Photoisomerization of Ethyl 2–(3–Acylselenoureido)thiophene– 3–carboxylates and Their Benzoanalogues

1
Department of Organic Chemistry, Faculty of Science, Masaryk University, CZ–611 37 Brno, Czech Republic
2
Department of Inorganic Chemistry, Faculty of Science, Masaryk University, CZ–611 37 Brno, Czech Republic
*
Author to whom correspondence should be addressed.
Molecules 1997, 2(9), 135-151; https://doi.org/10.3390/20900135
Submission received: 6 January 1997
/
Accepted: 24 May 1997
/
Published: 15 September 1997
Abstract
:Synthesis, isomerisation and structure elucidation of the title compounds 1–6 and its isomers 7–12 by FTIR, 1H, 13C, 15N, 77Se NMR spectroscopy is reported. Ethyl 2–(3–acylselenoureido)thiophene–3–carboxylates and their benzoanalogues (where acyl is benzoyl and pivaloyl) were prepared by addition of ethyl 2–aminothiophene–3–carboxylates and ethyl 2–aminobenzoate on benzoyl– or pivaloylisoselenocyanate in acetone solution. An isomerization of 1–6 to the corresponding 3–acylisoselenoureas 7–12 was obtained. The isomerisation proceeds either by irradiation with light (340–400 nm) or in the case of benzoylderivatives 1, 3, 5 by treatment with acetic acid. On the other hand the acid action in the pivaloyl set inhibited this isomerisation and evoked the retroisomerisation reaction of 8, 10, 12 to 2, 4, 6. Thermal analyses showed that isomerisation can be initiated also by heating. These changes proceed in the solid phase as an exothermic process at an elevated temperature but always below the temperature of melting. The structure 2 was supported by X–ray analysis. Molecular design of 2 and 8 was modeled during application of ab initio quantum chemistry calculation.
Introduction
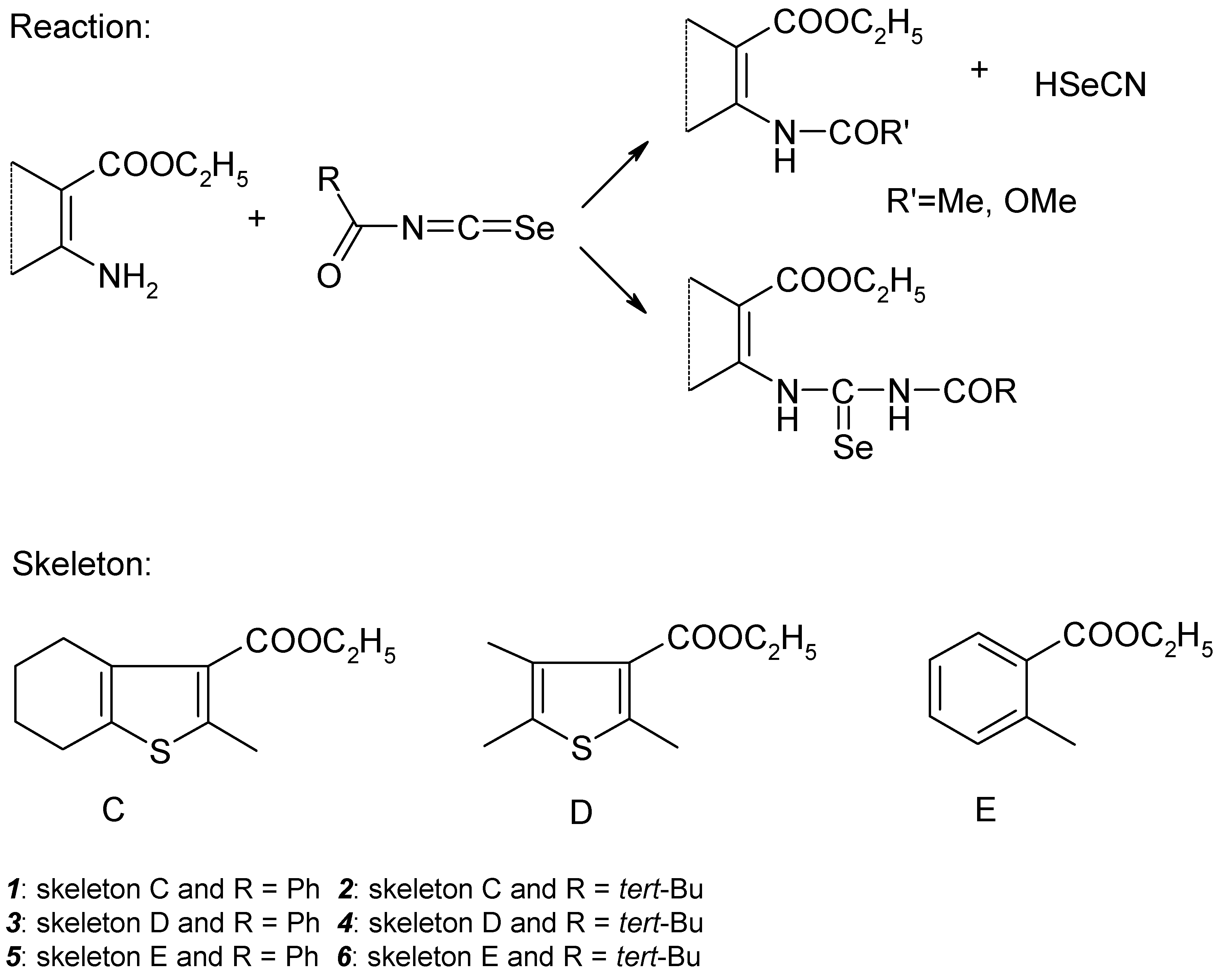
The research interest of our group for the past few years has lain in the synthesis of fused pyrimidine derivatives via cyclisation reactions of carbonic acid functional derivatives, primarily guanidines, ureas and thioureas obtained from aromatic and heterocyclic nitriles and esters of 2–aminocarboxylic acids [1]. In this connection it was necessary to prepare esters of some 2–(3–acylselenoureido)thiophene–3–carboxylic and benzoic acids.
1–Acyl–3–substituted selenoureas were prepared for the first time by Douglas via addition of amines to acylisoselenocyanates in anhydrous aprotic solvents [2]. Acylisoselenocyanates can be synthesized in situ by the reaction of acylchlorides with potassium selenocyanate in acetone solution. It is known that under these conditions there is competition between attack of the amino group on the C=Se double bond and on the C=O double bond of the acyl group. Depending on the nature of the acyl rest and on the type of amine (aliphatic or aromatic) one reaction can predominate. Good yields of selenourea products were obtained in the reaction of aroylisoselenocyanates with aromatic amines only [2].
Scheme 1.
Synthesis of acylselenoureas 1-6.
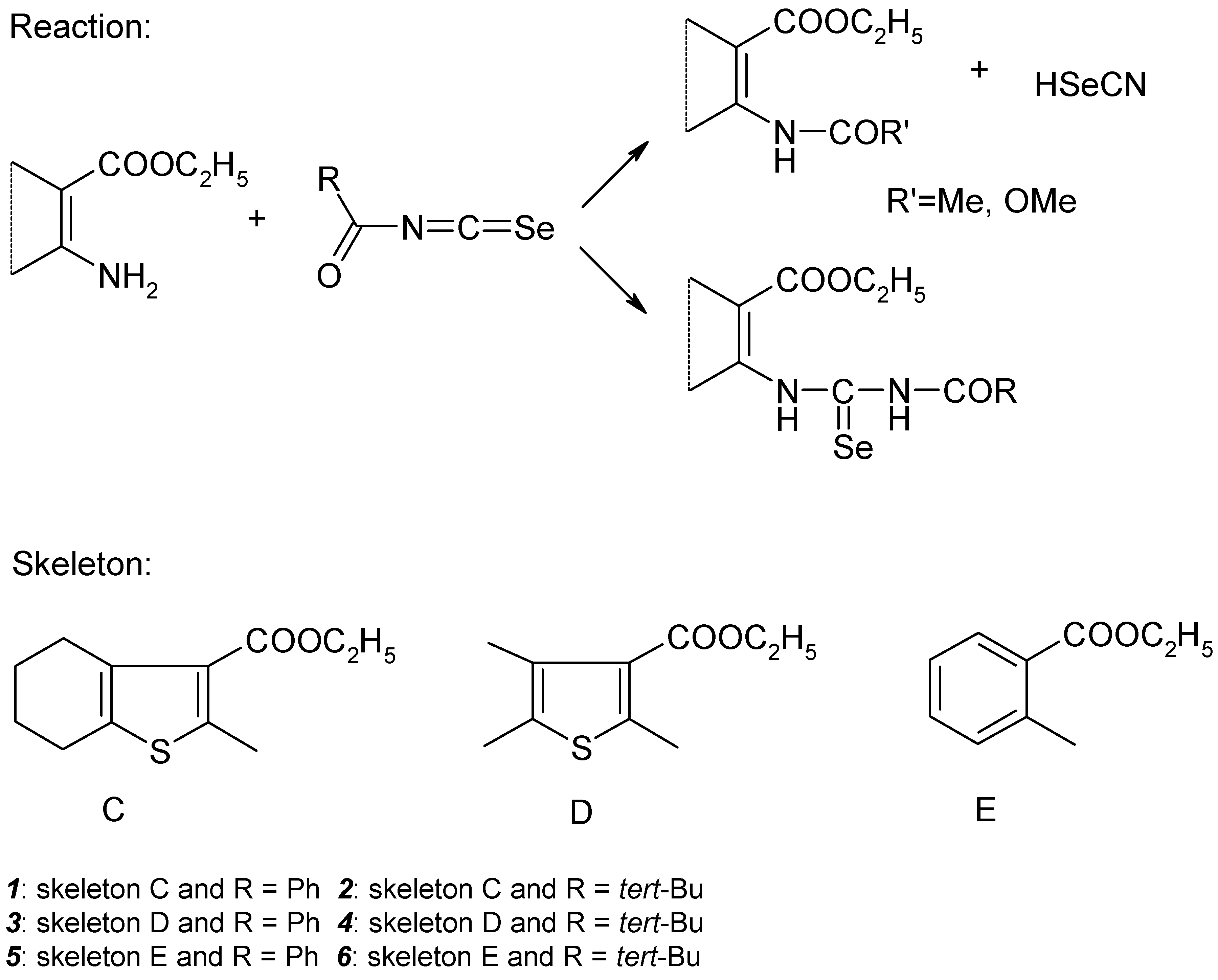
It is known that substituted thioureas [3,4] exist in several conformations in thermodynamic equilibrium due to restrictions of free rotation around the bonds N–(C=S) of the thioureido group. We propose the analogy between the behavior of thiourea and selenourea derivatives. X–ray structural analysis of 1–benzoyl–3–phenylselenourea (the compound structurally similar to the title compounds) was performed [5]. It was found that the molecular flexibility of this compound is further lowered by a hydrogen bond between the oxygen atom of the benzoyl group and the hydrogen atom on the atom N3. In the case of the title acylselenoureas the molecular flexibility decrease can be another explanation for further hydrogen bond existence.
The reason for the preparation of the mentioned acylselenoureas was also due to the information that selenium has been shown to be a trace element essential for animals. Its importance is connected with immunobiochemical activity of enzymatic selenoproteins [6]. It is also known [7] that the population of South Moravia showed serum selenium deficiency in blood with values similar to those found in the countries of China, Scandinavia and New Zealand [8].
We would like to study the acid– and base–initiated cyclization and retrocyclization reactions of the prepared acylselenoureas to fused 1,3–selenazines and fused 2–selenoxo–pyrimidines later.
Results and discussion
We have attempted to prepare acylselenoureas derived from ethylesters of 2–amino–4,5,6,7–tetrahydro–benzo[b]thiophene–3–carboxylic, 2–amino–5,6–dimethylthiophene–3–carboxylic and 2–aminobenzoic acids by an addition of the corresponding amines to acetyl–, benzoyl–, methoxycarbonyl– and pivaloylisoselenocyanate (Scheme 1), respectively. We have obtained good results for benzoyl– and pivaloylisoselenocyanates only. The others acylisoselenocyanates gave either the product of acylation of the starting aminoester, identified by acylation of the corresponding amines with acylchlorides, or a mixture of this with acylselenourea. We have found the same results for analogous addition of the corresponding nitriles [9]. Separation of acylselenoueas by column chromatography was unsuccessful for the reason of elemental selenium formation by oxidation of selenourea to urea derivatives. Therefore we focused our interest on the benzoyl– and pivaloylderivatives of the title compounds.
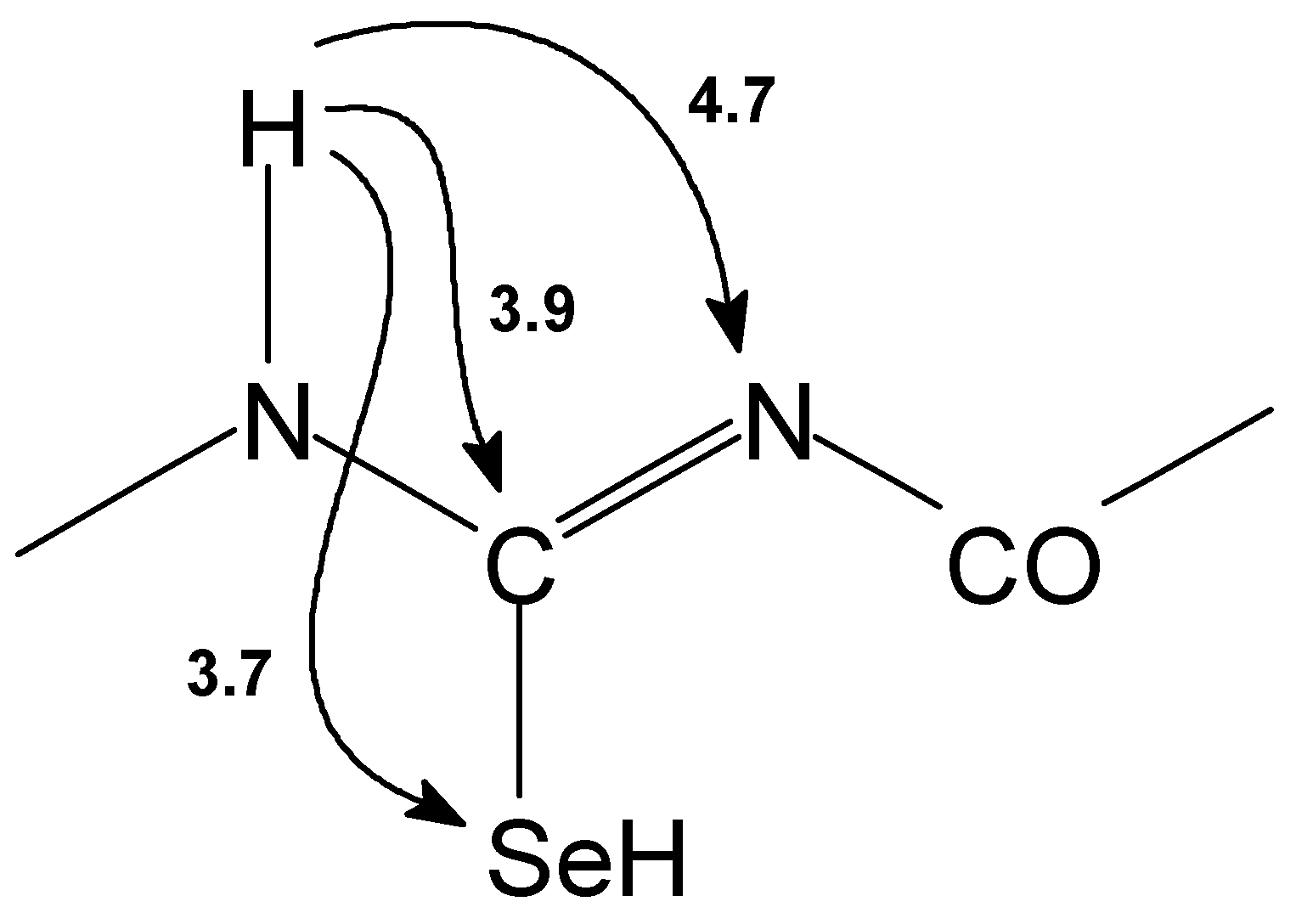
Figure 1.
The nonoptimized long range 1H-13C, 1H-15N and 1H-77Se interactions [Hz] observed for basic skeleton of acylselenourea 1.
Figure 1.
The nonoptimized long range 1H-13C, 1H-15N and 1H-77Se interactions [Hz] observed for basic skeleton of acylselenourea 1.
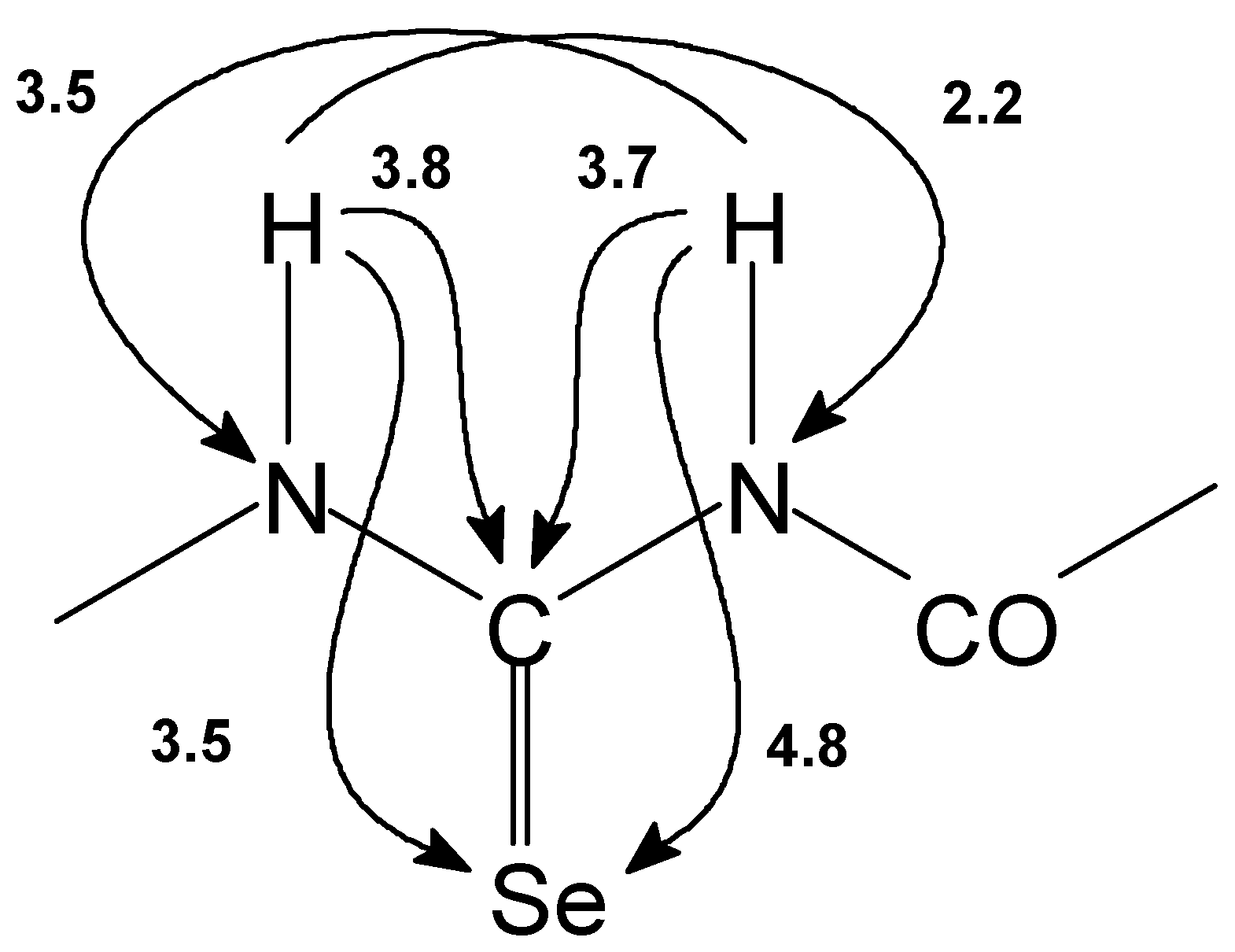
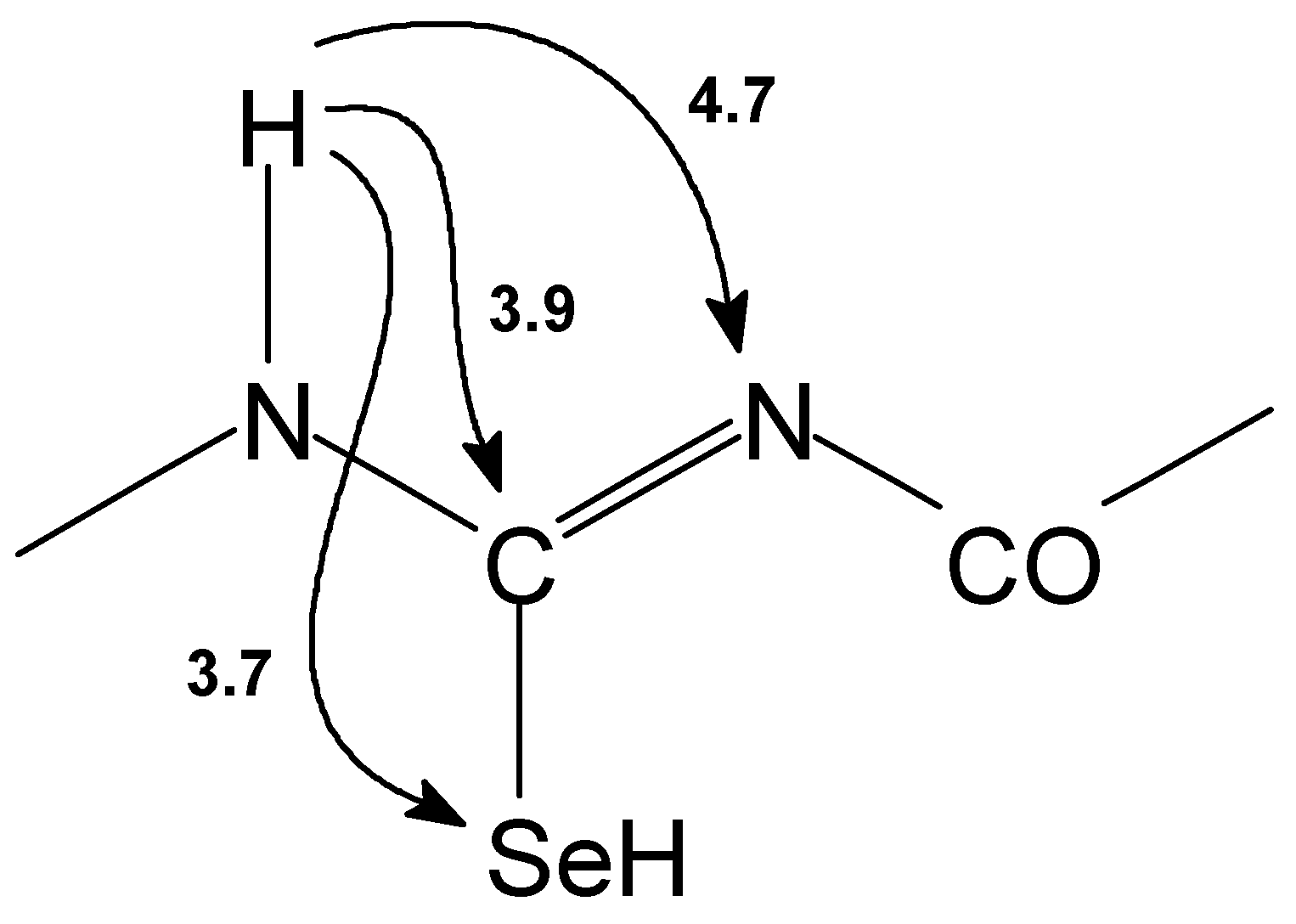
Structures of prepared selenoureas 1–6 were supported by CHNSe elemental analysis, FTIR spectra and 1H–, 13C–, 77Se– and in some cases also by 15N NMR spectroscopy. Assignment of the NMR signals was carried out on the basis of 2D chemical shift correlation experiments. 1H–1H dipolar interactions were determined by 2D NOESY experiment. For observation of 1H–13C interactions (both direct and long–range) HETCOR, COLOC, HSQC and HMBC pulse sequences were used. In HSQC and HMBC experiments pulsed field gradients were used for coherence selection. GHMBC and GSQMBC experiments [10] were also used for detection of 1H–15N and 1H–77Se long–range interactions. The nonoptimized long range 1H–13C, 1H–15N, 1H–77Se interactions observed for basic skeleton of acylselenourea 1 are presented on the Figure 1.
Structure type shown on Figure 1 is described by a few characteristic features (for atom numbering see Figure2). Thus signal of H(N–8) can be found around 9 ppm. The signal of H(N–6) at 14–15 ppm is shifted downfield due to the presence of two intramolecular hydrogen bonds with both carbonyl oxygen atoms. 13C NMR record displays C–7 carbon atom signal at ca. 175 ppm and its coupling with Se–9 atom is 220–225 Hz. N–6 and N–8 nitrogen atom signals can be found in the range of 150–170 ppm. Se–9 selenium atom signals can be found in the range of 383–480 ppm. Detail NMR study on heteronuclear long–range coupling constants will be presented elsewhere [11].
Also FTIR data, the first vibrational bands at 1685–1715 cm-1, 1250 cm-1 (COOC), 1650–1690 cm-1, 1550 cm-1 (bands amide I and II) and 1510–1530 cm-1, 960–980 cm-1 (bands selenoamide III and I), confirmed structure of the type 1–6.
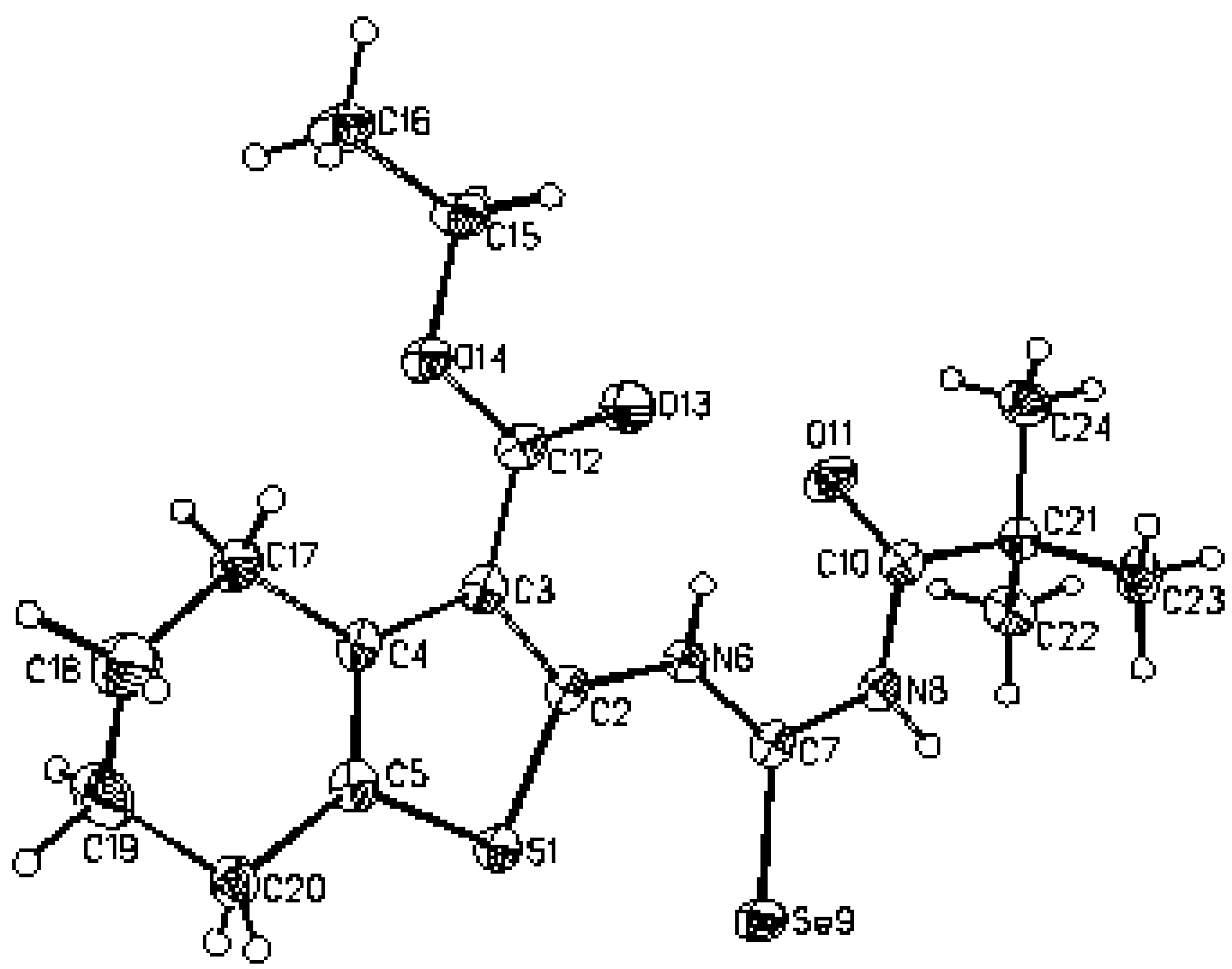
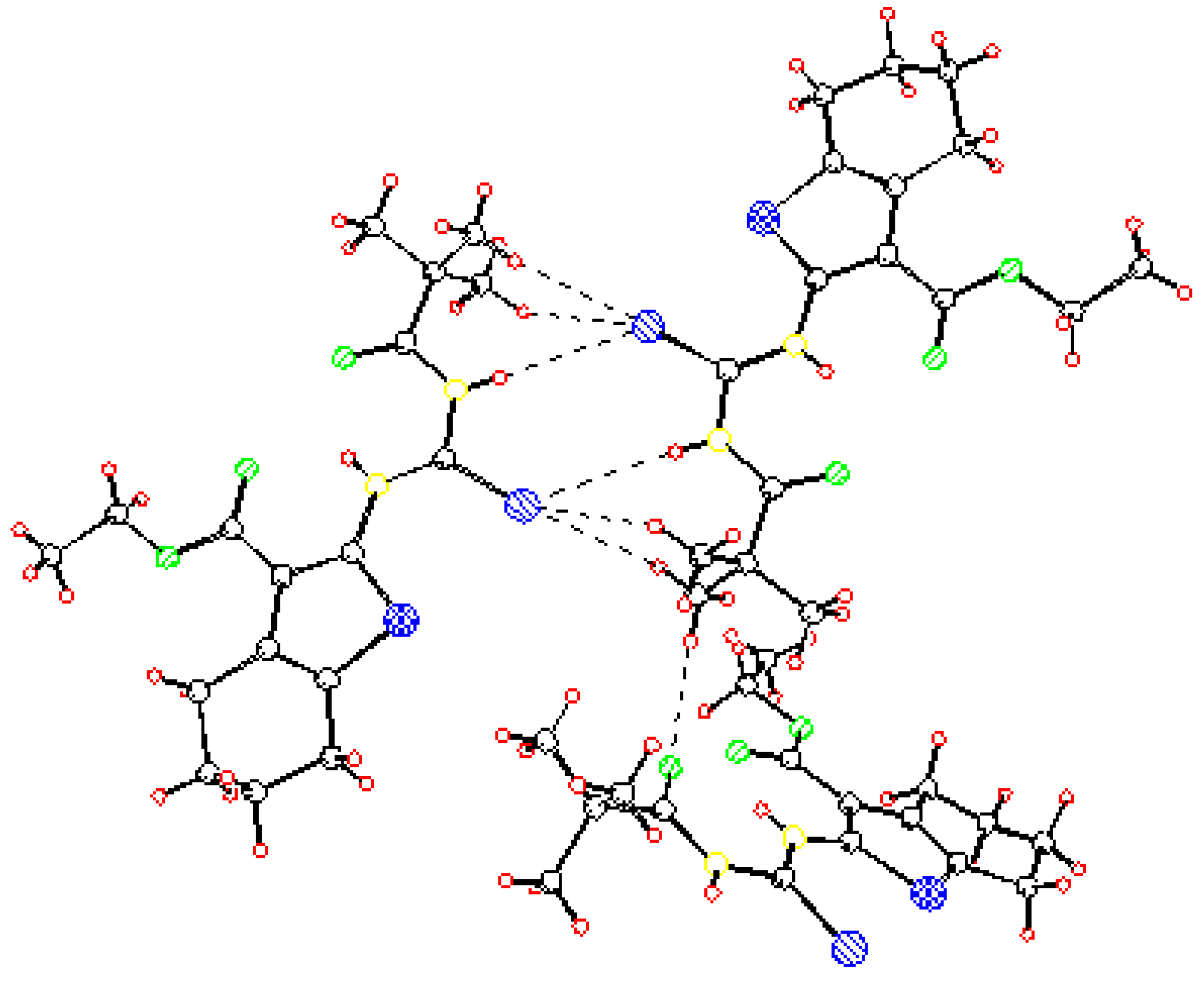
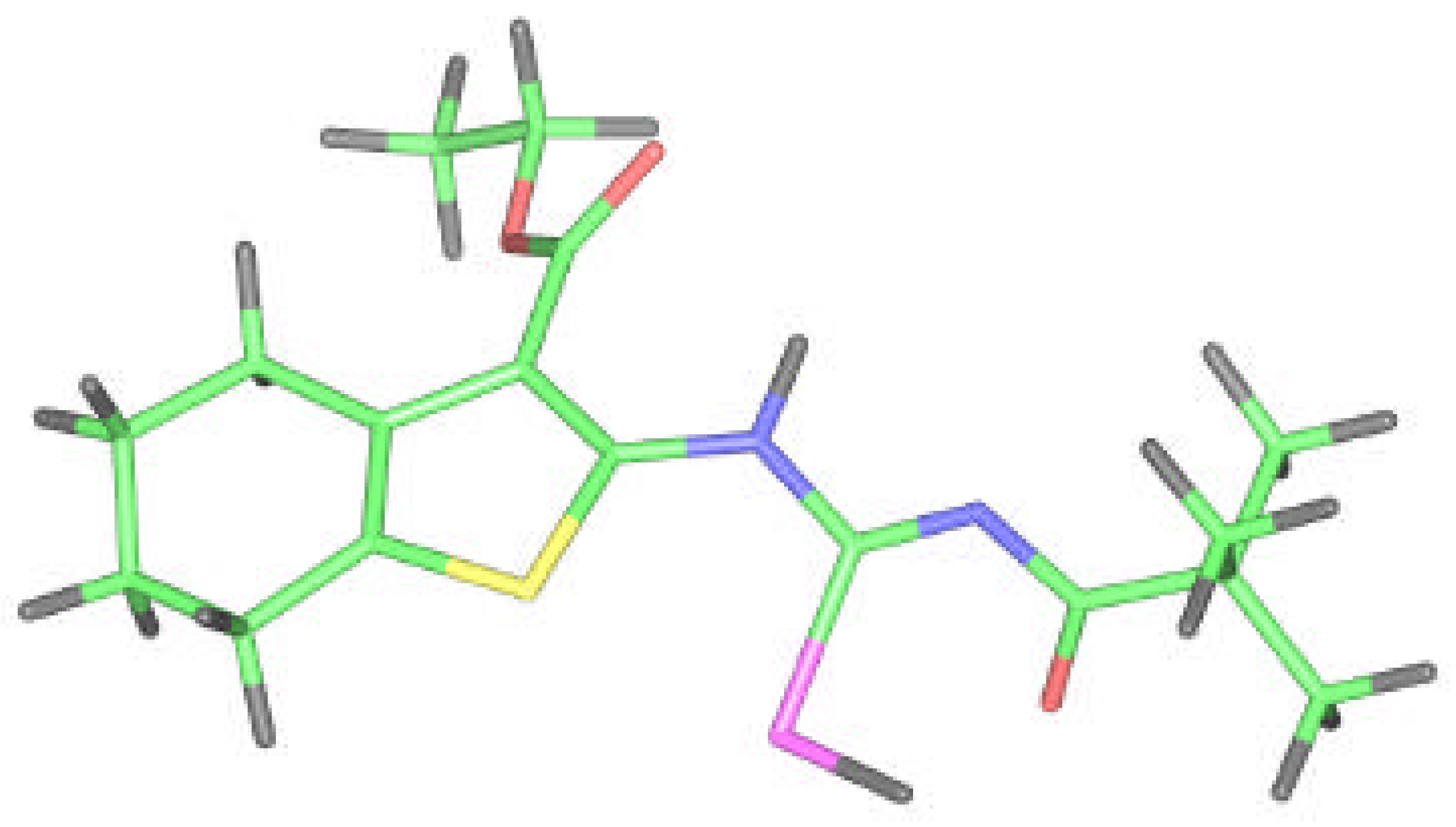
Ethyl 2–(3–pivaloylselenoureido)–4,5,6,7–tetrahydrobenzo[b]thiophene–3–carboxylate (2) was analyzed by X–ray structural analysis. The found structural data presented in Table 1, Table 2 and Table 3 correspond very well with the one calculated by ab initio DSF quantum chemistry calculations. This and the crystal structure (X–ray) of molecule 2 are presented in Figure 1, Figure 2 and Figure 3. It is shown that the flexibility of the molecule is restricted by H-bonding between hydrogen atom on N6 of selenoureido group and the oxygen atoms of both the carbonyl groups.
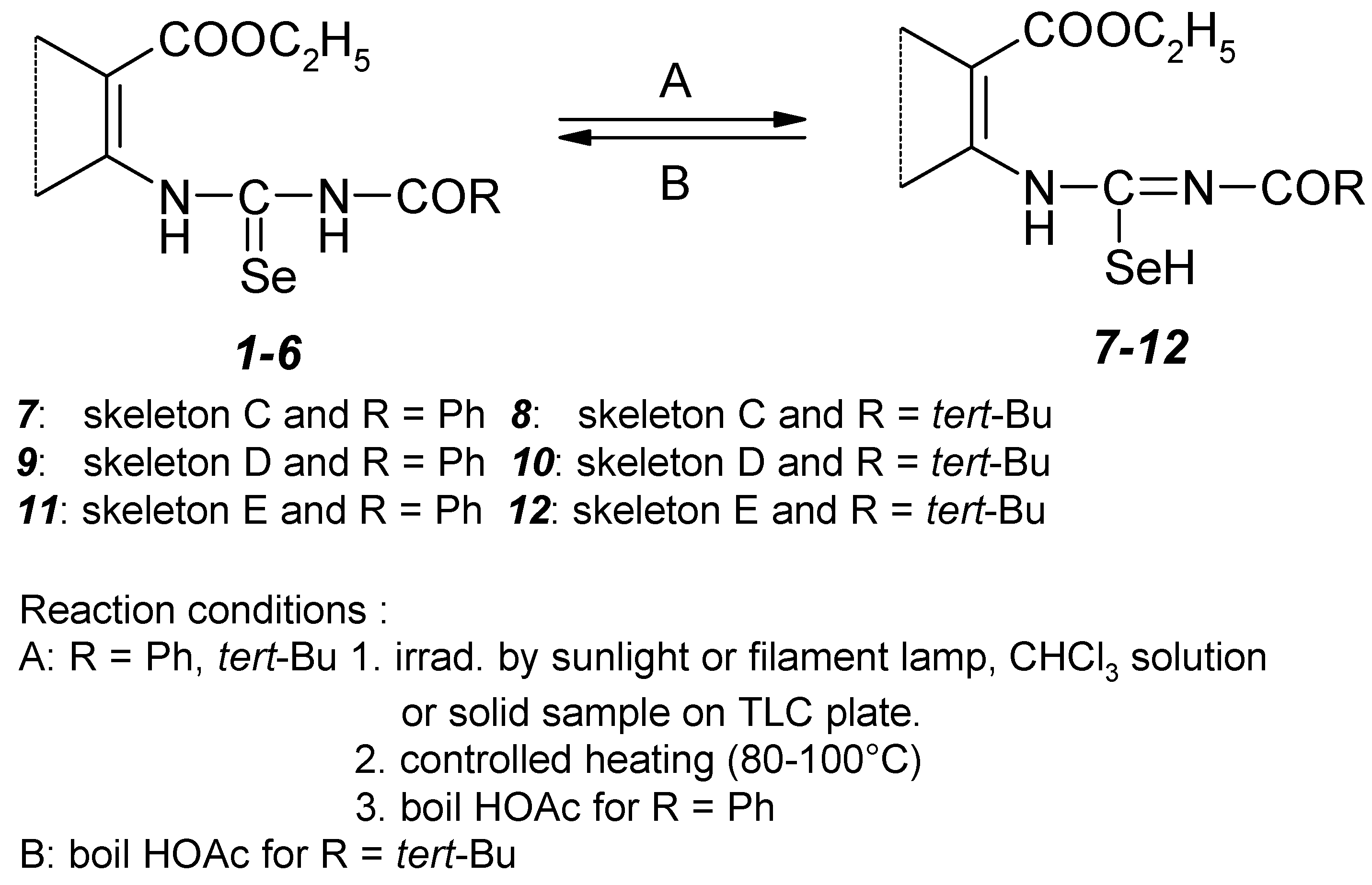
Because of the problems with oxidation of the prepared title selenoureas, we needed to work under an inert atmosphere. Besides but we met further problem. In the case of the individual selenoureas syntheses in a glass reaction vessel or during longer drying of TLC plates in the light (before TLC) we have observed the formation of a new compound. This could be obtained in a quantitative yield by extension of the irradiation time to sunlight or a filament lamp. Namely, UV–VIS spectra of starting selenoureas showed the presence of a long wave absorption band in the range of 340–400 nm. These new compounds were according to TLC more polar than the starting selenoureas and their solubility in the same solvents was also lower. The NMR spectra showed that these new compounds contain the same fragments as the starting selenoureas i.e. ethoxycarbonyl–, acyl–, selenocarbonyl groups but all of them with different chemical shifts.
These findings showed that the changes which proceeded were not fundamental. The values of elemental analyses and also melting points were identical for both series of compounds.
FTIR data, 1685–1715 cm-1, 1250 cm-1 (COOC) and 1650 cm-1 (NCO) also confirmed these facts. On the other hand we found the vibrational band of the C=N group at 1620–1630 cm-1 and observed an absence of the bands of NHCSe and NHCO groups (bands amide II and selenoamide I and III). All the presented facts led us to the conclusion that this transformation is a light initiated isomerization of the title 3–acylselenoureas 1–6 to corresponding 3–acylisoselenoureas 7–12.
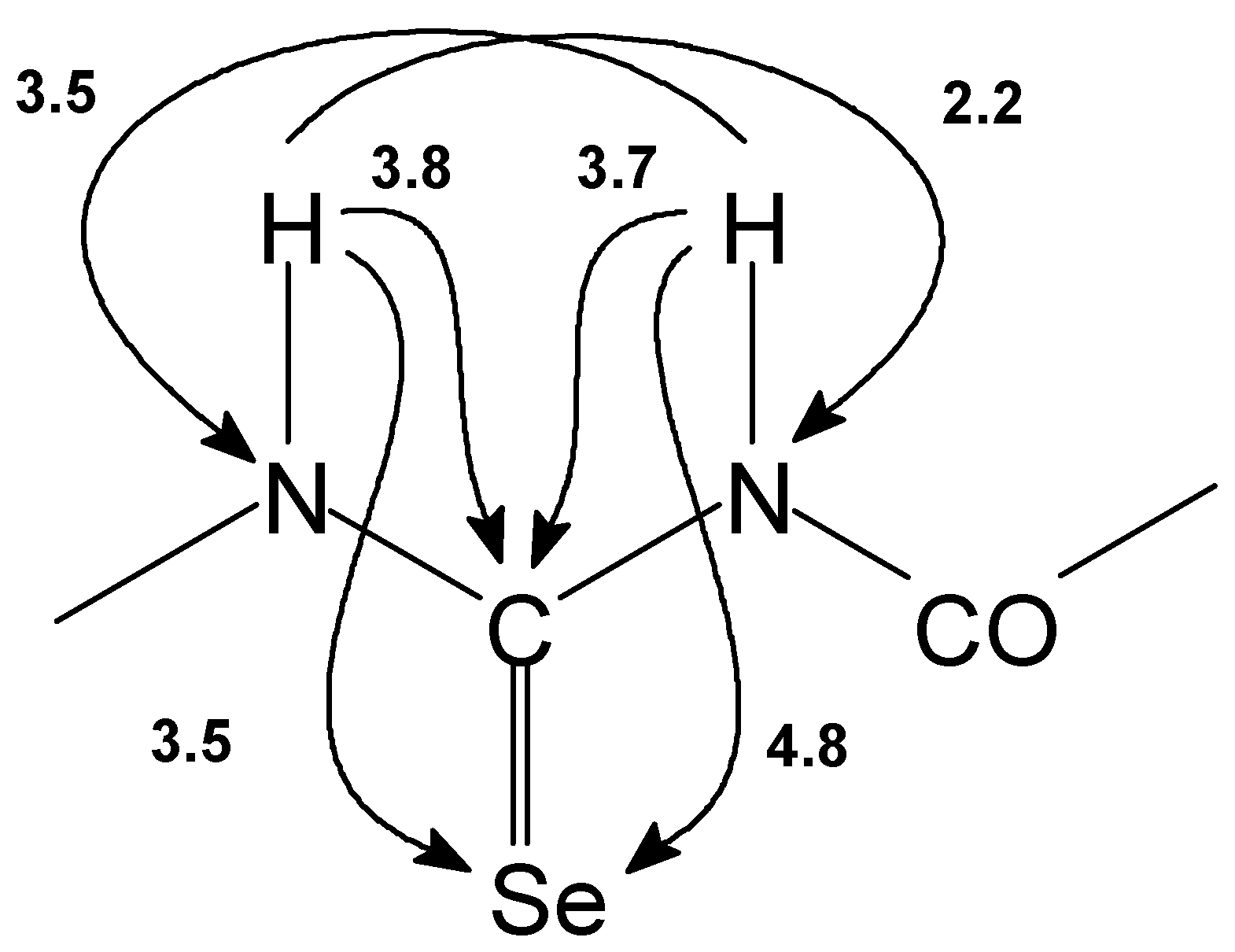
1H, 13C, 77Se and 15N NMR experiments supported our findings. We observed totally different chemical shifts for the basic skeleton part of the selenourea. The nonoptimized long range 1H–13C, 1H–15N and 1H–77Se interactions observed for the basic skeleton part of acylisoselenourea 7 are presented on the Figure 5.
Comparing the spectra with the structure of 1–6, H(N–8) signal disappeared in 1H NMR spectrum of structure 7–12. The signal of H(N–6) is shifted upfield by 2–3 ppm and appears approximately at 12 ppm. C–7 carbon atom signal is shifted from the original range 175–180 ppm and is situated near 160 ppm. Its coupling to Se–9 ranges between 192 Hz and 195 Hz. Se–9 selenium atom signal is shifted by approximately 50–150 ppm downfield compared with structure of 1–6. N–6 nitrogen atom signal one can find near 140 ppm but N–8 signal appears at 240 ppm in the range characteristic for nitrogen atom of C=N groups. However, the signal of suggested Se–H group was not detected in 1H NMR even at low temperature probably due to the fast chemical exchange. Detail NMR study on heteronuclear long–range coupling constants will be presented elsewhere [11].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interatomic distances (Å) | ||||
| Bond | 2 (X-ray) | 2 (calc.) | 8 (calc.) | |
| C2-N6 | 1.39 | 1.40 | 1.39 | |
| N6-C7 | 1.34 | 1.33 | 1.38 | |
| C7-N8 | 1.40 | 1.37 | 1.26 | |
| N8-C10 | 1.38 | 1.39 | 1.38 | |
| C10-C21 | 1.52 | 1.53 | 1.49 | |
| C7-Se9 | 1.81 | 1.84 | 1.96 | |
| Se9-HSe9 | - | - | 1.48 | |
| N6-HN6 | 0.99 | 1.01 | 1.03 | |
| N8-HN8 | 1.40 | 1.37 | - | |
| C10-O11 | 1.21 | 1.19 | 1.30 | |
| HSe9-O11 | - | - | 2.99 | |
| HN6-O13 | 1.95 | 1.93 | 2.63 | |
| Valency angles (deg.) | |||
| Angle | 2 (X-ray) | 2 (calc.) | 8 (calc.) |
| C2-N6-HN6 | 116.76 | 115.82 | 112.99 |
| HN6-N6-C7 | 116.76 | 116.72 | 116.83 |
| C2-N6-C7 | 126.48 | 128.85 | 130.14 |
| N6-C7-N8 | 119.18 | 116.62 | 116.83 |
| N6-C7-Se9 | 114.88 | 126.01 | 128.48 |
| Se9-C7-N8 | 125.94 | 117.37 | 114.68 |
| C7-N8-C10 | 125.69 | 128.64 | 131.24 |
| N8-C10-C21 | 115.87 | 115.83 | 114.54 |
| O11-C10-C21 | 112.07 | 129.79 | 123.45 |
| O11-C10-N8 | 122.00 | 124.38 | 122.01 |
| C7-Se9-HSe9 | 94.35 | - | - |
| Dihedral angles (deg.) | |||
| Angle | 2 (X-ray) | 2 (calc.) | 8 (calc.) |
| S1-C2-N6-HN6 | 170.28 | 171.44 | -130.53 |
| HN6-N6-C7-Se9 | 177.20 | 176.54 | 162.30 |
| HSe9-Se9-C7-N8 | - | - | 43.23 |
| Se9-C7-N8-C10 | 175.22 | 173.65 | 8.98 |
| C7-N8-C10-O11 | -176.47 | -0.90 | 36.87 |
| C7-N8-C10-C21 | 2.85 | 179.03 | -145.12 |
| C2-N6-C7-N8 | -177.38 | -179.79 | 162.44 |
| HSe9-Se9-C7-N8 | - | - | -136.62 |
Figure 2.
Molecular structure of 2 according to the X-ray analysis.
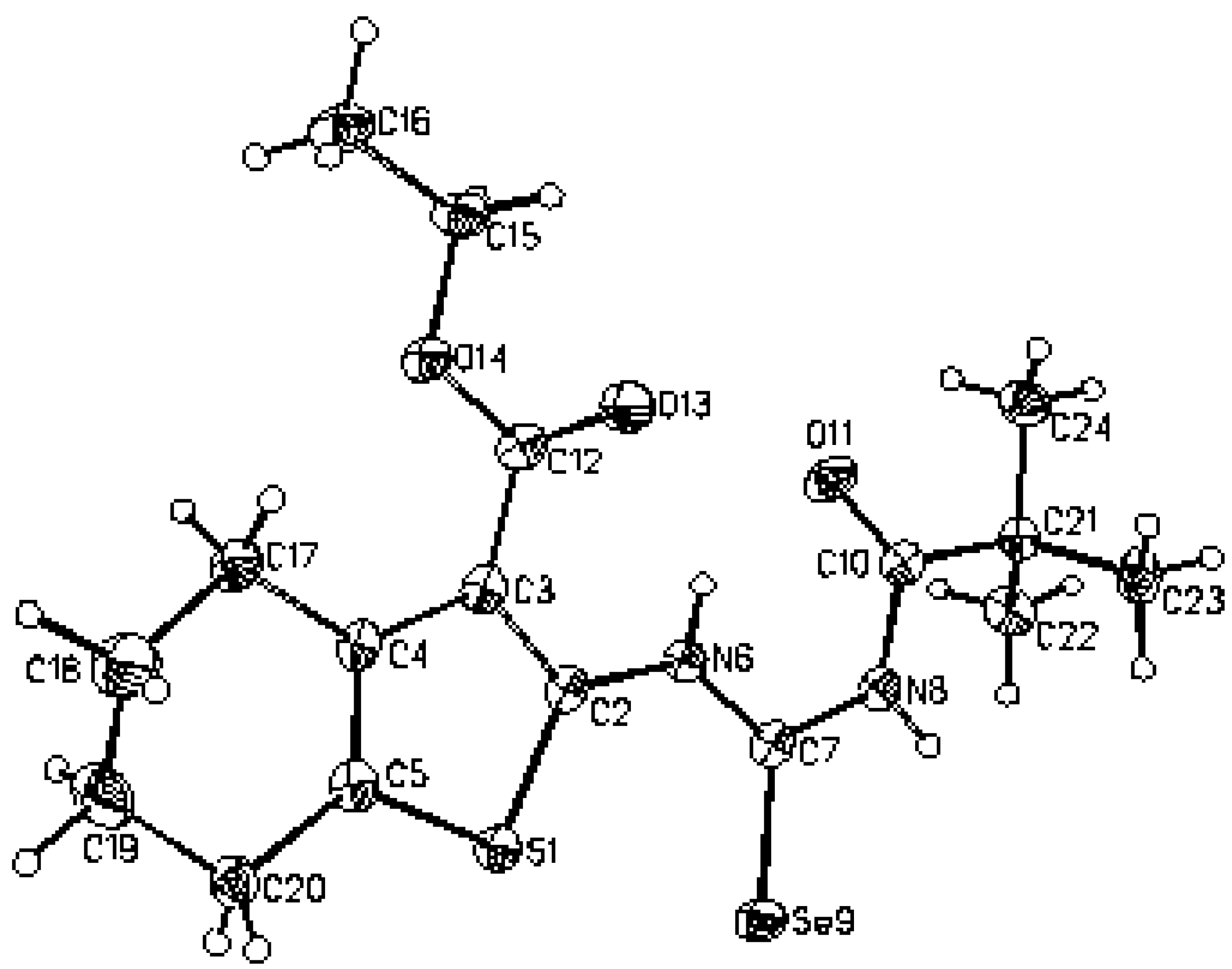
Figure 3.
Intermolecular interactions between two molecules of 2 according to the X-ray analysis.

Figure 4.
Molecular design of 2 according to the computational calculation.
Scheme 2.
Isomerization acylselenoureas 1-6 to acylisoselenoureas 7-12.
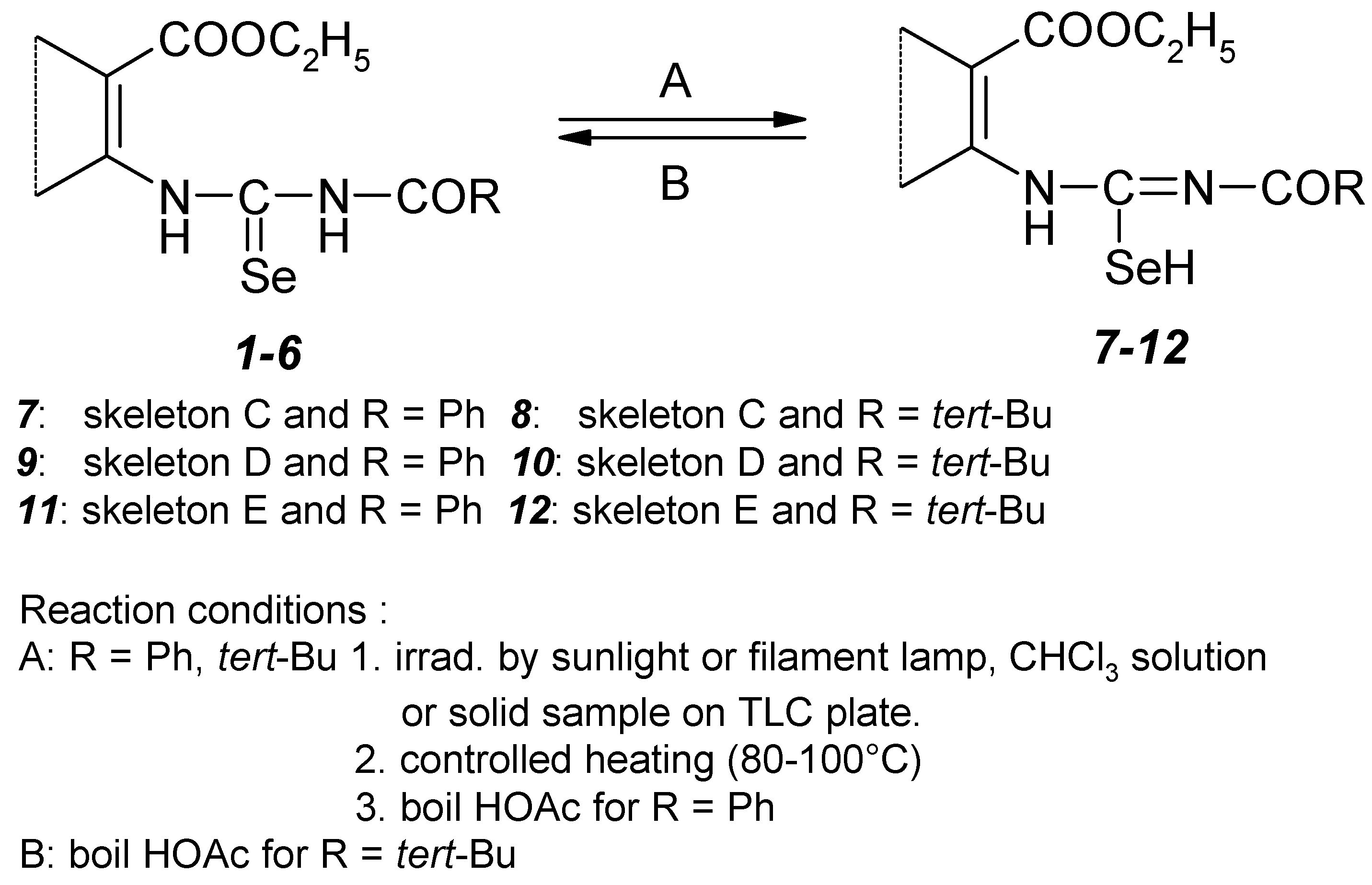
Figure 5.
The nonoptimized long range 1H-13C, 1H-15N and 1H-77Se interactions [Hz] observed for the basic skeleton part of acylisoselenourea 7.
Figure 5.
The nonoptimized long range 1H-13C, 1H-15N and 1H-77Se interactions [Hz] observed for the basic skeleton part of acylisoselenourea 7.
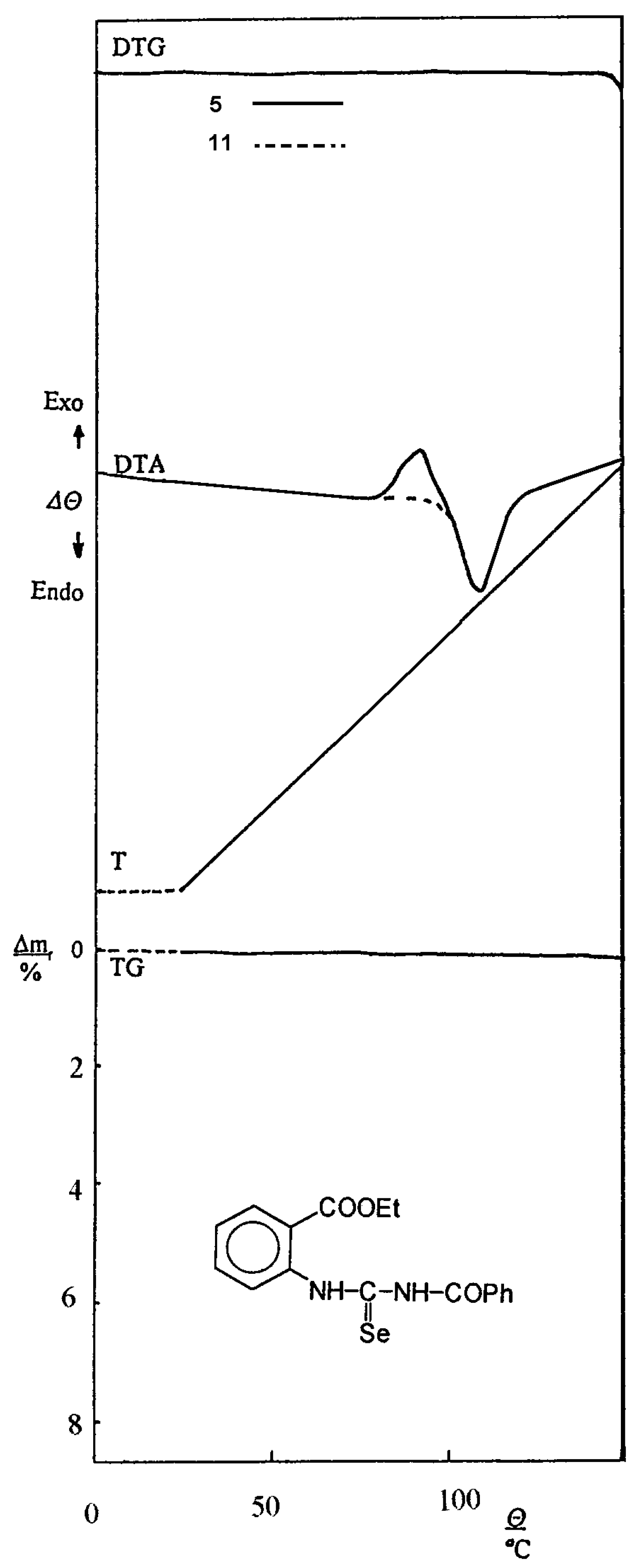
When melting points of the prepared acylselenoureas and their corresponding isomers were measured, we obtained the same values. This interesting observation was found by thermal analysis method in the temperature range of 25–150 °C. The characteristic records of DTA, DTG and TG curves of ethyl 2–(3–benzoyl-selenoureido)benzoate (5) and its isomer 11 are presented on Figure 6.
On the DTA curve of the starting acylselenourea 5 exothermic ΘE and endothermic ΘM peaks are observed, e.g. the changes proceed in the sample during the temperature rise.
The curve of its isomer is similar, the temperature values of exothermic ΘE and endothermic ΘM peaks of further compounds 1–6, 7–12 are presented in Table 4.
The differences are evident only in the range of the starting selenourea exothermic change. We suppose that the exothermic change is connected with the isomerization of the starting selenourea and with a reorganization of the crystalline sample. This assumption is supported by the character of TG and DTG curves, respectively. The course of both curves is in the above mentioned range without changes of masse and isomerization proceeds in the solid sample without melting. The endothermic peak on the DTA curve corresponds to the melting point. These actions could be also observed during the melting point measurement on a hot–stage microscope.
We wanted to support the conclusion about the detailed structure of the isoselenoureas by X–ray analyses. But our effort to prepare a convenient single crystal was unsuccessful. The crystals were prepared either in a thread form or as a microcrystalline sample.
On the other hand we found that treatment in hot anhydrous acetic acid or hydrogen chloride in an aprotic solvent caused the retroisomerization of the pivaloylisoselenoureas to the starting selenoureas. In the case of benzoylanalogues the change proceeds in the opposite way. Both changes in the presence of acid are irreversible. We must here present that we were not able to isomerize the cyanoanalogues of the title compounds 1–6 to isoselenoureas [9]. We found similar observation about the impossibility of isomerisation of the at thioureido– and ureido– analogues of studied compounds 1–6 [1].
Table 4.
Temperature values of exothermic ΘE and endothermic ΘM peaks of compounds 1-6, 7-12 and their melting points.
| Compound | ΘE / °C | ΘM / °C | m.p. / °C |
| 1 | 152 | 173 | 172-174 |
| 7 | - | 173 | 173-174 |
| 2 | 144 | 154 | 153-155 |
| 8 | - | 154 | 156 |
| 3 | 163 | 174 | 173-175 |
| 9 | - | 174 | 175-178 |
| 4 | 150 | 185 | 183-186 |
| 10 | - | 185 | 185-187 |
| 5 | 89 | 110 | 107-110 |
| 11 | - | 110 | 108-110 |
| 6 | 89 | 111 | 109-112 |
| 12 | - | 111 | 110-112 |
Figure 6.
The record of thermal analysis of 5 and 11.
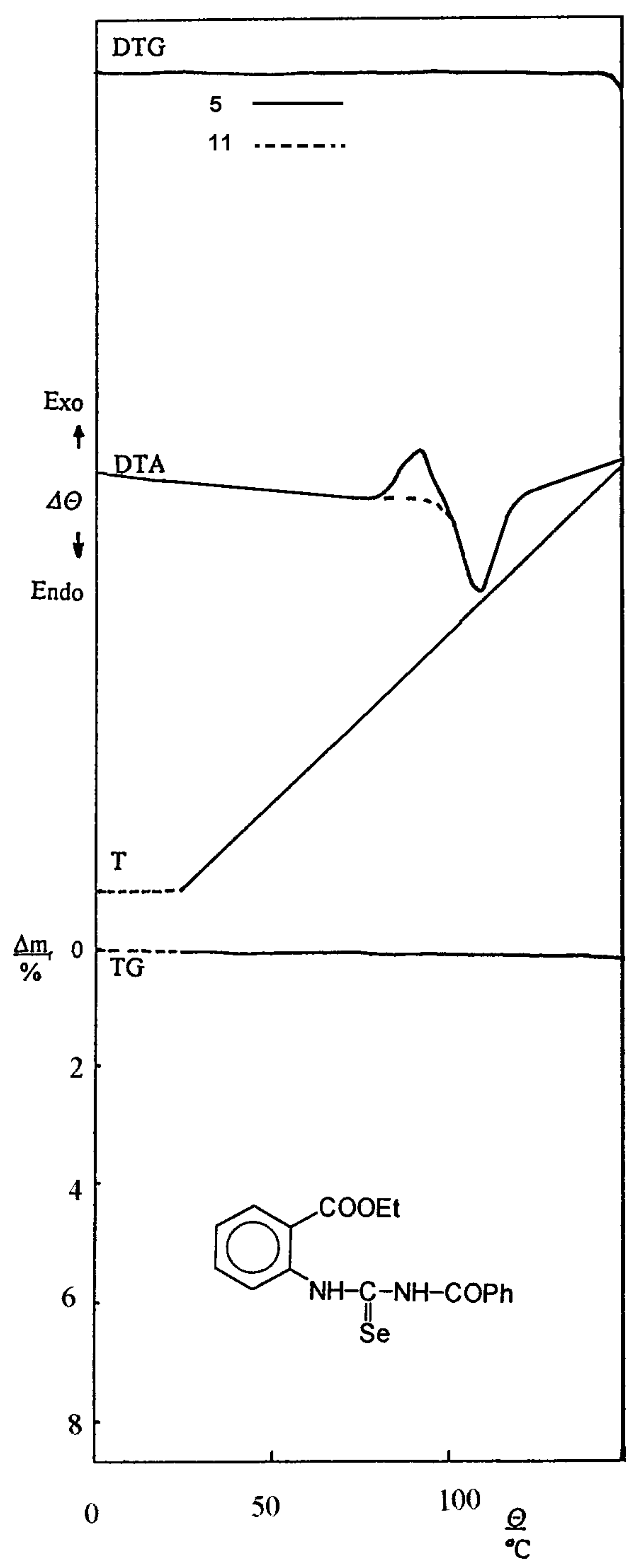
Figure 7.
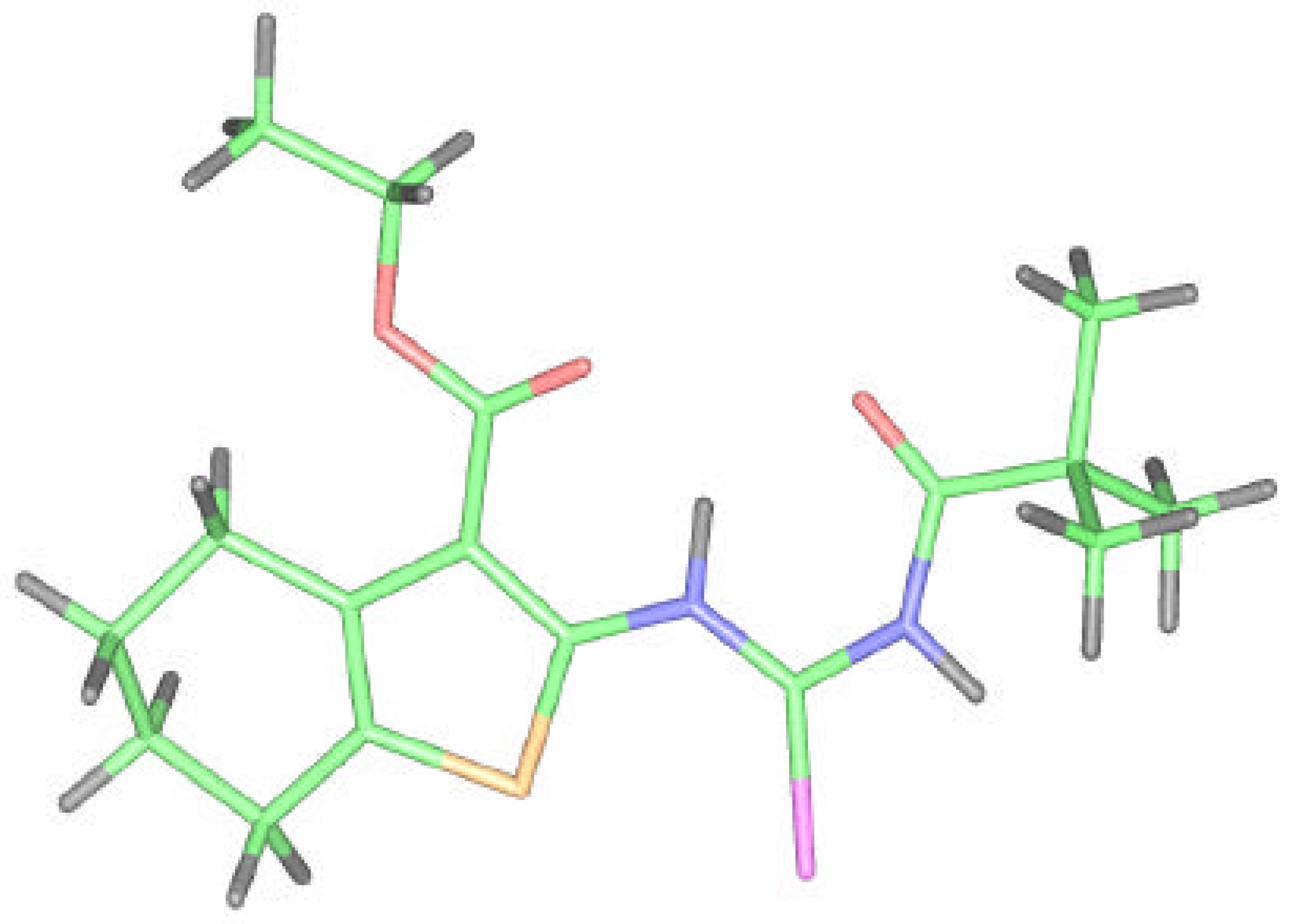
Molecular design of 8 according to the computational calculation.
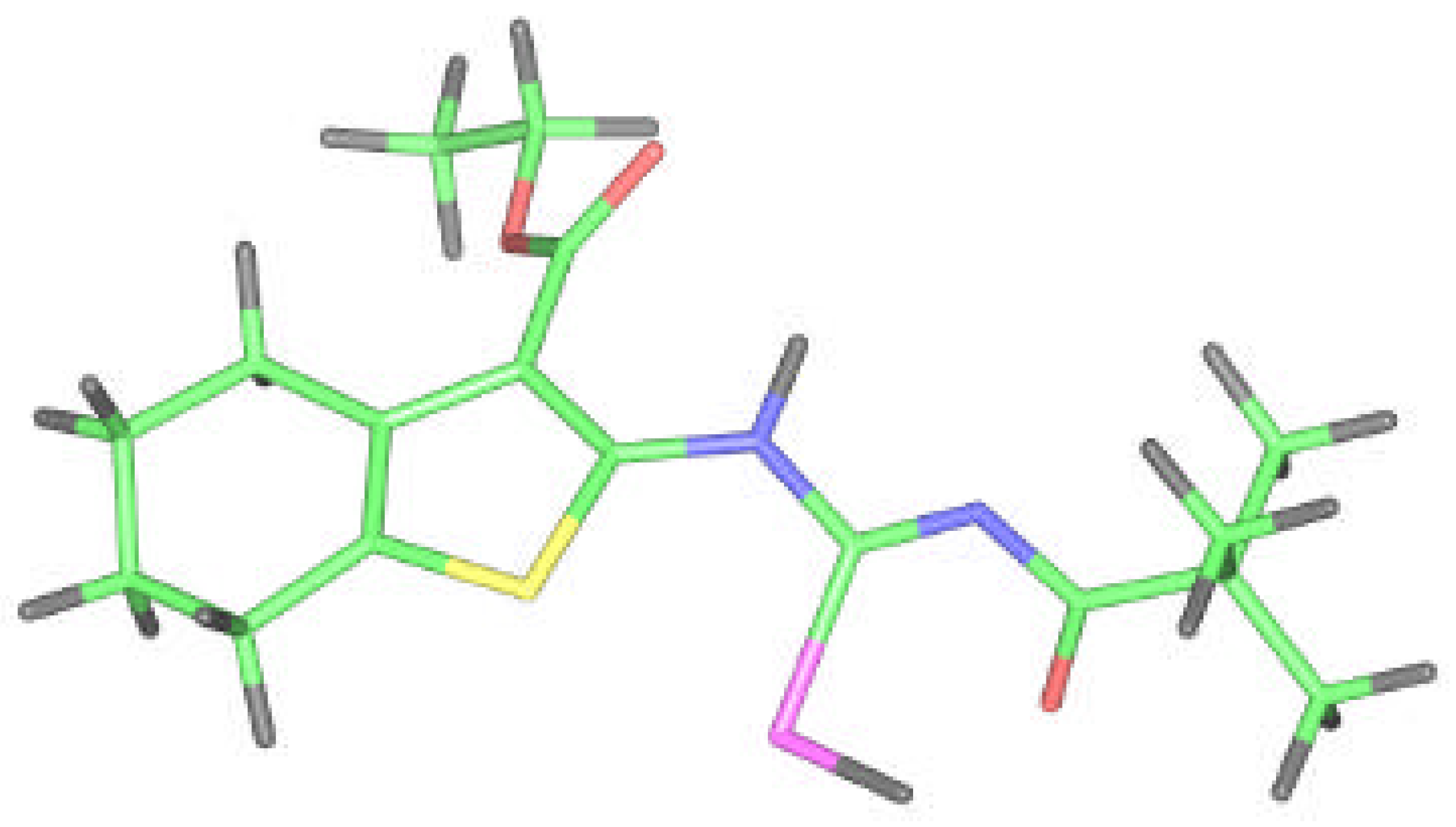
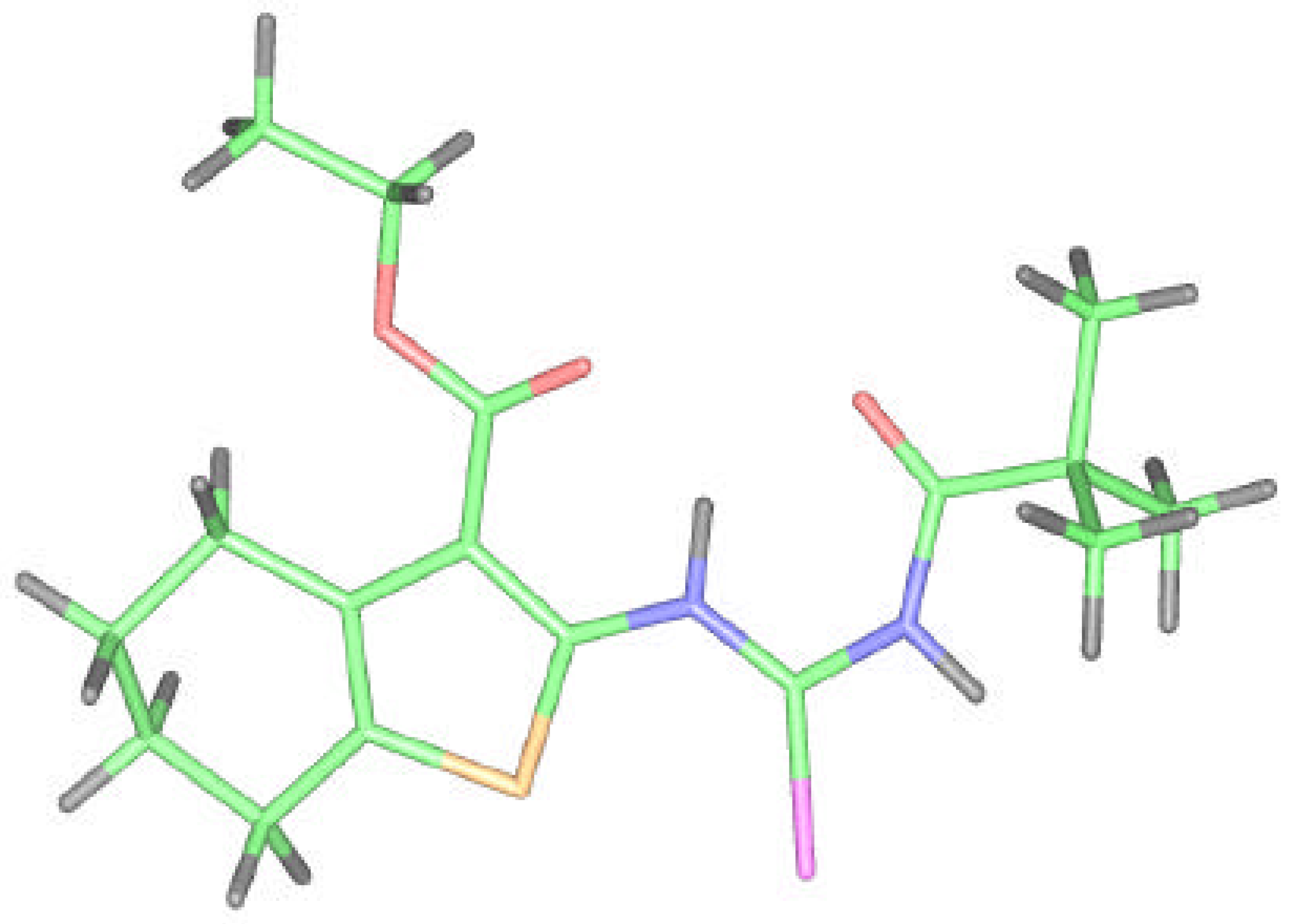
Because we had no convenient single crystal and the reported correspondence between X–ray and ab initio RHF data for compound 2 was very good, we tried to model and optimize the molecular structure of isoselenourea 8 by a quantum chemistry method. The results for compound 8 are presented on the Figure 7 and in the Table 1, Table 2 and Table 3.
The flexibility of the molecule is restricted again but in this case due to the presence of bonding between the hydrogen on N6 selenoureido group and the oxygen atom of ethoxycarbonyl group. An additional H-bond between the acyl oxygen and the hydrogen atom of Se–H group is formed.
Experimental section
General Details
Chemicals and reagent were purchased from Fluka Chemie Co. and used without further purification. The ethyl 2–aminothiophene–3–carboxylates were prepared by Gewald's method [12] . Melting points of prepared compounds 1–6, 7–12 were measured on a Boetius Rapido PHMK 79/2106 (Wägetechnik) instrument and are presented in the Table 4. The purity of compounds was controlled by CHN elemental analysis on an instrument 1102 (Erba), by determinations of selenium on spectrometer ICP AES 7500 (Unicam) and the found values correspond to the calculated ones. TLC was carried out on Silufol UV 254 plates (Kavalier, Votice) and the detection with Fluotes Universal (Qurtzlampen, Hanau) and with iodine vapors. Chloroform and diethylether in a container saturated with vapors of the used solvent was used as an eluent.
The thermal behaviour of compounds was followed with Derivatograph OD–102 (MOM, Budapest). The analyses were provided in about 100 mg samples in a platinum crucible without a lid in a stationary atmosphere of the furnace, as a standard material preglowed α–Al2O3 was used. The measurements were carried out at 150 °C, TG 100 mg, DTG 1/10 and DTA 1/5. The heating rate was 6 °C min-1.
FTIR spectra were taken on a spectrometer Genesis (UNICAm) in potassium bromide pellets.
NMR spectra were measured on a Bruker Avance DRX–500 spectrometer. The 13C and 1H spectra were referenced to tetramethylsilane used as an internal standard or to the solvent signals of CDCl3 and of residual CHCl3 at 77.00 ppm (13C) and 7.27 ppm (1H), respectively. 77Se chemical shifts were referenced to H2SeO3 (1282 ppm) and SeOCl2 (1479 ppm) and 15N chemical shifts to liquid ammonia (0 pppm) used as external standards. Spectral width : 9000 Hz for 1H, 27500 Hz for 13C and 38000 Hz for 77Se.
HETCOR and COLOC spectra: Bruker standard sequences; relaxation delay 2.5 s; delay for evolution of direct 3.45 ms and long range 40 ms 1H–13C couplings; WALTZ16 decoupling during acquisition, spectral widths were taken from the corresponding 1D spectra; data table 2 k x 0.5 k.
1H–15N HMBC spectra [10]: relaxation delay 2.5 s; delay for evolution of long range couplings 100 ms, postgradient recovery 100 μs, gradient pulses 1 ms, gradient ratios 42:18:30 G/cm; 1H–77Se GHMBC [10]; 1H–15N and 1H–77Se GSQMBC [10,11].
UV–VIS spectra were taken on a spectrophotometer SP 1800 (Unicam) in chloroform solutions.
Irradiation of reaction mixtures was performed with a filament lamp 60 W (Tesla).
X–Ray structural data of compound 2 were collected with a KUMA KM–4 kappa four–circles diffractometer. The structure was solved by direct methods using SHELXS–86 [13] and refined on F2 for all reflections using SHELXL–93 [14]. Crystal suitable for X–ray was obtained by recrystallization from chloroform in the form of yellow monoclinic needles.
Geometry optimization of structures 2, 8 was performed at ab initio level of quantum chemical calculation, RHF/DZVP and DFT/VWN/DZVP, respectively.
Acylselenoureas 1–6
Addition of aminoester to acylisoselenocyanate (a)
Potassium selenocyanate (7.63 g, 53 mmol) was dissolved in acetone (50 ml, dried 24 h with anhydrous calcium chloride and distilled) under stirring at room temperature. Benzoylchloride (7.45 g, 53 mmol) or pivaloylchloride (6.39 g, 53 mmol) dissolved in acetone (20 ml) was dropwise added. The reaction mixture was stirred under inert gas (argon, nitrogen) for 10 min, the precipitated potassium chloride was filtered off by suction and washed with acetone (10 ml). The corresponding aminoester (50 mmol) was poured by benzoyl– or pivaloylisoselenocyanate in acetone solution connected acetone part and the formed solution reacted at room temperature. After 15–30 min (TLC control) the reaction mixture was dried on an evaporator and the crude product dissolved in chloroform at a temperature of about 4 °C. The formed suspension was filtered with charcoal, the filtrate separated from colloid selenium and concentrated to 1/5 original volume and mixed with an equivalent of petroleum ether. The precipitated crystals were filtered off by suction, washed with petroleum ether and finally with cold methanol. The product was dried in vacuo.
Acid catalyzed retroisomerization of pivaloylisoselenoureas 8, 10, 12 (b)
Pivaloylisoselenoureas 8, 10, 12 (5 mmol) dissolved in acetic acid (50 ml) were stirred for 5–10 min (TLC monitoring). The pure product was obtained after removing of acetic acid on an evaporator.
Ethyl 2–(3–benzoylselenoureido)–4,5,6,7–tetrahydrobenzo[b]thiophene–3–carboxylate (1)
M.p. 172–174 °C; Yield 21.9 g (95%); FTIR 3410, 3330 (NH), 1680, 1250 (COOC), 1670, 1560 (NHCO, amide I, II), 1532, 965 (NHCSe, selenoamide III, I) cm-1; 1H NMR (CDCl3) 1.40 (3H, t, J 7.1 Hz, OCH2CH3), 1.78–1.81 (4H, m, 5–CH2 and 6–CH2), 2.65 (2H, t, J 5.0 Hz, 4–CH2), 2.84 (2H, t, J 6.0 Hz, 7–CH2), 4.46 (2H, q, J 7.1 Hz, OCH2CH3), 7.50–7.94 (5H, m, C6H5), 9.48 (1H, s, H(N–8)), 15.12 (1H, s, H(N–6)); 13C NMR (CDCl3) 14.41 (CH3, OCH2CH3), 22.81 (C–5 and C–6), 24.57 (C–4), 26.38 (C–7), 60.89 (CH2, OCH2CH3), 117.70 (C–3), 127.74 (C–2' and C–6', C6H5), 128.37 (C–4, thiophene), 129.04 (C–3' and C–5', C6H5), 131.36 (C–1', C6H5), 132.42 (C–5, thiophene), 133.57 (C–4', C6H5), 146.84 (C–2), 164.75 (C=O, COC6H5), 165.20 (C=O, COOCH2CH3), 174.47 (C=Se, 1JC,Se 223 Hz); 15N NMR (CDCl3) 156.20 (N–6), 164.80 (N–8); 77Se NMR (CDCl3) 480; UV–VIS (CHCl3, λmax, nm/log ε, log (m2 mol-1)) 266/4.23, 286/4.16, 386/4.08.
Ethyl 2–(3–pivaloylselenoureido)–4,5,6,7–tetrahydrobenzo[b]thiophene–3–carboxylate (2)
M.p. 153–155 °C; Yield (a) 20.5 g (93%), (b) 21.8 (99%); FTIR 3280, 3200 (NH), 1685, 1235 (COOC), 1660, 1555 (NHCO, amide I, II), 1525, 980 (NHCSe, selenoamide III, I) cm-1; 1H NMR (CDCl3) 1.33 (9H, s, CMe3), 1.38 (3H, t, J 7.1 Hz, OCH2CH3), 1.78–1.81 (4H, m, 5–CH2 and 6–CH2), 2.65 (2H, t, J 5.0 Hz, 4–CH2), 2.83 (2H, t, J 6.0 Hz, 7–CH2), 4.44 (2H, q, J 7.1 Hz, OCH2CH3), 8.89 (1H, s, H(N–8)), 14.93 (1H, s, H(N–6)); 13C NMR (CDCl3) 14.33 (CH3, OCH2CH3), 22.79 (C–5 and C–6), 24.54 (C–4), 26.36 (C–7), 26.98 (CH3, C(CH3)3, 39.75 (C(CH3)3, 60.82 (CH2, OCH2CH3), 117.74 (C–3), 128.42 (C–4, thiophene), 132.40 (C–5, thiophene), 146.76 (C–2, thiophene), 165.09 (C=O, COOCH2CH3), 176.98 (C=O, COCMe3), 174.83 (C=Se, 1JC,Se 220 Hz); 15N NMR (CDCl3) 156.1 (N–6), 163.9 (N–8); 77Se NMR (CDCl3) 459; UV–VIS (CHCl3, λmax, nm/log ε, log (m2 mol-1)) 250/4.70, 274/4.66, 370/4.99.
Ethyl 2–(3–benzoylselenoureido)–4,5–dimethylthiophene–3–carboxylate (3)
M.p. 173–175 °C; Yield 19.9 g (92%); FTIR 3220 (NH), 1685, 1245 (COOC), 1675, 1557 (NHCO, amide I, II), 1522, 985 (NHCSe, selenoamide III, I) cm-1; 1H NMR (CDCl3) 1.42 (3H, t, J 7.1 Hz, OCH2CH3), 2.29 (3H, s, CH3, C–4 thiophene), 2.32 (3H, s, CH3, C–5 thiophene), 4.50 (2H, q, J 7.1 Hz, OCH2CH3), 7.53–7.95 (5H, m, C6H5), 9.42 (1H, s, H(N–8)), 15.11 (1H, s, H(N–6)); 13C NMR (CDCl3) 12.70 (CH3, C–4 thiophene), 14.39 (CH3, OCH2CH3), 14.40 (CH3, C–5 thiophene), 61.06 (CH2, OCH2CH3), 119.02 (C–3), 125.22 (C–4), 127.74 (C–2' and C–6', C6H5), 129.16 (C–3' and C–5', C6H5), 130.77 (C–5), 131.45 (C–1', C6H5), 133.68 (C–4', C6H5), 145.82 (C–2), 164.83 (C=O, COC6H5), 165.26 (C=O, COOCH2CH3), 174.45 (C=Se, 1JC,Se 222 Hz); 77Se NMR (CDCl3) 474; UV–VIS (CHCl3, λmax, nm/log ε, log(m2 mol-1)) 270/4.21, 306/4.14, 386/4.06.
Ethyl 2–(3–pivaloylselenoureido)–4,5–dimethylthiophene–3–carboxylate (4)
M.p. 183–186 °C; Yield (a) 18.4 g (89%), (b) 20.3 (98%); FTIR 3348, 3250, 3182 (NH), 1688, 1239 (COOC), 1660, 1563 (NHCO, amide I, II), 1515, 942 (NHCSe, selenoamide III, I) cm-1; 1H NMR (CDCl3) 1.33 (9H, s, CMe3), 1.39 (3H, t, J 7.1 Hz, OCH2CH3), 2.27 (3H, s, CH3, C–4 thiophene), 2.29 (3H, s, CH3, C–5 thiophene), 4.46 (2H, q, J 7.1 Hz, OCH2CH3), 8.89 (1H, s, H(N–8)), 14.92 (1H, s, H(N–6)); 13C NMR (CDCl3) 12.67 (C–4), 14.32 (CH3, OCH2CH3), 14.41 (C–5), 27.00 (CH3, C(CH3)3, 39.77 (C(CH3)3, 60.97 (CH2, OCH2CH3), 118.87 (C–3), 125.10 (C–4), 130.64 (C–5, thiophene), 145.75 (C–2, thiophene), 165.11 (C=O, COOCH2CH3), 177.07 (C=O, COCMe3), 174.69 (C=Se); 77Se NMR (CDCl3) 458; UV–VIS (CHCl3, λmax, nm/log ε, log (m2 mol-1)) 250/3.98, 318/3.89, 382/3.68.
Ethyl 2–(3–benzoylselenoureido)benzoate (5)
M.p. 107–110 °C; Yield 16.9 g (85 %); FTIR 3380, 3220 (NH), 1700, 1265 (COOC), 1670, 1540 (NHCO, amide I, II), 1520, 980 (NHCSe, selenoamide III, I) cm-1; 1H NMR (CDCl3) 1.38 (3H, t, J 7.1 Hz, OCH2CH3), 4.40 (2H, q, J 7.1 Hz, OCH2CH3), 7.37–8.45 (9H, m, C6H5 and C6H4), 9.61 (1H, s, H(N–8)), 13.70 (1H, s, H(N–6)); 13C NMR (CDCl3) 14.22 (CH3, OCH2CH3), 61.59 (CH2, OCH2CH3), 123.49 (C–1), 126.87 (C–5), 127.18 (C–3), 127.77 (C–2' and C–6', C6H5), 129.08 (C–3' and C–5', C6H5), 130.86 (C–6), 131.36 (C–1', C6H5), 133.71 (C–4', C6H5), 139.27 (C–2), 165.75 (C=O, COOCH2CH3), 165.83 (C=O, COC6H5), 180.22 (C=Se, 1JC,Se 225 Hz); 77Se NMR (CDCl3) 383; UV–VIS (CHCl3, λmax, nm/log ε, log (m2 mol-1)) 247/4.41, 310/4.92, 362/4.70.
Ethyl 2–(3–pivaloylselenoureido)benzoate (6)
M.p. 109–112 °C; Yield (a) 15.4 g (82%), (b) 18.4 (98%); FTIR 3256, 3195 (NH), 1715, 1256 (COOC), 1680, 1546 (NHCO, amide I, II), 1520, 979 (NHCSe, selenoamide III, I) cm-1;1H NMR (CDCl3) 1.33 (9H, s, CMe3), 1.37 (3H, t, J 7.1 Hz, OCH2CH3), 4.39 (2H, q, J 7.1 Hz, OCH2CH3), 7.35–8.37 (4H, m, C6H4), 8.93 (1H, s, H(N–8)), 13.49 (1H, s, H(N–6)); 13C NMR (CDCl3) 14.09 (CH3, OCH2CH3), 26.84 (CH3, C(CH3)3, 39.80 (C(CH3)3, 61.43 (CH2, OCH2CH3), 123.56 (C–1), 126.76 (C–5), 127.10 (C–3), 130.76 (C–6), 132.17 (C–4), 139.07 (C–2), 165.55 (C=O, COOCH2CH3), 178.04 (C=O, COCMe3), 180.32 (C=Se); 77Se NMR (CDCl3) 384; UV–VIS (CHCl3, λmax, nm/log ε, log (m2 mol-1)) 276/4.73, 316/4.95, 379/4.82.
| Crystal data and structure refinement of 2 | ||
| Empirical formula | C17 H24 N2 O3 S Se | |
| Formula weight | 415.4 | |
| Temperature | 153(2) K | |
| Wave length | 0.71073 Å | |
| Crystal system | monoclinic | |
| Space group | P 2(1)/c | |
| Unit cell dimensions | a = 9.198(2) Å b = 10.006(2) Å c = 19.864(4) Å | alpha = 90 deg. beta = 99.98(3) deg. gamma = 90 deg. |
| Volume | 1800.5(6) Å3 | |
| Z | 4 | |
| Density (calculated) | 1.532 mg/m3 | |
| Absorption coefficient | 2.219 mm-1 | |
| F(000) | 856 | |
| Crystallize | 0.80 x 0.80 x 0.40 mm | |
| Theta range for data collection | 2.08 to 25.07 deg. | |
| Index ranges | 0<=h<=10, 0<=k<=11, -23<=l<=23 | |
| Reflections collected | 3386 | |
| Independent reflections | 3176 [R(int) = 0.0555] | |
| Refinement method | Full-matrix least-squares on F2 | |
| Data / restraints / parameters | 3176 / 0 / 303 | |
| Goodness-of-fit on F2 | 0.991 | |
| Final R indices [I> sigma(I)] | R1 = 0.0443, wR2 = 0.1134 | |
| R indices (all data) | R1 = 0.0574, wR2 = 0.1231 | |
| Largest diff. peak and hole | 1.467 and -0.902 eÅ-3 | |
Table 5.
Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2 x 103). U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
| Atom | x | y | z | U(eq) |
| S(1) | 1950(1) | 4842(1) | 576(1) | 19(1) |
| C(2) | 2349(4) | 4526(4) | -225(2) | 16(1) |
| C(3) | 3193(4) | 5540(4) | -441(2) | 18(1) |
| C(4) | 3518(4) | 6581(4) | 61(2) | 18(1) |
| C(5) | 2910(4) | 6330(4) | 622(2) | 19(1) |
| N(6) | 1895(3) | 3397(3) | -617(2) | 17(1) |
| C(7) | 1200(4) | 2309(4) | -444(2) | 17(1) |
| N(8) | 934(4) | 1293(3) | -932(2) | 19(1) |
| Se(9) | 566(1) | 2040(1) | 360(1) | 26(1) |
| C(10) | 1154(4) | 1308(4) | -1602(2) | 18(1) |
| O(11) | 1691(4) | 2268(3) | -1839(2) | 31(1) |
| C(12) | 3664(4) | 5480(4) | -1106(2) | 19(1) |
| O(13) | 3418(3) | 4544(3) | -1504(2) | 29(1) |
| O(14) | 4407(3) | 6563(3) | -1246(1) | 20(1) |
| C(15) | 4908(5) | 6558(5) | -1900(2) | 24(1) |
| C(16) | 5780(5) | 7806(5) | -1941(3) | 30(1) |
| C(17) | 4390(5) | 7842(4) | 5(2) | 25(1) |
| C(20) | 2996(5) | 7220(4) | 1236(2) | 24(1) |
| C(21) | 664(4) | 59(4) | -2019(2) | 18(1) |
| C(22) | -986(4) | -192(5) | -2023(2) | 24(1) |
| C(23) | 1588(5) | -1129(5) | -1715(3) | 28(1) |
| C(24) | 901(5) | 278(5) | -2753(2) | 29(1) |
| C(18) | 4728(10) | 8562(7) | 684(4) | 78(3) |
| C(19) | 3602(10) | 8565(7) | 1081(4) | 69(2) |
| S(1)-C(2) | 1.722(4) | C(2)-S(1)-C(5) | 91.1(2) |
| S(1)-C(5) | 1.725(4) | C(3)-C(2)-N(6) | 123.3(4) |
| C(2)-C(3) | 1.389(5) | C(3)-C(2)-S(1) | 111.8(3) |
| C(2)-N(6) | 1.394(5) | N(6)-C(2)-S(1) | 124.9(3) |
| C(3)-C(4) | 1.436(5) | C(2)-C(3)-C(4) | 112.1(3) |
| C(3)-C(12) | 1.461(6) | C(2)-C(3)-C(12) | 121.2(4) |
| C(4)-C(5) | 1.354(6) | C(4)-C(3)-C(12) | 126.8(4) |
| C(4)-C(17) | 1.510(5) | C(5)-C(4)-C(3) | 111.7(3) |
| C(5)-C(20) | 1.502(6) | C(5)-C(4)-C(17) | 121.1(4) |
| N(6)-C(7) | 1.338(5) | C(3)-C(4)-C(17) | 127.2(4) |
| C(7)-N(8) | 1.398(5) | C(4)-C(5)-C(20) | 126.0(4) |
| C(7)-Se(9) | 1.812(4) | C(4)-C(5)-S(1) | 113.3(3) |
| N(8)-C(10) | 1.380(5) | C(20)-C(5)-S(1) | 120.7(3) |
| C(10)-O(11) | 1.212(5) | C(7)-N(6)-C(2) | 128.8(4) |
| C(10)-C(21) | 1.523(5) | N(6)-C(7)-N(8) | 116.6(3) |
| C(12)-O(13) | 1.222(5) | N(6)-C(7)-Se(9) | 126.1(3) |
| C(12)-O(14) | 1.336(5) | N(8)-C(7)-Se(9) | 117.4(3) |
| O(14)-C(15) | 1.452(5) | C(10)-N(8)-C(7) | 128.7(3) |
| C(15)-C(16) | 1.494(6) | O(11)-C(10)-N(8) | 121.4(4) |
| C(17)-C(18) | 1.514(7) | O(11)-C(10)-C(21) | 122.8(4) |
| C(20)-C(19) | 1.508(7) | N(8)-C(10)-C(21) | 115.8(3) |
| C(21)-C(23) | 1.523(6) | O(13)-C(12)-O(14) | 122.0(4) |
| C(21)-C(24) | 1.528(6) | O(13)-C(12)-C(3) | 124.5(4) |
| C(21)-C(22) | 1.538(5) | O(14)-C(12)-C(3) | 113.5(3) |
| C(18)-C(19) | 1.406(9) | C(12)-O(14)-C(15) | 116.0(3) |
| O(14)-C(15)-C(16) | 107.6(4) | ||
| C(4)-C(17)-C(18) | 111.2(4) | ||
| C(5)-C(20)-C(19) | 109.5(4) | ||
| C(10)-C(21)-C(23) | 109.5(3) | ||
| C(10)-C(21)-C(24) | 108.8(3) | ||
| C(23)-C(21)-C(24) | 109.1(4) | ||
| C(10)-C(21)-C(22) | 109.6(3) | ||
| C(23)-C(21)-C(22) | 110.9(4) | ||
| C(24)-C(21)-C(22) | 108.8(3) | ||
| C(19)-C(18)-C(17) | 116.3(6) | ||
| C(18)-C(19)-C(20) | 116.5(6) |
Table 7.
Symmetry transformations used to generate equivalent atoms: anisotropy displacement parameters (Å2 x 103). The anisotropic displacement factor exponent takes the form: -2 pi2 [h2 a*2 U11 + ... + 2 h k a* b* U12].
| Atom | U11 | U22 | U33 | U23 | U13 | U12 |
| S(1) | 16(1) | 21(1) | 20(1) | -2(1) | 2(1) | -5(1) |
| C(2) | 9(2) | 19(2) | 19(2) | 1(2) | -4(1) | -1(2) |
| C(3) | 9(2) | 20(2) | 23(2) | 1(2) | -2(1) | -1(2) |
| C(4) | 10(2) | 17(2) | 23(2) | -1(2) | -3(2) | -3(2) |
| C(5) | 10(2) | 24(2) | 22(2) | -2(2) | -3(2) | -3(2) |
| N(6) | 11(2) | 20(2) | 18(2) | -2(1) | -1(1) | -4(1) |
| C(7) | 8(2) | 20(2) | 20(2) | 1(2) | -4(1) | -2(1) |
| N(8) | 16(2) | 18(2) | 22(2) | -2(1) | 1(1) | -6(1) |
| Se(9) | 30(1) | 26(1) | 23(1) | -2(1) | 7(1) | -13(1) |
| C(10) | 9(2) | 23(2) | 20(2) | 1(2) | -3(1) | -1(2) |
| O(11) | 44(2) | 25(2) | 23(2) | -1(1) | 6(1) | -18(1) |
| C(12) | 9(2) | 24(2) | 20(2) | 1(2) | -3(1) | -4(2) |
| O(13) | 31(2) | 30(2) | 25(2) | -6(1) | 5(1) | -14(1) |
| O(14) | 14(1) | 23(2) | 22(1) | -1(1) | 2(1) | -8(1) |
| C(15) | 18(2) | 32(2) | 22(2) | -3(2) | 5(2) | -9(2) |
| C(16) | 19(2) | 39(3) | 32(3) | 2(2) | 6(2) | -11(2) |
| C(17) | 25(2) | 25(2) | 24(2) | -3(2) | 2(2) | -11(2) |
| C(20) | 24(2) | 23(2) | 25(2) | -5(2) | 5(2) | -3(2) |
| C(21) | 9(2) | 20(2) | 23(2) | -1(2) | 1(2) | -2(2) |
| C(22) | 9(2) | 35(3) | 27(2) | -7(2) | -1(2) | -7(2) |
| C(23) | 20(2) | 23(2) | 41(3) | 1(2) | 4(2) | 3(2) |
| C(24) | 31(3) | 32(3) | 26(2) | 6(2) | 7(2) | 6(2) |
| C(18) | 133(7) | 55(4) | 57(4) | -29(3) | 50(4) | -69(5) |
| C(19) | 111(6) | 52(4) | 58(4) | -36(3) | 53(4) | -56(4) |
| Atom | x | y | z | U(eq) |
| H(6A) | 2101(53) | 3386(50) | -997(27) | 26(13) |
| H(8A) | 502(51) | 632(50) | -784(23) | 21(12) |
| H(15B) | 5501(58) | 5735(56) | -1927(26) | 37(14) |
| H(15A) | 4059(60) | 6476(54) | -2267(28) | 37(14) |
| H(16C) | 5182(64) | 8588(61) | -1873(28) | 45(16) |
| H(16B) | 6492(69) | 7793(59) | -1569(32) | 48(17) |
| H(16A) | 6190(53) | 7805(48) | -2354(26) | 27(12) |
| H(17B) | 3836(59) | 8512(57) | -338(28) | 41(15) |
| H(17A) | 5437(62) | 7591(57) | -97(27) | 40(14) |
| H(20B) | 3504(60) | 6831(53) | 1620(29) | 36(14) |
| H(20A) | 2063(65) | 7298(58) | 1361(28) | 44(15) |
| H(22C) | -1573(61) | 561(60) | -2217(28) | 43(15) |
| H(22B) | -1236(67) | -872(67) | -2327(33) | 58(19) |
| H(22A) | -1170(52) | -347(49) | -1582(26) | 27(12) |
| H(23C) | 1344(64) | -1916(60) | -1970(31) | 46(16) |
| H(23B) | 2636(70) | -960(61) | -1721(30) | 52(17) |
| H(23A) | 1434(53) | -1335(50) | -1244(26) | 30(13) |
| H(24B) | 426(65) | 1058(64) | -2935(29) | 47(16) |
| H(24A) | 1987(64) | 381(57) | -2768(27) | 43(15) |
| H(24C) | 524(60) | -487(60) | -3031(28) | 42(15) |
| H(18A) | 5627(55) | 8146(25) | 961(17) | 385(141) |
| H(18B) | 4979(18) | 9512(60) | 596(7) | 59(18) |
| H(19A) | 2729(43) | 9143(29) | 829(12) | 278(91) |
| H(19B) | 4005(22) | 9038(24) | 1545(23) | 40(14) |
Acylisoselenoureas 7–12
Photoisomerization (a)
Acylselenoureas 1–6 (5 mmol) dissolved in chloroform (30 ml) were irradiated by a filament lamp or by sunlight under an inert gas atmosphere for 12–20 h (course of the isomerization was monitored by TLC). Pure product was obtained after removing of chloroform on an evaporator.
Use of acetic acid (b)
Benzoylselenoureas 1, 3, 5 (5 mmol) dissolved in acetic acid (30 ml) were refluxed for 5–10 min (TLC monitoring). The pure product was obtained after removing of acetic acid on an evaporator.
Controlled melt (c)
Acylselenourea 1–6 (0.5 mmol) was heated on a microscope slide. This slide was placed on the hot–stage of a melting point apparatus. The temperature increase was stopped when it corresponded with the temperature of the exothermic peak on DTA curve. During examination the change in solid character was observed. After coling to sample to a room temperature the mixture of 1–6 and 7–12 was suspended in chloroform, the extract filtered off with silica gel and evaporated.
Ethyl 2–(3–benzoylisoselenoureido)–4,5,6,7–tetrahydrobenzo[b]thiophene–3–carboxylate (7)
M.p. 173–174 °C; Yield (a) 2.13 g (98%), (b) 2.07 (95%), (c) 0.130 (60%); FTIR 3237, 3125 (NH), 1709, 1275 (COOC), 1650 (NCO), 1624 (C=N) cm-1; 1H NMR (CDCl3) 0.96 (3H, t, J 7.1 Hz, OCH2CH3), 1.72–1.76 (4H, m, 5–CH2 and 6–CH2), 2.66 (2H, t, J 5.0 Hz, 4–CH2), 2.67 (2H, t, J 6.0 Hz, 7–CH2), 3.78 (2H, q, J 7.1 Hz, OCH2CH3), 7.50–8.45 (5H, m, C6H5), 12.22 (1H, s, H(N–6)); 13C NMR (CDCl3) 14.15 (CH3, OCH2CH3), 22.74 (C–5), 22.90 (C–6), 24.50 (C–4), 26.28 (C–7), 60.45 (CH2, OCH2CH3), 113.88 (C–3), 128.26 (C–3' and C–5', C6H5), 128.95 (C–4, thiophene), 130.66 (C–2' and C–6', C6H5), 131.64 (C–5, thiophene), 132.65 (C–4', C6H5), 135.35 (C–1', C6H5), 146.43 (C–2), 157.12 (C–Se, 1JC,Se 195 Hz), 165.43 (C=O, COOCH2CH3), , 176.99 (C=O, COC6H5); 15N NMR (CDCl3) 135.9 (N–6), 240.2 (N–8); 77Se NMR (CDCl3) 533; UV–VIS (CHCl3, λmax, nm/log ε, log (m2 mol-1)) 286/3.96, 324/4.24, 400/4.11.
Ethyl 2–(3–pivaloylisoselenoureido)–4,5,6,7–tetrahydrobenzo[b]thiophene–3–carboxylate (8)
M.p. 156 °C; Yield (a) 2.03 g (98%), (b) 1.97 (95%), (c) 0.135 (65%); FTIR 3282, 3179 (NH), 1711, 1241 (COOC), 1660 (NCO), 1630 (C=N) cm-1; 1H NMR (CDCl3) 1.24 (3H, t, J 7.1 Hz, OCH2CH3), 1.37 (9H, s, CMe3), 1.76–1.78 (4H, m, 5–CH2 and 6–CH2), 2.65 (2H, t, J 5.0 Hz, 4–CH2), 2.72 (2H, t, J 6.0 Hz, 7–CH2), 4.22 (2H, q, J 7.1 Hz, OCH2CH3), 11.95 (1H, s, H(N–6)); C NMR (CDCl3) 14.46 (CH3, OCH2CH3), 22.83 (C–5), 22.96 (C–6), 24.43 (C–4), 26.34 (C–7), 27.60 (CH3, C(CH3)3, 41.53 (C(CH3)3, 60.46 (CH2, OCH2CH3), 113.48 (C–3), 128.64 (C–4, thiophene), 131.48 (C–5, thiophene), 146.64 (C–2), 156.85 (C–Se, 1JC,Se 195 Hz), 165.34 (C=O, COOCH2CH3), 191.40 (C=O, COCMe3); 15N NMR (CDCl3) 133.8 (N–6), 241.4 (N–8); 77Se NMR (CDCl3) 537; UV–VIS (CHCl3, λmax, nm/log ε, log (m2 mol-1)) 286/3.98, 322/4.11, 395/4.12.
Ethyl 2–(3–benzoylisoselenoureido)–4,5–dimethylthiophene–3–carboxylate (9)
M.p. 175–178 °C; Yield (a) 2.00 g (98%), (b) 1.92 (96%), (c) 0.131 (64%); FTIR 3316, 3199 (NH), 1670, 1259 (COOC), 1655 (NCO), 1625 (C=N) cm-1; 1H NMR (CDCl3) 1.00 (3H, t, J 7.1 Hz, OCH2CH3), 2.17 (3H, s, CH3, C–4 thiophene), 2.30 (3H, s, CH3, C–5 thiophene), 3.85 (2H, q, J 7.1 Hz, OCH2CH3), 7.52–8.47 (5H, m, C6H5), 12.23 (1H, s, H(N–6)); 13C NMR (CDCl3) 12.50 (CH3, C–4 thiophene), 14.13 (CH3, OCH2CH3), 14.27 (CH3, C–5 thiophene), 60.60 (CH2, OCH2CH3), 115.05 (C–3), 125.59 (C–4), 128.30 (C–3' and C–5', C6H5), 129.88 (C–5), 130.68 (C–2' and C–6', C6H5), 132.70 (C–4', C6H5), 135.37 (C–1', C6H5), 145.59 (C–2), 157.09 (C–Se), 165.48 (C=O, COOCH2CH3), 177.05 (C=O, COC6H5); UV–VIS (CHCl3, λmax, nm/log ε, log (m2 mol-1)) 282/3.97, 322/4.27, 400/4.13.
Ethyl 2–(3–pivaloylisoselenoureido)–4,5–dimethylthiophene–3–carboxylate (10)
M.p. 185–187 °C; Yield (a) 1.91 g (98%), (b) 1.83 (96%), (c) 0.117 (64%); FTIR 3348, 3189 (NH), 1688, 1260 (COOC), 1652 (NCO), 1625 (C=N) cm-1; 1H NMR (CDCl3) 1.21 (3H, t, J 7.1 Hz, OCH2CH3), 1.36 (9H, s, CMe3), 2.16 (3H, s, CH3, C–4 thiophene), 2.27 (3H, s, CH3, C–5 thiophene), 3.87 (2H, q, J 7.1 Hz, OCH2CH3), 12.09 (1H, s, H(N–6)); UV–VIS (CHCl3, λmax, nm/log ε, log (m2 mol-1)) 270/4.37, 343/4.29, 410/4.21.
Ethyl 2–(3–benzoylisoselenoureido)benzoate (11)
M.p. 108–110 °C; Yield (a) 1.84 g (98%), (b) 1.74 (93%), (c) 0.113 (60%); FTIR 3433, 3181 (NH), 1703, 1256 (COOC), 1650 (NCO), 1625 (C=N) cm-1;1H NMR (CDCl3) 1.03 (3H, t, J 7.1 Hz, OCH2CH3), 3.89 (2H, q, J 7.1 Hz, OCH2CH3), 7.17–8.80 (9H, m, C6H5 and C6H4), 11.40 (1H, s, H(N–6)); 13C NMR (CDCl3) 13.95 (CH3, OCH2CH3), 61.34 (CH2, OCH2CH3), 118.95 (C–1), 124.02 (C–3), 124.11 (C–5), 128.27 (C–3' and C–5', C6H5), 130.21 (C–2' and C–6', C6H5), 130.57 (C–6), 132.60 (C–4', C6H5), 135.66 (C–1', C6H5), 140.61 (C–2), 160.46 (C–Se, 1JC,Se 192 Hz), 166.77 (C=O, COOCH2CH3), 176.83 (C=O, COC6H5), .77Se NMR (CDCl3) 544; UV–VIS (CHCl3, λmax, nm/log ε, log (m2 mol-1)) 270/4.34, 349/4.05, 397/4.64.
Ethyl 2–(3–pivaloylisoselenoureido)benzoate (12)
M.p. 110–112 °C; Yield (a) 1.74 g (98%), (b) 1.60 (90%), (c) 0.108 (61%); FTIR 3436, 3228, 3182 (NH), 1701, 1256 (COOC), 1652 (NCO), 1624 (C=N) cm-1; 1H NMR (CDCl3) 1.27 (9H, s, CMe3), 1.36 (3H, t, J 7.1 Hz, OCH2CH3), 4.44 (2H, q, J 7.1 Hz, OCH2CH3), 7.13–7.99 (4H, m, C6H4), 12.49 (1H, s, H(N–6)); UV–VIS (CHCl3, λmax, nm/log ε, log (m2 mol-1)) 270/3.95, 348/4.15, 396/4.52.
Acknowledgements
This work was supported by the Grant No. 203/93/0715 of the Grant Agency of the Czech Republic. We would like to thank Biosym/Molecular Simulations of San Diego for providing us with the academic license for the InsightII and Turbomole software. We thank the Academic supercomputer center in Brno for access to the computer facilities. The authors thank Dr. J. Jambor of the Department of Analytical Chemistry of our Faculty for determination of selenium by ICP AES, and the Analytical Department of Lachema Co., Brno, Czech Republic for elemental analyses.
References
- Pazdera, P.; Ondracek, D.; Novacek, E. Chem. Papers 1989, 43, 771. Pazdera, P.; Rezka, M. Chem. Papers 1990, 44, 229. Pazdera, P.; Potucek, V.; Kalvinsh, I.; Trapencieris, P.; Pugovits, O.; Novacek, E. Chem. Papers 1991, 45, 527. Pazdera, P.; Potucek, V. Chem. Papers 1991, 45, 677. Pazdera, P.; Preissova, I. Chem. Papers 1992, 46, 396. Pazdera, P.; Meindl, J.; Novacek, E. Chem. Papers 1992, 46, 322. Sibor, J.; Pazdera, P.; Pichler, J. Folia Pharm. Universitatis Carolinae XVIII 1995, 177.
- Douglas, I. B. J. Am. Chem. Soc. 1937, 59, 740.
- Walter, W.; Ruess, K.–P. Justus Liebigs Ann. Chem. 1971, 743, 167.
- Guiliani, A. M. J. Chem. Soc. Dalton Trans. 1972, 492.
- Hope, H. Acta Cryst. 1965, 18, 259.
- Fuchs, O. Chem. Listy 1996, 90, 444.
- Hertlova, M. Thesis, Faculty of Medicine, Masaryk University, Brno, 1996.
- International group of experts (Diplock, A. I., Chairman). Environmental Health Criteria 58, SELENIUM; World Health Organization: Geneva, 1987; p. 56. [Google Scholar] Geisler, K.; Bulka, E. Wissenschaftliche Z. der EMAU Greifswald 1976, 25, 93.
- Pazdera, P.; Sibor, J.; Zurek, D.; Marek, R.; Kuty, M.; Marek, J. Collect. Czech. Chem. Commun. to be published.
- Marek, R.; Kralik, L.; Sklenar, V. Tetrahedron Lett. 1997, 38, 655. Marek, R.; Dostal, J.; Slavik, J. Molecules 1996, 1, 166. Martin, B. E.; Crouch, R. C.; Sharaf, M. H. M.; Schiff, P. L., Jr. J. Nat. Prod 1966, 59, 2.
- Marek, R.; Tousek, J.; Kralik, L.; Humpa, O.; Sibor, J.; Pazdera, P.; Sklenar, V. to be published.
- Gewald, K.; Schinke, E.; Böttcher, H. Chem. Ber. 1966, 99, 94. Gewald, K.; Schinke, E. Chem. Ber. 1966, 99, 2712.
- Sheldrick, G. M. Acta Cryst. 1990, A46, 467.
- Sheldrick, G. M. SHELXL93: Program for structure refinement. 1993; University of Göttingen: Göttingen. [Google Scholar]
- Sample Availability: not available.
© 1997 MDPI. All rights reserved
Share and Cite
MDPI and ACS Style
Pazdera, P.; Sibor, J.; Marek, R.; Kuty, M.; Marek, J. Photoisomerization of Ethyl 2–(3–Acylselenoureido)thiophene– 3–carboxylates and Their Benzoanalogues. Molecules 1997, 2, 135-151. https://doi.org/10.3390/20900135
AMA Style
Pazdera P, Sibor J, Marek R, Kuty M, Marek J. Photoisomerization of Ethyl 2–(3–Acylselenoureido)thiophene– 3–carboxylates and Their Benzoanalogues. Molecules. 1997; 2(9):135-151. https://doi.org/10.3390/20900135
Chicago/Turabian StylePazdera, Pavel, Jiri Sibor, Radek Marek, Michal Kuty, and Jaromir Marek. 1997. "Photoisomerization of Ethyl 2–(3–Acylselenoureido)thiophene– 3–carboxylates and Their Benzoanalogues" Molecules 2, no. 9: 135-151. https://doi.org/10.3390/20900135