Combinatorial Libraries on Rigid Scaffolds: Solid Phase Synthesis of Variably Substituted Pyrazoles and Isoxazoles

Pharmaceuticals Division, Core Drug Discovery Technologies, Ciba-Geigy, CH-4002 Basel, Switzerland
*
Author to whom correspondence should be addressed.
Molecules 1997, 2(1), 17-30; https://doi.org/10.3390/jan97p5
Submission received: 22 September 1996
/
Accepted: 17 January 1997
/
Published: 29 January 1997
Abstract
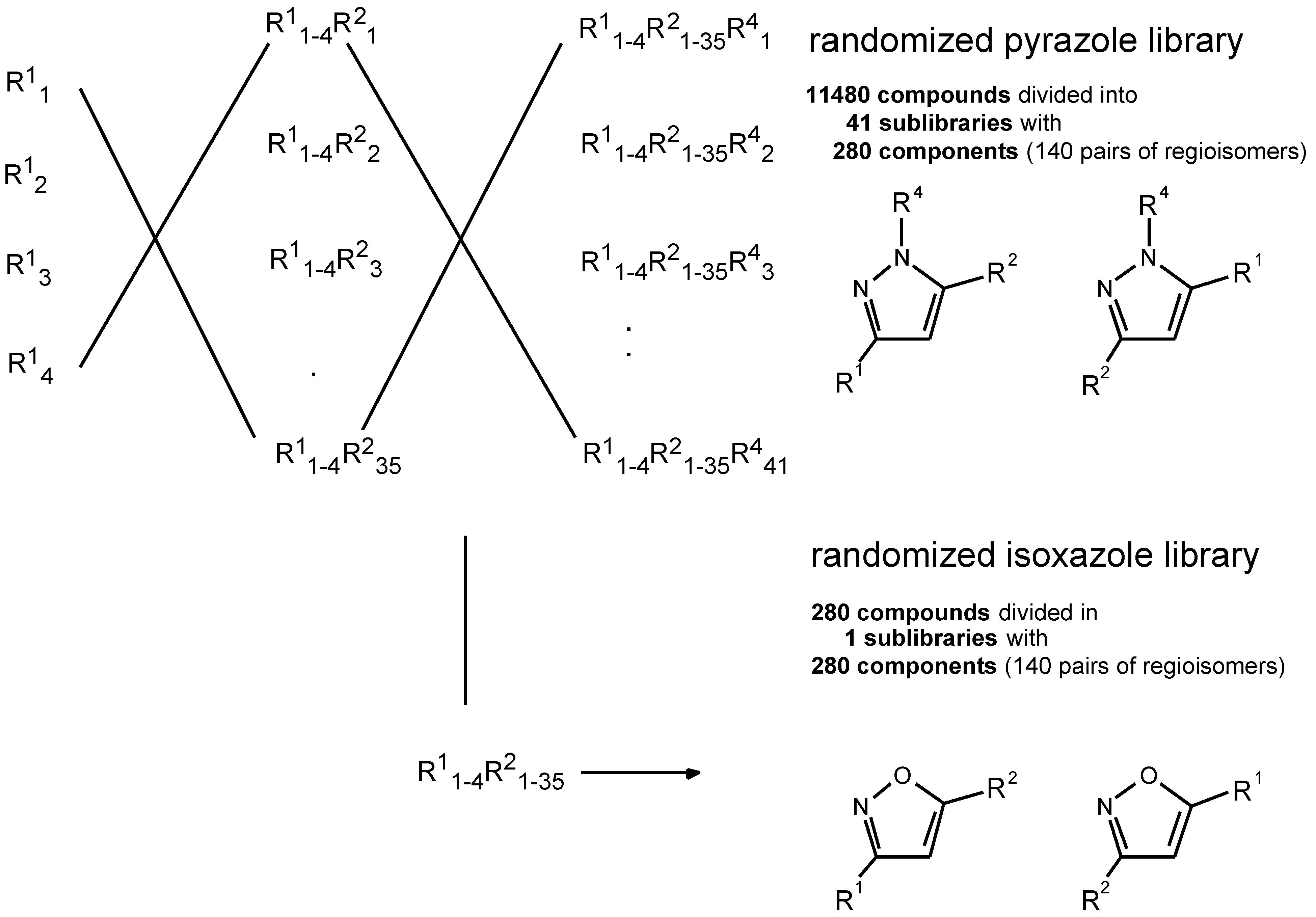
:The synthesis of combinatorial compound libraries has become a powerful lead finding tool in modern drug discovery. The ability to synthesize rapidly, in high yield, new chemical entities with low molecular weight on a solid support has a recognized strategic relevance (“small molecule libraries”). We designed and validated a novel solid phase synthesis scheme, suitable to generate diversity on small heterocycles of the pyrazole and isoxazole type. Appropriate conditions were worked out for each reaction, and a variety of more or less reactive agents (building blocks) was utilized for discrete conversions, in order to exploit the system’s breadth of applica- bility. Four sequential reaction steps were validated, including the loading of the support with an acetyl bearing moiety, a Claisen condensation, an α-alkylation and a cyclization of a β-diketone with monosubstituted hydrazines. In a second stage, the reaction sequence was applied in a split and mix approach, in order to prepare a combina- torial library built-up from 4 acetyl carboxylic acids (R1), 35 carboxylic esters (R2) and 41 hydrazines (R4) (and 1 hydroxylamine) to yield a total of 11,760 compounds divided into 41 pyrazole sublibraries with 140 pairs of regioisomers and 1 isoxazole sublibrary of equal size.
Introduction
The synthesis of combinatorial compound libraries has be- come a powerful lead finding tool in modern drug discovery [1]. The ability to synthesize rapidly, in high yield, new chemi- cal entities with low molecular weight on a solid support has a recognized strategic relevance (“small-molecule libraries”). The solid phase synthesis format greatly simplifies work-up procedures after each reaction and enables the application of combinatorial principles following the split-and-mix concept [2].
It is our aim to develop synthetic schemes on solid phase, which are broadly applicable to the generation of molecular diversity by combinatorial methods. We are particularly in-
![Molecules 02 00017 g001]() terested in the incorporation of a wide selection of commer- cially available building blocks and in the access to new origi- nal compounds by combination of known fragments. We re- port here on the design, validation and application of a novel solid phase strategy, suitable to create diversity on small heterocycles of the pyrazole and isoxazole type.
terested in the incorporation of a wide selection of commer- cially available building blocks and in the access to new origi- nal compounds by combination of known fragments. We re- port here on the design, validation and application of a novel solid phase strategy, suitable to create diversity on small heterocycles of the pyrazole and isoxazole type.
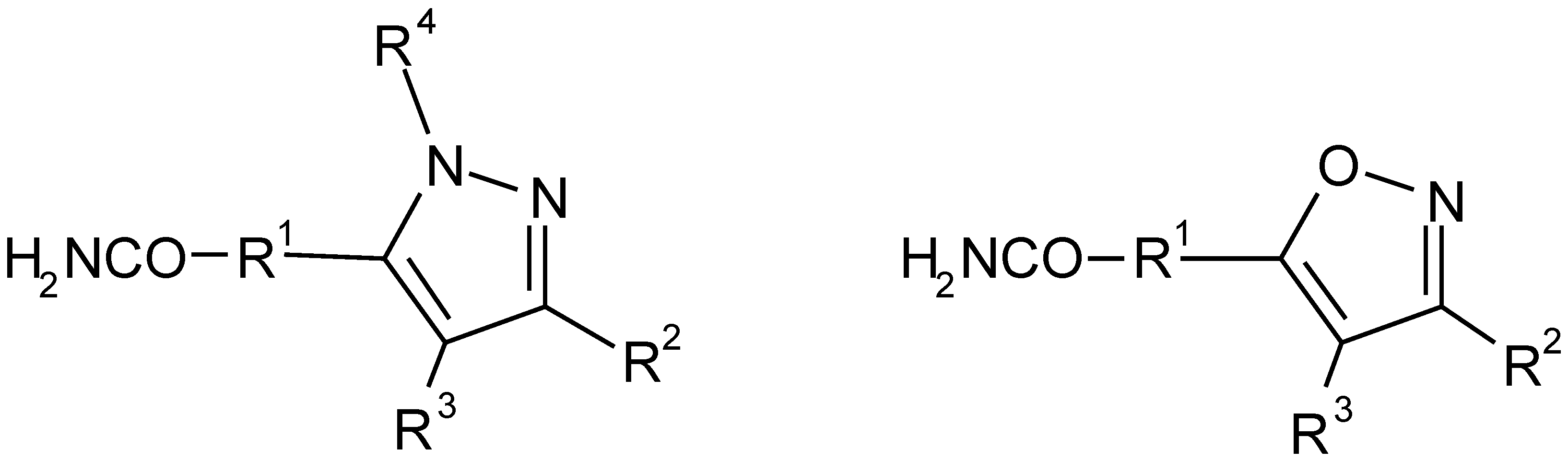
Figure 1.
Pursued substitution pattern on the pyrazole and isoxazole scaffolds.
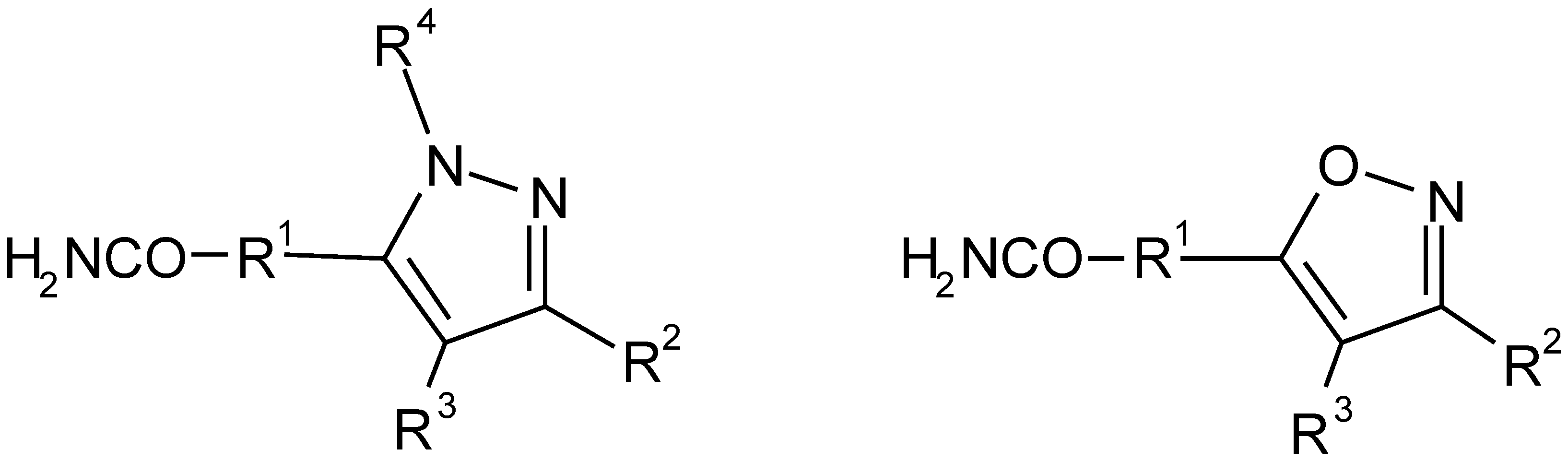
Our investigations concentrated, in the first stage, on the performance of discrete steps. For each reaction, appropriate conditions were worked out and a variety of more or less reactive agents (building blocks) was utilized, in order to explore the system’s breadth of applicability (validation). A concise account of the chemistry performed, and the results obtained, was recently published [3].
In the second stage, the reaction sequence was applied in a split-and-mix approach in order to prepare a combinatorial library of 11760 variably substituted heterocyclic compounds (Figure 1).
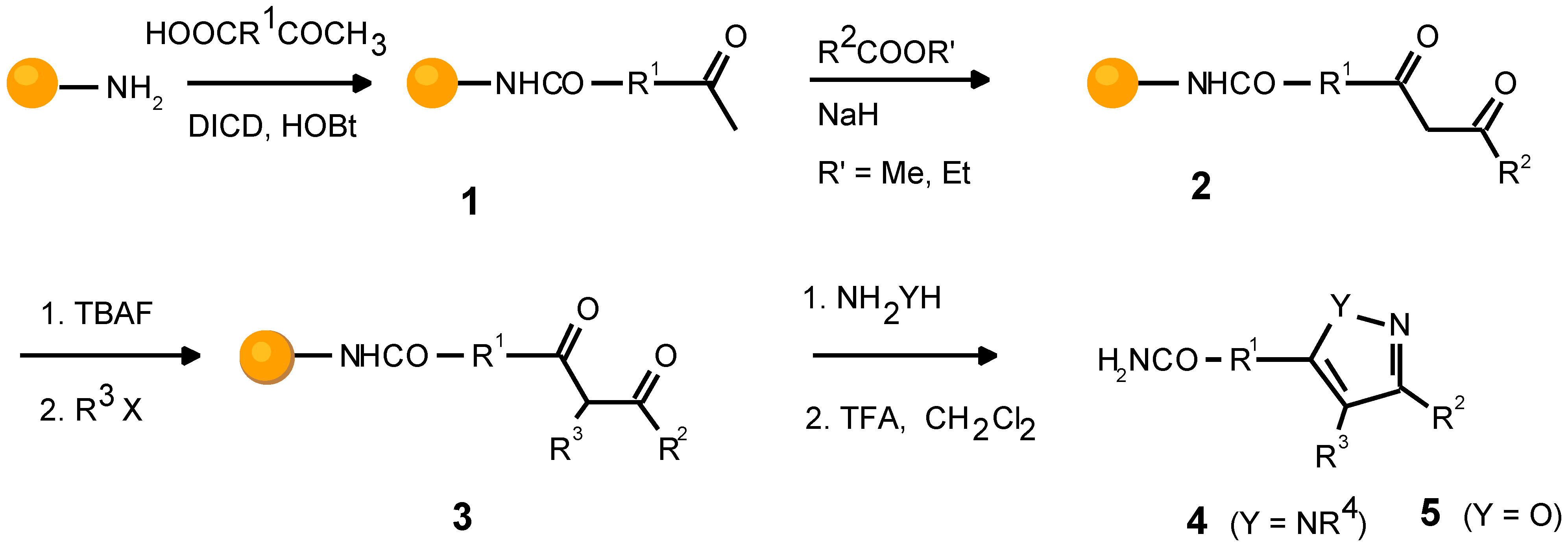
The sequence of reactions utilized comprises 4 steps, consisting of: a) the loading of the support with an acetyl bearing moiety; b) a Claisen condensation; c) an α-alkyla- tion; d) a cyclization of a β-diketone with monosubstituted hydrazines (Scheme 1).
Results and Discussion
Validation of reaction sequences
The objective of our validation study, by determining the scope and limitation of each reaction in the scheme, was to reach the necessary reliability level to obtain quality prod- ucts in the absence of purification steps, and without the pros- pect of having to characterize each library component with individual analyses. At times, rather complex mixtures of over 100 components per sublibrary arise from split-and-mix protocols. Our intention was to utilize the approach for broad lead finding with combinatorial libraries of original, semi- rigid molecules built-up from simple, commercially avail- able building blocks. The quality standards of complicated synthetic mixtures are critically dependent on the diligence of the previously run ‘chemical rehearsals’.
We handled the process analysis in two different steps: validation of all chemical steps by model syntheses of par-
![Molecules 02 00017 sch001]()
![Molecules 02 00017 g002]() ticular compounds (with building blocks that are representa- tive of larger classes of analogs), followed by spectroscopic analysis of a number of library mixtures. In practice, after each transformation that was part of the validation on the solid support, the reaction products were cleaved, identified by MS, and the purity was defined by HPLC. To that end, Table 1, Table 2, Table 3, Table 4 and Table 5 list the residues of the reagents and products, of which the conversions on solid phase were monitored by LCMS of starting materials, as well as product samples, cleaved from the solid support into solution. Occasionally, compounds were prepared on a larger scale and were char- acterized by NMR, after cleavage from the resin. The capa- bility for high resolution structural elucidations of com- pounds, while covalently attached to the solid support, was reported in the literature [4,5] but not available to us in time for this study.
ticular compounds (with building blocks that are representa- tive of larger classes of analogs), followed by spectroscopic analysis of a number of library mixtures. In practice, after each transformation that was part of the validation on the solid support, the reaction products were cleaved, identified by MS, and the purity was defined by HPLC. To that end, Table 1, Table 2, Table 3, Table 4 and Table 5 list the residues of the reagents and products, of which the conversions on solid phase were monitored by LCMS of starting materials, as well as product samples, cleaved from the solid support into solution. Occasionally, compounds were prepared on a larger scale and were char- acterized by NMR, after cleavage from the resin. The capa- bility for high resolution structural elucidations of com- pounds, while covalently attached to the solid support, was reported in the literature [4,5] but not available to us in time for this study.
Scheme 1.
Sequence of reactions generating molecular diversity.

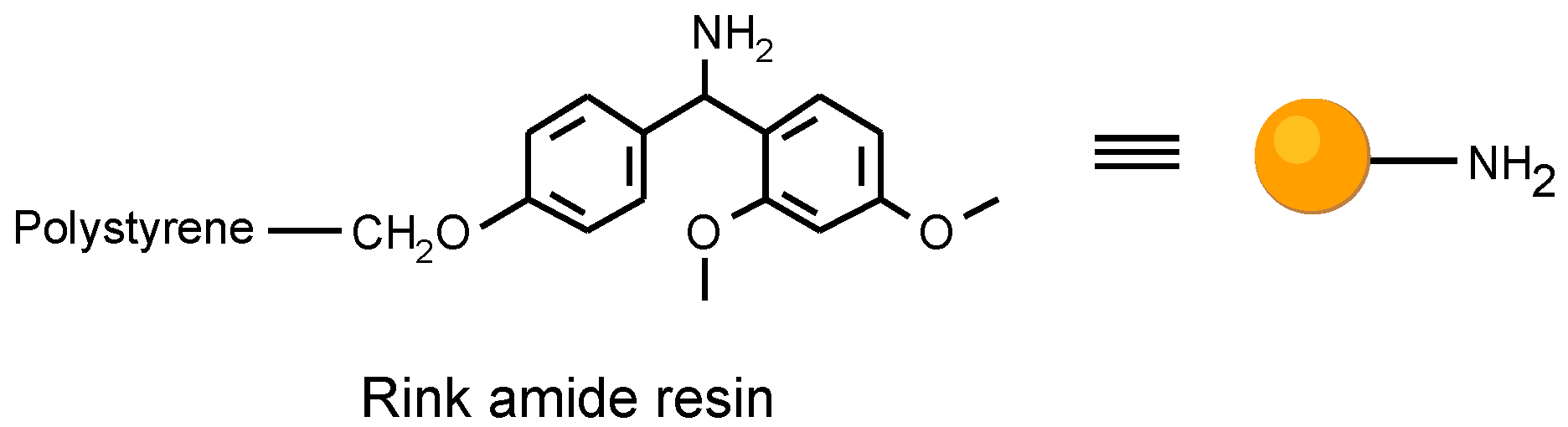
Figure 2.
The Rink amide resin utilized as solid support.
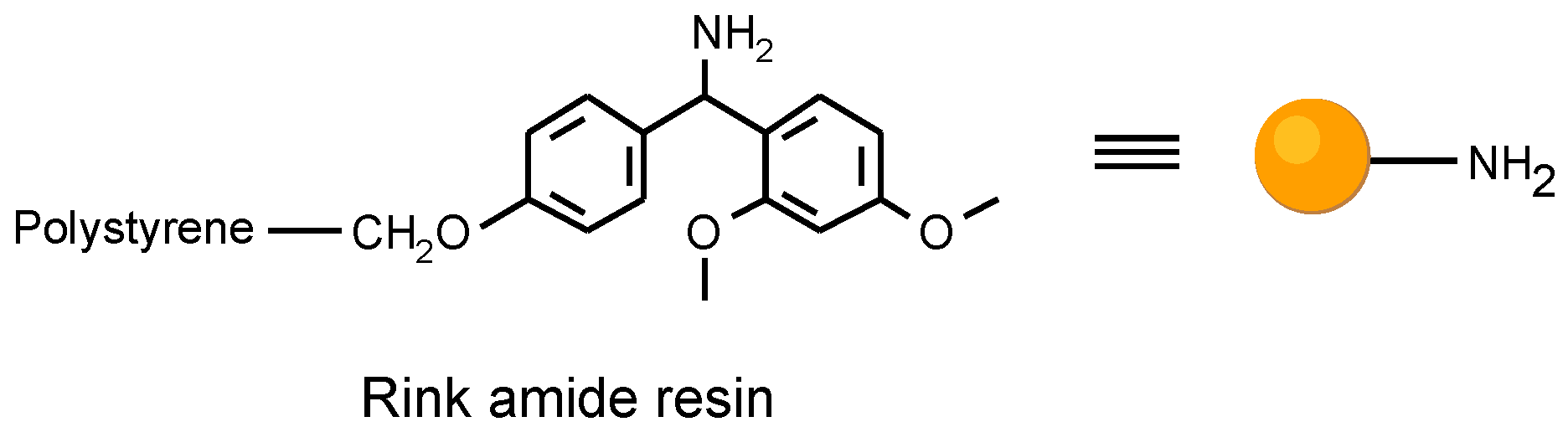
In planning the approach, we had to consider the resin matrix and the linkage to the solid support for the organic solid phase synthesis. The attachment to the solid support had to be compatible with the reaction sequence, and cleav- able after the template synthesis. We utilized a common poly- styrene resin with a trialkoxybenzhydryl linker (Rink amide resin) [6].
The fully substituted pyrazole scaffold is built from four different building block reagent classes, namely from car- boxylic acids with an acetyl function, carboxylic acid esters, alkylating agents, and monosubstituted hydrazines. A four step procedure with four different reagents leads to functionalities R1, R2, R3, and R4. However, the formation of regioisomers in the last reaction doubles the number of mol- ecules in a library.
In the first validation step the solid support was loaded with the R1 component bearing the acetyl function. We ob- served quantitative transformation, after one hour, for vari- ous heteroaromatic derivatives 1a, 1b, 1c, and 1e, unless ortho substituted bifunctional derivatives like o-acetophenone and acetylphthalanilidic acid (1d) were used (Table 1), which undergo ring closure side reactions [7]. After each transfor- mation the product was cleaved by treatment with 95% TFA and the quality was checked by HPLC (Table 1). The purity of each component was higher than 95 % throughout. Con- sidering that traces of impurity were caused by partial break- down products of the linker, it can be assumed that the com- pounds on the resin were of excellent quality.
For the Claisen condensation, optimization of the reac- tion protocol with the prototypic aromatic ester ethyl ben- zoate ( 2a ) led to conditions, which also ensure that desactivated benzoates, e.g. 2b, 2h and heteroaromatic car- boxylic esters 2i-k with widely differing electronic proper- ties condense without appreciable formation of side prod- ucts. As expected, carboxylic esters with α-hydrogens (2g) are unsuited. Since there are more than one hundred mono- substituted aromatic carboxylic acid esters commercially available, this limitation is acceptable, until a strategy suit- able to incorporate aliphatic residues is worked out. Intrigu- ingly, the employed reaction conditions gave rise to partial reduction of a nitroaromatic building block (2c). Also weakly acidic heteroaromatic compounds cannot be applied. The ethyl indole carboxylate (2k) forms the indolyl anion, which lacks sufficient electrophilicity. Noticeably, the series of suc- cessful conversions to the diketone included a bifunctional building block and a component with an additional electrophilic center (2e and 2f). The reactivity of further aro- matic carboxylic esters could be estimated by the Hammett equation.
In general, β-diketones undergo α-alkylationin the pres- ence of the phase transfer catalyst tetra-n-butylammonium
![Molecules 02 00017 i001]()
![Molecules 02 00017 i002]() hydrogen sulfate in water/methylene chloride. Substrate 2a was examined under these conditions. We used NaOH as the base and EtI as alkylating agent. After 16h at room temperature no starting material was detected. Although α-alkylation was the major product, we identified O-alkylation and various side products.
hydrogen sulfate in water/methylene chloride. Substrate 2a was examined under these conditions. We used NaOH as the base and EtI as alkylating agent. After 16h at room temperature no starting material was detected. Although α-alkylation was the major product, we identified O-alkylation and various side products.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Building blocks used for R1 validation and the corresponding values of loading capacity ob- tained on the resin.
Table 1.
Building blocks used for R1 validation and the corresponding values of loading capacity ob- tained on the resin.

Table 2.
Claisen condensation to β-diketone 2a-l (R1= 4-Ph).

For the α-alkylation step, the best results were obtained in the presence of TBAF, which shields the oxygen atoms of the β-dicarbonyl intermediate by complexation, hence inhibiting O-alkylation as a side reaction and furthermore increasing the nucleophilicity of the compound. Under strictly anhydrous conditions, yields of 90% in C-monoalkylated product can be obtained. To expand the diversity, we also tested alkylating agents other than the simple alkyl iodides described in analogous solution chemistry [8]. Ethyl bromoacetate (3c) and allyl bromide (3f) reacted without side reactions. The failure with benzyl bromide (3b) was rather unexpected. Iodoacetonitrile (3d) and bromoacetophenone (3e) were unsatisfactory.
A large number of nitrogen containing heteroaromatic
![Molecules 02 00017 i003]() compounds are commercially available for the R2 position in the previous Claisen condensation. Various side products could arise upon alkylation of these compounds. Experimentally, we confirmed that the alkylation step is incompatible with the presence of acidic or basic heteroaromatic R1 and R2 residues: with the phenyl pyridine diketone 2k several side products were observed upon alkylation with the alkylating agents 3a, 3c, 3d and 3f. Therefore, for the preparation of combinatorial libraries, a strategy that skips the alkylation step enables a broader choice of building blocks for the previous Claisen condensation, by allowing e.g. the inclusion of N-heterocyclic esters as an alternative source of diversity.
compounds are commercially available for the R2 position in the previous Claisen condensation. Various side products could arise upon alkylation of these compounds. Experimentally, we confirmed that the alkylation step is incompatible with the presence of acidic or basic heteroaromatic R1 and R2 residues: with the phenyl pyridine diketone 2k several side products were observed upon alkylation with the alkylating agents 3a, 3c, 3d and 3f. Therefore, for the preparation of combinatorial libraries, a strategy that skips the alkylation step enables a broader choice of building blocks for the previous Claisen condensation, by allowing e.g. the inclusion of N-heterocyclic esters as an alternative source of diversity.
Table 3.
aα-Alkylation of diketone 2a (R1 = 4-Ph, R2 = Ph).

The ring closure to form a heterocyclic scaffold progressed quantitatively with hydrazines 4a-c and with hydroxylamine (5a).
For further validation the reaction of structurally and electronically diverse substrates were synthesized on a larger scale, so that these compounds could be isolated and identified by NMR.
Compound 4d was prepared from hydralazine with the intention of exploring the limits of using building blocks with unfavourable electronic and steric properties (see Scheme 2). The reaction was sluggish and after one day, only traces of both regioisomers 4d were detectable. A 20% conversion yield was reached after four days. The presence of the unsubstituted analog 4a indicated that the non-alkylated diketone precursor ( a residual impurity) is far more reactive than the alkylated intermediate.
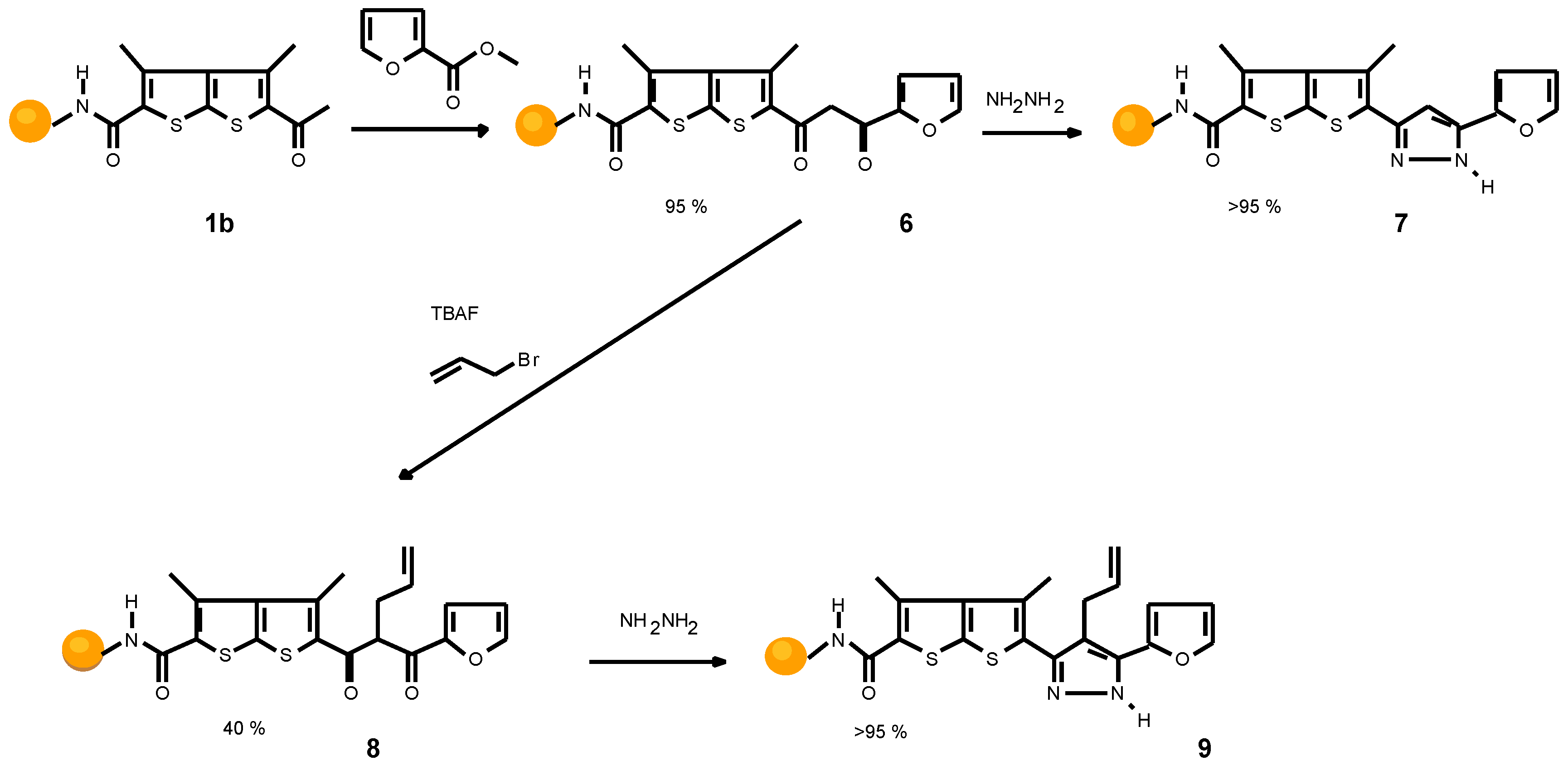
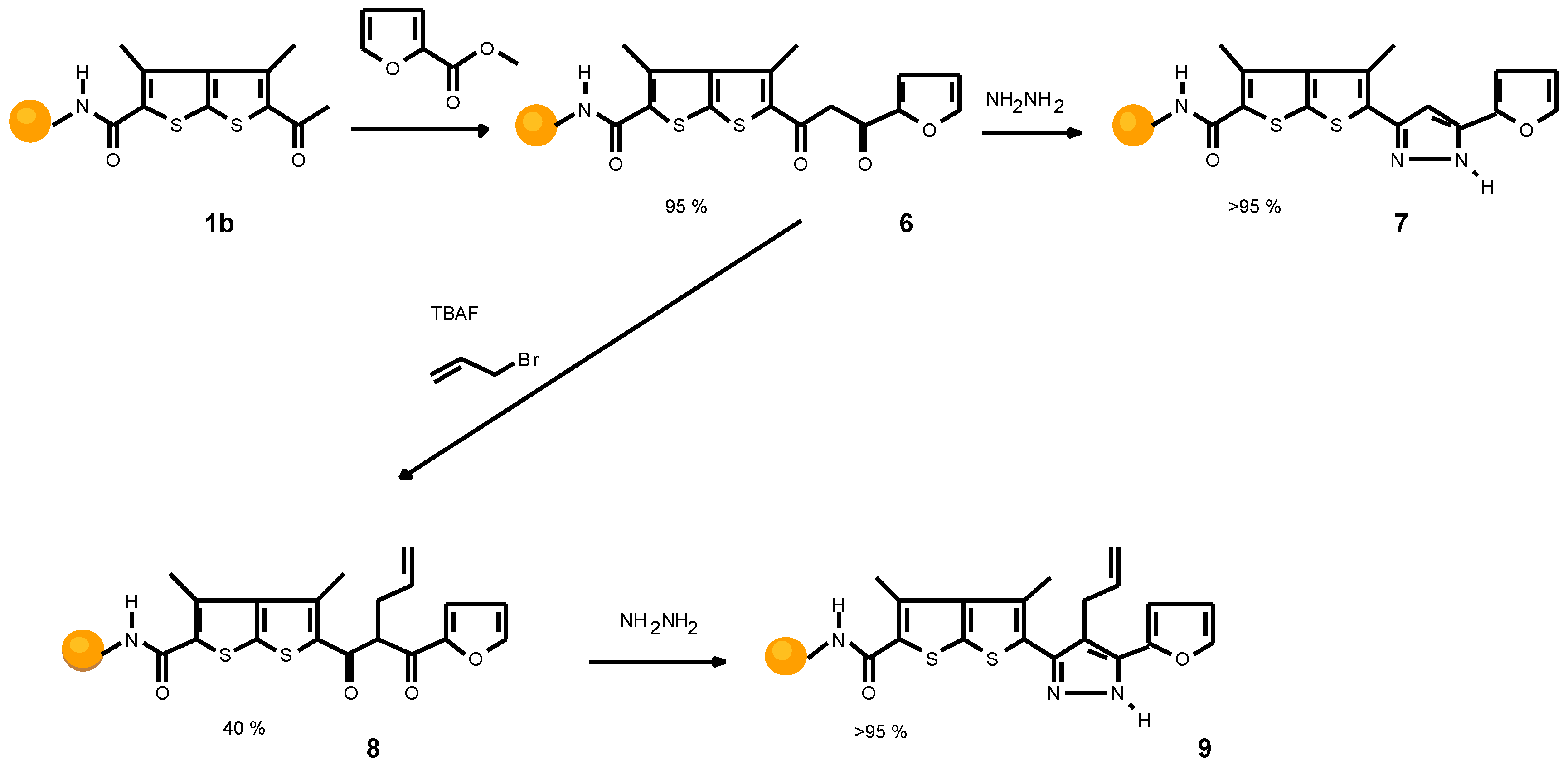
Scheme 3 outlines the synthesis of a furyl pyrazole (9). A quantitative transformation was observed for the coupling procedure and the following Claisen condensation which af- fords diketone 6. Unfortunately, alkylation of 6 leads to the alkylated diketone only in 40% yield. Bis-alkylated prod- ucts and also a small amount of starting material were iden- tified. In order to isolate and characterize the final product, hydrazine was chosen as cyclization reagent, to avoid the formation of regioisomers. Compound 9 was formed with- out side products.
The difficulties in predicting the influence of different substituents on the alkylation together with the broad devia- tions in yield prompted us to dispense with the alkylation for the preparation of a first, diverse library.
Our strategy was then to study other reagents which could be used in the cyclization reaction of non-alkylated diketones. We found (Table 5) that the twofold nucleophilic character of monosubstituted hydrazines, including desactivated hydrazines, and also hydroxyl amine, which leads to isoxazoles, allows them to react with each carbonyl group of a 1,3-diketone in satisfactory yield. It was found that the con- version of reactive hydrazines, like methyl hydrazine (4f) or benzylhydrazine (4h), is nearly quantitative, which means that there is no (or only a trace of) starting material and no impurities caused by the cyclization reaction. Also for aro- matic hydrazines with strong electron withdrawing groups,
![Molecules 02 00017 sch002]()
![Molecules 02 00017 i004]()
![Molecules 02 00017 sch003]() like pyrimidines or even phthalazines, we observed a satis- factory transformation. As expected, acid hydrazides like 4i are unsuited. The major product of this reagent is the N- unsubstituted pyrazole 4e, because the 1-acylpyrazoles are cleaved by nucleophilic attack.
like pyrimidines or even phthalazines, we observed a satis- factory transformation. As expected, acid hydrazides like 4i are unsuited. The major product of this reagent is the N- unsubstituted pyrazole 4e, because the 1-acylpyrazoles are cleaved by nucleophilic attack.
Scheme 2.
Introduction of the bulky phthalazine substituent.
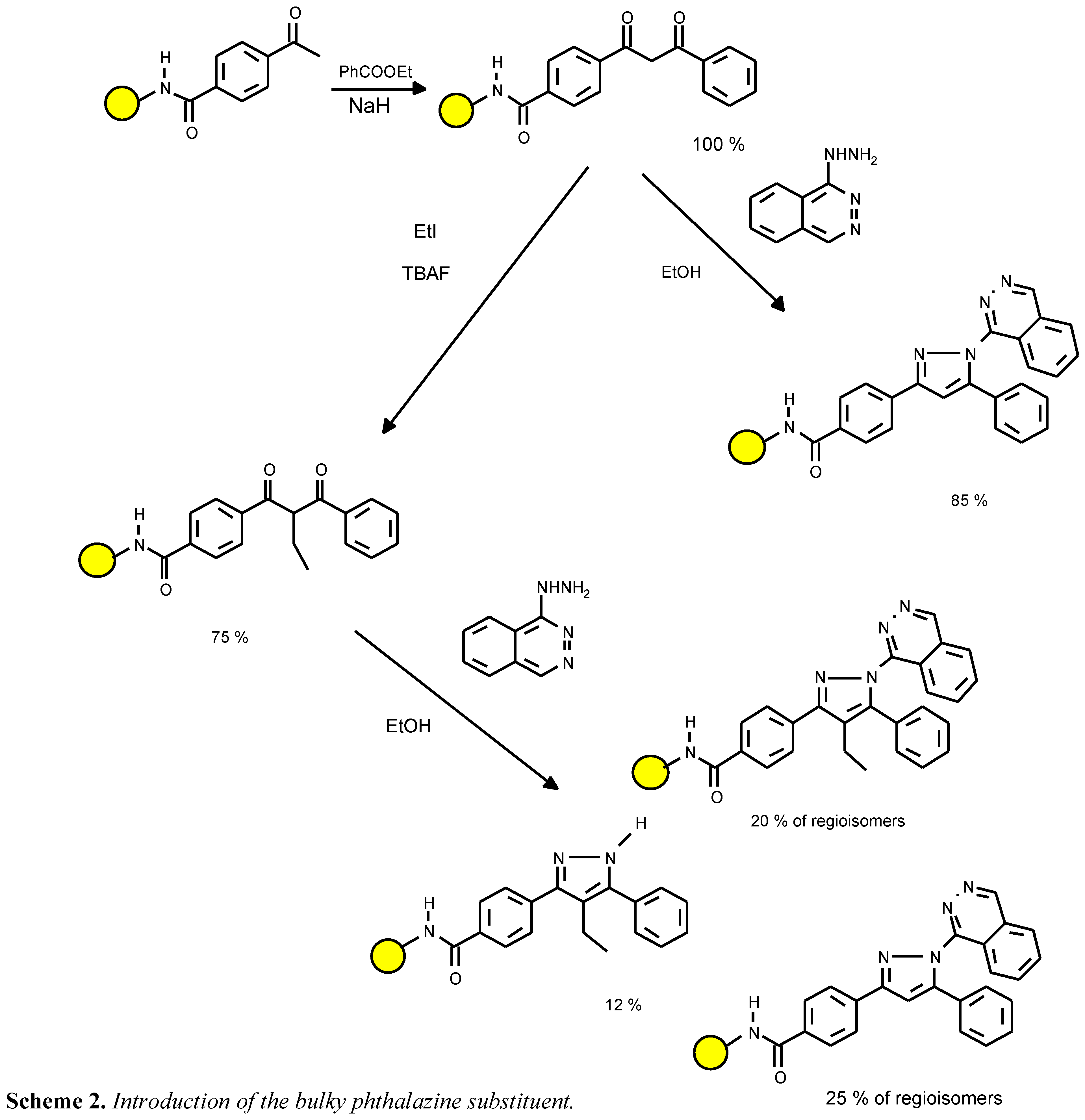
Table 4.
Formation of pyrazoles 4a-4d (R1 = 4-Ph, R2 = Ph, R3 = Et) and isoxazole 5a (R1 = 4-Ph, R2 = Ph, R3 = Et).
Table 4.
Formation of pyrazoles 4a-4d (R1 = 4-Ph, R2 = Ph, R3 = Et) and isoxazole 5a (R1 = 4-Ph, R2 = Ph, R3 = Et).

Scheme 3.
Synthesis of furyl pyrazole 9.
In the course of our investigations we found that, for rea- gents with low solubilities, saturated solutions are often sufficent to drive the reaction to completion. As many rea- gents as possible, particularly those with questionable reac- tivity (e.g. for steric or electronic reasons), should be tested in model reactions before they are positively selected for a library preparation, so as to minimize unpredictable results.
![Molecules 02 00017 i005]() For example it was found that some aromatic hydrazines contain the far more reactive hydrazine as an impurity. With a 5% contamination and usage of a 20-fold excess of the actual reagent, the reaction product obtained is almost ex- clusively the N-unsubstituted pyrazole. We found that the addition of acetylacetone is an excellent scavenger for hydrazine impurities. Another reason to examine the reac- tivity of reagents experimentally is to eliminate those rea- gents, which are not soluble or which decompose under the specific reaction conditions. It is known that hydrazines tend to decompose and loose nitrogen if kept at high temperature for prolonged periods of time.
For example it was found that some aromatic hydrazines contain the far more reactive hydrazine as an impurity. With a 5% contamination and usage of a 20-fold excess of the actual reagent, the reaction product obtained is almost ex- clusively the N-unsubstituted pyrazole. We found that the addition of acetylacetone is an excellent scavenger for hydrazine impurities. Another reason to examine the reac- tivity of reagents experimentally is to eliminate those rea- gents, which are not soluble or which decompose under the specific reaction conditions. It is known that hydrazines tend to decompose and loose nitrogen if kept at high temperature for prolonged periods of time.
Table 5.
Formation of pyrazoles 4e-n (R1 = 4-Ph, R2 = Ph, R3 = H).

The collected data on the scope and limitations of the various chemical reactions, that are part of the envisaged scheme for molecular diversity generation, provided the nec- essary information for a diligent planning of combinatorial libraries according to the split-and-mix principle [2]. The preparation of a first library with more than 10,000 compo- nents is described in the following sections.
Preparation of the combinatorial library
A list of building blocks, used for the preparation of the com- binatorial library, is provided in the Appendices.
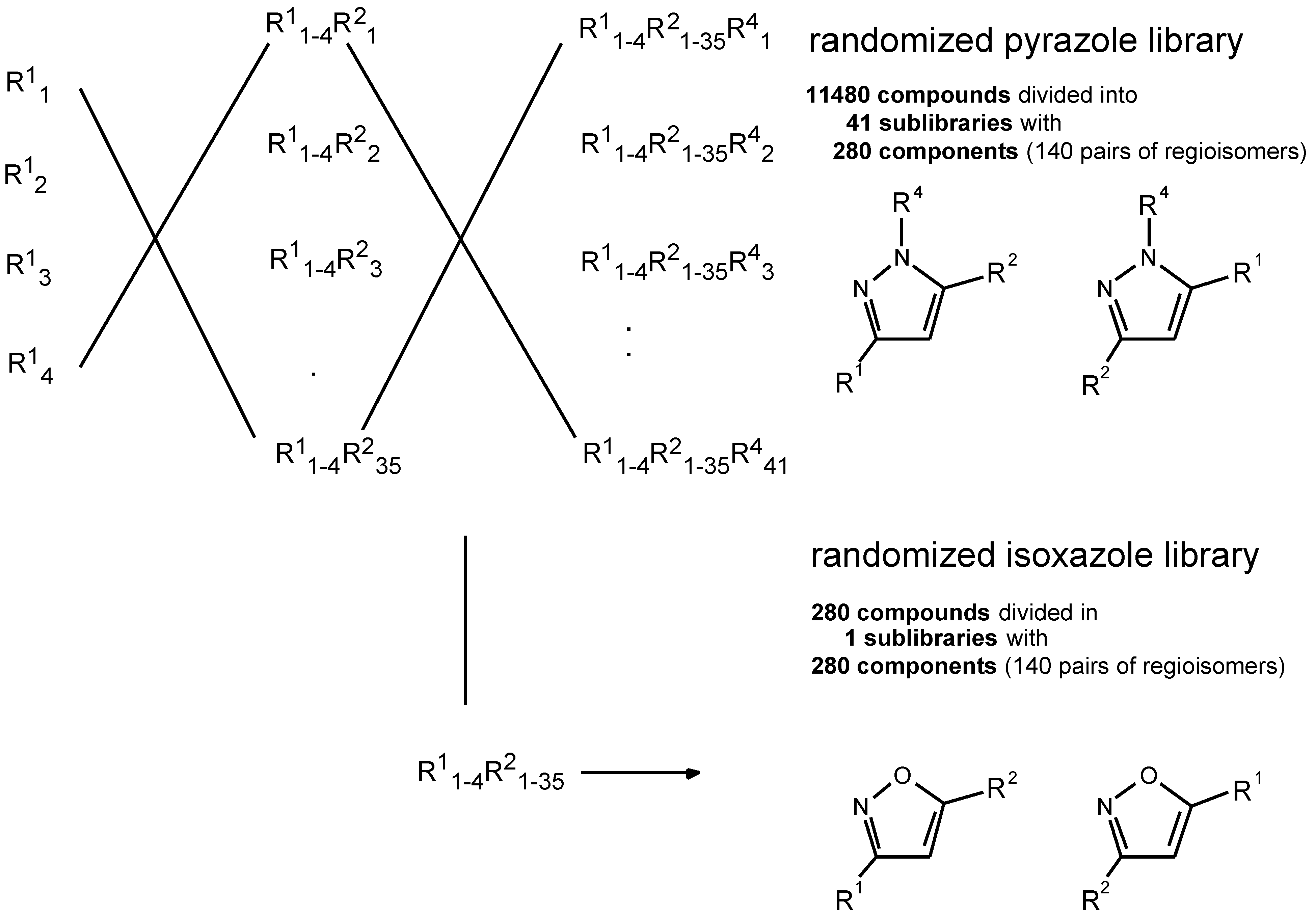
In a first step the carboxylic acids bearing the acetyl moiety were coupled to the polystyrene resin (functionalized with the Rink amide linker) in separate vessels. The four portions were then washed, mixed and redistributed into 35 vessels, where each portion reacted with a distinct R2 rea- gent, i.e. a carboxylic acid ester. This gave rise to a randomized R1 and a fixed R2 position. The beads were again mixed, and then divided into 42 equal portions, of which 41 were reacted with monosubstituted hydrazines R4. This pro- duced a pyrazole library with (potentially) 11,480 compounds, divided into 41 sublibraries with 280 compounds. The re- maining last portion of the beads was reacted with hydroxy- lamine to an isoxazole library comprising 280 compounds. The bulk of materials (approx. 15 µmol of each sublibrary) was kept linked to the solid beads for storage. From aliquots of the beads, the necessary amount of test material was cleaved into solution, as mixtures of partially randomized compounds (with one defined residue) for lead finding in functional assay systems.
The quality assessment of our library relied mostly on the validation work previously run (by individual test syn- theses) on the chemical reactions involved. To a limited ex- tent, a sampling of individual beads was carried out and the cleaved material was subjected to MS analysis in order to confirm the presence of only one major component (one bead one compound). Analyses of the sublibraries with LC-MS turned out to be difficult to interpret. From the 140 pairs of regioisomers in each sublibrary, some have the same mass or differ only by one Dalton. Another complication was the broad range of lipophilicity with the consequent difficulty of separating all the components in our reverse phase LC-sys- tem.
In the course of an iterative unrandomization [9] applied to one of our drug discovery targets, we had the opportunity to verify easily the quality of the prepared daughter libraries (with two defined residues and the randomized R1 position), since only four pairs of regioisomers are expected at that stage of the deconvolution (data not shown).
Conclusion
The methodology we reported here, is a validated process that leads rapidly to heterocyclic structures of the pyrazole and isoxazole type. The synthesis of individual model com- pounds was part of the concept to rehearse the reaction se- quences to an extent that acceptable quality of products is achieved in a reliable manner, also in the production phase of complex mixtures, where thorough analytical characteri- zation remains elusive with the currently available methods.
The specific value of our method resides in the way func- tional groups and potential pharmacophores are connected and presented in space, i.e. on a small and conformationally constrained template, as opposed to chain-like and flexible oligomers. Moreover, the general utility of the approach could be exploited to form additional ring types (e.g. pyrimidines from amidines) if the diketone intermediates were subjected to cyclization with other reagents bearing two nucleophilic
![Molecules 02 00017 sch004]() centers. The library we synthesized serves for lead finding and is not part of a thematic series of analogs in an optimiza- tion process.
centers. The library we synthesized serves for lead finding and is not part of a thematic series of analogs in an optimiza- tion process.
Scheme 4.
Combinatorial scheme of the library preparation.
Experimental Section
General
The Rink Amide resin (4-(2',4'-Dimethoxyphenyl-fmoc- aminomethyl)-phenoxy resin) was purchased from Novabiochem (loading of the resin approx. 450 μmolg-1).
A complete list of the utilized building block reagents is enumerated in the Appendices
All reagents used were commercially available from Aldrich, Fluka, Lancaster or Maybridge, except the R1-build- ing blocks No. 2 and No. 3, which were freshly prepared from their carboxylic esters by hydrolysis.
Unless otherwise specified, after each reaction, the resin was thoroughly washed by sequential treatments with DMA, DMSO and i-PrOH. Previous to each reaction, traces of iso- propanol were washed away with the corresponding dry sol- vent.
After each transformation that was part of the validation on the solid support, the reaction products were cleaved with 95% TFA and the purity was analyzed by HPLC and identi- fied by MS.
LCMS: HPLC analytical separation was achieved using a reverse phase nucleosil C18 5μ 250 mm x 4.6 mm column, 215 nm, 10-90% CH3CN/0.1% TFA over 30 min, 1 ml/min. A part of the eluate (split 1:25) was introduced into a Quattro- BQ mass spectrometer (VG Biotech, Altrincham, England), operated at a source temperature of 60°C and a cone voltage of 50 V, via an electrospray interface (EI). The mass range from 100 to 800 Dalton was scanned in 4 seconds.
Synthesis of individual compounds (Arrays)
Deprotection of the resin
4-(2',4'-Dimethoxyphenyl-fmoc-aminomethyl)phenoxy resin (Rink amide resin) was subjected to repeated washes with 20% piperidine/DMA until no UV absorption from Fmoc was detected in the eluate.
Coupling procedure
The NH2-linker group was acylated with a 0.3M solu- tion of a carboxylic acid (3 eq in DMA) at r.t. (preactivation 40min with 3.3eq DICD and 3.3eq HOBt) until the Kaiser test [10] was negative.
Claisen condensation
Resin (50 mg, 22.5 μmol) was suspended in a solution of 675 μmol carboxylic ester in 670 μl of DMA. Under inert gas 18 mg (450 μmol) of sodium hydride (60%) was added and the reaction mixture was well shaken for 1h at 90°C. The resin was filtered, washed (30% v/v acetic acid / H2O, DMA, DMSO, and i-PrOH), and dried under reduced pres- sure.
Alkylation
This resin (20 mg, 8.6 μmol) was treated with 86 μl 1M TBAF in THF for 2h at r.t. After addition of 150 μl of a 2.5M solution of the appropriate alkylating agent in CH2Cl2, the reaction was continued for another 2h. The resin was filtered off and washed well with CH2Cl2 and THF.
Cyclization
The resulting resin was heated with 500 μl of a 2.5 M solution of hydrazines or hydroxyl amine (HCl was neutral- ized by NEt3) in DMA at 80°C for 24 h.
Cleavage
Cleavage from the support was carried out with diluted TFA according to a procedure described by Rink6.
Pyrazole 4d
The compound 1a was prepared from 1.50 g of Rink amide resin by the standard coupling protocol; 100%; HPLC 10.2 min, MS (EI) m/z 163 (M+). Treatment of the resin 1a with 0.51g (3.4 mmol) PhCOOEt, 0.14 g (3.4 mmol) NaH (60% dispersion), and 10.5 ml DMA provided 2a; 95%; HPLC 26.3 min, MS (EI) m/z 267 (M+). Conversion of resin 2a with 6.75 ml (6.75 mmol) 1M TBAF in THF, 5.68 ml CH2Cl2, 2.11 g (13.5 mmol) EtI afforded 3a (75%; HPLC 22.7 min, MS (EI) m/z 294 (M+). Heating resin 3a 4 days under reflux with 1.09 g (6.75 mmol) hydralazine and 67 mg (0.67 mmol) acetyl acetone in EtOH gave 4d1 (10%; HPLC 25.3 min) and 4d2 (10%; HPLC 26.3 min) after cleavage with 20% v/v TFA/CH2Cl2. HPLC preparative separation was carried out using a reverse phase Nucleosil C18 5 20 mm x 250 mm column, 215 nm, 10-90% CH3CN/0.1% TFA over 90 min, 15 ml/min. 4d1 1H-NMR (DMSO-d6) 9.72 (s, 1H), 8.29 (m, 1H), 8.11 (m, 4H), 8.02 (d, J = 8.6 Hz, 2H), 7.90 (d, J = 8.6 Hz, 2H) 7.46 (bs, 1H), 7.32 (m, 2H), 7.24 (m, 1H), 2.77 (q, J = 7.4 Hz, 2H), 1.06 (t, J = 7.4 Hz, 3H), MS (EI) m/z 420 (M+). 4d2 1H-NMR (DMSO-d6) 9.71 (s, 1H), 8.29 (m, 1H), 8.20 (m, 1H), 8.15 (m, 2H), 7.99 (bs, 1H), 7.82 (m, 4H), 7.51 (m, 4H), 7.42 (bs, 1H), 7.31 (d, J = 8.5 Hz, 2H), MS (EI) m/z 420 (M+).
Pyrazole 9
The compound 1b was prepared from 3.0 g Rink amide resin by the standard coupling protocol; 95 %; HPLC 19.8 min. To a slurry of Rink resin-bound 1b (1.00 g, 0.46 mmol) and 8.7 ml dry DMA was added 0.18g (4.5 mmol) of NaH (60% dispersion) and the mixture was well shaken under ar- gon at 80°C for 1h. The resulting mixture was filtered, washed with 30% v/v acetic acid / H2O, DMA, DMSO, and i-PrOH and dried under vacuo; 95 %; HPLC 10.2 min, MS (EI) m/z 347 (M+). A 1M solution of TBAF in THF (4.5 ml, 4.5 mmol) was added to 1.08 g (0.45 mmol) of the resin bound diketone 1b at 23°C and the mixture was shaken at this temperature for 1h and then treated with 779 μl (9 mmol) allyl bromide. The mixture was shaken for another 2 hours, followed by filtration, washing with CH2Cl2, DMA, DMSO, and i-PrOH and air drying to yield the resin bound 8; 40%; HPLC 26.0 min, 1H-NMR (DMSO-d6) 8.02 (d, J = 1.0 Hz, 1H), 7.65 (bs, 2H), 7.54 (d, J = 1.8 Hz, 1H), 6.76 (m, 1H), 5.82 (m, 1H), 5.05 (m, 2H), 4.95 (d, 1H), 2.80 (s, 3H), 2.68 (s, 3H), MS (EI) m/z 388 (M+). Cyclization of 8 was performed us- ing 1 ml hydrazine hydrate in 4 ml DMA. The mixture was heated at 80°C for 24h. The resulting mixture was filtered, washed with DMA, DMSO, and i-PrOH and dried under vacuo to yield 9; 95 %; HPLC 26.4 min; 1H-NMR (DMSO- DMSOd6) 7.77 (s, 1H), 7.53 (bs, 2H, NH2), 6.71 (d, J = 1.8 Hz, 1H), 6.61 (d, J = 1.8 Hz, 1H), 5.83 (m, 1H), 4.85 (m, 2H), 3.35 (d, J= 5.4 Hz, 2H), 2.70 (s, 3H), 2.37 (s, 3H), MS (EI) m/z 383 (M+).
Synthesis of combinatorial library
Coupling of 4 acetyl caboxylic acids to Rink amide resin
Four portions of Rink Amide resin, each 0.5 g (208.5 mmol), were treated with 3eq of a 0.3 M solution of the appropriate carboxylic acid which had been preactivated with 3.3 eq DICD and HOBt for 40 min. Once the Kaiser test was negative the four portions were mixed and washed with DMA, DMSO, and i-PrOH and dried under vacuo.
Claisen Condensation
The resin from the former coupling procedure was divided into 35 separate reaction vessels. Under inert gas atmosphere 21 mg (521 μmol) sodium hydride and 782 μmol carboxylic acid ester in 770 μl of DMA were added to each resin portion (23.4 μmol). The vigorously mixed reaction mixtures were heated 75 min at 90°C. All portions were mixed and washed with 30 % v/v acetic acid/water, THF, DMA, DMSO, i-PrOH and dried under vacuo.
Cyclization
The starting material (1.6 g, 640 μmol) were separated in 42 reaction vessels and each resin was treated with 790 μl of a 0.5 M solution of the appropriate monosubstituted hydrazine in DMA. After heating the reaction mixtures for 3 days at 90°C each portion was washed separately with DMA, DMSO, and i-PrOH.
Cleavage
Approx. 1/3 of the library material, i.e. 12.07 mg (5 μmol) resin from each sublibrary were mixed with 300 μl 20 % v/v TFA/CH2Cl2 three times for 30 minutes. Then the resin was washed with 300 µl 1,2-dichloroethane and 300 μl trifluoroethanol. The solvents were evaporated in a microcentrifuge and the residue was dissolved in 500 μl DMSO.
Acknowledgements
We are indebted to C. Guenat and B. Inverardi, from the Central Research Services, MS Applica- tions & Services, for LCMS analytical data.
Appendices
Complete lists of utilized building block rea- gents (chemical structures).
Appendices





References
- Gallop, M. A.; Barrett, R. W.; Dower, W. J.; Fodor, S. P. A.; Gordon, E. M. J. Med. Chem. 1994, 37, 1233. [PubMed]Gordon, E. M.; Barrett, R. W.; Dower, W. J.; Fodor, S. P. A.; Gallop, M. A. J. Med. Chem. 1994, 37, 1385. [PubMed]Felder, E. R. Chimia 1994, 48, 531. Terrett, N. K.; Gardner, M.; Gordon, D. W.; Kobylecki, R. J.; Steele, J. Tetrahedron 1995, 51, 8135. Ellman, J. A. Acc. Chem. Res. 1996, 29, 132.
- Furka, A.; Sebestyen, F.; Asgedom, M.; Dibo, G. Abstr. 14th Int. Congr. Biochem., Prague 1988, 47. Lam, K. S.; Salmon, S. E.; Hersh, E. M.; Hruby, V. J.; Kazmierski, W. M.; Knapp, R. J. Nature 1991, 354, 82. [PubMed]
- Marzinzik, A. L.; Felder, E. R. Tetrahedron Lett. 1996, 37, 1003.
- Yan, B.; Kumaravel, G.; Anjaria, H.; Wu, A.; Petter, R. C.; Jewell, C. F.; Wareing, J. R. J. Org. Chem. 1995, 60, 5736.
- Sarkar, S. K.; Garigipati, R. S.; Adams, J. L.; Keifer, P. A. J. Am. Chem. Soc. 1996, 118, 2305.
- Rink, H. Tetrahedron Lett. 1987, 28, 3787.
- Nishio, T.; Yamamoto, H. J. Heterocycl. Chem. 1995, 32, 883.
- Clark, J. H.; Miller, J. M. J. Chem Soc., Perkin Trans. I 1977, 1743.
- Houghten, R. A.; Pinilla, C.; Blondelle, S. E.; Appel, J. R.; Dooley, C. T.; Cuervo, J. H. Nature 1991, 354, 84. [PubMed]
- Kaiser, E. Anal. Biochem. 1970, 34, 595. [PubMed]
- Sample Availability: Sample not available.
© 1997 MDPI. All rights reserved
Share and Cite
MDPI and ACS Style
Marzinzik, A.L.; Felder, E.R. Combinatorial Libraries on Rigid Scaffolds: Solid Phase Synthesis of Variably Substituted Pyrazoles and Isoxazoles. Molecules 1997, 2, 17-30. https://doi.org/10.3390/jan97p5
AMA Style
Marzinzik AL, Felder ER. Combinatorial Libraries on Rigid Scaffolds: Solid Phase Synthesis of Variably Substituted Pyrazoles and Isoxazoles. Molecules. 1997; 2(1):17-30. https://doi.org/10.3390/jan97p5
Chicago/Turabian StyleMarzinzik, Andreas L., and Eduard R. Felder. 1997. "Combinatorial Libraries on Rigid Scaffolds: Solid Phase Synthesis of Variably Substituted Pyrazoles and Isoxazoles" Molecules 2, no. 1: 17-30. https://doi.org/10.3390/jan97p5