon, R.A. Reaction intermediates in the Pd/tppts-catalyzed aqueous phase hydrocarboxylation of olefins monitored by NMR spectroscopy (tppts = P(C6H4-m-SO3Na)3). J. Organomet. Chem. 1999, 586, 196–199. [Google Scholar] [CrossRef]
- Jayasree, S.; Seayad, A.; Sarkar, B.R.; Chaudhari, R.V. Biphasic carbonylation of vinyl aromatic compounds using novel water-soluble Pd catalyst: Activity, selectivity and mechanistic studies. J. Mol. Catal. A Chem. 2002, 181, 221–235. [Google Scholar] [CrossRef]
- Vavasori, A.; Toniolo, L.; Cavinato, G. Carbon monoxide-ethylene copolymerisation catalysed by [PdCl2(dppp)] in methanol-water or in acetic acid-water as solvents (dppp = 1,3-bis(diphenylphosphine)propane). J. Mol. Catal. A Chem. 2004, 215, 63–72. [Google Scholar] [CrossRef]
- Cavinato, G.; Vavasori, A.; Toniolo, L.; Ronchin, L.; Dall’Acqua, F.; Dolmella, A. Synthesis, characterization and X-ray structure of [Pd(SO4)(dppp)]·H2O, a catalyst for the CO-ethene copolymerization [dppp = 1,3-bis(diphenylphosphino)propane]. Inorg. Chim. Acta 2005, 358, 4555–4562. [Google Scholar] [CrossRef]
- Vavasori, A.; Bellieni, A.; Ronchin, L.; Dall’Acqua, F.; Toniolo, L.; Cavinato, G. Selective alternating copolymerisation of carbon monoxide and ethene catalysed by [PdCl2(dppf)] in acetic acid-water as a solvent [dppf = 1,1'bis(diphenylphosphino)ferrocene]. J. Mol. Catal. A Chem. 2007, 263, 9–14. [Google Scholar] [CrossRef]
- Vavasori, A.; dall’Acqua, F.; Cavinato, G.; Toniolo, L. Influence of the operating conditions on the catalytic activity of [PdCl2(dapp)] in the CO-ethene copolymerization in the H2O-CH3COOH as a solvent (dapp = 1,3-bis(di(2-methoxyphenyl)phosphino)propane). J. Mol. Catal. A Chem. 2010, 332, 158–164. [Google Scholar] [CrossRef]
- Vavasori, A.; Ronchin, L.; Toniolo, L. Carbon monoxide-ethene alternating copolymerization catalyzed by [PdCl2(dppf)] in H2O-HCOOH [dppf = 1,1'-bis(diphenylphosphino)ferrocene]. J. Mol. Catal. A Chem. 2012, 363–364, 398–403. [Google Scholar]
- Vavasori, A.; Ronchin, L.; Toniolo, L. Terpolymerization of propene and ethene with carbon monoxide catalyzed by [PdCl2(dppf)] in HCOOH-H2O as a solvent [dppf = 1,1'-bis(diphenylphosphino)ferrocene]. Appl. Catal. A Gen. 2010, 389, 108–113. [Google Scholar] [CrossRef]
- Vavasori, A.; Ronchin, L.; Toniolo, L. Influence of formic acid and water on the [Pd(OAc)2(dppp)] catalyzed ethene-carbon monoxide copolymerization carried out in aprotic organic solvents. Appl. Catal. A Gen. 2012, 449, 198–202. [Google Scholar] [CrossRef]
- Bianchini, C.; Meli, A.; Oberhauser, W.; Segarra, A.M.; Claver, C.; Garcia Suarez, E.J. CO-ethylene copolymerization reactions in different reaction media catalyzed by palladium(II) complexes with chelating diphosphines bearing ortho-methoxy-substituted aryl groups. J. Mol. Catal. A Chem. 2007, 265, 292–305. [Google Scholar] [CrossRef]
- Bianchini, C.; Bruegeller, P.; Claver, C.; Czermak, G.; Dumfort, A.; Meli, A.; Oberhauser, W.; Garcia Suarez, E.J. Synthesis and characterization of palladium(II) complexes with new diphosphonium-diphosphine and diphosphine ligands. Production of low molecular weight alternating polyketones via catalytic CO/ethene copolymerisation. Dalton Trans. 2006, 2964–2973. [Google Scholar]
- Fessler, M.; Czermak, G.; Eller, S.; Trettenbrein, B.; Brueggeller, P.; Bettucci, L.; Bianchini, C.; Meli, A.; Ienco, A.; Oberhauser, W. A novel linkage-isomeric pair of dinuclear Pd(II) complexes bearing a bis-bidentate tetraphos ligand. Dalton Trans. 2009, 1859–1869. [Google Scholar] [CrossRef]
- Chepaikin, E.G.; Bezruchenko, A.P.; Leshcheva, A.A.; Boiko, G.N. Catalytic carbonylation of ethylene in the presence of the Pd(acac)2-m-Ph2PC6H4SO3Na(H)-AcOH system. Russ. Chem. Bull. 1994, 43, 360–363. [Google Scholar] [CrossRef]
- Chepaikin, E.G.; Bezruchenko, A.P.; Leschcheva, A.A. Catalityic mono- and polycarbonylation of ethylene into propionic acid derivatives and alternative polyketones. Kinet. Catal. 1999, 40, 313–321. [Google Scholar]
- Lindner, E.; Schmid, M.; Wald, J.; Queisser, J.A.; Geprägs, M.; Wegner, P.; Nachtigal, C. Catalytic activity of cationic diphospalladium(II) complexes in the alkene/CO copolymerization in organic solvents and water in dependence on the length of the alkyl chain at the phosphine ligands. J. Organomet. Chem. 2000, 602, 173–187. [Google Scholar] [CrossRef]
- Sunjuk, M.; Al-Noaimi, M.; Al-Degs, Y.; Al-Qirem, T.; Lindner, E.; Abu-Surrah, A.S. Higher α-Olefins Carbonylation in Aqueous Media by Pd(II) Catalysts Modified with Substituted Diphosphine Ligands: Aqueous Polyketone Latices with High Solid Contents and Molecular Weights. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 6715–6725. [Google Scholar] [CrossRef]
- Held, A.; Kolb, L.; Zuideveld, M.A.; Thomann, R.; Mecking, S.; Schmid, M.; Pietruschka, R.; Lindner, E.; Khanfar, M.; Sunjuk, M. Aqueous Polyketone Latices Prepared with Water-Insoluble Palladium(II) Catalysts. Macromolecules 2002, 35, 3342–3347. [Google Scholar] [CrossRef]
- Vavasori, A.; Ronchin, L.; Quartarone, G.; Tortato, C. The Catalytic Copolymerization of Ethene with Carbon Monoxide Efficiently Carried out in Water-Dichloromethane-Sodium Dodecylsulfate Emulsion. Mod. Res. Catal. 2013, 2, 93–99. [Google Scholar] [CrossRef]
- Zim, D.; de Souza, R.F.; Dupont, J.; Monteiro, A.L. Regioselective Synthesis of 2-Arylpropionic Esters by Palladium-Catalyzed Hydroesterification of Styrene Derivatives in Molten Salt Media. Tetrahedron Lett. 1998, 39, 7071–7074. [Google Scholar] [CrossRef]
- Garcia Suarez, E.J.; Khokarale, S.G.; van Buu, O.N.; Fehrmann, R.; Riisager, A. Pd catalysed ethylene methoxycarbonylation with Brønsted acid ionic liquids as promoter and phase-separable reaction media. Green Chem. 2014, 16, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Khokarale, S.G.; Garcia Suarez, E.J.; Xiong, J.; Mentzel, U.V.; Fehrmann, R.; Riisager, A. Zwitterion enhanced performance in palladium-phosphine catalysed ethylene methoxycarbonylation. Catal. Commun. 2014, 44, 73–75. [Google Scholar] [CrossRef]
- Chukanova, O.M.; Shul’ga, Y.M.; Belov, G.P. Alternating Copolymerization of Ethylene with Carbon Monoxide on a Supported Palladium Catalyst. Polym. Sci. Ser. B 2009, 51, 283–290. [Google Scholar] [CrossRef]
- Chukanova, O.M.; Alpherov, K.A.; Belov, G.P. Ethylene and carbon monoxide copolymerization catalyzed by supported palladium catalyst. J. Mol. Catal. A Chem. 2010, 325, 60–64. [Google Scholar] [CrossRef]
- Chukanova, O.M.; Alpherov, K.A.; Chernyak, A.V.; Perepelitsina, E.O.; Belov, G.P. Synthesis of Poly(carbon monoxide-co-ethylene-co-propylene) in the Presence of Supported Palladium Catalysts. Polym. Sci. Ser. B 2012, 54, 73–78. [Google Scholar] [CrossRef]
- Chukanova, O.M.; Alferov, K.A.; Belov, G.P. Synthesis of Poly(carbon monoxide-co-ethylene-co-hexene) with Supported Palladium Catalysts. Polym. Sci. Ser. B 2012, 54, 149–152. [Google Scholar] [CrossRef]
- Chukanova, O.M.; Alferov, K.A.; Chernyak, A.V.; Belov, G.P. Terpolymerization of Carbon Monoxide, Ethylene and Styrene in the Presence of Supported Palladium Catalyst. Polym. Sci. Ser. B 2013, 55, 14–21. [Google Scholar] [CrossRef]
- Chukanova, O.M.; Alferov, K.A.; Chernyak, A.V.; Smirnov, M.A.; Belov, G.P. Cooligomerization of Carbon Monoxide with Cyclic Olefins on Supported Palladium Catalysts. Polym. Sci. Ser. B 2013, 55, 546–550. [Google Scholar] [CrossRef]
- Severini, F.; Gallo, R.; Brambilla, L.; Castiglioni, C.; Ipsale, S. Outdoor ageing of ethylene-carbon monoxide alternating copolymer. Polym. Degrad. Stab. 2000, 69, 133–142. [Google Scholar] [CrossRef]
- De Vito, S.; Bronco, S. A thermogravimetric study of the effect of palladium catalyst residue on the degradation of ethylene-carbon monoxide alternating copolymer. Polym. Degrad. Stab. 1999, 63, 399–406. [Google Scholar] [CrossRef]
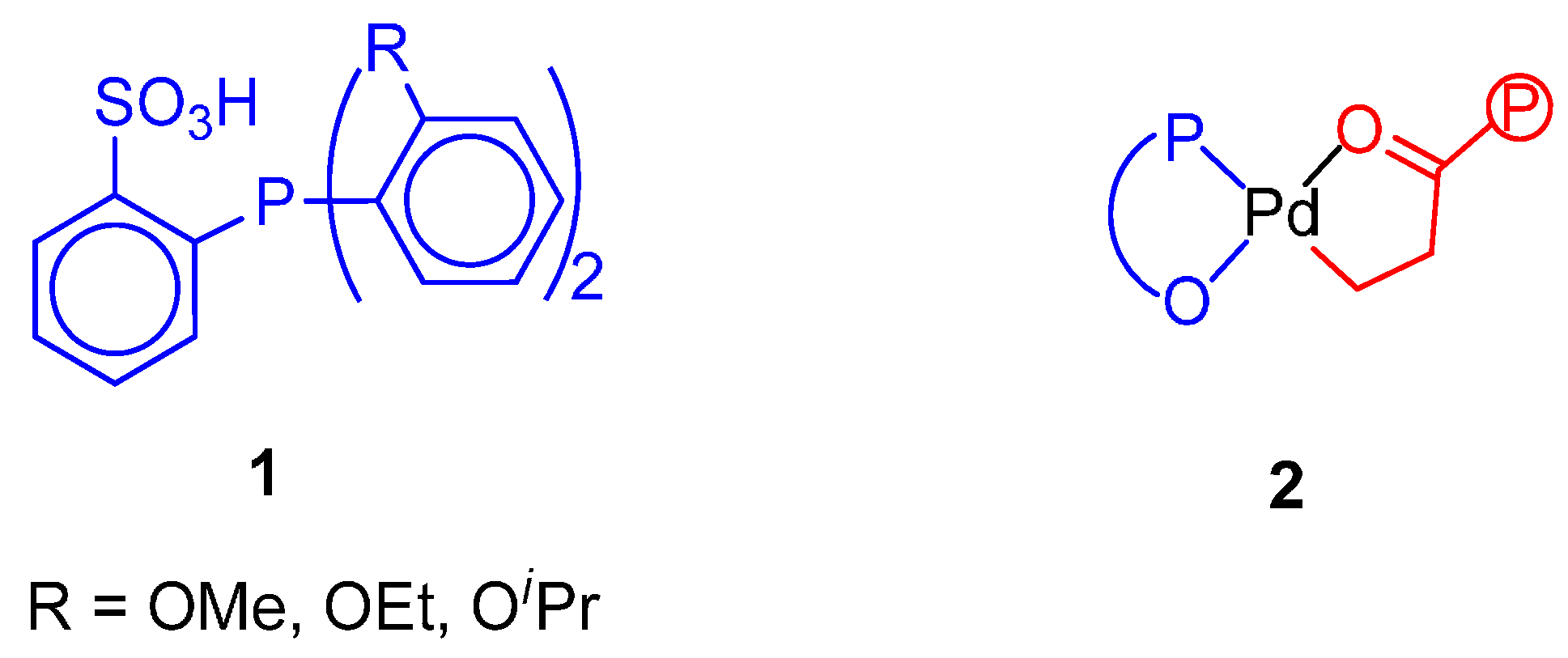
- Nakamura, A.; Anselment, T.M.J.; Claverie, J.; Goodall, B.; Jordan, R.J.; Mecking, S.; Rieger, B.; Sen, A.; van Leeuwen, P.W.N.M.; Nozaki, K. Ortho-Phosphinobenzene Sulfonate: A Superb Ligand for Palladium-Catalyzed Coordination-Insertion Copolymerization of Polar Vinyl Monomers. Acc. Chem. Res. 2013, 46, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Drent, E.; van Dijk, R.; van Ginkel, R.; van Oort, B.; Pugh, R.I. The first example of palladium catalysed non-perfectly alternating copolymerisation of ethene and carbon monoxide. Chem. Commun. 2002, 964–965. [Google Scholar] [CrossRef]
- Hearley, A.K.; Nowack, R.J.; Rieger, B. New Single-Site Palladium Catalysts for the Nonalternating Copolymerization of Ethylene and Carbon Monoxide. Organometallics 2005, 24, 2755–2763. [Google Scholar] [CrossRef]
- Bettucci, L.; Bianchini, C.; Claver, C.; Garcia Suarez, E.J.; Ruiz, A.; Meli, A.; Oberhauser, W. Ligand effects in the non-alternating CO-ethylene copolymerization by palladium(II) catalysis. Dalton Trans. 2007, 5590–5602. [Google Scholar] [CrossRef]
- Haras, A.; Michalak, A.; Rieger, B.; Ziegler, T. Theoretical Analysis of Factors Controlling the Nonalternating CO/C2H4 Copolymerization. J. Am. Chem. Soc. 2005, 127, 8765–8774. [Google Scholar] [CrossRef] [PubMed]
- Haras, A.; Michalak, A.; Rieger, B.; Ziegler, T. Comparative Study on Catalytic Systems for the Alternating and Nonalternating CO/Ethene Copolymerization. Organometallics 2006, 25, 946–953. [Google Scholar] [CrossRef]
- Newsham, D.K.; Borkar, S.; Sen, A.; Conner, D.M.; Goodall, B.L. Inhibitory Role of Carbon Monoxide in Palladium(II)-Catalyzed Nonalternating Ethene/Carbon Monoxide Copolymerization and the Synthesis of Polyethene-block-poly(ethene-alt-carbon monoxide). Organometallics 2007, 26, 3636–3638. [Google Scholar] [CrossRef]
- Luo, R.; Newsham, D.K.; Sen, A. Palladium-Catalyzed Nonalternating Copolymerization of Ethene and Carbon Monoxide: Scope and Mechanism. Organometallics 2009, 28, 6994–7000. [Google Scholar] [CrossRef]
- Bianchini, C.; Meli, A.; Oberhauser, W.; Segarra, A.M.; Passaglia, E.; Lamac, M.; Stepnicka, P. Palladium(II) Complexes with Phosphanylferrocenecarboxylate Ligands and Their Use as Catalyst Precursors for Semialternating CO-Ethylene Copolymerization. Eur. J. Inorg. Chem. 2008, 441–452. [Google Scholar] [CrossRef]
- Chen, C.; Anselment, T.M.J.; Frohlich, R.; Rieger, B.; Keher, G.; Erker, G. o-Diarylphosphinoferrocene Sulfonate Palladium Systems for Nonalternating Ethene-Carbon Monoxide Copolymerization. Organometallics 2011, 30, 5248–5257. [Google Scholar]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cavinato, G.; Toniolo, L. Carbonylation of Ethene Catalysed by Pd(II)-Phosphine Complexes. Molecules 2014, 19, 15116-15161. https://doi.org/10.3390/molecules190915116
Cavinato G, Toniolo L. Carbonylation of Ethene Catalysed by Pd(II)-Phosphine Complexes. Molecules. 2014; 19(9):15116-15161. https://doi.org/10.3390/molecules190915116
Chicago/Turabian StyleCavinato, Gianni, and Luigi Toniolo. 2014. "Carbonylation of Ethene Catalysed by Pd(II)-Phosphine Complexes" Molecules 19, no. 9: 15116-15161. https://doi.org/10.3390/molecules190915116