
Site-Specific Bioconjugation of an Organometallic Electron Mediator to an Enzyme with Retained Photocatalytic Cofactor Regenerating Capacity and Enzymatic Activity


Abstract
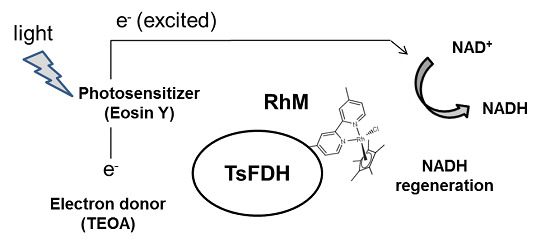
:
1. Introduction
2. Results and Discussion
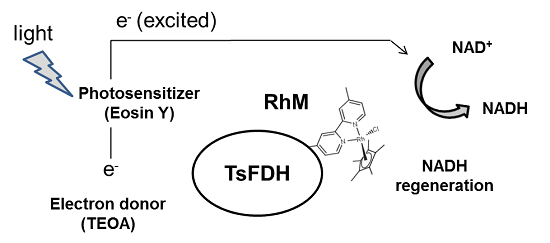
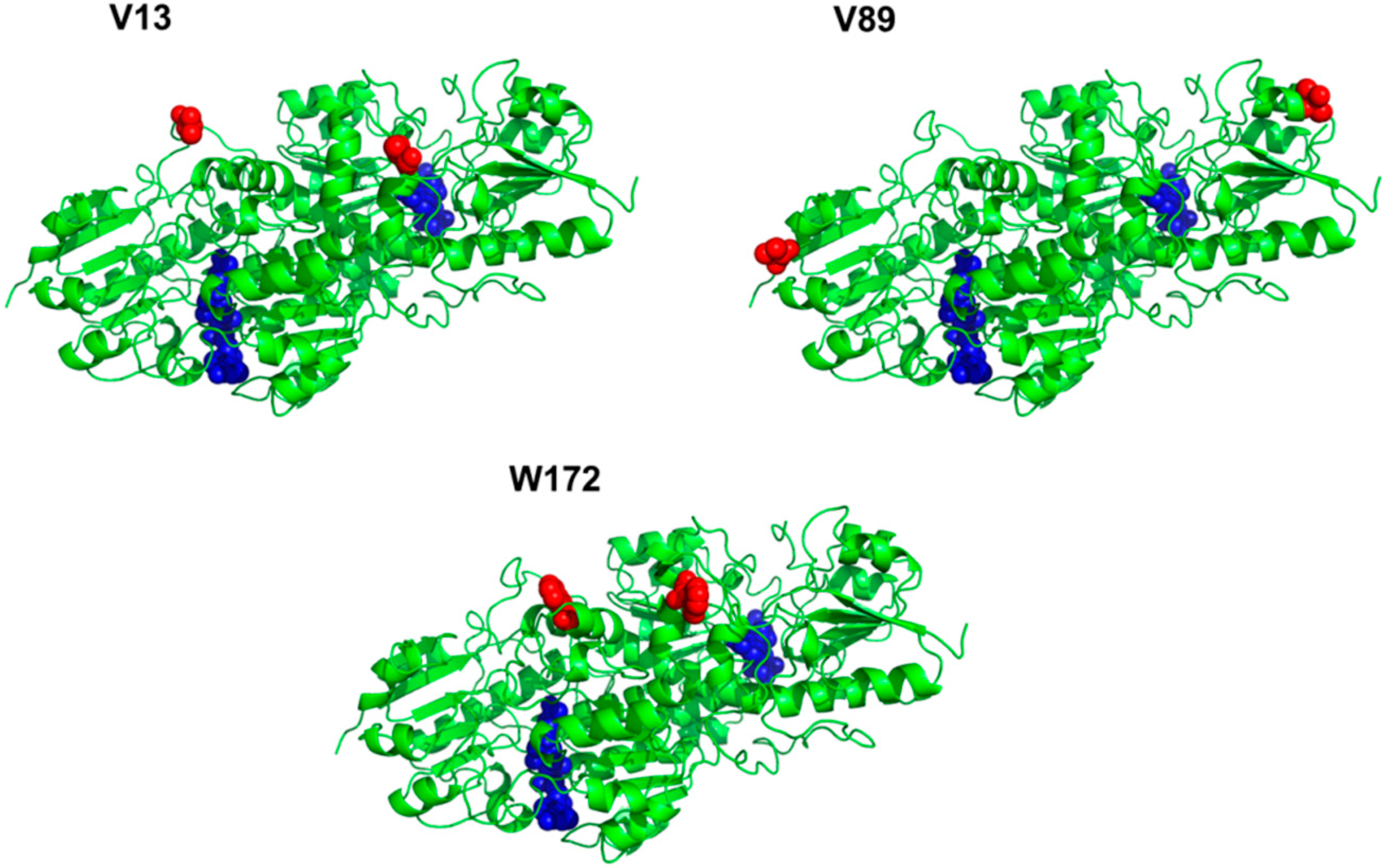
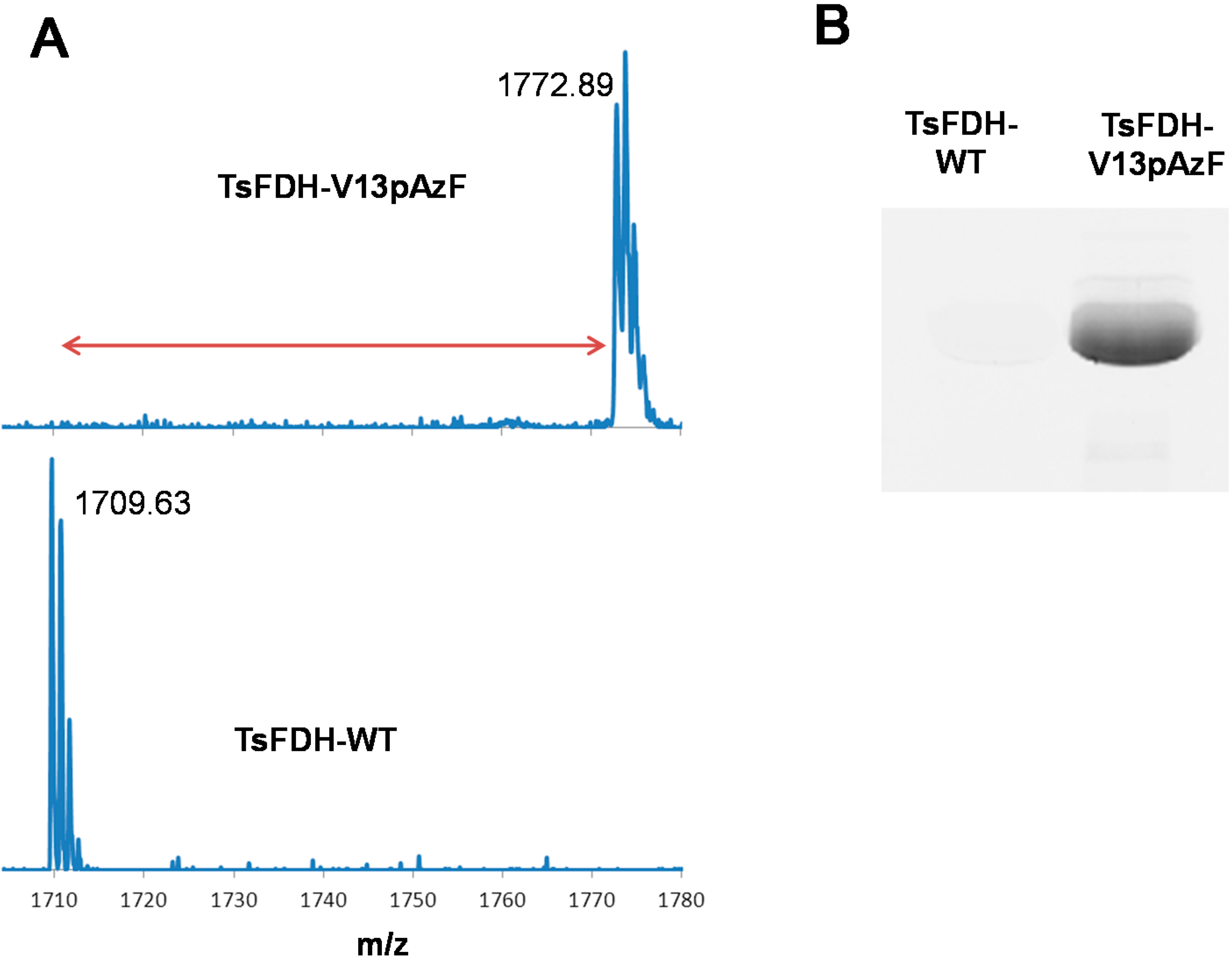
2.1. Site-Specific Incorporation of p-Azido-L-Phenylalanine (pAzF) into Formate Dehydrogenase

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | V13 | V89 | W172 |
|---|---|---|---|
| Solvent accessibility | 0.70 | 0.38 | 0.32 |


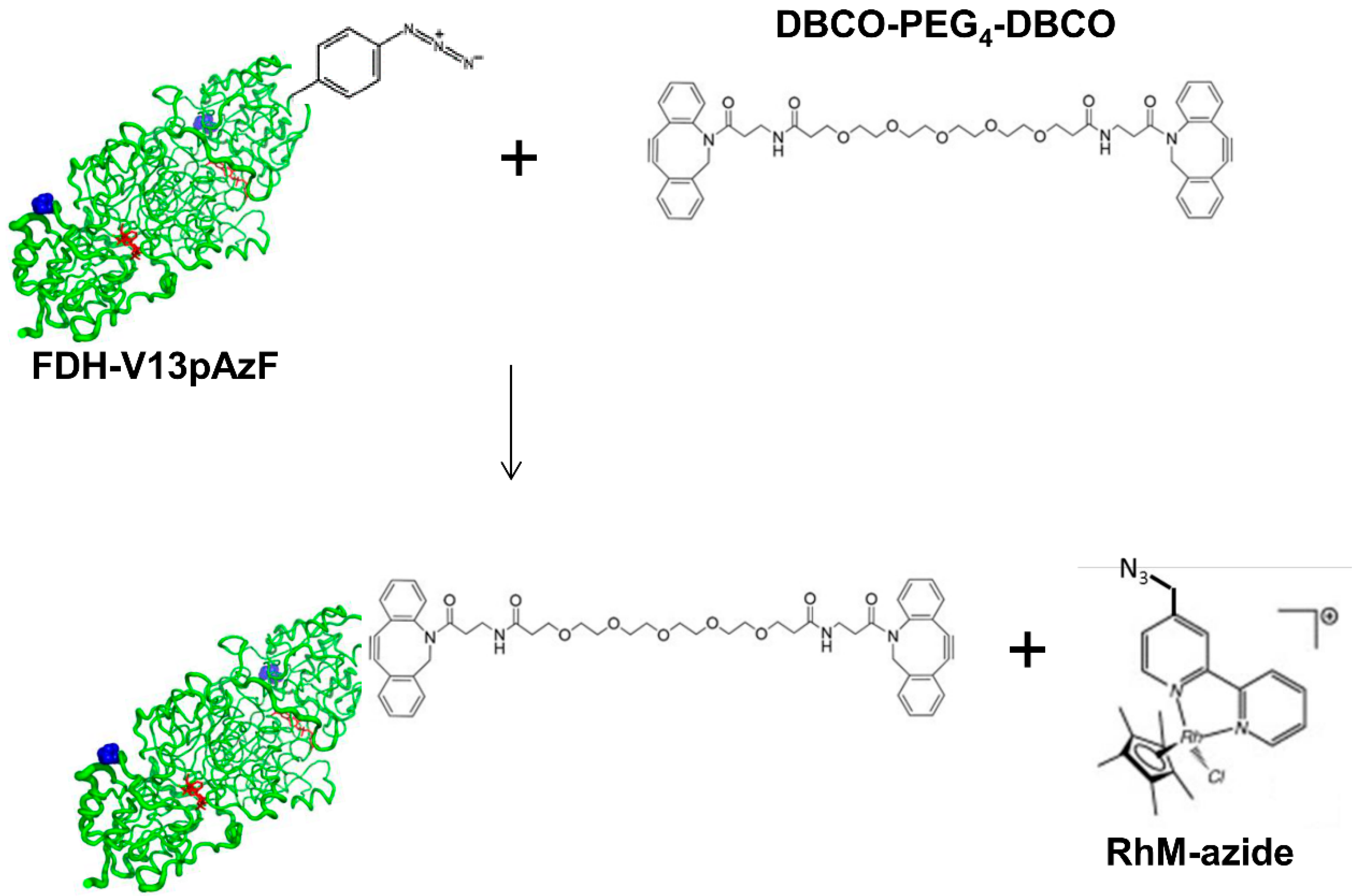
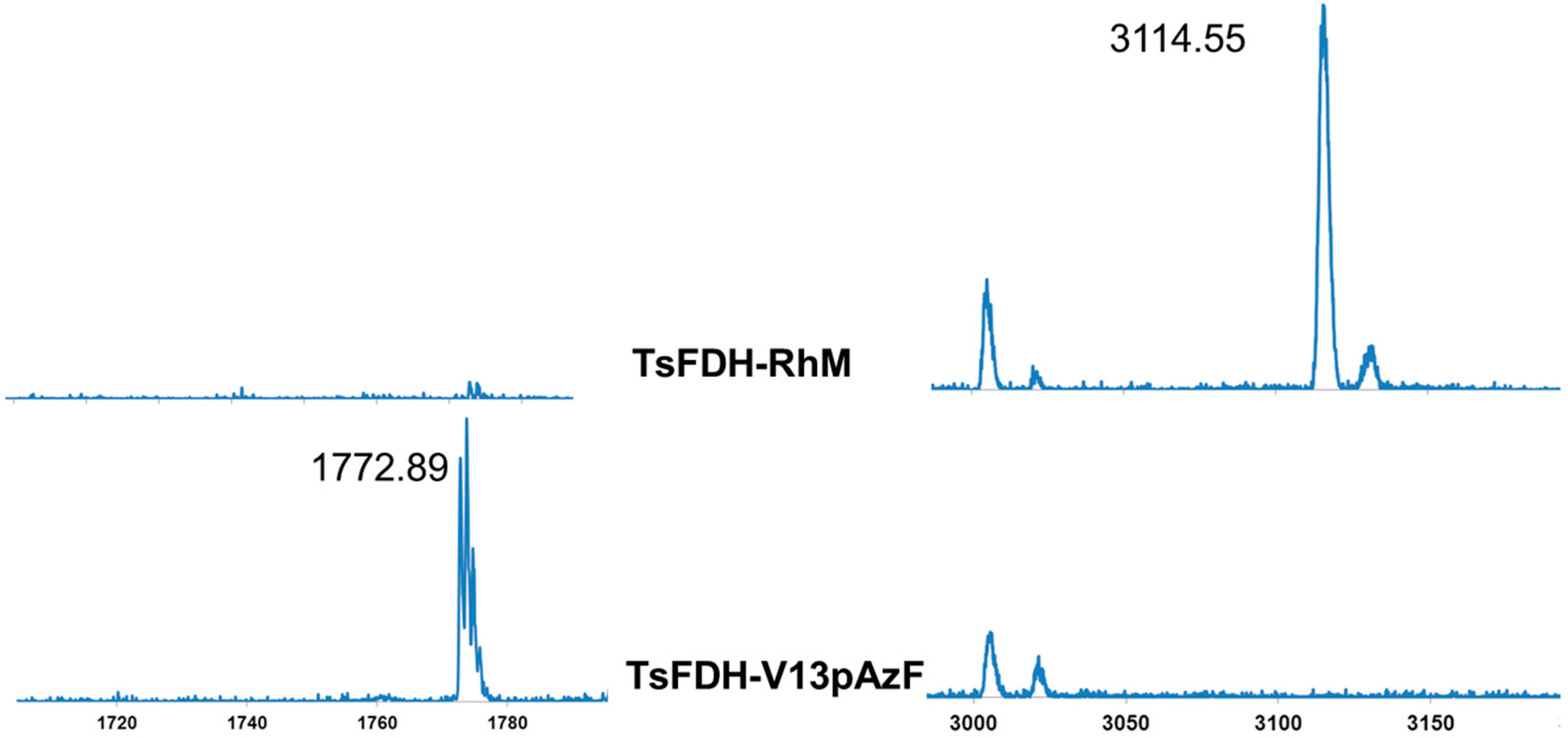
2.2. Conjugation of Rh-Based Electron Mediator to TsFDH-V13pAzF


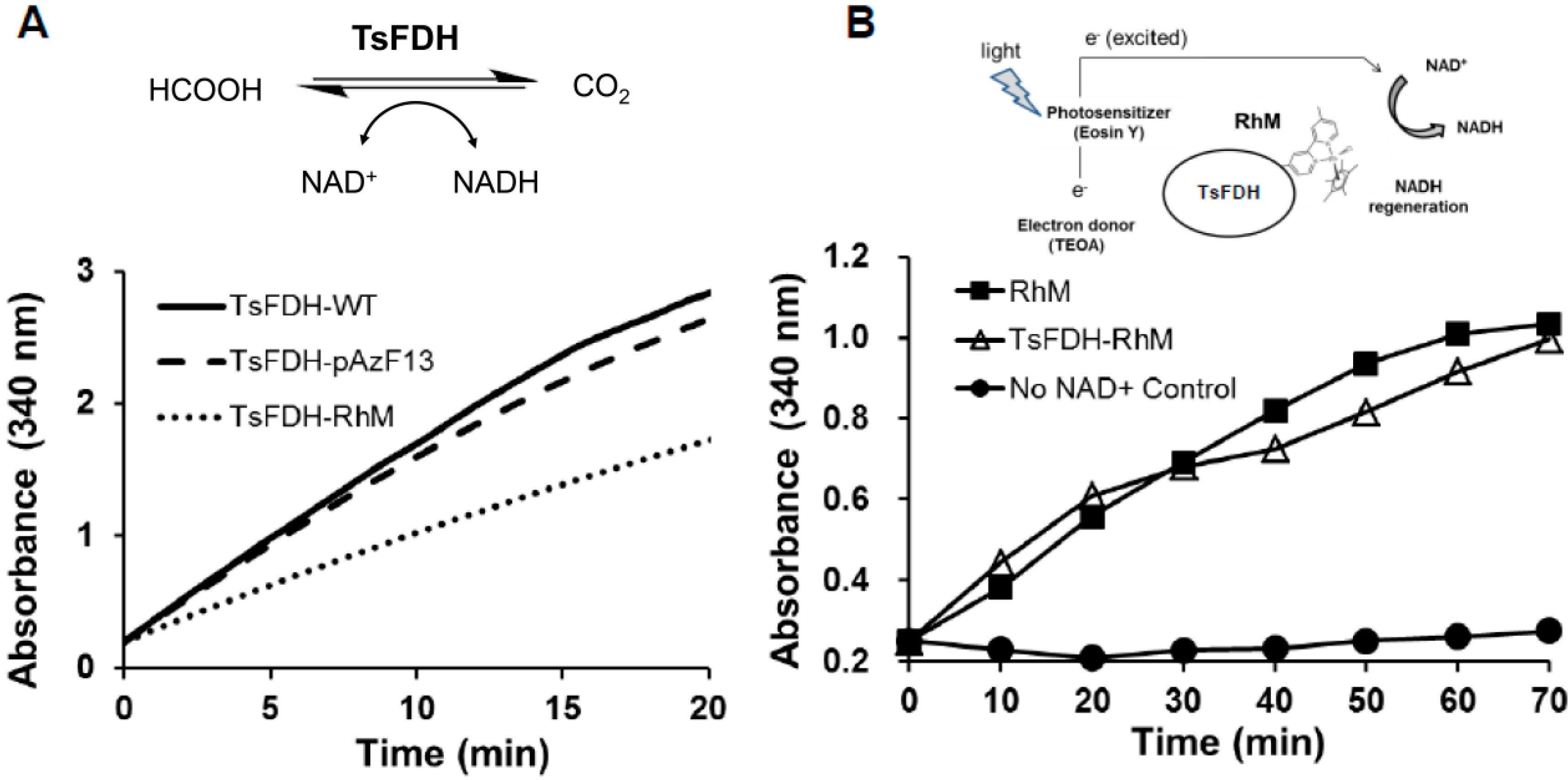
2.3. Characterization of the TsFDH-RhM Conjugate: Enzymatic Activity and Cofactor Generation

3. Experimental Section
3.1. General Information
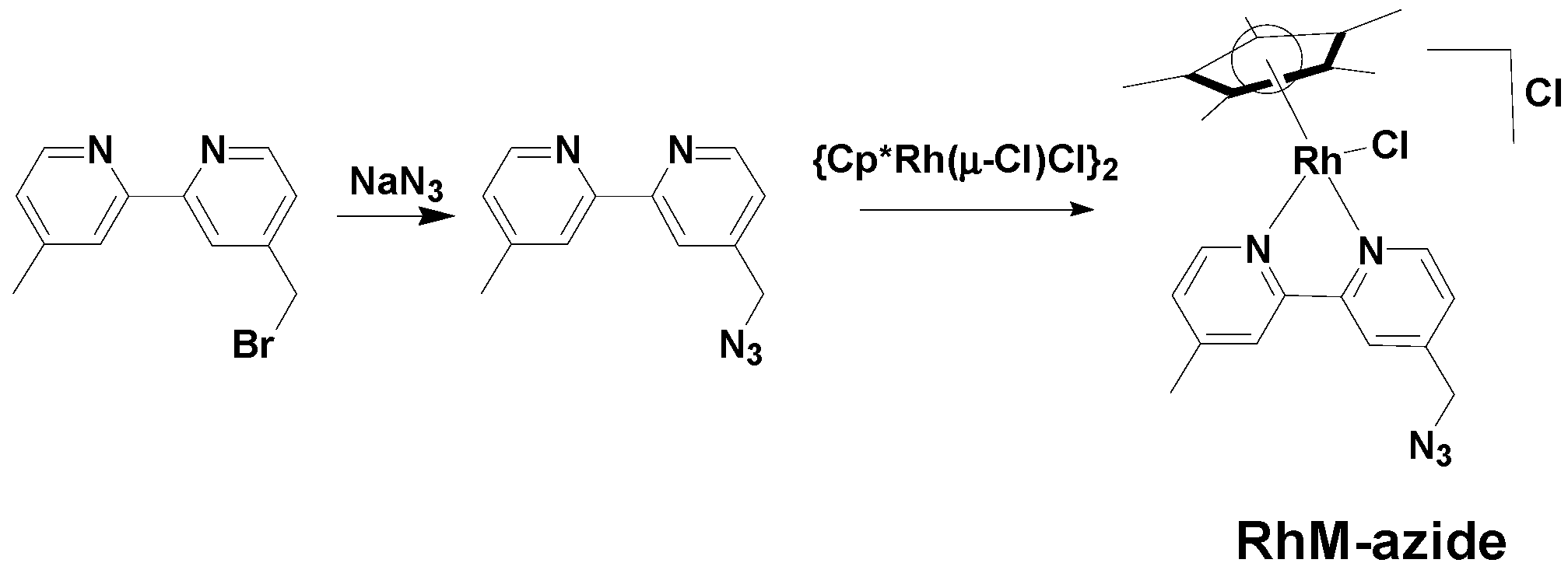
3.2. Synthesis of [Cp*Rh(4-(Azidomethyl)-4'-Methyl-2,2'-Bipyridine)Cl]Cl (RhM-azide)

3.3. Site-Specific Incorporation of pAzF into Formate Dehydrogenase
3.4. Site-Specific Conjugation of Dye or Rh-Based Electron Mediator to TsFDH-V13pAzF
3.5. Functional Assay of the TsFDH-RhM Conjugate
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Meyer, T.J. Catalysis: The art of splitting water. Nature 2008, 451, 778–779. [Google Scholar] [CrossRef] [PubMed]
- Sayama, K.; Mukasa, K.; Abe, R.; Abe, Y.; Arakawa, H. A new photocatalytic water splitting system under visible light irradiation mimicking a z-scheme mechanism in photosynthesis. J. Photochem. Photobiol. A-Chem. 2002, 148, 71–77. [Google Scholar] [CrossRef]
- Gabrielsson, A.; Lindsay Smith, J.R.; Perutz, R.N. Remote site photosubstitution in metalloporphyrin-rhenium tricarbonylbipyridine assemblies: Photo-reactions of molecules with very short lived excited states. Dalton Trans. 2008, 4259–4269. [Google Scholar] [CrossRef]
- Li, X.; Wang, M.; Zhang, S.; Pan, J.; Na, Y.; Liu, J.; Akermark, B.; Sun, L. Noncovalent assembly of a metalloporphyrin and an iron hydrogenase active-site model: Photo-induced electron transfer and hydrogen generation. J. Phys. Chem. B 2008, 112, 8198–8202. [Google Scholar] [PubMed]
- Hollmann, F.; Witholt, B.; Schmid, A. [cp*rh(bpy)(h2o)]2+: A versatile tool for efficient and non-enzymatic regeneration of nicotinamide and flavin coenzymes. J. Mol. Catal. B Enzym. 2002, 19–20, 167–176. [Google Scholar]
- Lo, H.C.; Leiva, C.; Buriez, O.; Kerr, J.B.; Olmstead, M.M.; Fish, R.H. Bioorganometallic chemistry. 13. Regioselective reduction of nad+ models, 1-benzylnicotinamde triflate and β-nicotinamide ribose-5'-methyl phosphate, with in situ generated [cp*rh(bpy)h]+: Structure-activity relationships, kinetics, and mechanistic aspects in the formation of the 1,4-nadh derivatives. Inorg. Chem. 2001, 40, 6705–6716. [Google Scholar]
- Choudhury, S.; Baeg, J.-O.; Park, N.-J.; Yadav, R.K. A photocatalyst/enzyme couple that uses solar energy in the asymmetric reduction of acetophenones. Angew. Chem. 2012, 124, 11792–11796. [Google Scholar] [CrossRef]
- Yadav, R.K.; Baeg, J.-O.; Oh, G.H.; Park, N.-J.; Kong, K.-J.; Kim, J.; Hwang, D.W.; Biswas, S.K. A photocatalyst–enzyme coupled artificial photosynthesis system for solar energy in production of formic acid from co2. J. Am. Chem. Soc. 2012, 134, 11455–11461. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, M.; Lee, J.S.; Park, C.B. Self-assembled light-harvesting peptide nanotubes for mimicking natural photosynthesis. Angew. Chem. Int. Ed. 2012, 51, 517–520. [Google Scholar] [CrossRef]
- Ryu, J.; Nam, D.H.; Lee, S.H.; Park, C.B. Biocatalytic photosynthesis with water as an electron donor. Chem.-Eur. J. 2014, 20, 12020–12025. [Google Scholar] [CrossRef] [PubMed]
- Basle, E.; Joubert, N.; Pucheault, M. Protein chemical modification on endogenous amino acids. Chem. Biol. 2010, 17, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Rusmini, F.; Zhong, Z.; Feijen, J. Protein immobilization strategies for protein biochips. Biomacromolecules 2007, 8, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.I.; Mizuta, Y.; Takasu, A.; Hahn, Y.S.; Kim, Y.H.; Kwon, I. Site-specific fatty acid-conjugation to prolong protein half-life in vivo. J. Control. Release 2013, 170, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.I.; Mizuta, Y.; Takasu, A.; Kim, Y.H.; Kwon, I. Site-specific bioconjugation of a murine dihydrofolate reductase enzyme by copper(I)-catalyzed azide-alkyne cycloaddition with retained activity. PLoS ONE 2014, 9, e98403. [Google Scholar] [CrossRef] [PubMed]
- Köhler, V.; Wilson, Y.M.; Dürrenberger, M.; Ghislieri, D.; Churakova, E.; Quinto, T.; Knörr, L.; Häussinger, D.; Hollmann, F.; Turner, N.J.; et al. Synthetic cascades are enabled by combining biocatalysts with artificial metalloenzymes. Nat. Chem. 2013, 5, 93–99. [Google Scholar]
- Poizat, M.; Arends, I.W.C.E.; Hollmann, F. On the nature of mutual inactivation between [cp*rh(bpy)(h2o)]2+ and enzymes—analysis and potential remedies. J. Mol. Catal. B Enzym. 2010, 63, 149–156. [Google Scholar] [CrossRef]
- De Torres, M.; Dimroth, J.; Arends, I.W.C.E.; Keilitz, J.; Hollmann, F. Towards recyclable nad(p)h regeneration catalysts. Molecules 2012, 17, 9835–9841. [Google Scholar]
- Tishkov, V.I.; Popov, V.O. Protein engineering of formate dehydrogenase. Biomol. Eng. 2006, 23, 89–110. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Gromiha, M.; Fawareh, H.; Sarai, A. Asaview: Database and tool for solvent accessibility representation in proteins. BMC Bioinform. 2004, 5, 51. [Google Scholar] [CrossRef] [Green Version]
- Choe, H.; Ha, J.M.; Joo, J.C.; Kim, H.; Yoon, H.-J.; Kim, S.; Son, S.H.; Gengan, R.M.; Jeon, S.T.; Chang, R.; et al. Structural insights into the efficient co2-reducing activity of an nad-dependent formate dehydrogenase from thiobacillus sp. Knk65ma. Acta. Crystallogr. D 2015, 71, 313–323. [Google Scholar]
- Guillaneuf, Y.; Dufils, P.E.; Autissier, L.; Rollet, M.; Gigmes, D.; Bertin, D. Radical chain end chemical transformation of sg1-based polystyrenes. Macromolecules 2010, 43, 91–100. [Google Scholar] [CrossRef]
- Li, Y.; Hoskins, J.N.; Sreerama, S.G.; Grayson, S.M. Maldi-tof mass spectral characterization of polymers containing an azide group: Evidence of metastable ions. Macromolecules 2010, 43, 6225–6228. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.F.; Borner, H.G.; Weichenhan, K. Combining atom transfer radical polymerization and click chemistry: A versatile method for the preparation of end-functional polymers. Macromol. Rapid Commun. 2005, 26, 514–518. [Google Scholar] [CrossRef]
- Raynaud, J.; Absalon, C.; Gnanou, Y.; Taton, D. N-heterocyclic carbene-induced zwitterionic ring-opening polymerization of ethylene oxide and direct synthesis of alpha, omega-difunctionalized poly(ethylene oxide)s and poly(ethylene oxide)-b-poly(epsilon-caprolactone) block copolymers. J. Am. Chem. Soc. 2009, 131, 3201–3209. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Nam, D.H.; Kim, J.H.; Baeg, J.-O.; Park, C.B. Eosin y-sensitized artificial photosynthesis by highly efficient visible-light-driven regeneration of nicotinamide cofactor. ChemBioChem 2009, 10, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Nam, D.H.; Park, C.B. Screening xanthene dyes for visible light-driven nicotinamide adenine dinucleotide regeneration and photoenzymatic synthesis. Adv. Synth. Catal. 2009, 351, 2589–2594. [Google Scholar] [CrossRef]
- Choe, H.; Joo, J.C.; Cho, D.H.; Kim, M.H.; Lee, S.H.; Jung, K.D.; Kim, Y.H. Efficient co2-reducing activity of nad-dependent formate dehydrogenase from thiobacillus sp. Knk65ma for formate production from co2 gas. PLoS ONE 2014, 9, e103111. [Google Scholar]
- Bond, S.R.; Naus, C.C. Rf-cloning.Org: An online tool for the design of restriction-free cloning projects. Nucleic Acids Res. 2012, 40, W209–W213. [Google Scholar]
- Lajoie, M.J.; Rovner, A.J.; Goodman, D.B.; Aerni, H.R.; Haimovich, A.D.; Kuznetsov, G.; Mercer, J.A.; Wang, H.H.; Carr, P.A.; Mosberg, J.A.; et al. Genomically recoded organisms expand biological functions. Science 2013, 342, 357–360. [Google Scholar]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995, 4, 2411–2423. [Google Scholar] [CrossRef] [PubMed]
- Grimsley, G.R.; Pace, C.N. Unit 3.1 Spectrophotometric determination of protein concentration. In Current Protocols in Protein Science; Taylor, G.P., Ed.; Wiley: Hoboken, NJ, USA, 2004. [Google Scholar]
- Sample Availability: RhM-azide sample is not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, S.I.; Yoon, S.; Kim, Y.H.; Kwon, I. Site-Specific Bioconjugation of an Organometallic Electron Mediator to an Enzyme with Retained Photocatalytic Cofactor Regenerating Capacity and Enzymatic Activity. Molecules 2015, 20, 5975-5986. https://doi.org/10.3390/molecules20045975
Lim SI, Yoon S, Kim YH, Kwon I. Site-Specific Bioconjugation of an Organometallic Electron Mediator to an Enzyme with Retained Photocatalytic Cofactor Regenerating Capacity and Enzymatic Activity. Molecules. 2015; 20(4):5975-5986. https://doi.org/10.3390/molecules20045975
Chicago/Turabian StyleLim, Sung In, Sungho Yoon, Yong Hwan Kim, and Inchan Kwon. 2015. "Site-Specific Bioconjugation of an Organometallic Electron Mediator to an Enzyme with Retained Photocatalytic Cofactor Regenerating Capacity and Enzymatic Activity" Molecules 20, no. 4: 5975-5986. https://doi.org/10.3390/molecules20045975