Hydrogenations without Hydrogen: Titania Photocatalyzed Reductions of Maleimides and Aldehydes


Abstract
:
1. Introduction
2. Results and Discussion
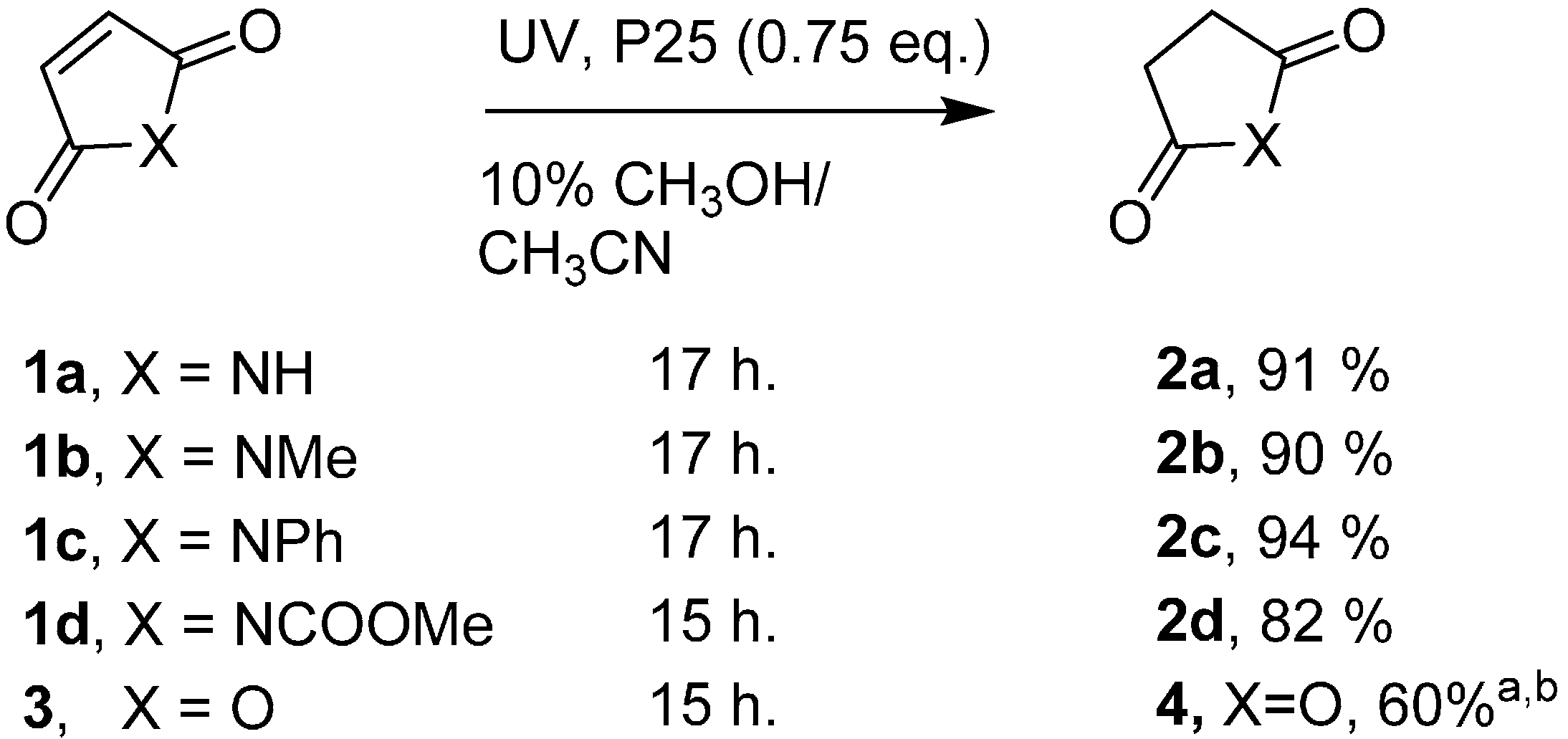
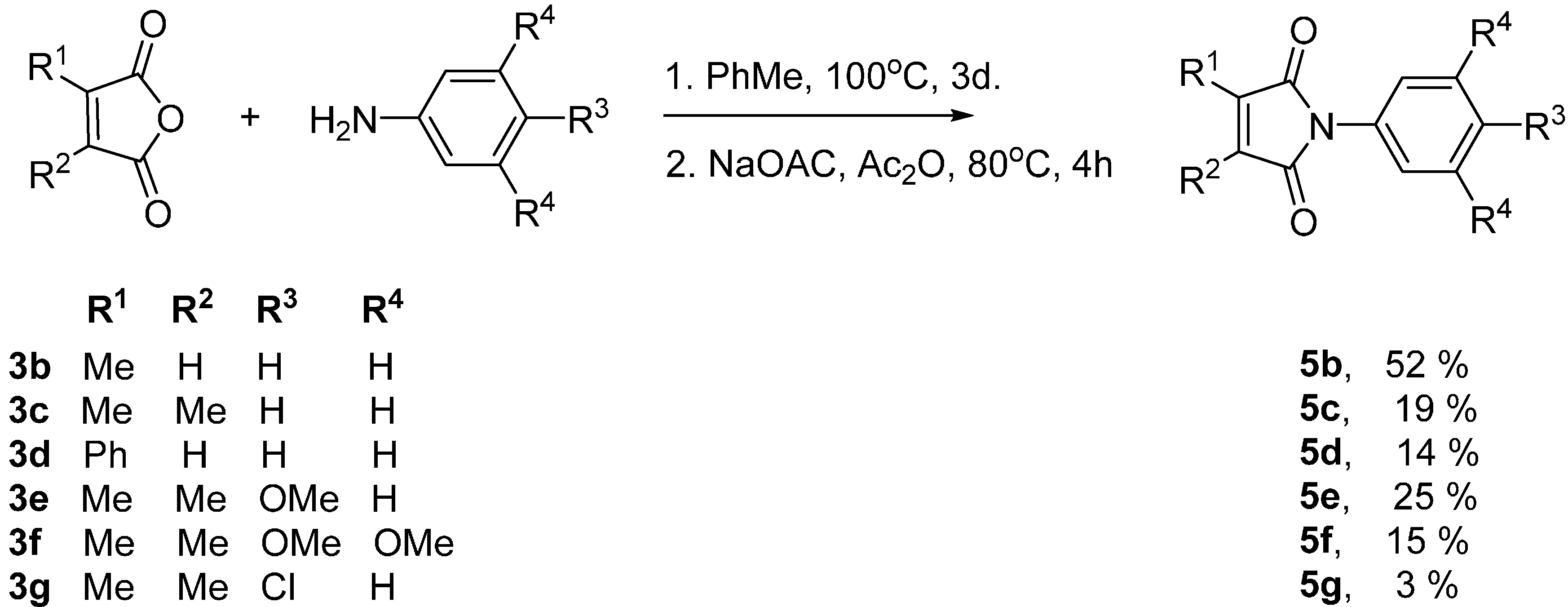
2.1. SCPC Reductions of Maleimides



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
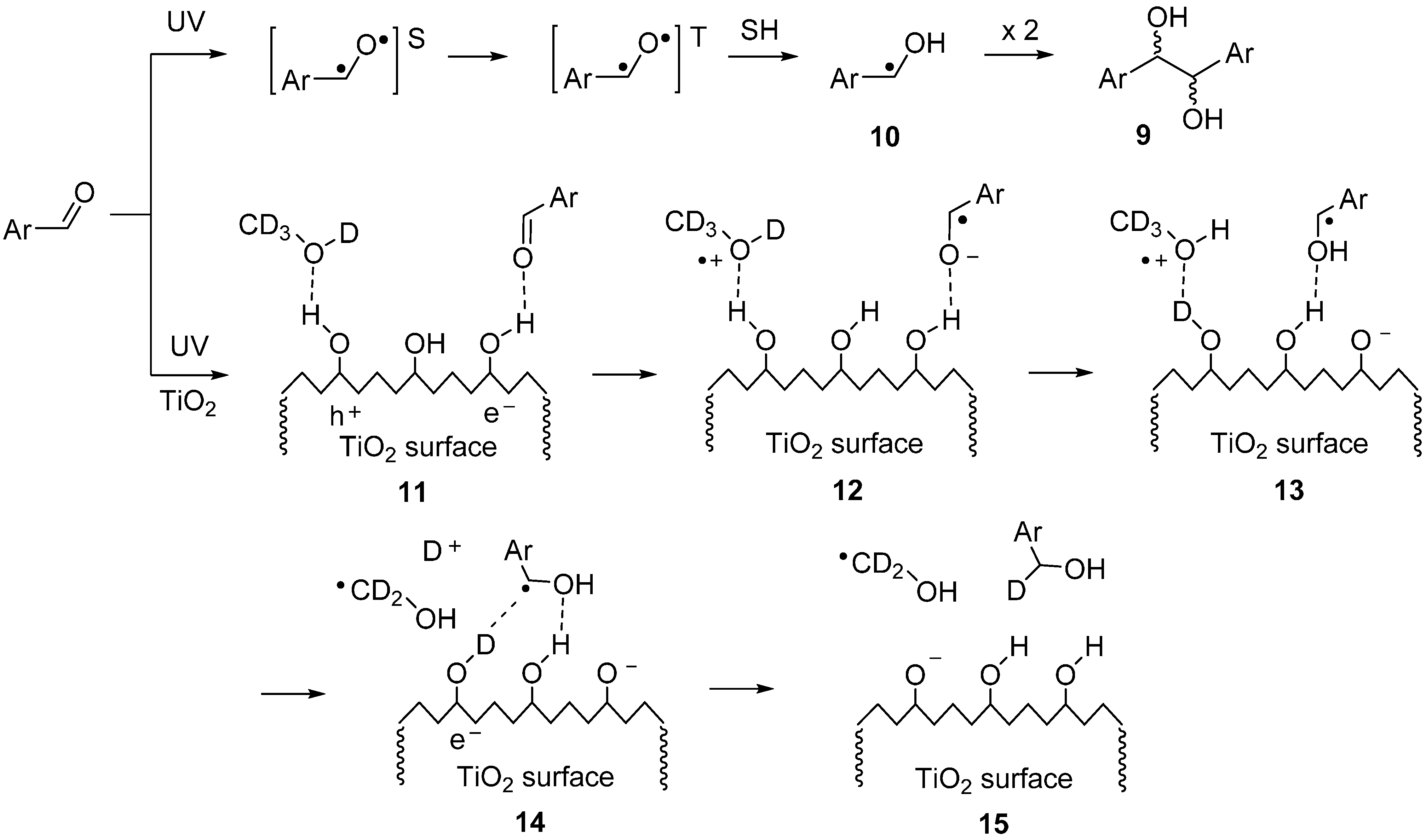{kind=link}

| Substrate | R1, R2, R3, R4 | Irrad. Time (h) | Yield of 6 (%) |
|---|---|---|---|
| 5b | Me, H, H, H | 20 | ≤5 |
| 5c | Me, Me, H, H | 20 | 17 |
| 5d | Ph, H, H, H | 20 | 9 |
| 5e | Me, Me, OMe, H | 20 | 58 |
| 5f | Me, Me, OMe, OMe | 72 | 79 |
| 5g | Me, Me, Cl, H | 20 | 19 |
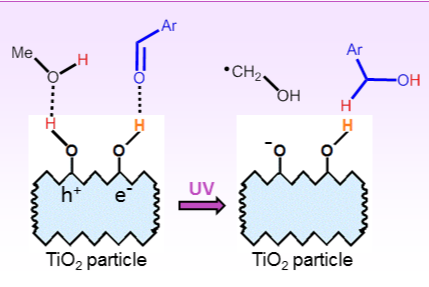
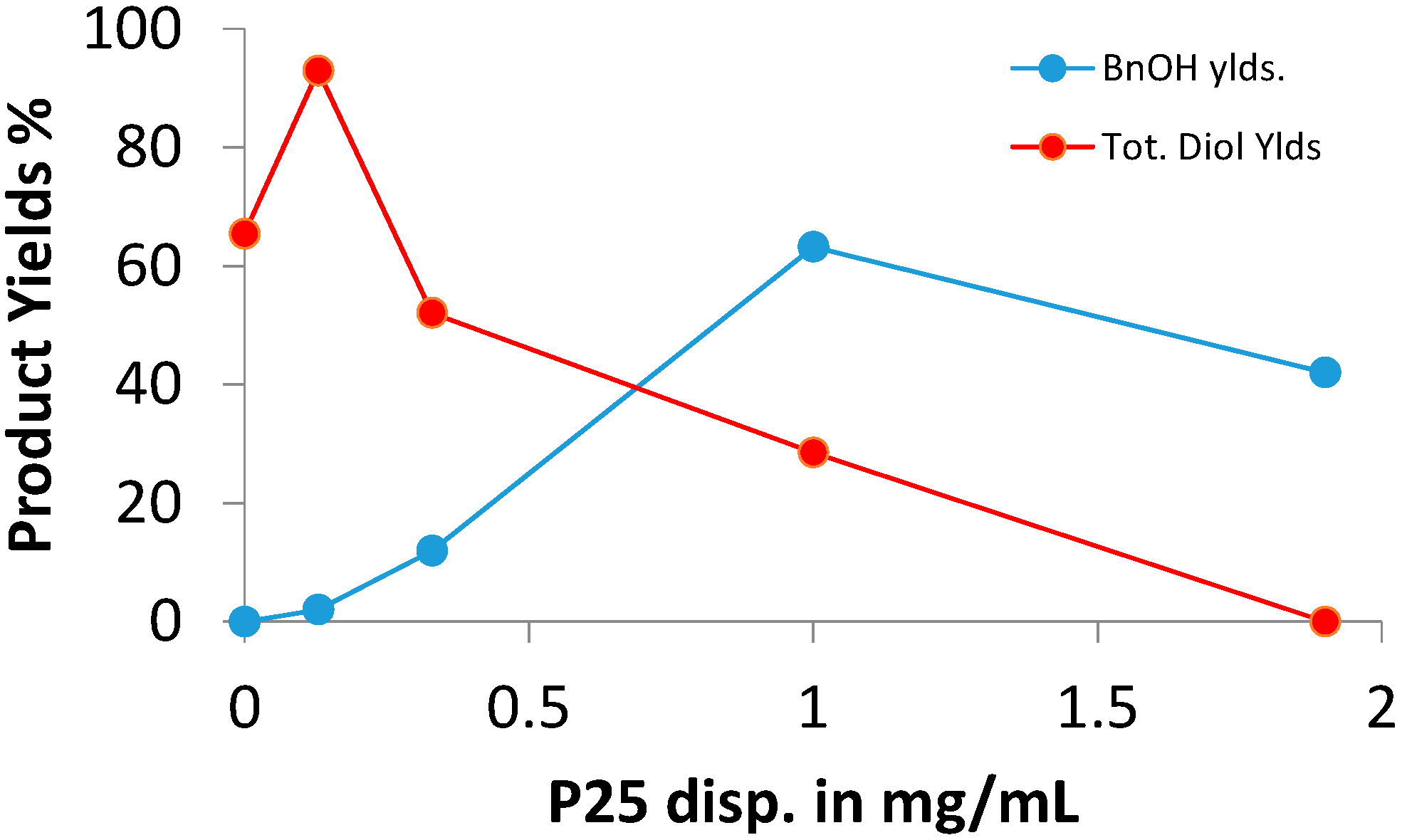
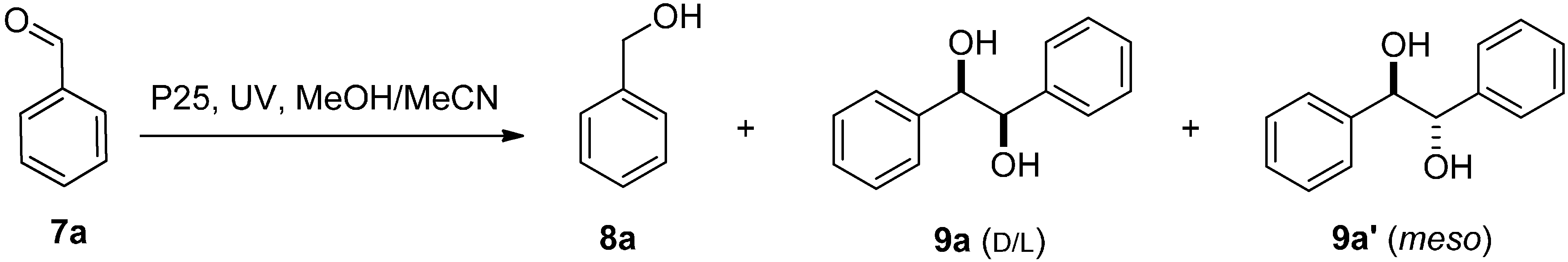
2.2. SCPC Reductions of Carbonyl Compounds


| Catalyst | BnOH (%) | Diols (9a + 9a') (%) | Ratio D/L: meso | Conversion (%) |
|---|---|---|---|---|
| P25 | 63 | 29 | 1.0 | 99 |
| Pt-P25 b | 10 | 65 | 1.1 | 98 |
| PC500 | 19 | 35 | 0.73 | 98 |
| Coated Tube c | 22 | 49 | 1.0 | 99 |
| ZnS d | 0 | 34 | 0.50 | n.d. |
| ZnS e | 0 | 65 | 0.51 | 99 |
| Carbonyl | Alcohol (%) with P25 | Alcohol (%) UVA Only | Diol (%) with P25 b | Diol (%) UVA Only |
|---|---|---|---|---|
| Benzaldehyde | 0 | 0 | 93 {75} | 65 |
| 2-Naphthaldehyde | 0 | 0 | 0 | 0 |
| 4-Me-Benzaldehyde | 7 | 0 | 43 {43} | 72 |
| 4-MeO-Benzaldehyde | 8 | 0 | 12 | 30 |
| 4-Cl-Benzaldehyde | 5 | 0 | 54 {50} | 58 |
| 4-CF3-Benzaldehyde | 4 | 0 | 58 {49} | 83 |
| 2-Thiophene-CHO | 0 | 0 | 13 | 0 |
| 2-Furan-CHO | 0 | 0 | 0 | 0 |
| 3-Benzofuran-CHO | 8 | 0 | 0 | 0 |
| Acetophenone | trace | - | 66 {92} | - |
| Carbonyl | Alcohol (%) | Diol (%) |
|---|---|---|
| benzaldehyde | 63 | 29 |
| 2-naphthaldehyde | 95 | 0 |
| 4-Me-benzaldehyde | 36 | 16 |
| 4-MeO-benzaldehyde | 56 | 0 |
| 4-Cl-benzaldehyde | 78 | 21 |
| 4-CF3-benzaldehyde | 76 | 0 |
| 2-thiophene-CHO | 63 | 29 |
| 3-benzofuran-CHO | 96 | 0 |
| Acetophenone | 0 | 6 |

3. Experimental Section
3.1. General Procedures
3.2. General Procedure for Preparation of Maleimides
3.3. General Procedure for SCPC Hydrogenation of Maleimides
3.4. General Procedure for SCPC Reduction of Carbonyl Compounds
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Linsebigler, A.L.; Lu, G.; Yates, J.T. Photocatalysis on TiO2 surfaces: Principles, mechanisms, and selected results. Chem. Rev. 1995, 95, 735–758. [Google Scholar]
- Ravelli, D.; Dondi, D.; Fagnoni, M.; Albini, A. Photocatalysis. A multi-faceted concept for green chemistry. Chem. Soc. Rev. 2009, 38, 1999–2011. [Google Scholar]
- Mills, A.; O’Rourke, C. Photocatalytic oxidation of toluene in an NMR tube. J. Photochem. Photobiol. A 2012, 233, 34–39. [Google Scholar]
- Künneth, R.; Feldmer, C.; Knoch, F.; Kisch, H. Heterogeneous photocatalysis. XIII. Semiconductor-catalyzed photoaddition of olefins and enol ethers to 1, 2-diazenes: A new route to allylhydrazines. Chem. Eur. J. 1995, 1, 441–448. [Google Scholar]
- Keck, H.; Schindler, W.; Knoch, F.; Kisch, H. Heterogeneous photocatalysis. XVI. Type B semiconductor photocatalysis: The synthesis of homoallyl amines by cadmium sulfide-catalyzed linear photoaddition of olefins and enol/allyl ethers to N-phenylbenzophenoneimine. Chem. Eur. J. 1997, 3, 1638–1645. [Google Scholar]
- Marinković, S.; Hoffmann, N. Efficient radical addition of tertiary amines to electron-deficient alkenes using semiconductors as photochemical sensitizers. Chem. Commun. 2001, 1576–1578. [Google Scholar]
- Marinković, S.; Hoffmann, N. Semiconductors as sensitizers for the radical addition of tertiary amines to electron deficient alkenes. Int. J. Photoenergy 2003, 5, 175–182. [Google Scholar]
- Marinković, S.; Hoffmann, N. Diastereoselective radical tandem addition-cyclization reactions of aromatic tertiary amines by semiconductor-sensitized photochemical electron transfer. Eur. J. Org. Chem. 2004, 3102–3107. [Google Scholar]
- Manley, D.W.; McBurney, R.T.; Miller, P.; Howe, R.F.; Rhydderch, S.; Walton, J.C. Unconventional titania photocatalysis: Direct deployment of carboxylic acids in alkylations and annulations. J. Am. Chem. Soc. 2012, 134, 13580–13583. [Google Scholar]
- Manley, D.W.; McBurney, R.T.; Miller, P.; Walton, J.C.; Mills, A.; O’Rourke, C. Titania-promoted carboxylic acid alkylations of alkenes and cascade addition-cyclizations. J. Org. Chem. 2014, 79, 1386–1398. [Google Scholar]
- Fles, D.; Vukovic, R.; Kuzmic, A.E.; Bogdanic, G.; Pilizota, V.; Karlovic, D.; Markus, K.; Wolsperger, K.; Vikic-Topic, D. Synthesis and spectroscopic evidences of N-arylmaleimides and N-aryl-2,3-dimethylmaleimides. Croat. Chem. Acta 2003, 76, 69–74. [Google Scholar]
- Mills, A. Platinization of semiconductor particles. J. Chem. Soc. Chem. Commun. 1982, 6, 367–368. [Google Scholar]
- Mills, A.; Elliott, N.; Hill, G.; Fallis, D.; Durrant, J.R.; Willis, R.L. Preparation and characterization of novel thick sol-gel titania film photocatalysts. Photochem. Photobiol. Sci. 2003, 2, 591–596. [Google Scholar]
- Hu, J.; Ren, L.; Guo, Y.; Liang, H.; Cao, A.; Wan, L.; Bai, C. Mass production and high photocatalytic activity of ZnS nanoporous nanoparticles. Angew. Chem. Int. Ed. 2005, 44, 1269–1273. [Google Scholar]
- Ciamician, G.; Silber, P. Chemical actions of light. Ber. Dtsch. Chem. Ges. 1900, 33, 2911–2913. [Google Scholar]
- Turro, N.J. Modern Molecular Photochemistry; Benjamin: NY, USA, 1978; p. 363, et seq. [Google Scholar]
- Nagwanshi, R.; Bakhru, M.; Jain, S. Photodimerization of heteroaryl chalcones: Comparative antimicrobial activities of chalcones and their photoproducts. Med. Chem. Res. 2012, 21, 1587–1596. [Google Scholar]
- McMurry, J.E. Carbonyl-coupling reactions using low-valent titanium. Chem. Rev. 1989, 89, 1513–1524. [Google Scholar]
- Okamoto, S.; He, J.-Q.; Ohno, C.; Oh-iwa, Y.; Kawaguchi, Y. McMurry coupling of aryl aldehydes and imino pinacol coupling mediated by Ti(O-i-Pr)4/Me3SiCl/Mg reagent. Tetrahedron Lett. 2010, 51, 387–390. [Google Scholar]
- Kim, Y.; Do, Y.; Park, S. Mechanistic study of half-titanocene-based reductive pinacol coupling reaction. Bull. Korean Chem. Soc. 2011, 32, 3973–3978. [Google Scholar]
- Scheffler, U.; Mahrwald, R. A one-pot cross-pinacol coupling/rearrangement procedure. Helv. Chim. Acta 2012, 95, 1970–1975. [Google Scholar]
- Deiana, C.; Fois, E.; Coluccia, S.; Martra, G. Surface structure of TiO2 P25 nanoparticles: Infrared study of hydroxy groups on coordinative defect sites. J. Phys. Chem. C 2010, 114, 21531–21538. [Google Scholar]
- Salazar, C.; Nanny, M.A. Influence of hydrogen bonding upon the TiO2 photooxidation of isopropanol and acetone in aqueous solution. J. Catal. 2010, 269, 404–410. [Google Scholar]
- Matuszak, N.; Muccioli, G.G.; Labar, G.; Lambert, D.M. Synthesis and in vitro evaluation of N-substituted maleimide derivatives as selective monoglyceride lipase inhibitors. J. Med. Chem. 2009, 52, 7410–7420. [Google Scholar]
- Bestmann, H.J.; Schade, G.; Luetke, H.; Moenius, T. Cumulated ylides. XVII. Synthesis of cyclic compounds from triphenyl[(phenylimino)ethenylidene]phosphorane and oxocarboxylic acids—A novel method for anellation. Chem. Ber. 1985, 118, 2640–2658. [Google Scholar]
- Hegasy, M.F.; Shishido, K.; Hirata, T. Asymmetric hydrogenation of the C-C double bond of 1- and 1,2-methylated maleimides with cultured suspension cells of Marchantia polymorpha. Tetrahedron-Asymmetry 2006, 17, 1859–1862. [Google Scholar]
- Walling, C.; El-Taliawi, G.M.; Zhao, C. Radical chain carriers in N-bromosuccinimide brominations. J. Am. Chem. Soc. 1983, 105, 5119–5124. [Google Scholar]
- Ito, R.; Umezawa, N.; Higuchi, T. Unique oxidation reaction of amides with pyridine-N-oxide catalyzed by ruthenium porphyrin: Direct oxidative conversion of N-acyl-l-proline to N-acyl-l-glutamate. J. Am. Chem. Soc. 2004, 127, 834–835. [Google Scholar]
- Kantak, A.A.; Potavathri, S.; Barham, R.A.; Romano, K.M.; Deboef, B. Metal-free intermolecular oxidative C-N bond formation via tandem C-H and N-H bond functionalization. J. Am. Chem. Soc. 2011, 133, 19960–19965. [Google Scholar]
- Kocz, R.; Roestamadji, J.; Mobashery, S. A convenient triphosgene-mediated synthesis of symmetric carboxylic acid anhydrides. J. Org. Chem. 1994, 59, 2913–2914. [Google Scholar]
- Stueckler, C.; Reiter, T.C.; Baudendistel, N.; Faber, K. Nicotinamide-independent asymmetric bioreduction of C=C-bonds via disproportionation of enones catalyzed by enoate reductases. Tetrahedron 2010, 66, 663–667. [Google Scholar]
- García-Muñoza, A.; Ortega-Arizmendia, A.; García-Carrillob, M.; Díazb, E.; Gonzalez-Rivasa, N.; Cuevas-Yañez, E. Direct, metal-free synthesis of benzyl alcohols and deuterated benzyl alcohols from p-toluenesulfonylhydrazones using water as solvent. Synthesis 2012, 44, 2237–2242. [Google Scholar]
- Murai, N.; Yonaga, M.; Tanaka, K. Palladium-catalyzed direct hydroxymethylation of aryl halides and trifates with potassium acetoxymethyltrifuoroborate. Org. Lett. 2012, 145, 1278–1281. [Google Scholar]
- Ashikari, Y.; Nokami, T.; Yoshida, J. Oxidative hydroxylation mediated by alkoxysulfonium ions. Org. Lett. 2012, 14, 938–941. [Google Scholar]
- Uchiyama, M.; Matsumoto, Y.; Nakamura, S.; Ohwada, T.; Kobayashi, N.; Yamashita, N.; Matsumiya, A.; Sakamoto, T. Development of a catalytic electron transfer system mediated by transition metal ate complexes: Applicability and tunability of electron-releasing potential for organic transformations. J. Am. Chem. Soc. 2004, 126, 8755–8759. [Google Scholar]
- Li, Y.G.; Tian, Q.S.; Zhao, J.; Feng, Y.; Li, M.J.; You, T.P. Asymmetric pinacol coupling of aromatic aldehydes catalyzed by a new titanium Schiff base complex. Tetrahedron-Asymmetry 2004, 15, 1707–1710. [Google Scholar]
- Wei, Y.; Xue, D.; Lei, Q.; Wang, C.; Xiao, J. Cyclometalated iridium complexes for transfer hydrogenation of carbonyl groups in water. Green Chem. 2013, 15, 629–634. [Google Scholar]
- Lacour, J.; Londez, A. Diastereoselective synthesis of C2-symmetric hexacoordinated phosphate anions (Hyphats) with predetermined chirality from 1,2-diaryl-ethane-1,2-diols. J. Organomet. Chem. 2002, 643, 392–403. [Google Scholar]
- Chen, Y.Z.; Ni, C.W.; Teng, F.L.; Ding, Y.S.; Lee, T.H.; Ho, J.H. Construction of polyaromatics via photocyclization of 2-(fur-3-yl)ethenylarenes, using a 3-furyl group as an isopropenyl equivalent synthon. Tetrahedron 2014, 70, 1748–1762. [Google Scholar]
- Wan, W.C.; Chen, W.; Liu, L.X.; Li, Y.; Yang, L.J.; Deng, X.Y.; Zhang, H.B.; Yang, X.D. Synthesis and cytotoxic activity of novel hybrid compounds between 2-alkylbenzofuran and imidazole. Med. Chem. Res. 2014, 23, 1599–1611. [Google Scholar]
- Sample Availability: Samples of some compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Manley, D.W.; Buzzetti, L.; MacKessack-Leitch, A.; Walton, J.C. Hydrogenations without Hydrogen: Titania Photocatalyzed Reductions of Maleimides and Aldehydes. Molecules 2014, 19, 15324-15338. https://doi.org/10.3390/molecules190915324
Manley DW, Buzzetti L, MacKessack-Leitch A, Walton JC. Hydrogenations without Hydrogen: Titania Photocatalyzed Reductions of Maleimides and Aldehydes. Molecules. 2014; 19(9):15324-15338. https://doi.org/10.3390/molecules190915324
Chicago/Turabian StyleManley, David W., Luca Buzzetti, Andrew MacKessack-Leitch, and John C. Walton. 2014. "Hydrogenations without Hydrogen: Titania Photocatalyzed Reductions of Maleimides and Aldehydes" Molecules 19, no. 9: 15324-15338. https://doi.org/10.3390/molecules190915324