Synthesis of Spiroisoxazolines by 1,3-Dipolar Cycloaddition

1
Department of Organic Chemistry, Slovak Technical University, SK-812 37 Bratislava, Slovak Republic
2
Chemical Institute, PFUK, Mlynská dolina, SK-842 15 Bratislava, Slovak Republic
*
Author to whom correspondence should be addressed.
Molecules 1997, 2(3), 57-61; https://doi.org/10.3390/20300057
Submission received: 31 August 1996
/
Accepted: 3 December 1996
/
Published: 8 April 1997
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
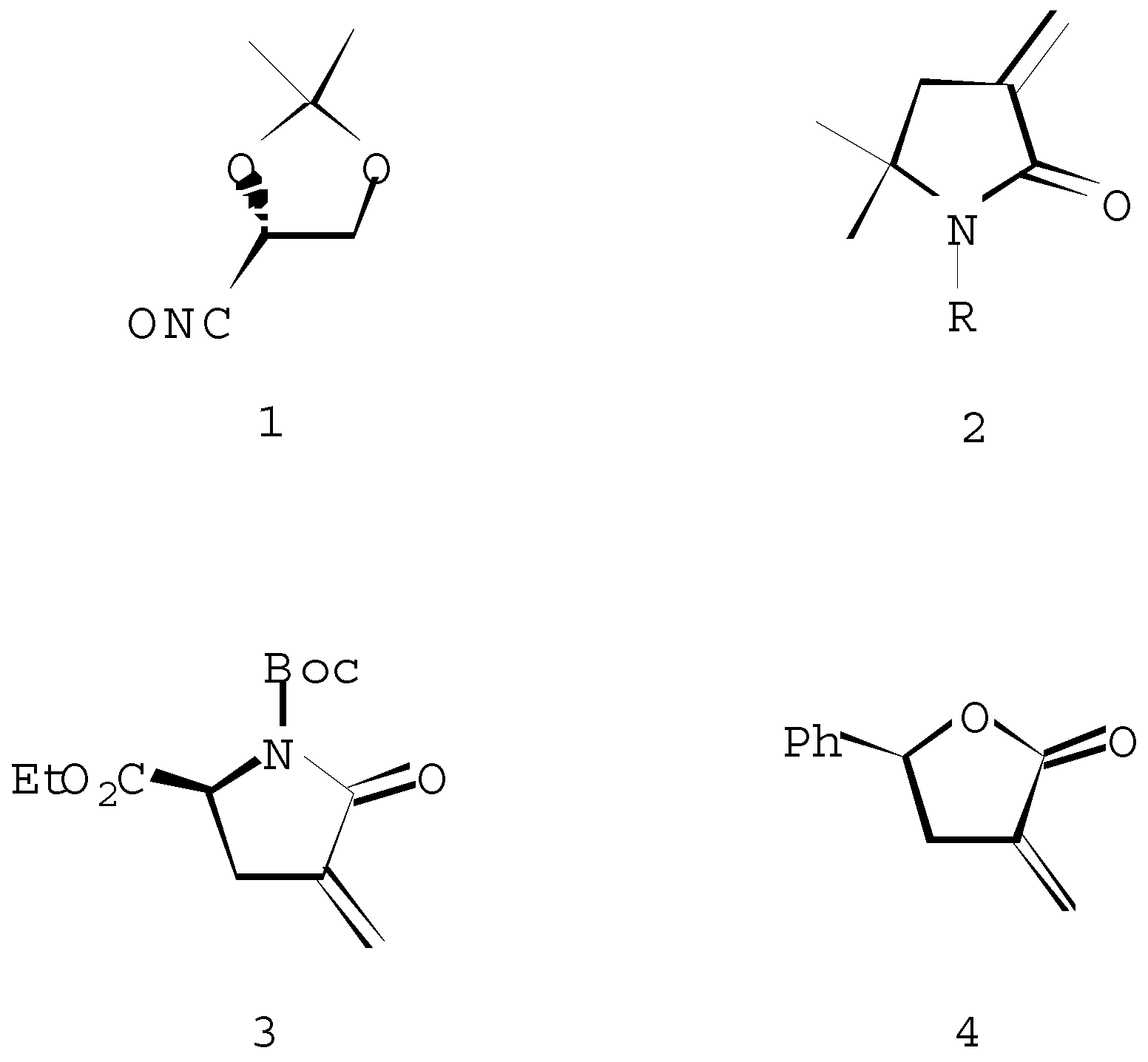
:The cycloaddition of the chiral nitrile oxide 1 to 1-R-substituted 3,3-methylene-5,5-dimethyl-2-pyrrolidinones 2 (where R is H, n-butyl-, 1,1-dimethylethoxycarbonyl-, 1-methylethenyl- and acetyl-) proceeds regioselectively under the formation of spiroisoxazolines, namely 7-R-substituted-6-oxo-8,8-dimethyl-1-oxa-2,7-diazaspiro[4,4]non-2-enes 5 and 6. The asymmetric induction expected by the α-chiral centre of the nitrile oxide 1 was not very effective, diastereoisomers 5 and 6 were formed in an approximate 50:50 ratio. The stereoselectivity of the 1,3-dipolar cycloaddition of the arylnitrile oxide 7 with the chiral lactam 3 and the achiral lactone 4 are investigated. The attack of the 1,3-dipole occurred from the less hindered face of the dipolarophile 3 and 4, giving the major isomer 8 and 10, respectively.
Introduction
The recent observation of the strong herbicidal activity of spiro cyclic lactams, coupled with the absence of toxicity to microorganisms [1] and also that some spiroisoxazolines occur naturally (Araplysillins are inhibitors of ATPase [2]) stimulated our interest in the synthesis of other spirocyclic derivatives. 2-Isoxazolines (4,5-dihydroisoxazoles) are versatile sources of the functionality present in natural products [3] and there is renewed interest in their synthesis via 1,3-dipolar cycloaddition of nitrile oxides to alkenes, particularly in the factors that influence stereo- and regio-selectivity [4]. As a continuation of our effort to utilize heterocyclic compounds as dipolarophiles in 1,3-dipolar cycloaddition reactions [5], we report the cycloaddition of chiral and achiral nitrile oxides with achiral and chiral α-methylene-γ-lactams and lactones (Scheme 1).
Results and Discussion
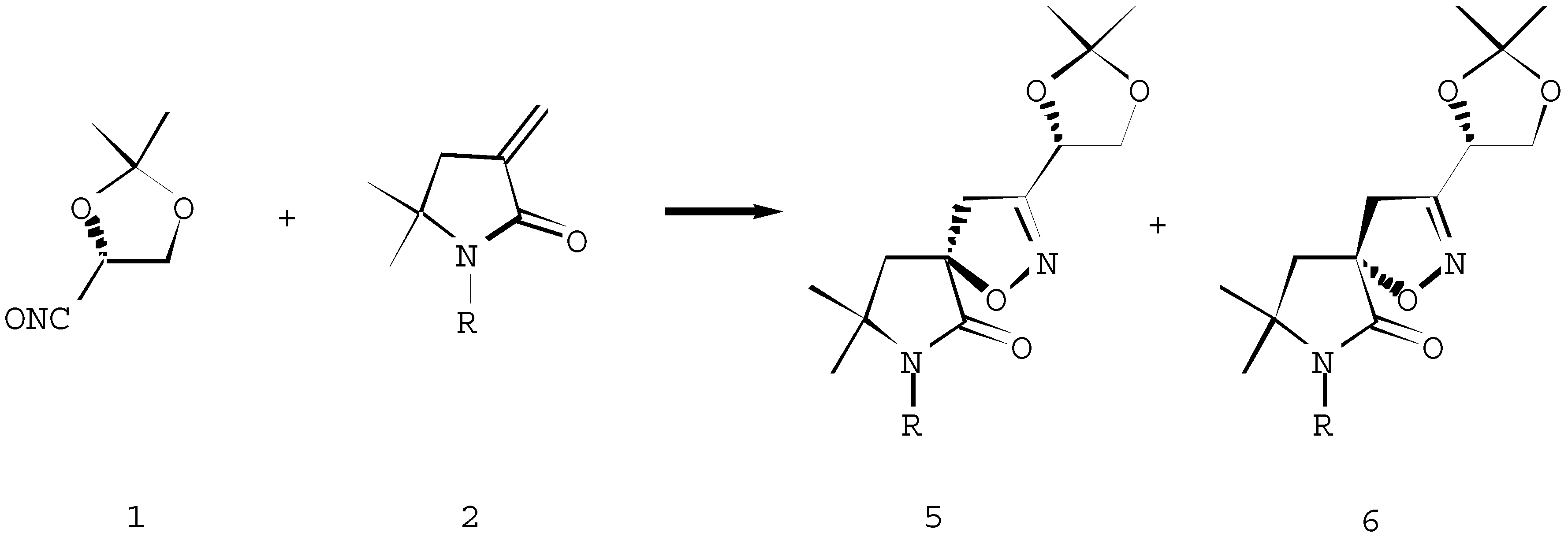
The 1,3-dipolar cycloaddition of the chiral nitrile oxide 1 and achiral α-methylene-γ-lactams 2 affords a mixture of spiroisoxazolines 5 and 6 in 50-65% yield (Scheme 2). The asymmetric induction expected by the α-chiral centre of the nitrile oxide 1 has not been very effective, as has been indicated by AM1 modeling of the respective transition states (The results will be presented elsewhere). While product 5a (R=H) was preferred kinetically (the difference in transition states energies is 0,80 kcal/mol), presumably product 6a was more thermodynamically stable (ΔfH=-0,22 kcal/mol). Indeed, diastereoisomers 5a and 6a were formed in an approximate 50:50 ratio.
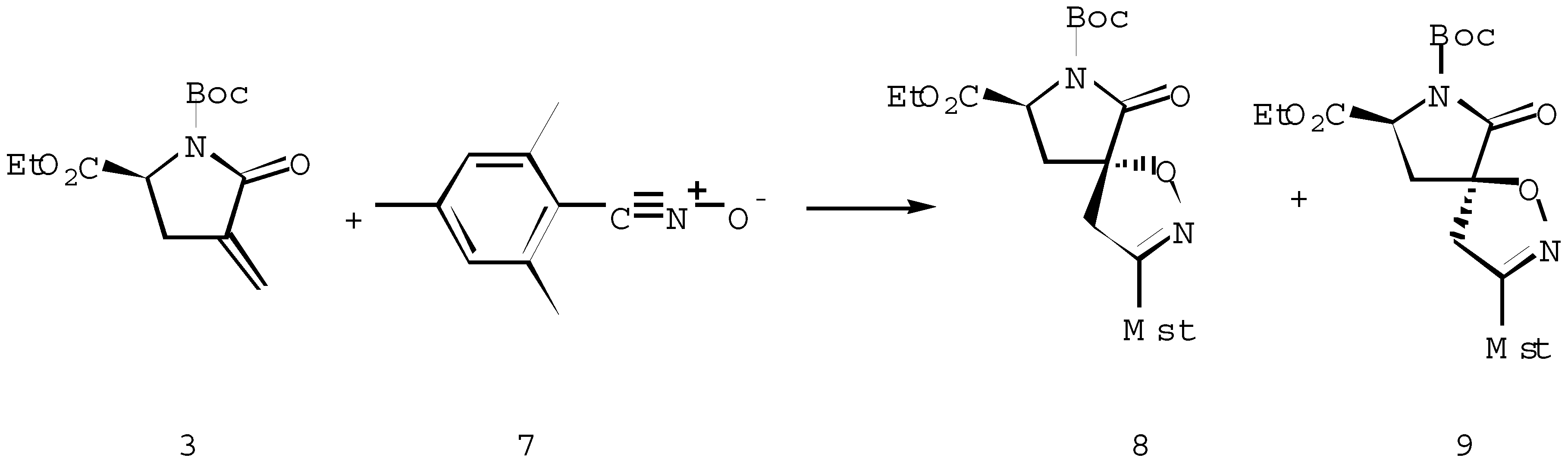
Further we envisaged the chiral α-methylene-γ-lactam 3 [7] to be a useful heterocycle for the study of the factors controlling π-facial selectivity since the substituents can be systematically varied. Moreover, the regioselective elaboration of the latent amino functionality of spiroisoxazolines can be used for the preparation of chiral amino acids derivatives. The reaction of chiral α- methylene-γ-lactam 3 and the stable nitrile oxide 7 proceeded with the formation of diastereoisomers 8 and 9 in the ratio of 67 :33, in favour of diastereoisomer 8 (Scheme 3). The attack of the 1,3-dipole occurred from the less hindered face of the dipolarophile 3 giving the major isomer 8. Stereochemical assignments, made on the basis of 13C NMR, supported the structure of the major isomer 8, arising from the predominant approach of the dipole to the “bottom” of the α-methylene-γ-lactam 3.
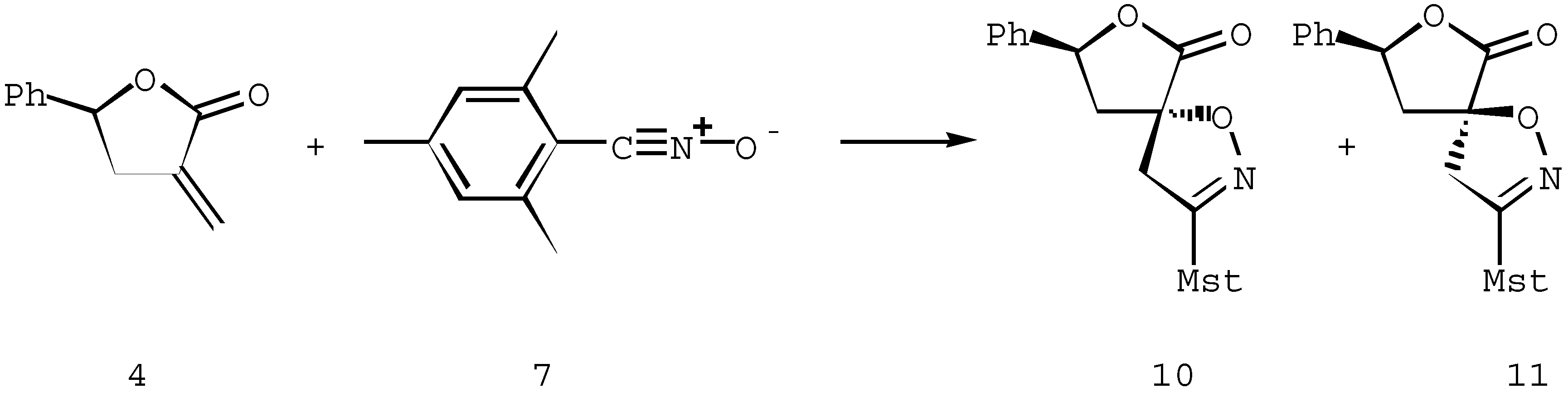
The cycloaddition to the achiral α-methylene-γ-lactone 4 [8] proceeded fully analogously. Also in this case the predominant approach of the dipole occurs at the anti-face to the phenyl substituent in the dipolarophile 4. Thus, the reaction of the nitrile oxide 7 with methylenelactone 4 afforded a 90 : 10 mixture of cycloadducts 10 and 11 (Scheme 4)
The stereochemical assignments for the cycloadducts 10 and 11 derived from the lactone 4 are based upon 13C NMR chemical shift correlations.
Conclusion
Evidence for a predictive anti-diastereoselective 1,3-dipolar cycloaddition of an arylnitrile oxide to a substituted chiral methylenelactam and an achiral methylenelactone has been presented.
Experimental
General
1H NMR spectra were recorded at 300 MHz on a Varian VXR 300 or at 80 MHz on a Tesla BS 487 at 293 K in CDCl3. Spectra were internally referenced to TMS. Peaks are reported in ppm downfield of TMS. Multiplicities are reported as singlet (s), doublet (d), triplet (t), quartet (q), some combination of these, broad (br), or multiplet (m). 13C NMR spectra were recovered at 75.0 MHz on the same spectrometer as 1H NMR spectra at 293 K in CDCl3. NMR analysis of the crude original mixture permitted a determination of ratio of the diastereoisomers. Flash chromatography was carried out on 63-200 μm or 40-60 μm silica gel. Thin layer chromatography was carried out on aluminium backed silica plates containing UV254 by Lachema and plates were visualized with UV light and Mostaine solution as appropriate. All yields refer to isolated, spectroscopically pure material, and have not been optimized.
Spiroisoxazolines 5a-e, 6a-e. General procedure
A solution of Et3N (8.8 mmol) in chloroform (25 ml) was added during 24 hours to a stirred solution of the dipolarophile 2 (8.0 mmol) and the unstable nitrile oxide 1 (8.0 mmol, prepared in situ from the corresponding hydroxymoyl chloride) in chloroform (25 ml). After evaporation of the solvent, the products were isolated as an inseparable mixture of diastereoisomers 5 and 6.
(1´S, 5R/S) 8,8-Dimethyl-3-(1´,2´-di-O-isopropylidene-1´,2´-dihydroxyethyl)-6-oxo-1-oxa-2,7-diazaspiro[4,4]- non-2-ene (5a + 6a)
1H NMR (CDCl3, 300 MHz): 5.0-5.3 (1H, m), 4.1-4.5 (2H, m), 2.0-3.8 (4H, m), 1.4, 1.5, 1.7, 1.8, 1.9 (12H, s, Me). 13C-NMR (CDCl3, 300 MHz): 171.66, 171.60, 156.49, 156.41 (C, 4-C, 9-C), 111.17, 110.93 (C, OCO), 87.17, 86.40 (C, 5-C), 69.78, 69.73 (CH, 1´-C), 65.80, 65.73, 53.13 (C, CH2, 8-C, 2´-C), 47.52, 47.26, 40.58, 40.44 (CH2, 4-C, 9-C), 28.30, 28.26, 25.12, 25.08, 24.95, 24.37, 24.08, 24.00 (CH3, Me).
(1´S, 5R/S) 7-Butyl-8,8-dimethyl-3-(1´,2´-di-O-isopropy-lidene-1´,2´-dihydroxyethyl)-6-oxo-1-oxa-2,7-diazaspiro-[4,4]non-2-ene (5b + 6b)
1H-NMR (CDCl3, 300 MHz): 4.5-4.9 (3H, m), 3.8-3.9 (2H, m), 1.3-3.3 (20H, m), 0.9-1.0 (3H, m). 13C-NMR (CDCl3, 300 MHz): 170.31, 158.74, 158.63 (C, 4-C, 9-C), 113.84 (C, OCO), 85.37 (C, 5-C), 72.58, 72.56 (CH, 1´-C), 62.69, 63.40 (CH2, 2´-C), 57.60, 57.49 (C, 8-C), 46.88, 46.66, 45.57, 42.07, 41.68, 40.44, 38.93, 38.7, 19.37 (CH2, CH2), 30.55, 30.25, 28.76, 26.73, 26.58, 26.39, 12.71 (CH3, Me).
(1´S, 5R/S) 7-Acetyl-8,8-dimethyl-3-(1´,2´-di-O-isopropy-lidene-1´,2´-dihydroxyethyl) -6-oxo-1-oxa-2,7-diazaspiro-[4,4]non-2-ene (5c + 6c)
1H-NMR (CDCl3, 300 MHz): 3.5-4.9 (5H, m), 3.1-3.3 (2H, m), 2.5 (3H, s), 2.0-2.4 (2H, m), 1.4, 1.5, 1.6 (12H, s, Me). 13C-NMR (CDCl3, 300 MHz): 172.88, 172.43, 171.26. 171.20, 157.31, 157.09 (C, 6-C, 3-C, N-CO), 109.83, 109.76 (C, OCO), 85.86, 85.78 (C, 5-C), 70.12, 70.04 (CH, 1´-C), 66.26, 66.12 (CH2,2´-C), 60.20, 60.11(C, 8-C), 42.68, 42.42, 41.53, 40.93 (CH2, 4-C, 9-C), 27.07, 25.75, 25.71, 25.55, 25.50, 25.37, 24.43, 24.36 (CH3, Me).
(1´S, 5R/S) 7-(1,1-Dimethylethoxycarbonyl)-8,8-dimethyl-3-(1´,2´-di-O-isopropylidene-1´,2´-dihydroxyethyl)-6-oxo-1-oxa-2,7-diazaspiro[4,4]non-2-ene (5d + 6d)
1H-NMR (CDCl3, 300 MHz): 4.7-4.9 (1H, m), 4.0-4.3 (2H, m), 3.0-3.6 (2H, m), 2.0-2.4 (2H, m), 1.4, 1.5, 1.6 (21H, s, Me). 13C-NMR (CDCl3, 300 MHz): 177.40, 170.50, 170.34, 157.09, 156.82 (C, 3-C, 6-C), 109.74, 109.66 (C, OCO), 85.77, 85.66 (C, 5-C), 82.80 (CMe3), 70.17, 70.09 (CH, 1´-C), 66.30, 66.27 (CH2, 2´-C), 59.47, 59.43 (C, 8-C), 46.53, 46.09, 41.61, 40.82 (CH2, 4-C, 9-C), 27.68, 27.33, 26.42, 26.40, 25.58, 25.53, 24.49, 24.40 (CH3, Me).
(1´S, 5R/S) 7-(1-Methylethenyl)-8,8-dimethyl-3-(1´,2´-di-O-isopropylidene-1´,2´-dihydroxyethyl)-6-oxo-1-oxa-2,7-diazaspiro[4,4]non-2-ene (5e + 6e)
1H-NMR (CDCl3, 300 MHz): 4.9-5.2 (1H, m), 4.0-4.2 (1H, m), 3.4-3.7 (1H, m), 3.1-3.3 (2H, m), 2.3-2.5 (1H, m), 1.3, 1.4, 1.5, 1.7 (15H, s, Me). 13C-NMR (CDCl3, 300 MHz): 168.77, 168.60, 156.56, 156.37 (C, 4-C, 9-C), 138.58, 138.53 (C, C=CH2), 114.24, 113.42, 109.93, 109.15 (C, CH2, OCO, C=CH2), 85.82, 85.74 (C, 5-C), 69.91, 69.89 (CH, 1´-C), 65.93, 65.85 (CH2, 2´-C), 52.87 (C, 8-C), 47.44, 47.02, 40.97, 40.29 (CH2, 4-C, 9-C), 30.34, 27.37, 25.17, 27.12, 25.26, 25.21, 24.89, 24.19, 24.12, 20.73 (CH3, Me).
Spiroisoxazolines 8-11. General procedure
The dipolarophile 3 or 4 (1.43 mmol) and mesityl nitrile oxide 7 (1.43 mmol) were dissolved in 5 ml of benzene and warmed (70°C) with stirring until the reaction was complete (TLC). After evaporation of the solvent, pure diastereoisomers were obtained by crystallization or silica gel column chromatography, as described below.
(8S,5S)-7-(1,1-Dimethylethoxycarbonyl)-8-ethoxycarbonyl-3-(2,4,6-trimethylphenyl)-6-oxo-1-oxa-2,7- diazaspiro[4,4]non-2-ene (8)
Yield: 88%. Reaction time, 26h. Pure diastereoisomer 8 was obtained by silica gel chromatography (i-hexane/ethyl acetate 6:1).
1H-NMR (CDCl3, 300 MHz): 6.8 (2H, s, Har), 4.6 (1H, dd, J = 8.4, 5.7 Hz, 8-H), 4.2 (2H, q, J = 7.1 Hz, CH2CH3), 3.8 (1H, d, J = 17.8 Hz, 4-HA), 2.9(1H, d, J = 17.8 Hz, 4- HB), 2.8 (1H, dd, J = 13.9, 8.4 Hz, 9-HA), 2.2 (3H, s, Ph-pMe), 2.2 (6H, s, 2xPh-oMe), 2.1 (1H, dd, J = 13.9, 5.7 Hz, 9-HB), 1.5 (9H, s, CMe3), 1.2 (3H, t, J = 7.1 Hz, CH2Me). 13C-NMR (CDCl3, 300 MHz): 14.1 (CH3, CH2Me), 19.7 (CH3, 2xPh-oMe), 21.1 (CH3, Ph-pMe), 27.9 (CH3, CMe3), 35.1 (CH2, 9-C), 47.0 (CH2, 4-C), 55.8 (CH, 8-C), 62,0 (CH2, CH2Me), 84.6 (C, CMe3), 86.0 (C, 5-C), 124,7 (C, Car), 128.5 (CH, Car), 136.8, 139.2 (C, Car), 149.0 (C, N-CO2), 157.0 (C, 3-C), 170.0 (C, 6-C), 170.8 (C, C-CO2).
Some relevant signals corresponding to a minor isomer (8S,5R)-7- (1,1-dimethylethoxycarbonyl)-8-ethoxycarbon- yl 3-(2,4,6-trimethylphenyl)-6-oxo-1-oxa-2,7-diazaspiro-[4,4]non-2-ene 9 were also clearly observed in the crude original mixture, and characterized by 13C-NMR (CDCl3, 300 MHz): 46.0 (CH2, 4-C), 56.1 (CH, 8-C), 84.1 (C, CMe3), 128.7 (C, Car), 149.2 (C, N-CO2), 169.8 (C, 6-C), 170.0 (C, C-CO2).
3-(2,4,6-Trimethylphenyl)-8-phenyl-6-oxo-1,7-dioxa-2-azaspiro[4,4]non-2-ene (10)
Yield: 90%. Reaction time, 3,5 h. Pure diastereoisomer 10 was obtained by crystallization from hexane/ethanol 2:1. 1H-NMR (CDCl3, 300 MHz): 7.3-7.4 (5H, m, Har), 6.9 (2H, m, Har), 5.7 (1H, dd, J = 9.7 , 5.5 Hz, 8-H), 3.8 (1H, d, J = 17.6 Hz, 4-HA), 3.1 (1H, d, J= 17.6 Hz, 4-HB), 3.1 (1H, dd, J = 13.8, 5.5 Hz, 9-HA), 2.3 (1H, dd, J = 13.8, 9.7 Hz, 9-HB), 2.3 (s, 3H, Me), 2.3 (s, 6H, 2xMe). 13C-NMR (CDCl3, 300 MHz): 177.3 (6-C), 157.4 (3-C), 139.2, 137.7, 136.7, 128.8, 128.4, 125.8, 125.4, 124.6 (Car), 85.4, 79.3 (5-C, 8-C), 45.3, 43.6 (4-C, 9-C), 21.0, 19.6 (Me).
Some relevant signals corresponding to a minor isomer 11 were also clearly observed in the crude original mixture. 1H-NMR (CDCl3,300 MHz): 3.7 (1H, d, J = 17.4 Hz, 4-HA), 3.3 (1H, d, J = 17.4 Hz, 4-HB). 13C-NMR (CDCl3, 300 MHz): 84.6, 77.9 (5-C, 8-C), 47.8, 42.9 (4-C, 9-C).
Acknowledgments
The authors are grateful to the Slovak Grant Agency for receiving financial support (No. 1/1416/94) and the VW-Stiftung in Hannover for a research grant and receiving financial support.
References
- Kobayashi, J.; Tsuda, M.; Agemi, K.; Shigemori, H.; Ishibashi, M.; Sasaki, T.; Mikami, Y. Tetrahedron 1991, 47, 6617.
- Longeon, A.; Guoyot, M.; Vacelet, J. Experimentia 1990, 46, 548.
- Torssell, K. B. G. Use of Nitrile Oxides, Nitrones and Silyl Nitronates in Organic Synthesis. Novel Strategies in Synthesis; Verlag Chemie: New York, 1988. [Google Scholar]
- Annunziata, R.; Cinquini, M.; Cozzi, F.; Raimondi, L. Gazz. Chim. Ital. 1992, 119, 253.
- Fisera, L.; Jarosková, L.; Matejková, I.; Heimgartner, H. Heterocycles 1995, 40, 271.
- Larsen, K. E.; Torssell, K. B. G. Tetrahedron 1984, 40, 2985.
- Panday, S. K.; Brunet, D. G.; Langlois, N. Tetrahedron Lett. 1994, 35, 6673. Eyquerra, J.; Pedregal, C. Tetrahedron: Asymmetry 1994, 5, 921.
- Petragnani, N.; Ferraz, H. M. C.; Silvia, G. V. J. Synthesis 1986, 157. Belaud, C.; Russakis, C.; Letourneux, Y.; Alami, N. E.; Villieras, J. Synth. Commun. 1985, 15, 1233.
- Sample Availability: Available from the authors.
Scheme 1.
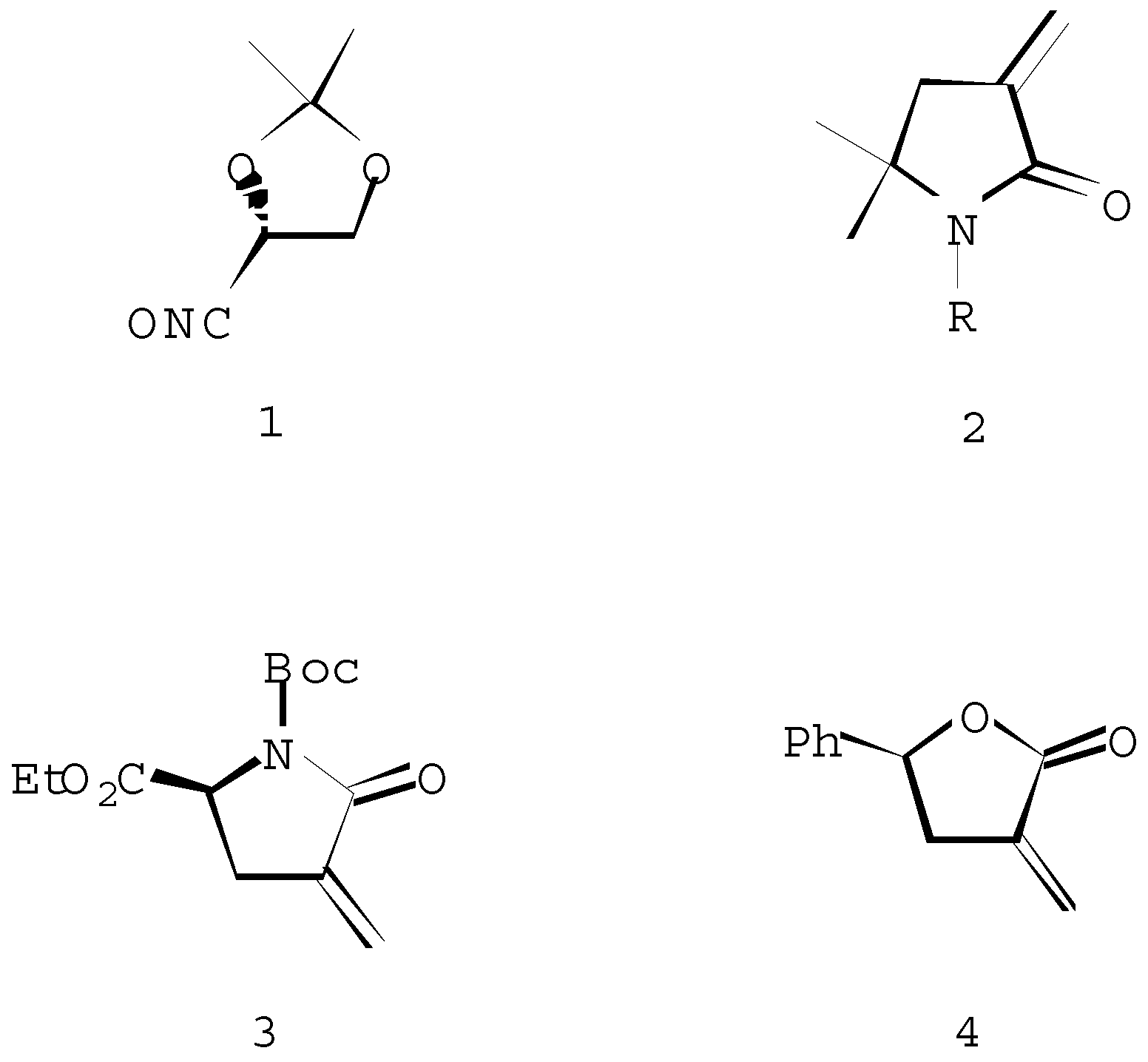
Scheme 2.
For a, R=H; b, R=n-Bu; c, R=COCH3; d, R=Boc, e, R=-C(CH3)=CH2.
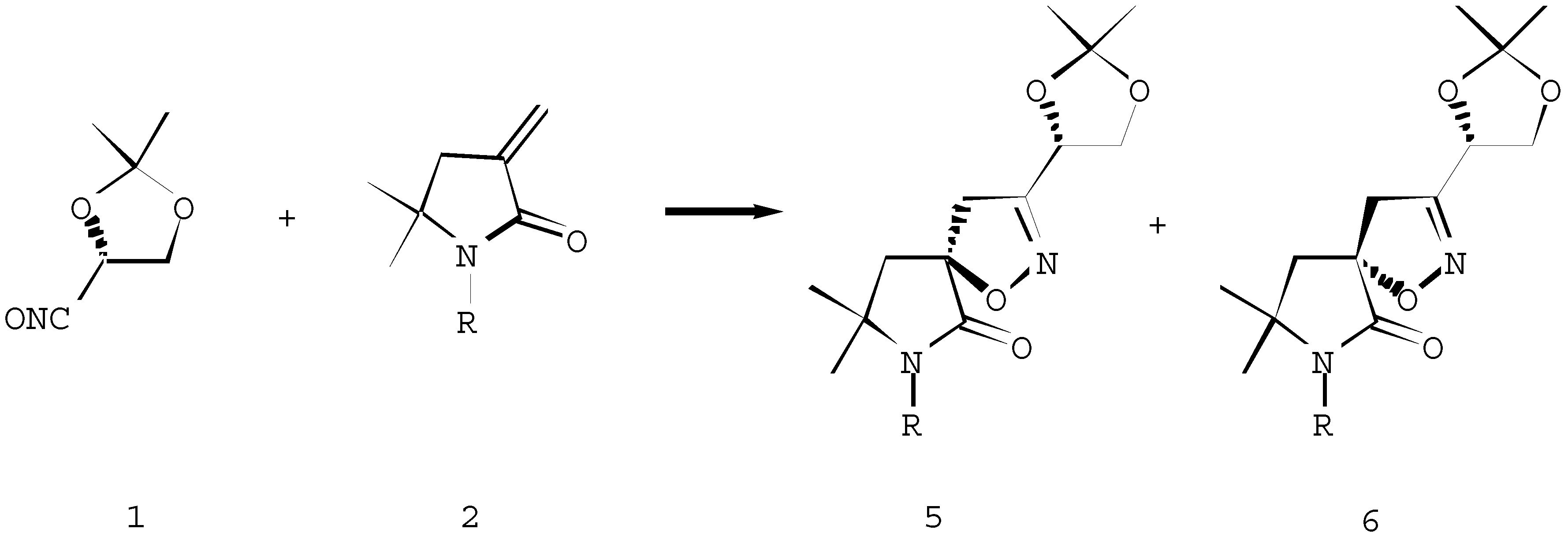
Scheme 3.
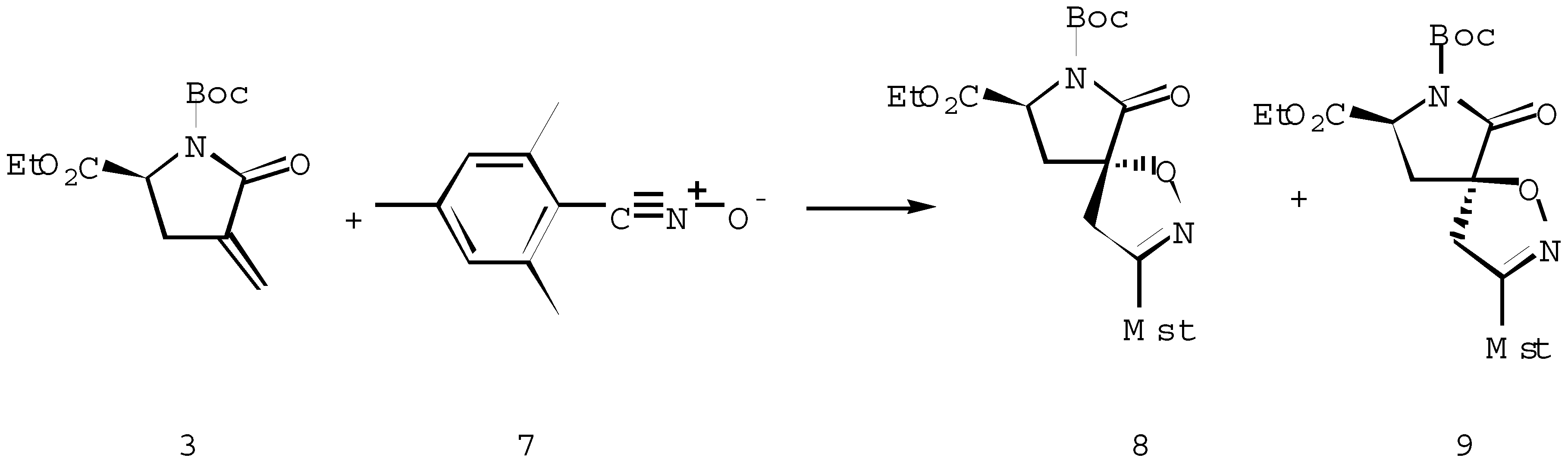
Scheme 4.
© 1997 MDPI. All rights reserved
Share and Cite
MDPI and ACS Style
Micuch, P.; Fisera, L.; Ondrus, V.; Ertl, P. Synthesis of Spiroisoxazolines by 1,3-Dipolar Cycloaddition. Molecules 1997, 2, 57-61. https://doi.org/10.3390/20300057
AMA Style
Micuch P, Fisera L, Ondrus V, Ertl P. Synthesis of Spiroisoxazolines by 1,3-Dipolar Cycloaddition. Molecules. 1997; 2(3):57-61. https://doi.org/10.3390/20300057
Chicago/Turabian StyleMicuch, Peter, Lubor Fisera, Vladimír Ondrus, and Peter Ertl. 1997. "Synthesis of Spiroisoxazolines by 1,3-Dipolar Cycloaddition" Molecules 2, no. 3: 57-61. https://doi.org/10.3390/20300057