2.1. Fabrication and Characterization of Polyelectrolyte Nanoparticles
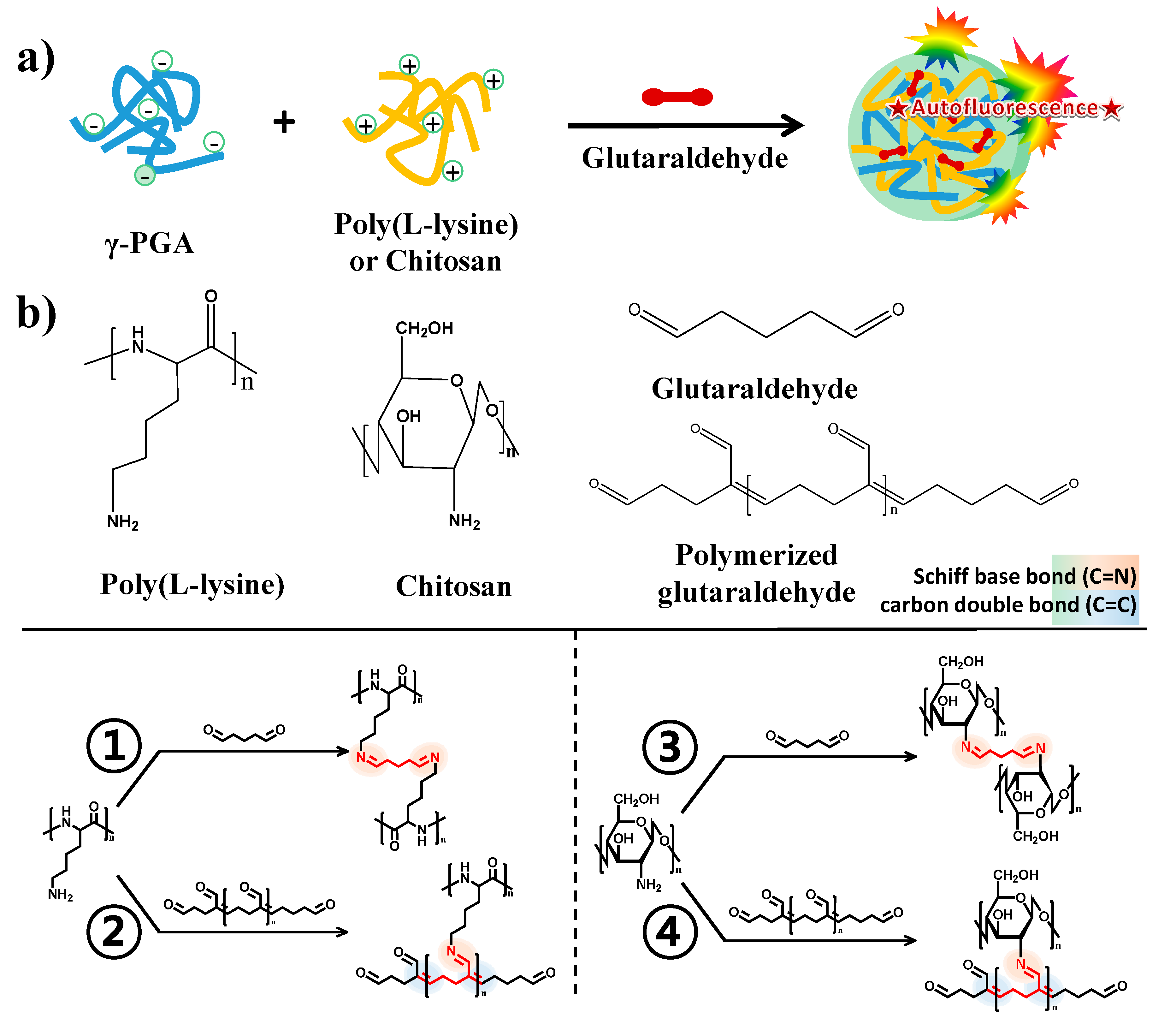
The synthesis of functional polyelectrolyte nanoparticles that provided optical imaging modalities was performed via electrostatic assembly and crosslinking without any fluorescent materials. The self-assembled polyelectrolyte nanoparticles were prepared via electrostatic interactions between the negatively charged carboxyl groups of poly(γ-glutamic acid) (γ-PGA) and the positively charged amino groups of two different polymers: poly-L-lysine (PLL) or chitosan. The crosslinking of amine moieties by glutaraldehyde in the self-assembled nanoparticles imparted higher stability and optical properties to the nanoparticles (
Figure 1a). Crosslinking of the amine moiety of cationic polymers with glutaraldehyde provided structural integrity to the nanogels and involved the formation of a autofluorescent chemical bond. Fluorescent Schiff base bonds (C=N) and double bonds (C=C) could be generated simultaneously by crosslinking the cationic polymer with monomeric glutaraldehyde or by polymerizing glutaraldehyde into oligomeric glutaraldehyde followed by a reaction with the cationic polymer, respectively. The chemical structures that result from the Schiff base bonds (C=N) and carbon double bonds (C=C) of the polymerized glutaraldehyde provide autofluorescence properties for biological applications (
Figure 1b). As shown in the transmission electron microscopy (TEM) images, the two types of synthesized nanoparticles exhibited a spherical shape (
Figure 1c). The mean size of the nanoparticles was approximately 151.3 ± 39.0 nm in the case of γ-PGA/PLL and 167.3 ± 44.6 nm in the case of γ-PGA/chitosan (
Figure 1d). The γ-PGA/PLL and γ-PGA/chitosan nanoparticles both possessed strong positive zeta potentials (+34.48 mV and +31.26 mV, respectively) in deionized water (DW).
Figure 1.
(a) Schematic illustration for the fabrication of autofluorescent nanocomposites based on electrostatic assembly and cross-linking by glutaraldehyde; (b) Chemical structure of cross-linked amino groups by glutaraldehyde monomer (PLL: ①, Chitosan: ③) and polymerized glutaraldehyde (PLL: ②, Chitosan: ④); (c) TEM images of γ-PGA/PLL (left) and γ-PGA/Chitosan (right) nanocomposites, respectively; (d) DLS analysis of γ-PGA/PLL (left) and γ-PGA/Chitosan (right) nanocomposites, respectively.
Figure 1.
(a) Schematic illustration for the fabrication of autofluorescent nanocomposites based on electrostatic assembly and cross-linking by glutaraldehyde; (b) Chemical structure of cross-linked amino groups by glutaraldehyde monomer (PLL: ①, Chitosan: ③) and polymerized glutaraldehyde (PLL: ②, Chitosan: ④); (c) TEM images of γ-PGA/PLL (left) and γ-PGA/Chitosan (right) nanocomposites, respectively; (d) DLS analysis of γ-PGA/PLL (left) and γ-PGA/Chitosan (right) nanocomposites, respectively.
2.2. Optical Properties of Self-Assembled Polyelectrolyte Nanoparticles
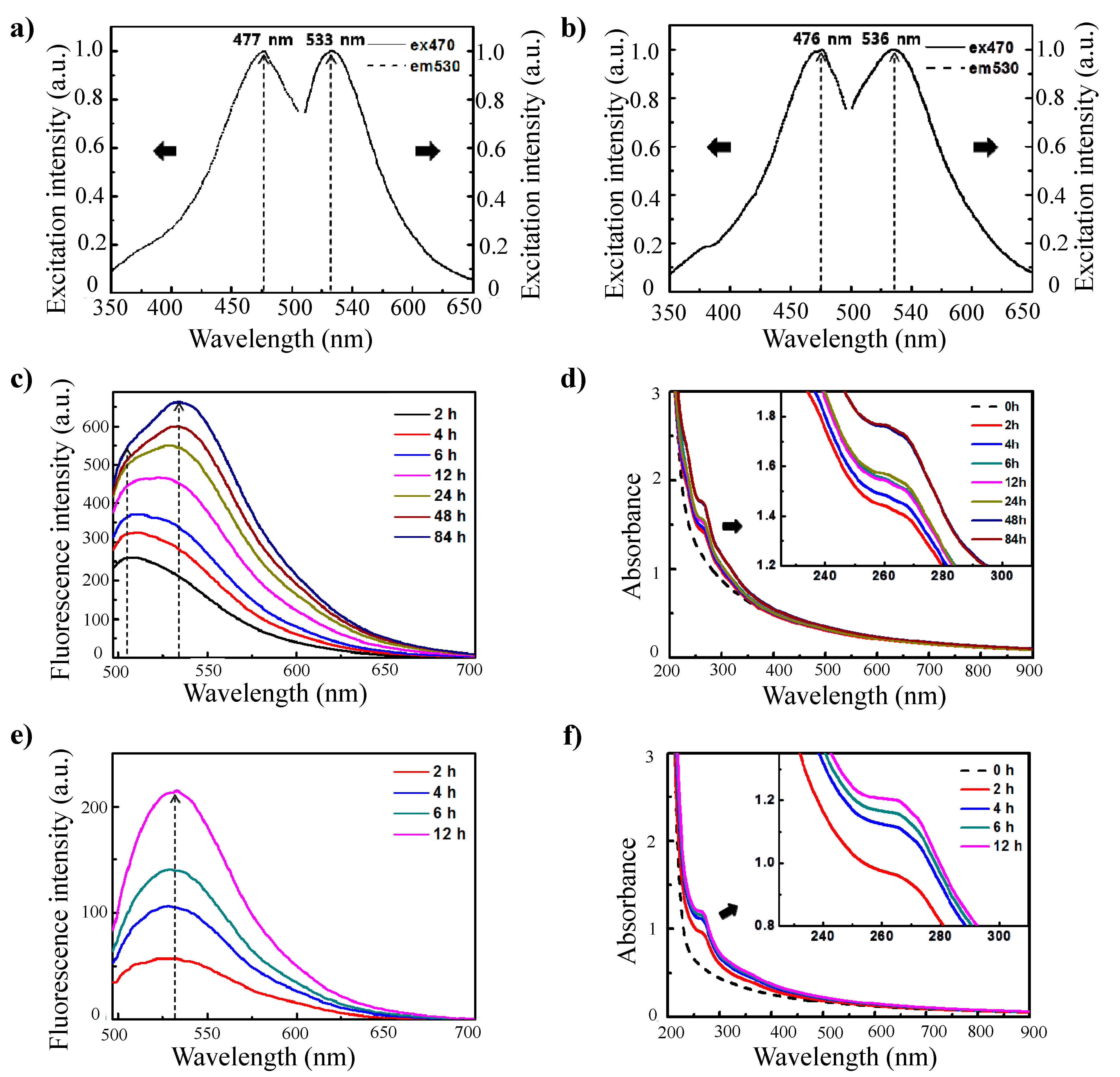
These nanoparticles exhibited unique optical properties. The nanoparticles generated a strong fluorescence signal, and the shapes of the excitation and emission curves of these nanoparticles were similar to those of conventional fluorophores. The fluorescence spectrum of the γ-PGA/PLL nanoparticles revealed that emission occurs at 533 nm upon excitation at 470 nm (
Figure 2a). When the γ-PGA/chitosan nanoparticles were excited at 470 nm, the emission peak was observed at 536 nm (
Figure 2b). The maximum peak in the emission spectrum was highly dependent on the crosslinking reaction time, and the behavior of this peak was considerably different in the two nanoparticle systems. The required crosslinking reaction time to exhibit distinct emission was longer for the γ-PGA/chitosan nanoparticles than for the γ-PGA/PLL nanoparticles. The different dependencies of the emission spectra on the crosslinking reaction time in the two nanoparticle systems may be a result of the different chemical structures of the cationic polymers. As shown in
Figure 1b, conjugation of the imine bond (C=N) and carbon double bond (C=C) causes an apparent fluorescence curve for their optical properties.
Figure 2.
(a) Fluorescence excitation (λem: 530 nm) and emission (λex: 470 nm) curves of γ-PGA/PLL cross-linked with glutaraldehyde for 12 h; (b) Fluorescence excitation (λem: 530 nm) and emission (λex: 470 nm) curves of γ-PGA/Chitosan cross-linked with glutaraldehyde for 96 h; (c) Fluorescence emission spectra and (d) UV-vis spectra of γ-PGA/Chitosan cross-linked with 10 μL glutaraldehyde for different times (λex: 470 nm). (e) Fluorescence emission spectra and (f) UV-vis spectra of γ-PGA/PLL cross-linked with 10 μL glutaraldehyde for different times (λex: 470 nm).
Figure 2.
(a) Fluorescence excitation (λem: 530 nm) and emission (λex: 470 nm) curves of γ-PGA/PLL cross-linked with glutaraldehyde for 12 h; (b) Fluorescence excitation (λem: 530 nm) and emission (λex: 470 nm) curves of γ-PGA/Chitosan cross-linked with glutaraldehyde for 96 h; (c) Fluorescence emission spectra and (d) UV-vis spectra of γ-PGA/Chitosan cross-linked with 10 μL glutaraldehyde for different times (λex: 470 nm). (e) Fluorescence emission spectra and (f) UV-vis spectra of γ-PGA/PLL cross-linked with 10 μL glutaraldehyde for different times (λex: 470 nm).
The fluorescence intensity of the γ-PGA/chitosan nanoparticles at λ
em = 533 nm improved with an increase in the crosslinking reaction time (
Figure 2c). For the γ-PGA/chitosan nanoparticles, the primary emission occurred at λ
em = 510 nm for crosslinking reaction times up to 6 h. For reaction times longer than 6 h, peaks at λ
em = 510 nm and λ
em = 531 nm were observed with approximately equal intensities. This result suggested that the imine bonds coexist equally with carbon double bonds. The emission peaks measured after 12 h were clearly dominant at 531–534 nm. From this result, we could infer that Schiff base bonds are gradually formed with an increase in the reaction time and that the effects of C=N bonds and C=C bonds on the improvement in fluorescence intensity are different depending on the crosslinking reaction time. Furthermore, the formation of Schiff base bonds (C=N) was confirmed by measuring the absorbance of the particles (
Figure 2) [
20]. The gradual increase in absorbance by the γ-PGA/chitosan nanoparticles within the range of 250–270 nm indicated that the increase in fluorescence intensity was caused by the formation of imine bonds. This result,
i.e., the steady increase in fluorescence intensity was caused by the formation of imine bonds, was consistent with the spectra obtained from the γ-PGA/PLL nanoparticles (
Figure 2e,f).
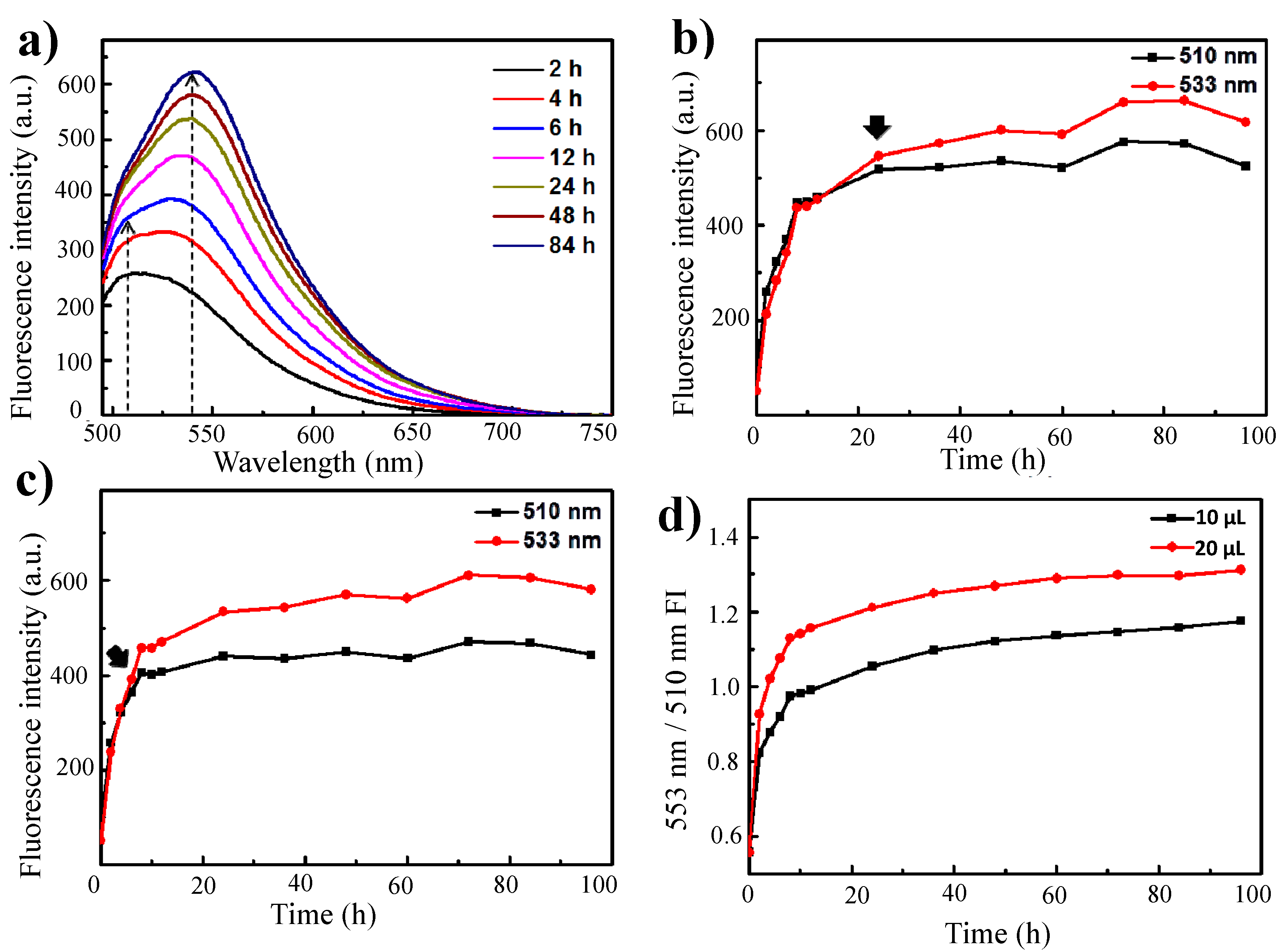
Notably, the shapes of the fluorescence emission curves were dependent on the crosslinker volume and on the crosslinking reaction time. When more than 10 μL of glutaraldehyde was added to the γ-PGA/chitosan reaction mixture, the emission peaks were dominant within 6 h at 533 nm (
Figure 3a). For γ-PGA/chitosan crosslinked with 10 μL of glutaraldehyde, the emission peak at 533 nm was more intense than the peak at 510 nm after 24 h of reaction, whereas the main emission peak of γ-PGA/chitosan crosslinked with 20 μL of glutaraldehyde decreased in intensity within 6 h (
Figure 3b,c). As indicated by the ratios between the fluorescence intensity at 533 nm and that at 510 nm for γ-PGA/chitosan crosslinked with 10 μL and 20 μL of glutaraldehyde, the formation of Schiff base bonds was faster in γ-PGA/chitosan crosslinked with more glutaraldehyde (
Figure 3d).
Figure 3.
(a) Fluorescence emission spectra of γ-PGA/Chitosan cross-linked with 20 μL glutaraldehyde for different times (λex: 470 nm); The fluorescence intensity of (b) γ-PGA/Chitosan cross-linked with 10 μL glutaraldehyde and (c) γ-PGA/Chitosan cross-linked with 20 μL glutaraldehyde at 510 nm and 533 nm as a function of time; (d) Relationship between rates of formation of Schiff base, showed by calculating the ratio between fluorescence intensity at 533 nm and 510 nm.
Figure 3.
(a) Fluorescence emission spectra of γ-PGA/Chitosan cross-linked with 20 μL glutaraldehyde for different times (λex: 470 nm); The fluorescence intensity of (b) γ-PGA/Chitosan cross-linked with 10 μL glutaraldehyde and (c) γ-PGA/Chitosan cross-linked with 20 μL glutaraldehyde at 510 nm and 533 nm as a function of time; (d) Relationship between rates of formation of Schiff base, showed by calculating the ratio between fluorescence intensity at 533 nm and 510 nm.
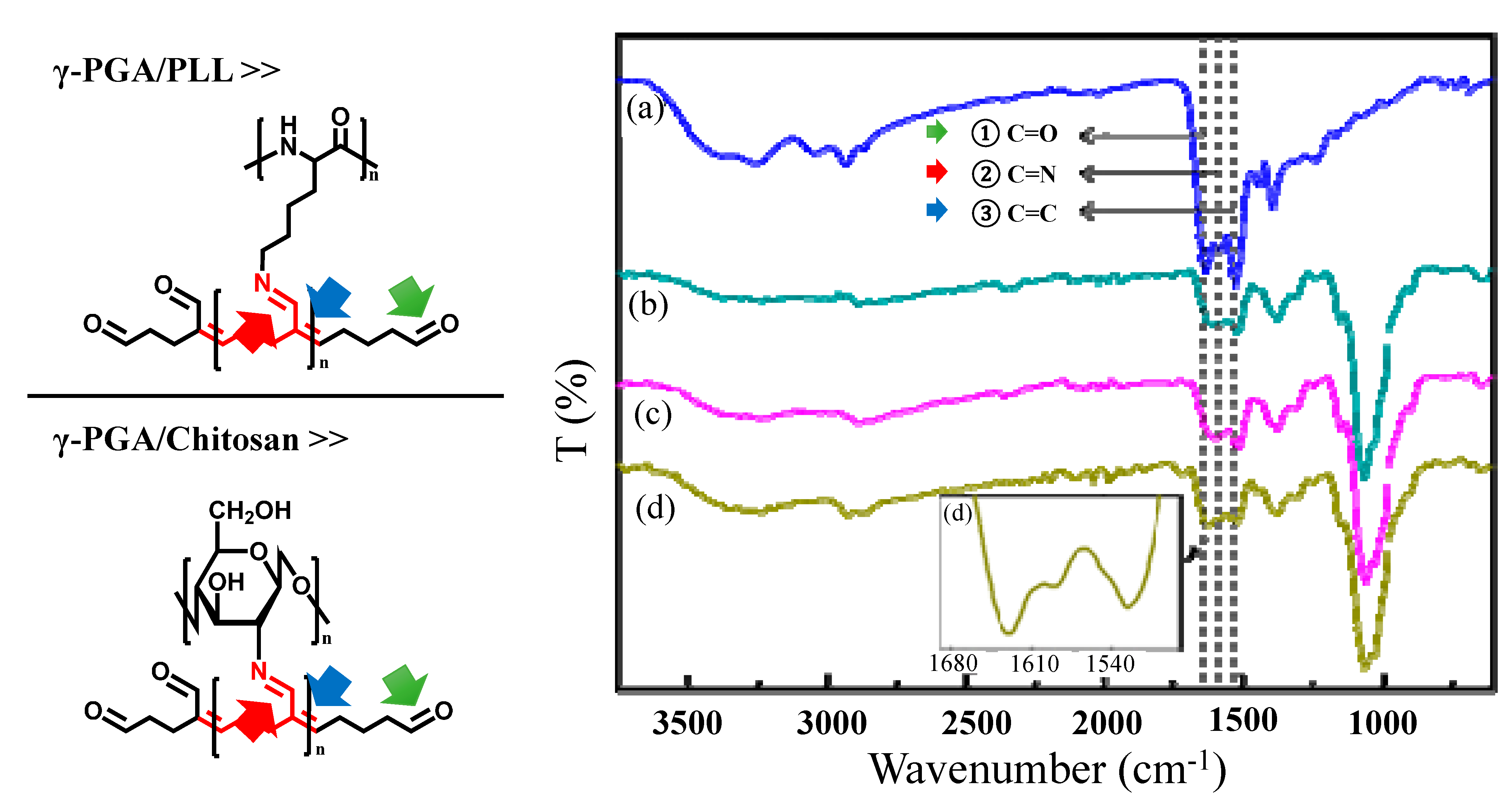
These results indicated that both the crosslinking reaction time and the crosslinker volume are important factors in the formation of chemical bonds that induce the final autofluorescence. In addition, the presence of C=N and C=C bonds was confirmed from FT-IR spectra (
Figure 4). We could analyze the peaks in the FT-IR spectra that corresponded to chemical bonds that induce autofluorescence. The peak at 1629–1640 cm
−1 was attributed to the aldehyde groups of glutaraldehyde (
Figure 4①), and the peak at 1514–1530 cm
−1 was assigned to C=C bonds conjugated with aldehyde groups of polymerized glutaraldehyde (
Figure 4③).
Figure 4.
FT-IR spectra of (a) γ-PGA/PLL cross-linked with glutaraldehyde for 24 h, γ-PGA/Chitosan cross-linked with glutaraldehyde for (b) 30 min, (c) 24 h, and (d) 96 h. (① C=O (1629–1640 cm−1), ② C=N (1589 cm−1), ③ C=C (1514–1530 cm−1)).
Figure 4.
FT-IR spectra of (a) γ-PGA/PLL cross-linked with glutaraldehyde for 24 h, γ-PGA/Chitosan cross-linked with glutaraldehyde for (b) 30 min, (c) 24 h, and (d) 96 h. (① C=O (1629–1640 cm−1), ② C=N (1589 cm−1), ③ C=C (1514–1530 cm−1)).
The peak at 1589 cm
−1 was attributed to the C=N bonds of the Schiff base (
Figure 4②), which indicated that both γ-PGA/PLL and γ-PGA/chitosan nanoparticles exhibit autofluorescence as a result of the formation of these bonds. In the case of γ-PGA/chitosan nanoparticles, the gradual increase in the intensity of the peak at 1589 cm
−1 was attributed to the formation of imine bonds with increasing crosslinking reaction time (
Figure 4d). The principle of fluorescence is based on the transition of electrons between different orbitals. It is well-known that C=N bonds are associated with n-π* transitions and that C=C bonds are related to π-π* transitions [
14]. Fluorescence images of the γ-PGA/PLL and γ-PGA/chitosan nanoparticles crosslinked with glutaraldehyde were obtained using suitable excitation and emission filters from the visible to near-infrared region (
Figure 5). The γ-PGA/PLL and γ-PGA/chitosan nanoparticles both appeared to autofluorescence, without any fluorescent probes, depending on the optical filters used.
Figure 5.
Fluorescence images of (a) γ-PGA/PLL cross-linked with glutaraldehyde for 12 h and (b) γ-PGA/Chitosan cross-linked with 10 μL glutaraldehyde for 96 h. (vertical axis: emission wavelength, horizontal axis: excitation wavelength, exposure time: 0.2 s).
Figure 5.
Fluorescence images of (a) γ-PGA/PLL cross-linked with glutaraldehyde for 12 h and (b) γ-PGA/Chitosan cross-linked with 10 μL glutaraldehyde for 96 h. (vertical axis: emission wavelength, horizontal axis: excitation wavelength, exposure time: 0.2 s).
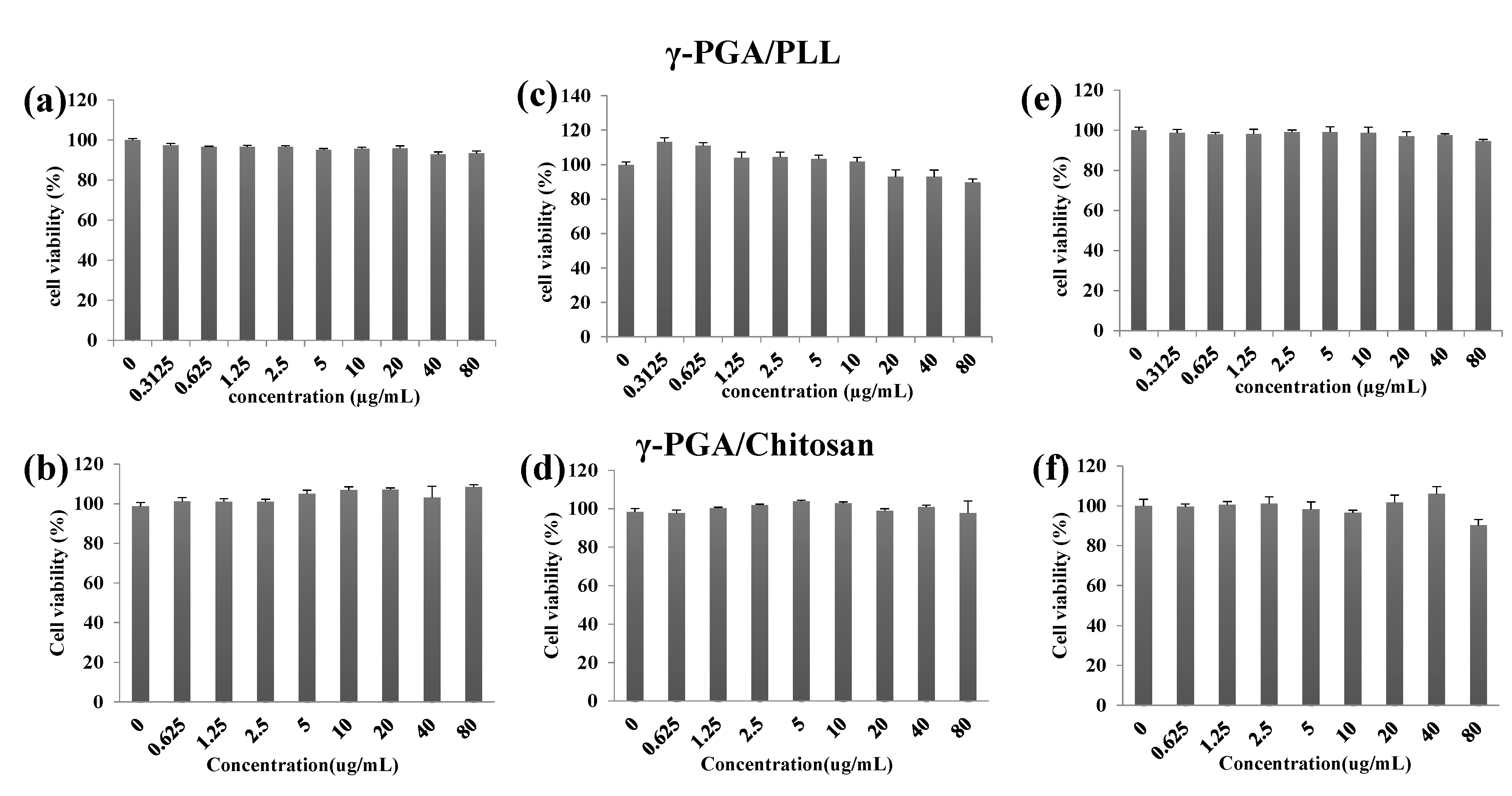
2.3. Cell Viability and Cellular Uptake
The cell viabilities of the γ-PGA/PLL and γ-PGA/chitosan nanoparticles were evaluated using an MTS assay
in vitro (
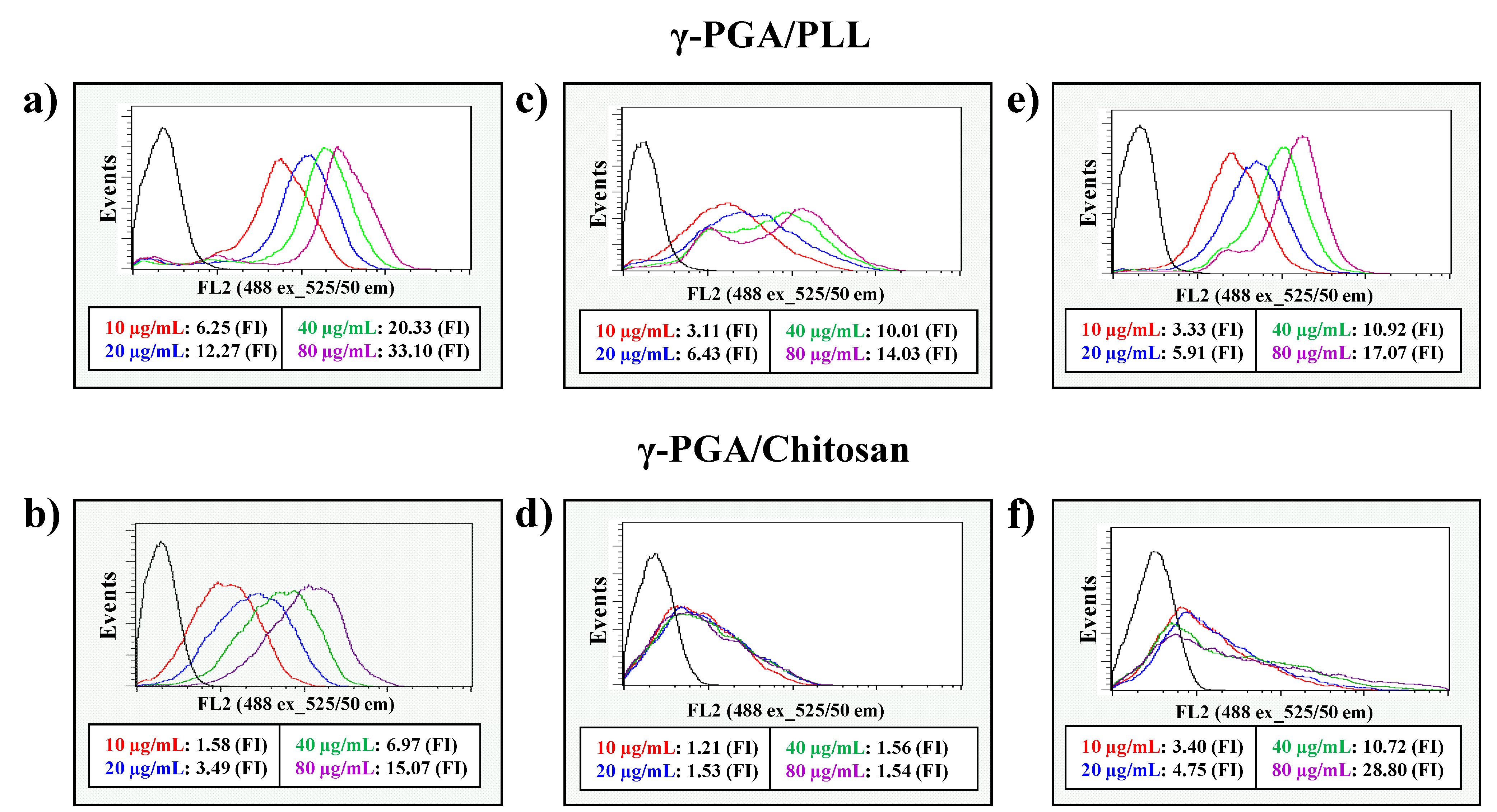
Figure 6). According to the results, the two types of nanoparticles were non-toxic to the cells. The intracellular delivery capacities of the two different nanoparticles were investigated using flow cytometry with various concentrations of the nanoparticles (
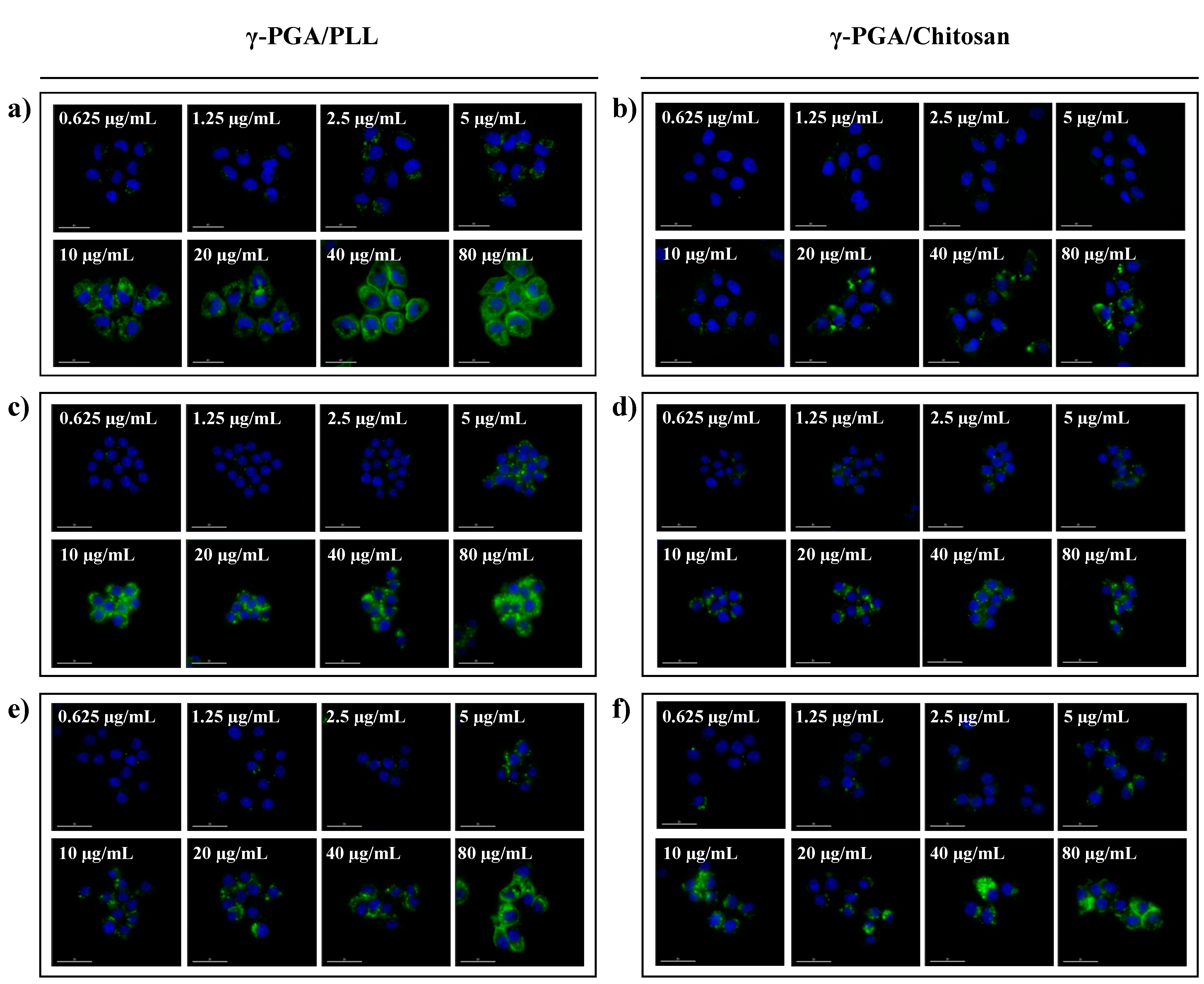
Figure 7). The effects of the nanoparticles on cellular uptake were also investigated using fluorescence microscopy imaging (
Figure 8). The intracellular uptake efficiency increased with increasing concentrations of nanoparticles. Regarding the charge effect on intracellular uptake, positively charged particles are more effective than negatively charged particles
in vitro [
21,
22,
23]. In the case of the γ-PGA/chitosan nanoparticles, the cellular uptake capacity was relatively lower because chitosan is deprotonated at neutral pH due to the pKa (pH 6.3) of the polymer [
24]. As shown in
Figure 8, the γ-PGA/PLL nanoparticles exhibited higher intracellular uptake efficiency than the γ-PGA/chitosan nanoparticles.
Figure 6.
The cell viability of γ-PGA/PLL (up) and γ-PGA/Chitosan (down), determined by the MTS assay ((a,b) HeLa cells, (c,d) RAW264.7 cells, (e,f) DC2.4 cells). The two types of nanocomposites were incubated with the displayed concentrations (from 0.3125 µg/mL to 80 µg/mL) for 24 h. All experiments were performed in triplicate.
Figure 6.
The cell viability of γ-PGA/PLL (up) and γ-PGA/Chitosan (down), determined by the MTS assay ((a,b) HeLa cells, (c,d) RAW264.7 cells, (e,f) DC2.4 cells). The two types of nanocomposites were incubated with the displayed concentrations (from 0.3125 µg/mL to 80 µg/mL) for 24 h. All experiments were performed in triplicate.
Figure 7.
The flow cytometry analysis of γ-PGA/PLL (up) and γ-PGA/Chitosan (down), respectively ((a,b) HeLa cells, (c,d) RAW264.7 cells, (e,f) DC2.4 cells). The two types of nanocomposites were incubated with various concentrations (from 10 µg/mL to 80 µg/mL) for 24 h.
Figure 7.
The flow cytometry analysis of γ-PGA/PLL (up) and γ-PGA/Chitosan (down), respectively ((a,b) HeLa cells, (c,d) RAW264.7 cells, (e,f) DC2.4 cells). The two types of nanocomposites were incubated with various concentrations (from 10 µg/mL to 80 µg/mL) for 24 h.
Figure 8.
Fluorescence microscope images of γ-PGA/PLL (left) and γ-PGA/Chitosan (right), respectively ((a,b) HeLa cells, (c,d) RAW264.7 cells, (e,f) DC2.4 cells). The two types of nanocomposites were incubated with various concentrations (from 0.625 µg/mL to 80 µg/mL) for 24 h. (Ex 490/20; Em 526/36).
Figure 8.
Fluorescence microscope images of γ-PGA/PLL (left) and γ-PGA/Chitosan (right), respectively ((a,b) HeLa cells, (c,d) RAW264.7 cells, (e,f) DC2.4 cells). The two types of nanocomposites were incubated with various concentrations (from 0.625 µg/mL to 80 µg/mL) for 24 h. (Ex 490/20; Em 526/36).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








