Results and Discussion
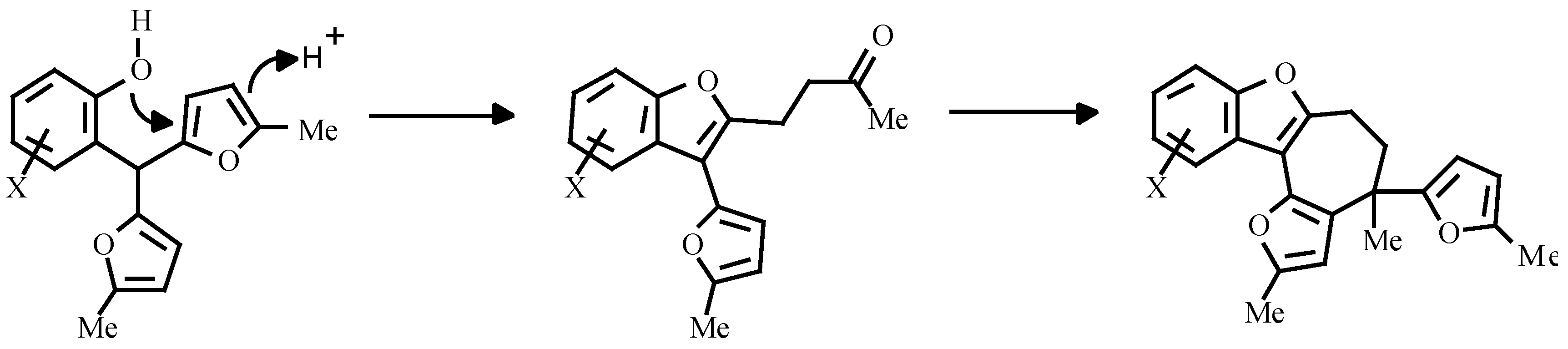
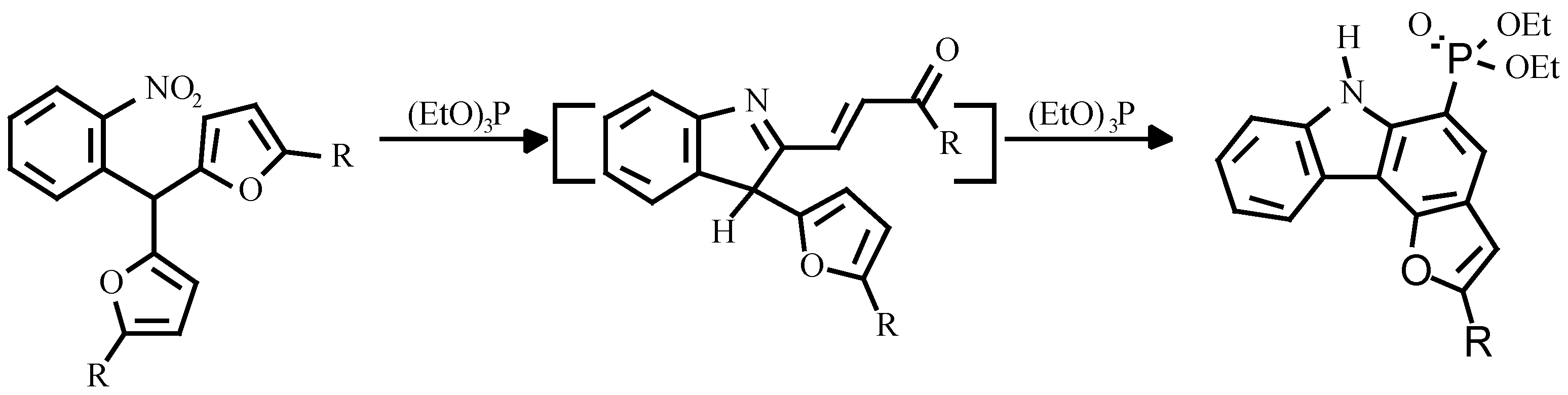
Earlier, the treatment of o-nitroarylbis-(5-alkylfur-2-yl)methanes with triethylphosphite was shown to give carbazole. The authors believed, that the reaction proceeded via nitrene attack on the furan ring with an unsaturated ketone as intermediate (
Scheme 2) [
7].
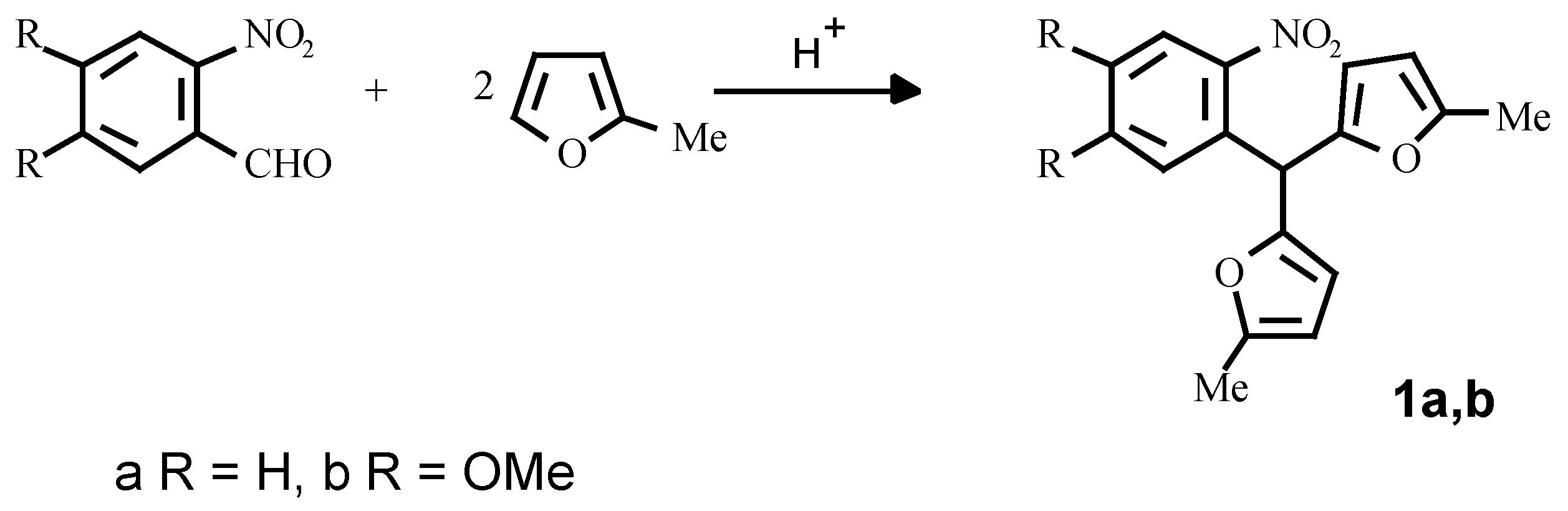
It appears that the optimum method for the synthesis of the starting o-nitroaryldifurylmethanes
1a, 1b is the perchloric acid catalysed condensation of the appropriate o-nitrobenzaldehydes and 2-methylfuran in dioxane (
Scheme 3) [
8].
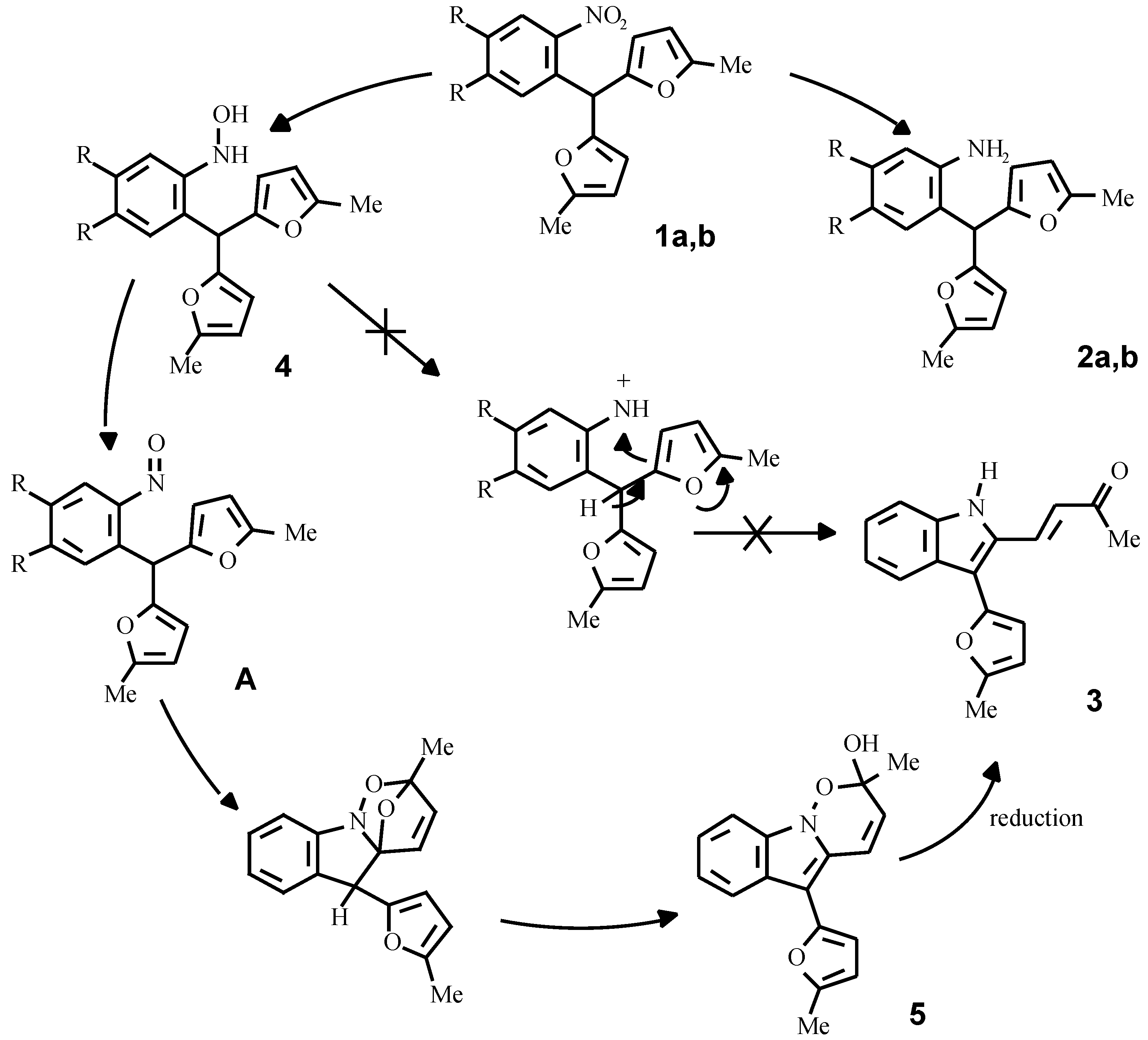
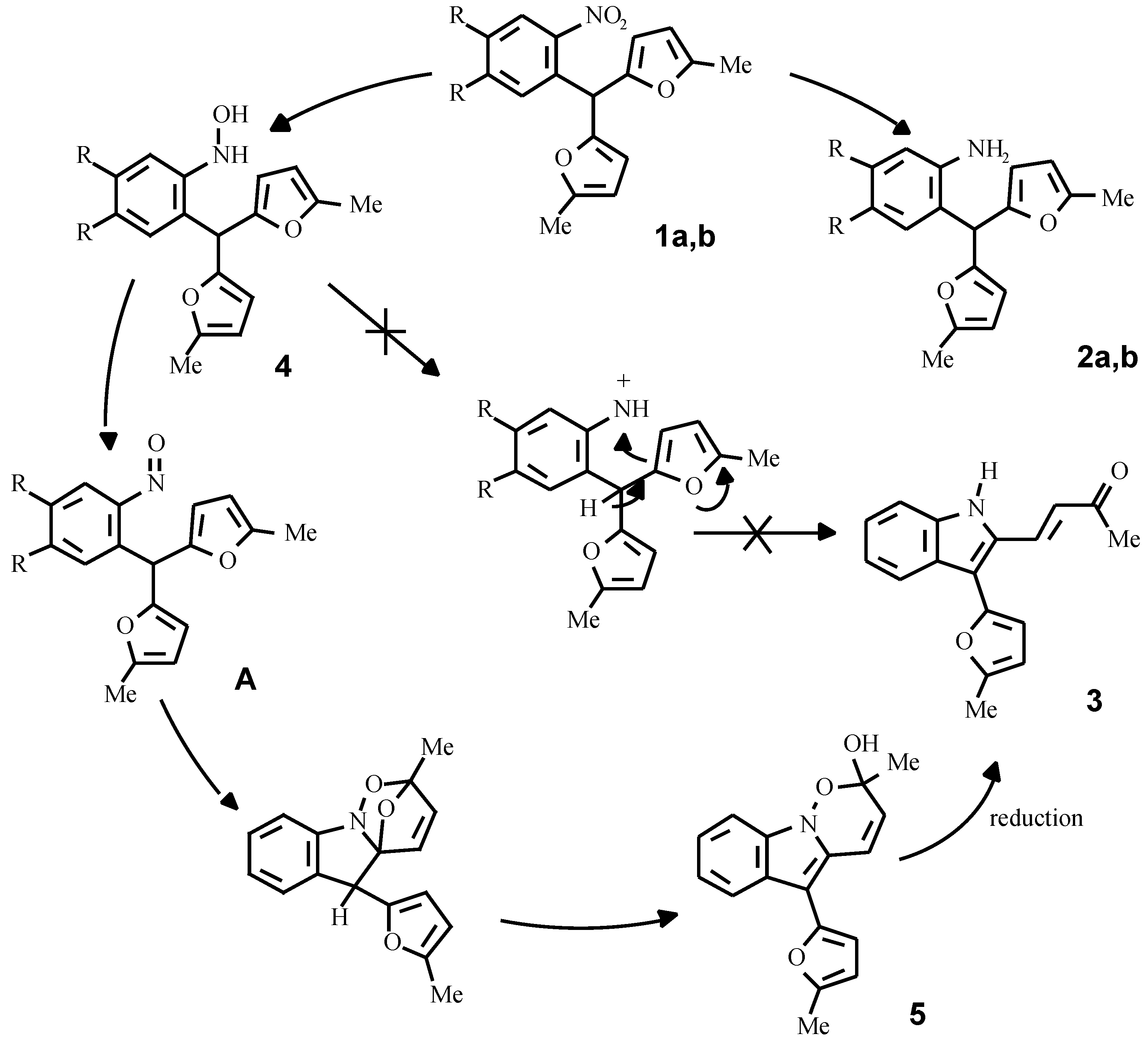
By the reduction of compounds 1a, 1b in methanol with zinc dust in the presence of hydrochloric acid, or with a hydrazine hydrate - Pd/C system, the corresponding anilines 2a, 2b were obtained in good yields. Different results were observed in the SnCl
2 promoted reduction of the compounds 1a, 1b in acidic media. Compound 1b under this conditions gave the corresponding aniline 2b, wherwas the reduction of o-nitrophenyldifurylmethane 1a led to the formation of an unsaturated ketone 3. It was originally supposed that a key intermediate in this conversion is the hydroxylamine 4 [
9], which under acid conditions forms a nitrenium cation or its equivalent with subsequent oxidative furan ring opening (
Scheme 4).
It appears, however, that the authentic hydroxylamine
4, obtained from the nitrocompound
1a by Zn reduction in the presence of NH
4Cl or complex Sn thiophenolates [
10], does not form the ketone
3 under acid conditions. On the other hand, we have established, that the purification of the hydroxylamine
4 by column chromatography and storage at room temperature gave compound
5, contami-nated with a small quantity of ketone
3.
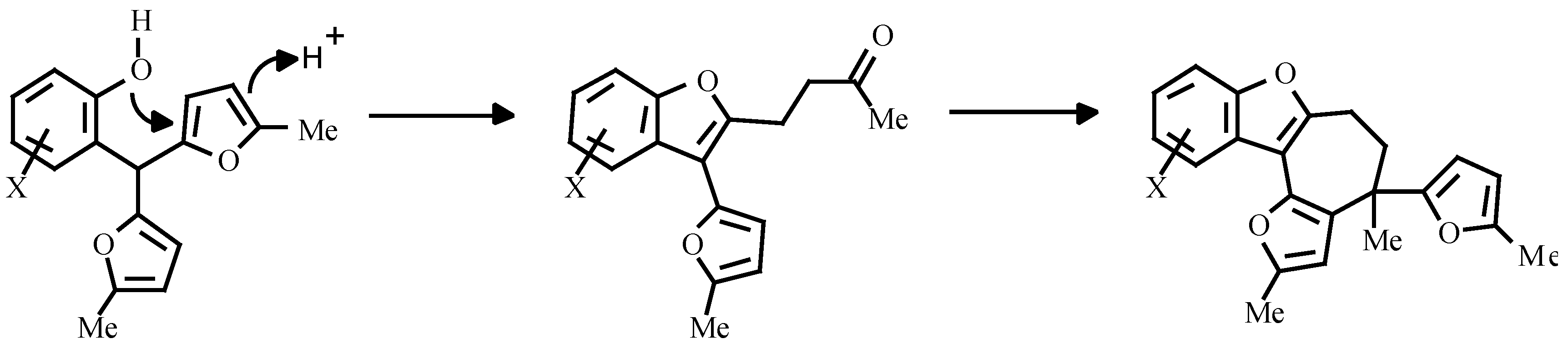
These facts have enabled us to assume, that a true intermediate of the reaction is the corresponding nitroso compound A, formed by the slow oxidation of the hydroxylamine 4 with oxygen from the air. To check this idea, compound 4 was refluxed in toluene and the tricyclic compound 5 was isolated from the reaction mixture; its structure was supported by IR- and NMR-spectroscopy and mass-spectrometry data. The same compound was obtained in an 80 % yield by the oxidation of the hydroxylamine 4 with K2Cr2O7 at a temperature of 0-5 °C for 10 minutes.
The mechanism of the formation of compound
5 probably includes an intramolecular Diels-Alder reaction between the furan ring and the nitroso-group with subsequent opening of the ether bridge in the initial strained adduct. Diels-Alder heterocycloaddition is well known and recently this methodology was exploited as the key step in the synthesis of mitomycin, as reported by Danishefsky [
11].
Since cycloadduct 5 and ketone 3 have different oxidation levels, a reductant is required for the conversion of 5 into 3. Thus, compound 5 can be reduced by the system SnCl2 + HCl rather smoothly to give ketone 3. It is noteworthy, that in the case of FeCl3 promoted oxidation of the hydroxylamine 4, ketone 3 was directly formed. It seems probable that under these conditions the in situ formed FeCl2 serves itself as the reducing agent. It is also possible, that the cycloadduct 5 can oxidize the hydroxylamine 4 under acidic conditions and the conversion as a whole proceeds catalytically.
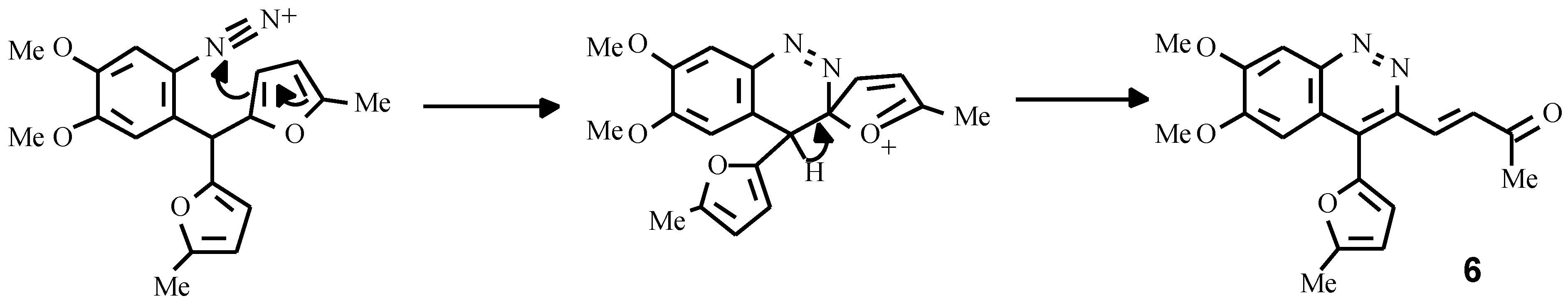
The other example is the synthesis of a cinnoline derivative as a result of an intramolecular electrophilic attack of diazonium group on the furan ring. Earlier attempts to obtain cinnolines failed due to strong resinification of the reaction mixture caused by the action of aqueous solutions of NaNO
2 and HCl [
11]. By carrying out the diazotisation reaction under mild conditions (isoamylnitrite, Me
3SiCl, acetonitrile), we succeeded in obtaining the cinnoline derivative
6 in high yield (
Scheme 5).
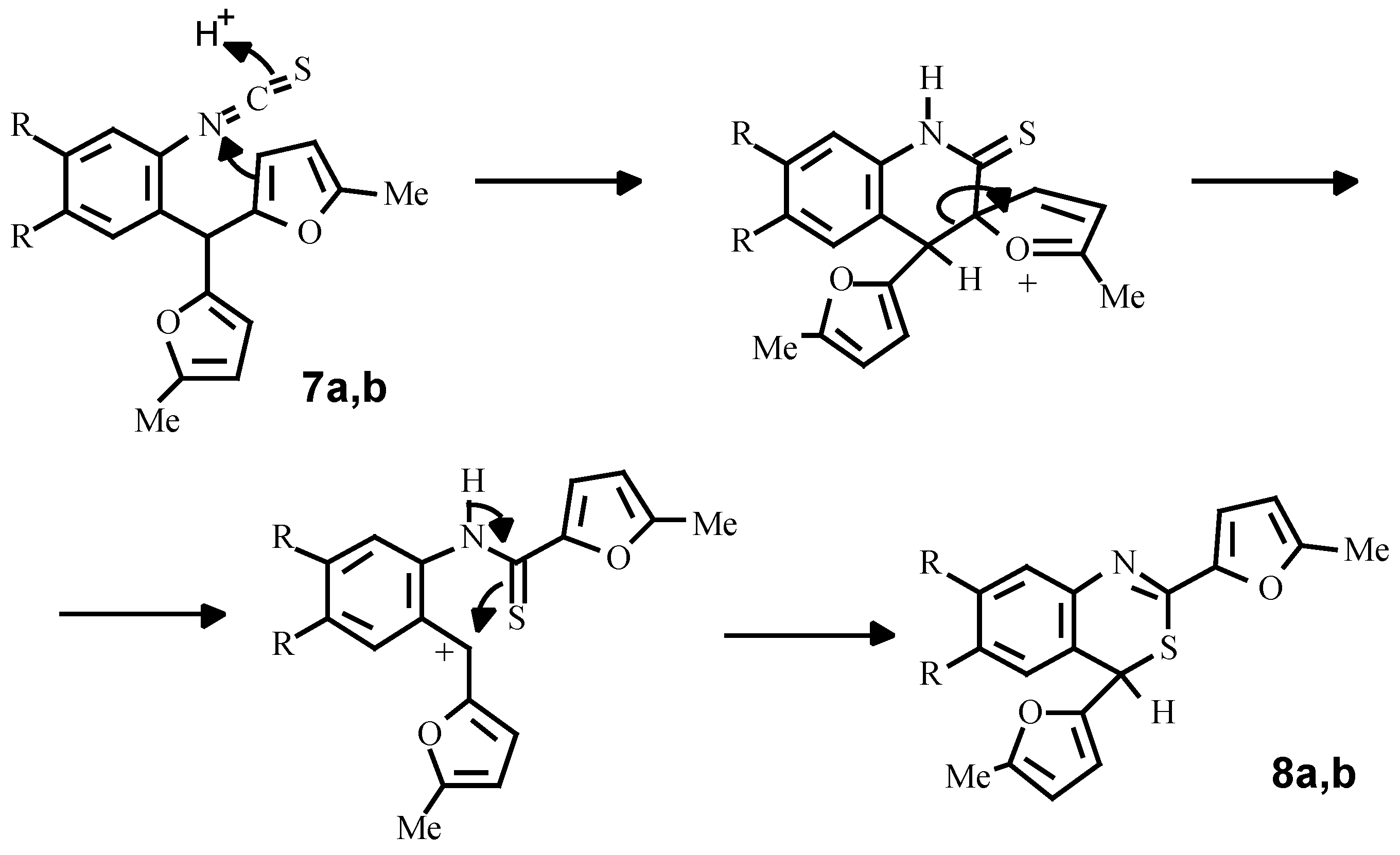
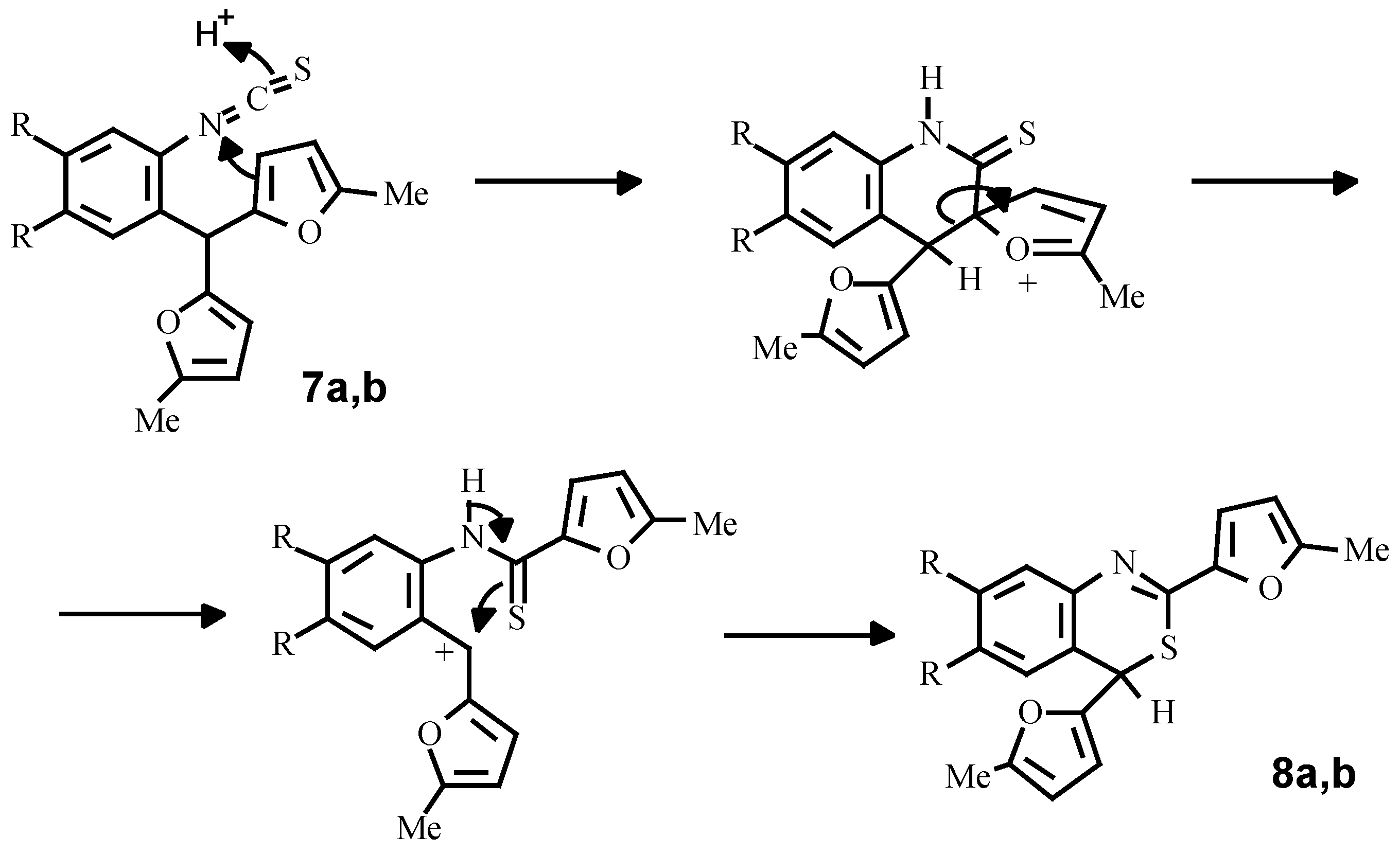
Finally, using a standard procedure, we have obtained isothiocyanates
7a, 8b which under the action of perchloric acid are transformed into 4
H-bensothiazines-3,1 (
8a, 8b). A possible mechanism of this conversion consists of intramolecular ipso-substitution of one of the furan rings by the protonated isothiocyano-group with subsequent alkylation of the sulphur-atom by a carbenium cation, arising from C-C bond fission (
Scheme 6).
The described results open up new possibilities in the application of difurylarylalkanes as synthons for condensed polycyclic heterocyclic compound synthesis.
Experimental
General
1H NMR spectra were recorded on a Bruker AMX-400 400 MHz and Tesla 80 80 MHz spectrometers. IR spectra were obtained with a Specord M80 spectrometer. Low resolution mass-spectra were recorded on a Varian Model 112M mass spectrometer.
Bis(5-methylfur-2-yl)-2-nitrophenylmethane (1a)
To a solution of 2-nitrobenzaldehyde (7.55 g, 50 mmol) and 2-methylfuran (15 mL) in dioxane (70 mL), perchloric acid (0.5 mL) was added. The mixture was left overnight then poured into water (0.5 L) and stirred until the brown oil was crystallised. The crystalline product was filtered off, air dried and recrystallised from hexane. Yield 11 g (74 %). Mp 83 °C (hexane/CH2Cl2). 1H NMR (CDCl3): δ 7.71-7.50 (m, 1H, 3-HAr), 7.48-7.18 (m, 3H, 4-,5-,6-HAr ), 6.17 (s, 1H, CH), 5.95-5.77 (m, 4H, Fur), 2.18 (s, 6H, 2 CH3). Anal. Calcd. for C17H15NO4: C, 68.68; H, 5.09; N, 4.71. Found: C, 68.71; H, 5.12; N, 4.69.
Bis(5-methylfur-2-yl)-4,5-dimethoxy-2-nitrophenylmethane (1b)
Yield 69 %. Mp 95 °C (methanol). 1H NMR (CDCl3): δ 7.48 (s, 1H, 3-HAr), 6.65 (s, 1H, 6-HAr), 5.90-5.69 (m, 5H, CH+Fur), 3.82 ( s, 3H, OCH3), 3.68 (s, 3H, OCH3), 2.18 (s, 6H, 2CH3). Anal. Calcd. for C19H19NO6: C, 63.86; H, 5.36; N, 3.92. Found: C, 63.89; H, 5.40; N, 3.91.
Bis(5-methylfur-2-yl)-2-aminophenylmethane (2a)
To a mixture of zinc dust (10 g), methanol (50 mL) and trimethylchlorosilane (4 mL) a solution of compound 1a (2.97 g, 10 mmol) in dioxane (15 mL) was added dropwise. The reaction mixture was stirred at room temperature until the starting compound was consumed (TLC - check), then filtered from the inorganic salts and excess of zinc dust. The filtrate was poured into water and extracted with ether. The ethereal layer was separated, dried over anhydrous sodium sulphate and evaporated to dryness. The residue was purified by column chromatography (silica gel, CH2Cl2/hexane) to give the pure amine 2a (1.87 g, yield 70 %). 1H NMR (CDCl3): δ 7.12 (ddd, J=1.3, 8.0, 8.5Hz, 1H, 4-HAr), 6.90 (dd, J = 1.3, 7.5 Hz, 1H, 6-H1-Fur), 6.77 (ddd, J=0.9, 7.5, 8.5 Hz, 1H, 5-HAr), 6.71 (dd, J=0.9, 8.0, 1H, 3-HAr), 5.93 (s, 4H, HFur), 5.41 (s, 1H, CH), 3.67 (br. s, 1H, NH), 2.28 (s, 6H, CH3). Anal. Calcd. for C17H17NO2: C, 76.41; H, 6.38; N, 5.27. Found: C, 76.38; H, 6.41; N, 5.24.
Bis(5-methylfur-2-yl)-2-amino-4,5-dimethoxyphenylmethane (2b)
Yield 73 %. Mp 81 °C (hexane/CH2Cl2). IR (vaseline oil, NaCl): 3450, 3380 cm-1 (NH2). 1H NMR (CDCl3): δ 6.41 (s, 1H, 6-HAr), 6.23 (s, 1H, 3-HAr), 5.83 (s, 4H, Fur), 5.27 (s, 1H, CH), 3.73 (s, 3H, OCH3), 3.62 (s, 3H, OCH3), 3.20 (s, 2H, NH2), 2.18 (s, 6H, CH3). Anal. Calcd. for C19H21NO4: C, 69.71; H, 6.47; N, 4.28. Found: C, 69.75; H, 6.51; N, 4.26.
2-(3-Oxobut-1-enyl)-3-(5-methylfur-2-yl) indole (3)
A solution of 2-nitrophenylmethane 1a (2.97 g, 10 mmol ) in ether (50 mL) was vigorously stirred with a solution of SnCl2·2H2O (10 g) in 5N HCl (40 mL) until the starting compound was consumed (TLC, 6 h). The organic layer was separated, washed with NaHCO3 solution, filtered through alumina and evaporated to dryness. The recrystallisation of the residue from toluene gave 1.4 g (yield 53 %) of yellow crystals. Mp 216 °C (toluene). IR (vaseline oil, NaCl): 3320 (NH), 1680 cm-1 (CO). 1H NMR (acetone-D6): δ 10.92 (b, 1H, NH), 8.12 (d, J = 16 Hz, 1H, β-H), 7.91 (dd, J = 8.0, 0.5 Hz, 1H, 4-H), 7.42 (dd, J = 8.0, 0.5 Hz, 1H, 7-H), 7.32-7.24 (m, 1H, 5-H), 7.18-7.09 (m, 1H, 6-H), 6.78 (d, J = 16.0 Hz, α-H), 6.68 (d, J = 3.2 Hz, 1H, 3-HFur), 6.25 ( d, J = 3.2 Hz, 1H, 4-HFur), 2.43 (s, 3H, CH3), 2.37 (s, 3H, COCH3). Anal. Calcd. for C17H15NO2: C, 76.96; H, 5.70; N, 5.28. Found: C, 76.91; H, 5.67; N, 5.32.
Bis(5-methylfur-2-yl)-2-hydroxylaminophenylmethane (4)
To a stirred solution of 2-nitrophenyldifurylmethane 1a ( 0.5 g 1.68 mmol) in THF (6 mL), a solution of NH4Cl (0.1 g) in water (3.5 mL) was added followed by zinc dust (0.46 g). The mixture was stirred at room temperature until the starting nitrocompound was consumed (TLC - check). The reaction mixture was filtered from inorganic salts, the filter cake was washed with toluene (5 mL) and the water layer was extracted with toluene (10 mL). The combined organic fractions were dried over sodium sulphate, evaporated in vacuo to yield 0.38 g ( 80 %) of the hydroxylamine 4 as an oil. 1H NMR (CDCl3): δ 7.32 - 6.87 (m, 4H, Ar), 5.82 (s, 4H, Fur.), 5.33 (s, 1H, CH), 2.19 (s, 6H, 2CH3).
As compound 4 is very unstable and easily destroyed by the action of acids and oxygen from the air, we consider it is not necessary to isolate the hydroxylamine 4. Compound 5 can be prepared from 4 in a one-pot procedure.
2-Hydroxy-2-methyl-5-(5-methylfur-2-yl)-2H-1.2-oxazyno[2,3-a]indole (5)
To a mixture of H2SO4 (0.125 mL) and water (1.65 mL), cooled in an ice-bath, the solution of hydroxylamine 4 (0.38 g, 1.34 mmol) in THF (10 mL) (the previous reaction mixture) was added immediately followed by an addition of K2Cr2O7 (0.12 g, 0.4 mmol) in water (3.5 mL). After 10 min the reaction mixture was extracted with ether, and the combined ethereal solutions were washed with NaHCO3 solution, dried over Na2SO4 and evaporated to dryness to give compound 5 (0.31 g, yield 82 % ) as an oil. IR (film, NaCl): 3380 cm-1 (b, OH). 1H NMR (CDCl3): δ 7.86 (dd, J = 8.0, 0.5 Hz, 1H, 6-H), 7.50 (dd, J = 8.0, 0.5 Hz, 1H, 9-H), 7.32-7.28 (m, 1H, 7-H), 7.22 (d, J = 10 Hz, 1H, 3-H), 7.20-7.16 (m, 1H, 8-H), 6.50 (d, J = 3.2 Hz, 1H, 3-HFur), 6.12 (d, J = 3.2 Hz, 1H, 4-HFur), 6.06 (d, J = 10 Hz, 1H, 4-H), 3.49 (b, 1H, OH), 2.42 (s, 3H, CH3), 1.81 (s, 3H, CH3). MS: m/e 281 (M+). Anal. Calcd. for C17H15NO3: C, 72.58; H, 5.37; N, 4.98. Found: C, 72.62; H, 5.38; N, 5.01.
3-(3-Oxobut-1-enyl)-4-(5-methylfur-2-yl)-6,7-dimethoxycynnoline (6)
To a stirred solution of amine 2b (3.27 g, 10 mmol) in acetonitrile (15 mL), trimethylchlorosilane (2 mL) and isoamylnitrite (1.5 mL) were added consecutively. The mixture was then stirred for an additional 15 min, poured into water (200 mL) and made alkaline with sodium carbonate. The crude cinnoline having separated from the reaction mixture on standing was filtered off, washed with methanol on the filter and air dried. The recrystallisation of this from ethylacetate gave 2.9 g (yield 86 % ) of yellow needles. Mp 216 °C (acetone). IR (vaseline oil, NaCl): 1680 cm-1 (CO). 1H NMR (CDCl3): δ 7.74 (s, 1H, 5-H), 7.65 (s, 1H, 8-H), 7.15 (d, J = 12.2 Hz, 1H, β-H), 6.94 (d, J = 3.2 Hz, 1H, 3-HFur), 6.43 (d, J = 3.2 Hz, 1H, 4-HFur), 6.41 (d, J = 12.2 Hz, 1H, α-H), 4.12 (s, 3H, OCH3), 4.03 (s, 3H, OCH3), 2.50 (s, 3H, CH3), 2.13 (s, 3H, COCH3). Anal. Calcd. for C19H18N2O4: C, 67.45; H, 5.36; N,8.28. Found: C, 67.47; H, 5.40; N, 8.30.
Bis(5-methylfur-2-yl)-2-isothiocyanatophenylmethane (7a)
To a stirred mixture of amine 2b (2.65 g, 10 mmol), CHCl3 (5 mL) and water (3 mL) a solution of thiophosgene (1mL in 3 mL CHCl3) was added simultaneously with a saturated aqueous sodium carbonate (3 g) solution. After stirring for 20 minutes at room temperature the mixture was quenched with excess of water and extracted with chloroform. The extract was dried over CaCl2, evaporated to dryness and the residue recrystallised from hexane. Yield 2.9 g (78 %). Mp 62 °C (hexane). 1H NMR (CDCl3): δ 7.21-7.05 (m, 4H, HAr), 5.88-5.79 (m, 4H, Fur), 5.59 (s, 1H, CH), 2.17 (s, 6H, 2CH3). Anal. Calcd. for C18H15NO2S: C, 69.88; H, 4.89; N, 4.53; S, 10.36. Found: C, 69.85; H, 4.86; N, 4.51; S, 10.40.
Bis(5-methylfur-2-yl)-2-isothiocyanato-4,5-dimethoxyphenylmethane (7b)
Yield 75 %. Mp 90-91 °C (hexane). 1H NMR (CDCl3): δ 6.69 (s, 1H, 3-HAr), 6.61 (s, 1H, 6-HAr), 5.91-5.84 (m, 4H, HFur), 5.54 (s, 1H, CH), 3.79 (s, 3H, OCH3), 3.72 (s, 3H, OCH3), 2.19 (s, 6H, 2CH3). Anal. Calcd. for C20H19NO4S: C, 65.02; H, 5.18; N, 3.79, S, 8.68. Found: C, 65.07; H, 5.22; N, 3.81; S, 8.65.
2,4-Bis(5-methylfur-2-yl)-4H-bensothiazine-3,1 (8a)
A mixture of isothiocyanate 7a (5 g, 16 mmol), perchloric acid (6 mL) and dioxane (50 mL) was left to stand at room temperature for 5h and then poured into a 1% sodium carbonate solution (300 mL). The water layer was decanted from the precipitated tar and the latter was dissolved in 100 mL of benzene, dried over CaCl2, filtered through Al2O3 and evaporated to a quarter of the original volume. Hexane was added and the solution was left to stand overnight. Yellow crystalls was filtered off, washed with hexane and air dried. Yield 3.5 g ( 70 %). Mp 112 °C (hexane/benzene). 1H NMR (CDCl3): δ 7.47-7.11 (m, 4H, HAr), 6.97 (d, J = 3.2 Hz, 1H, 3-H1-Fur), 6.07 (d, J = 3.2 Hz, 1H, 4-H1-Fur), 5.68 (d, J = 3.2 Hz, 1H, 4-H2-Fur), 5.62 (d, J = 3.2 Hz, 1H, 3-H2-Fur), 5.24 (s, 1H, CH), 2.37 (s, 3H, CH3), 2.18 (s, 3H, CH3). Anal. Calcd. for C18H15NO2S: C, 68.88; H, 4.89; N, 4.53; S, 10.36. Found: C, 69.90; H, 4.93; N, 4.55; S, 10.33.
2,4-Bis(5-methylfur-2-yl)-6,7-dimethoxy-4H-bensothiazine-3,1 (8b)
Yield 75 %. Mp 160 °C (hexane/benzene). 1H NMR (CDCl3): δ 7.07 (s, 1H, 3-HAr), 6.89 (d, J = 3.2 Hz, 1H, 3-H1-Fur), 6.56 (s,1H,6-HAr), 6.03 (d, J = 3.2 Hz, 1H, 4-H1-Fur), 5.71 (d, J = 3.2 Hz, 1H, 4-H2-Fur), 5.59 (d, J = 3.2 Hz, 1H, 3-H2-Fur), 5.16 (s, 1H, CH), 3.87 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 2.34 (s, 3H, CH3), 2.15 (s, 3H, CH3). Anal. Calcd. for C20H19NO4S: C, 65.02; H, 5.18; N, 3.79; S, 8.68. Found: C, 64.99; H, 5.17; N, 3.75; S, 8.69.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}