Small Molecule Tyrosine Kinase Inhibitors of ErbB2/HER2/Neu in the Treatment of Aggressive Breast Cancer

Abstract
:1. Introduction

{kind=link}
{kind=link}
| Growth Factor | EGFR | HER2 | HER3 | HER4 |
|---|---|---|---|---|
| Amphiregulin | + | − | − | − |
| Betacellulin | + | − | − | + |
| Epiregulin | + | − | − | + |
| Epigen | + | − | − | − |
| Epidermal Growth Factor (EGF) | + | − | − | − |
| Heparin-binding EGF-like Growth Factor (HB-EGF) | + | − | − | + |
| Transforming Growth Factor alpha (TGF-α) | + | − | − | − |
| Neuregulin-1 (NRG1) | − | − | + | + |
| Neuregulin-2 (NRG2) | − | − | + | + |
| Neuregulin-3 (NRG3) | − | − | − | + |
| Neuregulin-4 (NRG4) | − | − | ‒ | + |
2. Monoclonal Antibodies in HER2 Inhibition
2.1. Trastuzumab
2.2. Pertuzumab
3. Small Molecules as HER2 Kinase Inhibitors
3.1. HER2 Tyrosine Kinase Inhibitors
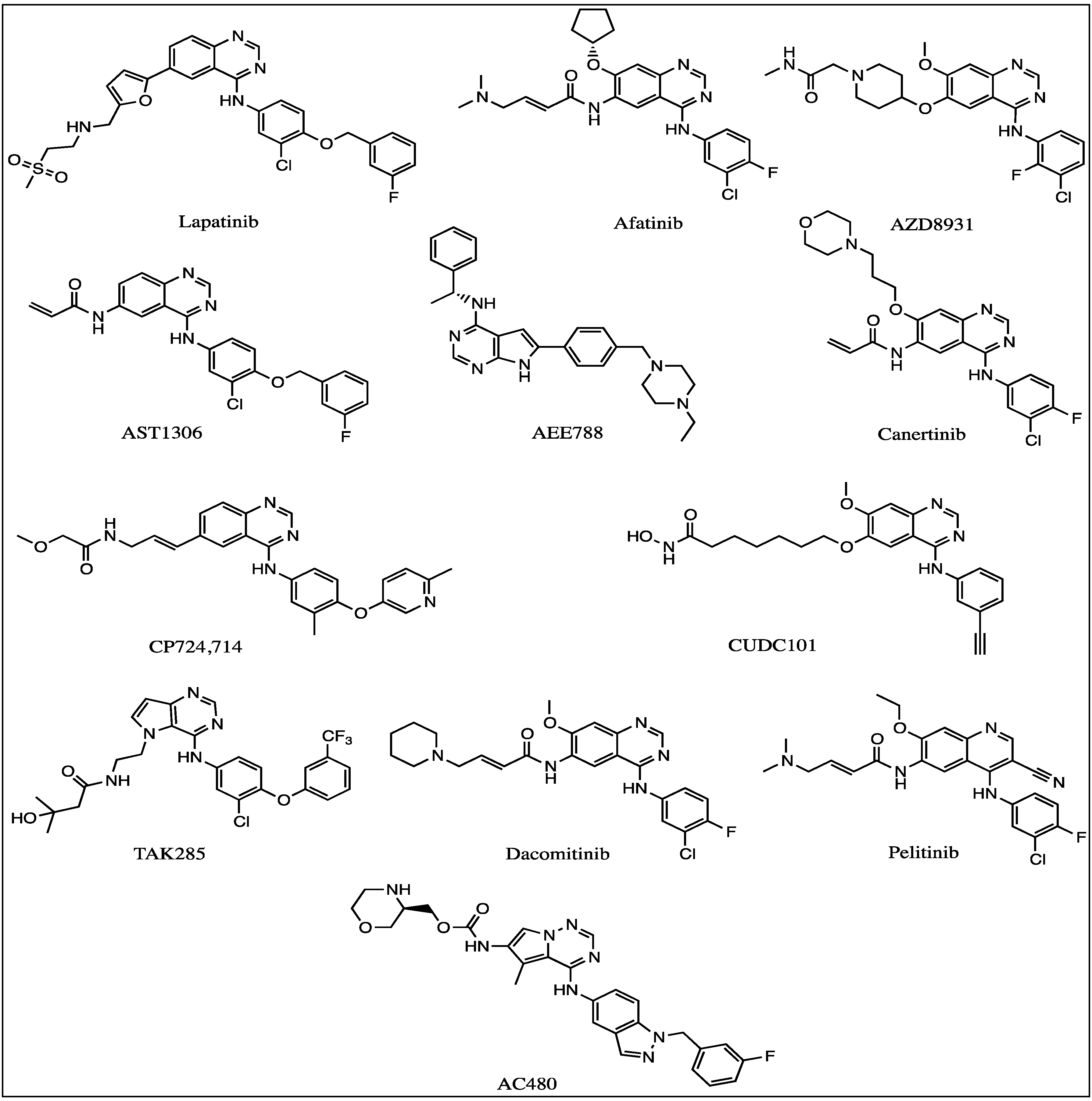
3.1.1. Lapatinib

3.1.2. Afatinib
3.1.3. AZD8931
3.2. Emerging HER2 Tyrosine Kinase Inhibitors
3.2.1. AST-1306
3.2.2. AEE-788
3.2.3. CI-1033 (Canertinib)
3.2.4. CP-724714
3.2.5. CUDC-101
3.2.6. TAK-285
3.2.7. AC-480 (BMS-599626)
3.2.8. PF299804, PF299 (Dacomitinib)
3.2.9. EKB-569 (Perlitinib)
| Inhibitor | EGFR IC50 (nM) | HER2 IC50 (nM) | HER3 IC50 (nM) | HER4 IC50 (nM) | Characteristics |
|---|---|---|---|---|---|
| Lapatinib [74] | 10.2 | 9.8 | - | 367 | Reversible |
| Afatinib [75] | 0.5 | 14 | - | - | Irreversible |
| AZD8931 [50] | 4 | 3 | 4 | - | Irreversible |
| AST-1306 [54] | 0.5 | 3.0 | - | 0.8 | Reversible |
| AEE-788 [76] | 2 | 6 | - | 160 | Reversible |
| CI-1033 (Canertinib) [77] | 1.5 | 9.0 | - | - | Irreversible |
| CP724,714 [60] | - | 9.8 | - | - | Reversible |
| CUDC-101 [78] | 2.4 | 15.7 | - | - | Irreversible |
| TAK-285 [79] | 24 | 36 | - | 260 | Irreversible |
| AC-480 (BMS-599626) [80] | 22 | 32 | - | 190 | Reversible |
| PF299804 PF299 (Dacomitinib) [75] | 6.0 | 45.7 | - | 73.7 | Irreversible |
| EKB-569 (Pelitinib) [73] | 38.5 | 1.26 | - | - | Irreversible |

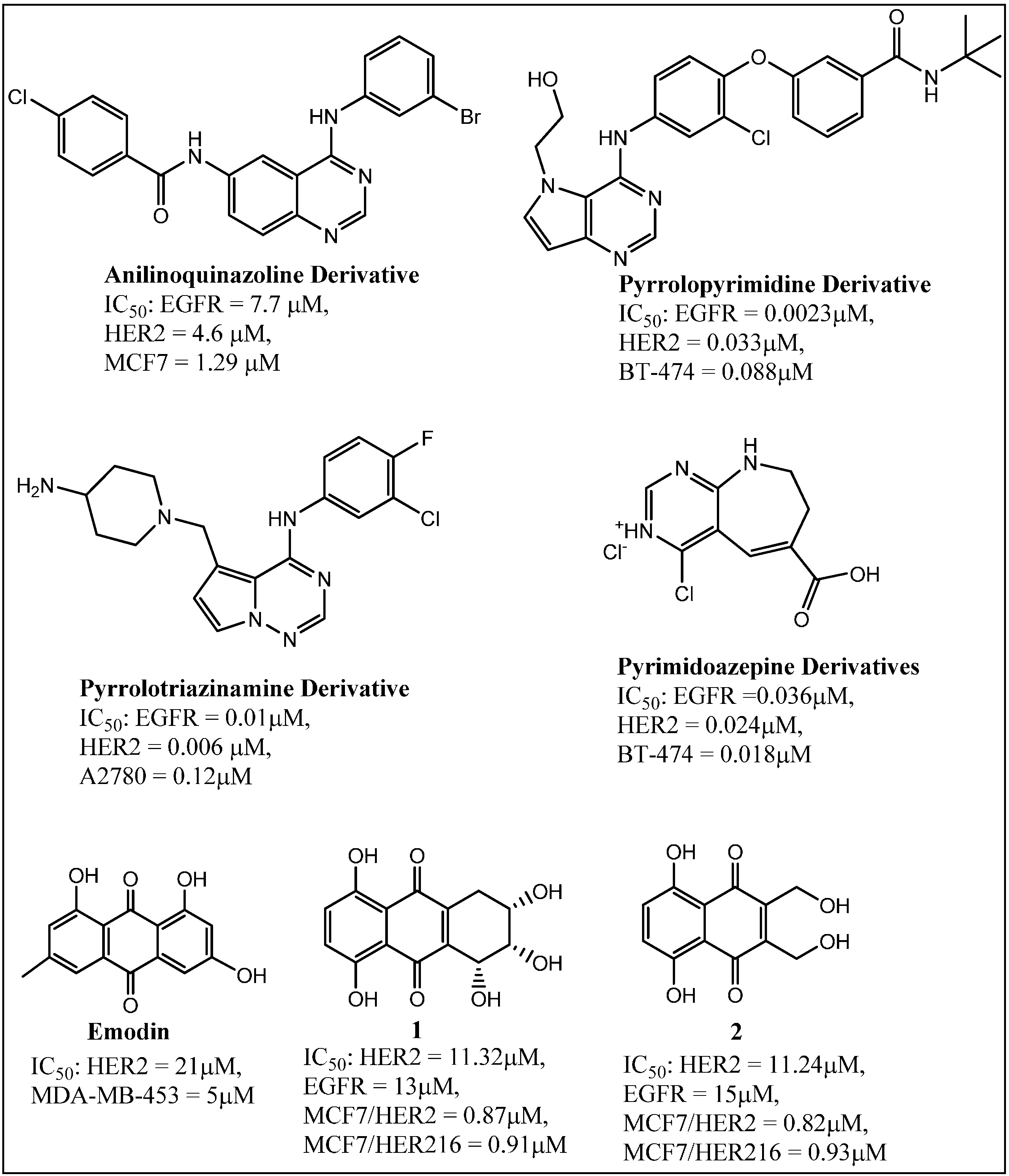
3.3. Ongoing Search for New Inhibitors
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef]
- Bacus, S.S.; Zelnick, C.R.; Plowman, G.; Yarden, Y. Expression of the erbb-2 family of growth factor receptors and their ligands in breast cancers. Implication for tumor biology and clinical behavior. Am. J. Clin. Pathol. 1994, 102, S13–S24. [Google Scholar]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef]
- Pegram, M.D.; Lipton, A.; Hayes, D.F.; Weber, B.L.; Baselga, J.M.; Tripathy, D.; Baly, D.; Baughman, S.A.; Twaddell, T.; Glaspy, J.A.; et al. Phase II study of receptor-enhanced chemosensitivity using recombinant humanized anti-p185HER2/neu monoclonal antibody plus cisplatin in patients with HER2/neu-overexpressing metastatic breast cancer refractory to chemotherapy treatment. J. Clin. Oncol. 1998, 16, 2659–2671. [Google Scholar]
- Arteaga, C.L. Progress in breast cancer: Overview. Clin. Cancer Res. 2013, 19, 6353–6359. [Google Scholar] [CrossRef]
- Zhang, X.H.; Giuliano, M.; Trivedi, M.V.; Schiff, R.; Osborne, C.K. Metastasis dormancy in estrogen receptor-positive breast cancer. Clin. Cancer Res. 2013, 19, 6389–6397. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the erbb signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Hynes, N.E.; Lane, H.A. Erbb receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef]
- Olayioye, M.A.; Graus-Porta, D.; Beerli, R.R.; Rohrer, J.; Gay, B.; Hynes, N.E. Erbb-1 and erbb-2 acquire distinct signaling properties dependent upon their dimerization partner. Mol. Cell. Biol. 1998, 18, 5042–5051. [Google Scholar]
- Lenferink, A.E.; Pinkas-Kramarski, R.; van de Poll, M.L.; van Vugt, M.J.; Klapper, L.N.; Tzahar, E.; Waterman, H.; Sela, M.; van Zoelen, E.J.; Yarden, Y. Differential endocytic routing of homo- and hetero-dimeric erbb tyrosine kinases confers signaling superiority to receptor heterodimers. EMBO J. 1998, 17, 3385–3397. [Google Scholar] [CrossRef]
- Tural, D.; Akar, E.; Mutlu, H.; Kilickap, S. P95 HER2 fragments and breast cancer outcome. Expert Rev. Anticancer Ther. 2014, 1–8. [Google Scholar]
- Arribas, J.; Baselga, J.; Pedersen, K.; Parra-Palau, J.L. P95HER2 and breast cancer. Cancer Res. 2011, 71, 1515–1519. [Google Scholar] [CrossRef]
- Hojjat-Farsangi, M. Small-molecule inhibitors of the receptor tyrosine kinases: Promising tools for targeted cancer therapies. Int. J. Mol. Sci. 2014, 15, 13768–13801. [Google Scholar] [CrossRef]
- Norman, P. Inducible tyrosine kinase inhibitors: A review of the patent literature (2010–2013). Expert Opin. Ther. Pat. 2014, 24, 979–991. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Erbb/HER protein-tyrosine kinases: Structures and small molecule inhibitors. Pharmacol. Res. 2014, 87C, 42–59. [Google Scholar] [CrossRef]
- Cui, J.J. Targeting receptor tyrosine kinase met in cancer: Small molecule inhibitors and clinical progress. J. Med. Chem. 2014, 57, 4427–4453. [Google Scholar] [CrossRef]
- Anandappa, G.; Turner, N.C. Targeting receptor tyrosine kinases in HER2-negative breast cancer. Curr. Opin. Oncol. 2013, 25, 594–601. [Google Scholar] [CrossRef]
- Warnault, P.; Yasri, A.; Coisy-Quivy, M.; Cheve, G.; Bories, C.; Fauvel, B.; Benhida, R. Recent advances in drug design of epidermal growth factor receptor inhibitors. Curr. Med. Chem. 2013, 20, 2043–2067. [Google Scholar] [CrossRef]
- Ocana, A.; Pandiella, A. Targeting HER receptors in cancer. Curr. Pharm. Des. 2013, 19, 808–817. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Sliwkowski, M.X.; Osborne, C.K.; Perez, E.A.; Puglisi, F.; Gianni, L. Treatment of HER2-positive breast cancer: Current status and future perspectives. Nat. Rev. Clin. Oncol. 2011, 9, 16–32. [Google Scholar] [CrossRef]
- Saini, K.S.; Azim, H.A., Jr.; Metzger-Filho, O.; Loi, S.; Sotiriou, C.; de Azambuja, E.; Piccart, M. Beyond trastuzumab: New treatment options for HER2-positive breast cancer. Breast 2011, 20 (Suppl. 3), S20–S27. [Google Scholar] [CrossRef]
- Nielsen, D.L.; Kümler, I.; Palshof, J.A.; Andersson, M. Efficacy of HER2-targeted therapy in metastatic breast cancer. Monoclonal antibodies and tyrosine kinase inhibitors. Breast 2013, 22, 1–12. [Google Scholar]
- Baselga, J.; Tripathy, D.; Mendelsohn, J.; Baughman, S.; Benz, C.C.; Dantis, L.; Sklarin, N.T.; Seidman, A.D.; Hudis, C.A.; Moore, J.; et al. Phase II study of weekly intravenous recombinant humanized anti-p185HER2 monoclonal antibody in patients with HER2/neu-overexpressing metastatic breast cancer. J. Clin. Oncol. 1996, 14, 737–744. [Google Scholar]
- Cobleigh, M.A.; Vogel, C.L.; Tripathy, D.; Robert, N.J.; Scholl, S.; Fehrenbacher, L.; Wolter, J.M.; Paton, V.; Shak, S.; Lieberman, G.; et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J. Clin. Oncol. 1999, 17, 2639–2648. [Google Scholar]
- Piccart-Gebhart, M.J.; Procter, M.; Leyland-Jones, B.; Goldhirsch, A.; Untch, M.; Smith, I.; Gianni, L.; Baselga, J.; Bell, R.; Jackisch, C.; et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N. Engl. J. Med. 2005, 353, 1659–1672. [Google Scholar] [CrossRef]
- Romond, E.H.; Perez, E.A.; Bryant, J.; Suman, V.J.; Geyer, C.E., Jr.; Davidson, N.E.; Tan-Chiu, E.; Martino, S.; Paik, S.; Kaufman, P.A.; et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N. Engl. J. Med. 2005, 353, 1673–1684. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Esteva, F.J.; Valero, V.; Booser, D.; Guerra, L.T.; Murray, J.L.; Pusztai, L.; Cristofanilli, M.; Arun, B.; Esmaeli, B.; Fritsche, H.A.; et al. Phase II study of weekly docetaxel and trastuzumab for patients with HER-2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002, 20, 1800–1808. [Google Scholar] [CrossRef]
- O’Shaughnessy, J.A.; Vukelja, S.; Marsland, T.; Kimmel, G.; Ratnam, S.; Pippen, J.E. Phase II study of trastuzumab plus gemcitabine in chemotherapy-pretreated patients with metastatic breast cancer. Clin. Breast Cancer 2004, 5, 142–147. [Google Scholar] [CrossRef]
- Pegram, M.D.; Konecny, G.E.; O’Callaghan, C.; Beryt, M.; Pietras, R.; Slamon, D.J. Rational combinations of trastuzumab with chemotherapeutic drugs used in the treatment of breast cancer. J. Natl. Cancer Inst. 2004, 96, 739–749. [Google Scholar] [CrossRef]
- Seidman, A.D.; Fornier, M.N.; Esteva, F.J.; Tan, L.; Kaptain, S.; Bach, A.; Panageas, K.S.; Arroyo, C.; Valero, V.; Currie, V.; et al. Weekly trastuzumab and paclitaxel therapy for metastatic breast cancer with analysis of efficacy by HER2 immunophenotype and gene amplification. J. Clin. Oncol. 2001, 19, 2587–2595. [Google Scholar]
- Baselga, J.; Albanell, J. Mechanism of action of anti-HER2 monoclonal antibodies. Ann. Oncol. 2001, 12, S35–S41. [Google Scholar] [CrossRef]
- Dokmanovic, M.; Wu, J.W. Mechanisms of trastuzumab resistance and opportunities to overcome therapeutic resistance. J. Mol. Biomark. Diagn. 2013, 4. [Google Scholar] [CrossRef]
- Abraham, J.; Stenger, M. Pertuzumab in neoadjuvant treatment of HER2-positive early breast cancer. J. Community Support. Oncol. 2014, 12, 84–86. [Google Scholar]
- Kang, Y.K.; Rha, S.Y.; Tassone, P.; Barriuso, J.; Yu, R.; Szado, T.; Garg, A.; Bang, Y.J. A phase IIa dose-finding and safety study of first-line pertuzumab in combination with trastuzumab, capecitabine and cisplatin in patients with HER2-positive advanced gastric cancer. Br. J. Cancer 2014, 111, 660–666. [Google Scholar] [CrossRef]
- Boix-Perales, H.; Borregaard, J.; Jensen, K.B.; Ersboll, J.; Galluzzo, S.; Giuliani, R.; Ciceroni, C.; Melchiorri, D.; Salmonson, T.; Bergh, J.; et al. The european medicines agency review of pertuzumab for the treatment of adult patients with HER2-positive metastatic or locally recurrent unresectable breast cancer: Summary of the scientific assessment of the committee for medicinal products for human use. Oncologist 2014, 19, 766–773. [Google Scholar] [CrossRef]
- Noble, M.E.; Endicott, J.A.; Johnson, L.N. Protein kinase inhibitors: Insights into drug design from structure. Science 2004, 303, 1800–1805. [Google Scholar] [CrossRef]
- Aertgeerts, K.; Skene, R.; Yano, J.; Sang, B.C.; Zou, H.; Snell, G.; Jennings, A.; Iwamoto, K.; Habuka, N.; Hirokawa, A.; et al. Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J. Biol. Chem. 2011, 286, 18756–18765. [Google Scholar] [CrossRef]
- Qiu, C.; Tarrant, M.K.; Choi, S.H.; Sathyamurthy, A.; Bose, R.; Banjade, S.; Pal, A.; Bornmann, W.G.; Lemmon, M.A.; Cole, P.A.; et al. Mechanism of activation and inhibition of the HER4/erbb4 kinase. Structure 2008, 16, 460–467. [Google Scholar] [CrossRef]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef]
- Zhang, X.; Pickin, K.A.; Bose, R.; Jura, N.; Cole, P.A.; Kuriyan, J. Inhibition of the egf receptor by binding of mig6 to an activating kinase domain interface. Nature 2007, 450, 741–744. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, S.; Hu, Y.P.; Wang, J.; Hauser, J.; Conway, A.N.; Vinci, M.A.; Humphrey, L.; Zborowska, E.; Willson, J.K.; et al. Blockade of EGFR and erbB2 by the novel dual EGFR and erbB2 tyrosine kinase inhibitor GW572016 sensitizes human colon carcinoma geo cells to apoptosis. Cancer Res. 2006, 66, 404–411. [Google Scholar] [CrossRef]
- Moulder, S.L.; Yakes, F.M.; Muthuswamy, S.K.; Bianco, R.; Simpson, J.F.; Arteaga, C.L. Epidermal growth factor receptor (HER1) tyrosine kinase inhibitor ZD1839 (Iressa) inhibits HER2/neu (erbB2)-overexpressing breast cancer cells in vitro and in vivo. Cancer Res. 2001, 61, 8887–8895. [Google Scholar]
- Piccart-Gebhart, M.J.; Holmes, A.P.; Baselga, J.; Azambuja, E.D.; Dueck, A.C.; Viale, G.; Zujewski, J.A.; Goldhirsch, A.; Santillana, S.; Pritchard, K.I.; et al. First results from the phase III ALTTO trial (BIG 2-06; NCCTG [alliance] N063D) comparing one year of anti-HER2 therapy with lapatinib alone (L), trastuzumab alone (T), their sequence (T→L), or their combination (T+L) in the adjuvant treatment of HER2-positive early breast cancer (EBC). J. Clin. Oncol. 2014, 32 (Suppl.), 5s. Abstract LBA4. [Google Scholar]
- Geuna, E.; Montemurro, F.; Aglietta, M.; Valabrega, G. Potential of afatinib in the treatment of patients with HER2-positive breast cancer. Breast Cancer (Dove Med. Press) 2012, 4, 131–137. [Google Scholar]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.R.; Padera, R.F.; Shapiro, G.I.; Baum, A.; Himmelsbach, F.; et al. Bibw2992, an irreversible egfr/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702–4711. [Google Scholar] [CrossRef]
- Katakami, N.; Atagi, S.; Goto, K.; Hida, T.; Horai, T.; Inoue, A.; Ichinose, Y.; Koboyashi, K.; Takeda, K.; Kiura, K.; et al. Lux-lung 4: A phase II trial of afatinib in patients with advanced non-small-cell lung cancer who progressed during prior treatment with erlotinib, gefitinib, or both. J. Clin. Oncol. 2013, 31, 3335–3341. [Google Scholar] [CrossRef]
- Eskens, F.A.; Mom, C.H.; Planting, A.S.; Gietema, J.A.; Amelsberg, A.; Huisman, H.; van Doorn, L.; Burger, H.; Stopfer, P.; Verweij, J.; et al. A phase I dose escalation study of bibw 2992, an irreversible dual inhibitor of epidermal growth factor receptor 1 (egfr) and 2 (HER2) tyrosine kinase in a 2-week on, 2-week off schedule in patients with advanced solid tumours. Br. J. Cancer 2008, 98, 80–85. [Google Scholar] [CrossRef]
- Yap, T.A.; Vidal, L.; Adam, J.; Stephens, P.; Spicer, J.; Shaw, H.; Ang, J.; Temple, G.; Bell, S.; Shahidi, M.; et al. Phase I trial of the irreversible egfr and HER2 kinase inhibitor bibw 2992 in patients with advanced solid tumors. J. Clin. Oncol. 2010, 28, 3965–3972. [Google Scholar] [CrossRef]
- Barlaam, B.; Anderton, J.; Ballard, P.; Bradbury, R.H.; Hennequin, L.F.; Hickinson, D.M.; Kettle, J.G.; Kirk, G.; Klinowska, T.; Lambert-van der Brempt, C.; et al. Discovery of azd8931, an equipotent, reversible inhibitor of signaling by egfr, HER2, and HER3 receptors. ACS Med. Chem. Lett. 2013, 4, 742–746. [Google Scholar] [CrossRef]
- Mu, Z.; Klinowska, T.; Dong, X.; Foster, E.; Womack, C.; Fernandez, S.V.; Cristofanilli, M. AZD8931, an equipotent, reversible inhibitor of signaling by epidermal growth factor receptor (EGFR), HER2, and HER3: Preclinical activity in HER2 non-amplified inflammatory breast cancer models. J. Exp. Clin. Cancer Res. 2014, 33, 47. [Google Scholar] [CrossRef]
- Kurata, T.; Tsurutani, J.; Fujisaka, Y.; Okamoto, W.; Hayashi, H.; Kawakami, H.; Shin, E.; Hayashi, N.; Nakagawa, K. Inhibition of egfr, HER2 and HER3 signaling with azd8931 alone and in combination with paclitaxel: Phase I study in japanese patients with advanced solid malignancies and advanced breast cancer. Investig. New Drugs 2014. [Google Scholar] [CrossRef]
- Tjulandin, S.; Moiseyenko, V.; Semiglazov, V.; Manikhas, G.; Learoyd, M.; Saunders, A.; Stuart, M.; Keilholz, U. Phase I, dose-finding study of AZD8931, an inhibitor of EGFR (erbB1), HER2 (erbB2) and HER3 (erbB3) signaling, in patients with advanced solid tumors. Investig. New Drugs 2014, 32, 145–153. [Google Scholar] [CrossRef]
- Xie, H.; Lin, L.; Tong, L.; Jiang, Y.; Zheng, M.; Chen, Z.; Jiang, X.; Zhang, X.; Ren, X.; Qu, W.; et al. Ast1306, a novel irreversible inhibitor of the epidermal growth factor receptor 1 and 2, exhibits antitumor activity both in vitro and in vivo. PLoS One 2011, 6, e21487. [Google Scholar]
- Zhang, J.; Cao, J.; Li, J.; Zhang, Y.; Chen, Z.; Peng, W.; Sun, S.; Zhao, N.; Wang, J.; Zhong, D.; et al. A phase I study of ast1306, a novel irreversible egfr and HER2 kinase inhibitor, in patients with advanced solid tumors. J. Hematol. Oncol. 2014, 7, 22. [Google Scholar] [CrossRef]
- Berezowska, S.; Diermeier-Daucher, S.; Brockhoff, G.; Busch, R.; Duyster, J.; Grosu, A.L.; Schlegel, J. Effect of additional inhibition of human epidermal growth factor receptor 2 with the bispecific tyrosine kinase inhibitor aee788 on the resistance to specific egfr inhibition in glioma cells. Int. J. Mol. Med. 2010, 26, 713–721. [Google Scholar] [CrossRef]
- Evans, A.H.; Pancholi, S.; Farmer, I.; Thornhill, A.; Evans, D.B.; Johnston, S.R.; Dowsett, M.; Martin, L.A. EGFR/HER2 inhibitor AEE788 increases ER-mediated transcription in HER2/ER-positive breast cancer cells but functions synergistically with endocrine therapy. Br. J. Cancer 2010, 102, 1235–1243. [Google Scholar] [CrossRef]
- Ako, E.; Yamashita, Y.; Ohira, M.; Yamazaki, M.; Hori, T.; Kubo, N.; Sawada, T.; Hirakawa, K. The pan-erbB tyrosine kinase inhibitor CI-1033 inhibits human esophageal cancer cells in vitro and in vivo. Oncol. Rep. 2007, 17, 887–893. [Google Scholar]
- Rixe, O.; Franco, S.X.; Yardley, D.A.; Johnston, S.R.; Martin, M.; Arun, B.K.; Letrent, S.P.; Rugo, H.S. A randomized, phase II, dose-finding study of the pan-erbB receptor tyrosine-kinase inhibitor CI-1033 in patients with pretreated metastatic breast cancer. Cancer Chemother. Pharmacol. 2009, 64, 1139–1148. [Google Scholar] [CrossRef]
- Jani, J.P.; Finn, R.S.; Campbell, M.; Coleman, K.G.; Connell, R.D.; Currier, N.; Emerson, E.O.; Floyd, E.; Harriman, S.; Kath, J.C.; et al. Discovery and pharmacologic characterization of CP-724,714, a selective ErbB2 tyrosine kinase inhibitor. Cancer Res. 2007, 67, 9887–9893. [Google Scholar] [CrossRef]
- Munster, P.N.; Britten, C.D.; Mita, M.; Gelmon, K.; Minton, S.E.; Moulder, S.; Slamon, D.J.; Guo, F.; Letrent, S.P.; Denis, L.; et al. First study of the safety, tolerability, and pharmacokinetics of CP-724,714 in patients with advanced malignant solid HER2-expressing tumors. Clin. Cancer Res. 2007, 13, 1238–1245. [Google Scholar] [CrossRef]
- Cai, X.; Zhai, H.X.; Wang, J.; Forrester, J.; Qu, H.; Yin, L.; Lai, C.J.; Bao, R.; Qian, C. Discovery of 7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDC-101) as a potent multi-acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer. J. Med. Chem. 2010, 53, 2000–2009. [Google Scholar] [CrossRef]
- Lai, C.J.; Bao, R.; Tao, X.; Wang, J.; Atoyan, R.; Qu, H.; Wang, D.G.; Yin, L.; Samson, M.; Forrester, J.; et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 2010, 70, 3647–3656. [Google Scholar] [CrossRef]
- Jimeno, A.; Galloway, T.; Wirth, L.; Gilbert, J.; Saba, N.; Bauman, J.; Colevas, D.; Mehra, R.; Raben, D.; Lai, C.-J.; et al. A phase I study of CUDC-101, a multitarget inhibitor of HDACs, EGFR, and HER2, in combination with chemoradiation in patients with intermediate/high risk locally advanced squamous cell carcinoma of the head and neck. Mol. Cancer Ther. 2013, 12 (Suppl. 11). [Google Scholar] [CrossRef]
- Ishikawa, T.; Seto, M.; Banno, H.; Kawakita, Y.; Oorui, M.; Taniguchi, T.; Ohta, Y.; Tamura, T.; Nakayama, A.; Miki, H.; et al. Design and synthesis of novel human epidermal growth factor receptor 2 (HER2)/epidermal growth factor receptor (EGFR) dual inhibitors bearing a pyrrolo[3,2-d]pyrimidine scaffold. J. Med. Chem. 2011, 54, 8030–8050. [Google Scholar] [CrossRef]
- Erdo, F.; Gordon, J.; Wu, J.T.; Sziraki, I. Verification of brain penetration of the unbound fraction of a novel HER2/EGFR dual kinase inhibitor (TAK-285) by microdialysis in rats. Brain Res. Bull. 2012, 87, 413–419. [Google Scholar] [CrossRef]
- Doi, T.; Takiuchi, H.; Ohtsu, A.; Fuse, N.; Goto, M.; Yoshida, M.; Dote, N.; Kuze, Y.; Jinno, F.; Fujimoto, M.; et al. Phase I first-in-human study of tak-285, a novel investigational dual HER2/egfr inhibitor, in cancer patients. Br. J. Cancer 2012, 106, 666–672. [Google Scholar] [CrossRef]
- Torres, M.A.; Raju, U.; Molkentine, D.; Riesterer, O.; Milas, L.; Ang, K.K. Ac480, formerly bms-599626, a pan HER inhibitor, enhances radiosensitivity and radioresponse of head and neck squamous cell carcinoma cells in vitro and in vivo. Investig. New Drugs 2011, 29, 554–561. [Google Scholar] [CrossRef]
- Axelrod, M.J.; Mendez, R.E.; Khalil, A.; Leimgruber, S.S.; Sharlow, E.R.; Capaldo, B.; Conaway, M.; Gioeli, D.G.; Weber, M.J.; Jameson, M.J. Synergistic apoptosis in head and neck squamous cell carcinoma cells by co-inhibition of insulin-like growth factor-1 receptor signaling and compensatory signaling pathways. Head Neck 2014. [Google Scholar] [CrossRef]
- Janne, P.A.; Boss, D.S.; Camidge, D.R.; Britten, C.D.; Engelman, J.A.; Garon, E.B.; Guo, F.; Wong, S.; Liang, J.; Letrent, S.; et al. Phase I dose-escalation study of the pan-HER inhibitor, pf299804, in patients with advanced malignant solid tumors. Clin. Cancer Res. 2011, 17, 1131–1139. [Google Scholar] [CrossRef]
- Kalous, O.; Conklin, D.; Desai, A.J.; O’Brien, N.A.; Ginther, C.; Anderson, L.; Cohen, D.J.; Britten, C.D.; Taylor, I.; Christensen, J.G.; et al. Dacomitinib (pf-00299804), an irreversible pan-HER inhibitor, inhibits proliferation of HER2-amplified breast cancer cell lines resistant to trastuzumab and lapatinib. Mol. Cancer Ther. 2012, 11, 1978–1987. [Google Scholar] [CrossRef]
- Reckamp, K.L.; Giaccone, G.; Camidge, D.R.; Gadgeel, S.M.; Khuri, F.R.; Engelman, J.A.; Koczywas, M.; Rajan, A.; Campbell, A.K.; Gernhardt, D.; et al. A phase 2 trial of dacomitinib (pf-00299804), an oral, irreversible pan-HER (human epidermal growth factor receptor) inhibitor, in patients with advanced non-small cell lung cancer after failure of prior chemotherapy and erlotinib. Cancer 2014, 120, 1145–1154. [Google Scholar] [CrossRef]
- Erlichman, C.; Hidalgo, M.; Boni, J.P.; Martins, P.; Quinn, S.E.; Zacharchuk, C.; Amorusi, P.; Adjei, A.A.; Rowinsky, E.K. Phase I study of ekb-569, an irreversible inhibitor of the epidermal growth factor receptor, in patients with advanced solid tumors. J. Clin. Oncol. 2006, 24, 2252–2260. [Google Scholar] [CrossRef]
- Rusnak, D.W.; Lackey, K.; Affleck, K.; Wood, E.R.; Alligood, K.J.; Rhodes, N.; Keith, B.R.; Murray, D.M.; Knight, W.B.; Mullin, R.J.; et al. The effects of the novel, reversible epidermal growth factor receptor/erbb-2 tyrosine kinase inhibitor, gw2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol. Cancer Ther. 2001, 1, 85–94. [Google Scholar]
- Barf, T.; Kaptein, A. Irreversible protein kinase inhibitors: Balancing the benefits and risks. J. Med. Chem. 2012, 55, 6243–6262. [Google Scholar] [CrossRef]
- Traxler, P.; Allegrini, P.R.; Brandt, R.; Brueggen, J.; Cozens, R.; Fabbro, D.; Grosios, K.; Lane, H.A.; McSheehy, P.; Mestan, J.; et al. AEE788: A dual family epidermal growth factor receptor/erbB2 and vascular endothelial growth factor receptor tyrosine kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2004, 64, 4931–4941. [Google Scholar] [CrossRef]
- Smaill, J.B.; Showalter, H.D.; Zhou, H.; Bridges, A.J.; McNamara, D.J.; Fry, D.W.; Nelson, J.M.; Sherwood, V.; Vincent, P.W.; Roberts, B.J.; et al. Tyrosine kinase inhibitors. 18. 6-substituted 4-anilinoquinazolines and 4-anilinopyrido[3,4-d]pyrimidines as soluble, irreversible inhibitors of the epidermal growth factor receptor. J. Med. Chem. 2001, 44, 429–440. [Google Scholar]
- Morphy, R. Selectively nonselective kinase inhibition: Striking the right balance. J. Med. Chem. 2010, 53, 1413–1437. [Google Scholar] [CrossRef]
- Kawakita, Y.; Seto, M.; Ohashi, T.; Tamura, T.; Yusa, T.; Miki, H.; Iwata, H.; Kamiguchi, H.; Tanaka, T.; Sogabe, S.; et al. Design and synthesis of novel pyrimido[4,5-b]azepine derivatives as HER2/EGFR dual inhibitors. Bioorg. Med. Chem. 2013, 21, 2250–2261. [Google Scholar] [CrossRef]
- Gavai, A.V.; Fink, B.E.; Fairfax, D.J.; Martin, G.S.; Rossiter, L.M.; Holst, C.L.; Kim, S.H.; Leavitt, K.J.; Mastalerz, H.; Han, W.C.; et al. Discovery and preclinical evaluation of [4-[[1-(3-fluorophenyl)methyl]-1h-indazol-5-ylamino]-5-methylpyrrolo[2,1-f][1,2,4]triazin-6-yl]carbamic acid, (3s)-3-morpholinylmethyl ester (bms-599626), a selective and orally efficacious inhibitor of human epidermal growth factor receptor 1 and 2 kinases. J. Med. Chem. 2009, 52, 6527–6530. [Google Scholar] [CrossRef]
- Fink, B.E.; Norris, D.; Mastalerz, H.; Chen, P.; Goyal, B.; Zhao, Y.; Kim, S.H.; Vite, G.D.; Lee, F.Y.; Zhang, H.; et al. Novel pyrrolo[2,1-f][1,2,4]triazin-4-amines: Dual inhibitors of egfr and HER2 protein tyrosine kinases. Bioorg. Med. Chem. Lett. 2011, 21, 781–785. [Google Scholar] [CrossRef]
- Sadek, M.M.; Serrya, R.A.; Kafafy, A.H.; Ahmed, M.; Wang, F.; Abouzid, K.A. Discovery of new HER2/egfr dual kinase inhibitors based on the anilinoquinazoline scaffold as potential anti-cancer agents. J. Enzym. Inhib. Med. Chem. 2014, 29, 215–222. [Google Scholar] [CrossRef]
- Li, D.D.; Qin, Y.J.; Sun, J.; Li, J.R.; Fang, F.; Du, Q.R.; Qian, Y.; Gong, H.B.; Zhu, H.L. Optimization of substituted 6-salicyl-4-anilinoquinazoline derivatives as dual egfr/HER2 tyrosine kinase inhibitors. PLoS One 2013, 8, e69427. [Google Scholar] [CrossRef]
- Kawakita, Y.; Miwa, K.; Seto, M.; Banno, H.; Ohta, Y.; Tamura, T.; Yusa, T.; Miki, H.; Kamiguchi, H.; Ikeda, Y.; et al. Design and synthesis of pyrrolo[3,2-d]pyrimidine HER2/egfr dual inhibitors: Improvement of the physicochemical and pharmacokinetic profiles for potent in vivo anti-tumor efficacy. Bioorg. Med. Chem. 2012, 20, 6171–6180. [Google Scholar] [CrossRef]
- Kawakita, Y.; Banno, H.; Ohashi, T.; Tamura, T.; Yusa, T.; Nakayama, A.; Miki, H.; Iwata, H.; Kamiguchi, H.; Tanaka, T.; et al. Design and synthesis of pyrrolo[3,2-d]pyrimidine human epidermal growth factor receptor 2 (HER2)/epidermal growth factor receptor (EGFR) dual inhibitors: Exploration of novel back-pocket binders. J. Med. Chem. 2012, 55, 3975–3991. [Google Scholar] [CrossRef]
- Zhou, W.; Ercan, D.; Janne, P.A.; Gray, N.S. Discovery of selective irreversible inhibitors for EGFR-T790M. Bioorg. Med. Chem. Lett. 2011, 21, 638–643. [Google Scholar] [CrossRef]
- Ok, S.; Kim, S.M.; Kim, C.; Nam, D.; Shim, B.S.; Kim, S.H.; Ahn, K.S.; Choi, S.H. Emodin inhibits invasion and migration of prostate and lung cancer cells by downregulating the expression of chemokine receptor cxcr4. Immunopharmacol. Immunotoxicol. 2012, 34, 768–778. [Google Scholar] [CrossRef]
- Sridhar, J.; Sfondouris, M.E.; Bratton, M.R.; Nguyen, T.L.; Townley, I.; Klein Stevens, C.L.; Jones, F.E. Identification of quinones as HER2 inhibitors for the treatment of trastuzumab resistant breast cancer. Bioorg. Med. Chem. Lett. 2013, 24, 126–131. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schroeder, R.L.; Stevens, C.L.; Sridhar, J. Small Molecule Tyrosine Kinase Inhibitors of ErbB2/HER2/Neu in the Treatment of Aggressive Breast Cancer. Molecules 2014, 19, 15196-15212. https://doi.org/10.3390/molecules190915196
Schroeder RL, Stevens CL, Sridhar J. Small Molecule Tyrosine Kinase Inhibitors of ErbB2/HER2/Neu in the Treatment of Aggressive Breast Cancer. Molecules. 2014; 19(9):15196-15212. https://doi.org/10.3390/molecules190915196
Chicago/Turabian StyleSchroeder, Richard L., Cheryl L. Stevens, and Jayalakshmi Sridhar. 2014. "Small Molecule Tyrosine Kinase Inhibitors of ErbB2/HER2/Neu in the Treatment of Aggressive Breast Cancer" Molecules 19, no. 9: 15196-15212. https://doi.org/10.3390/molecules190915196