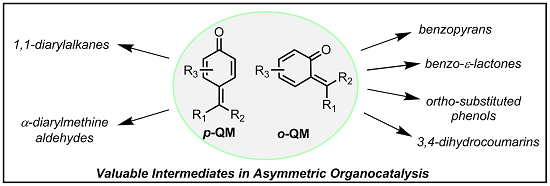
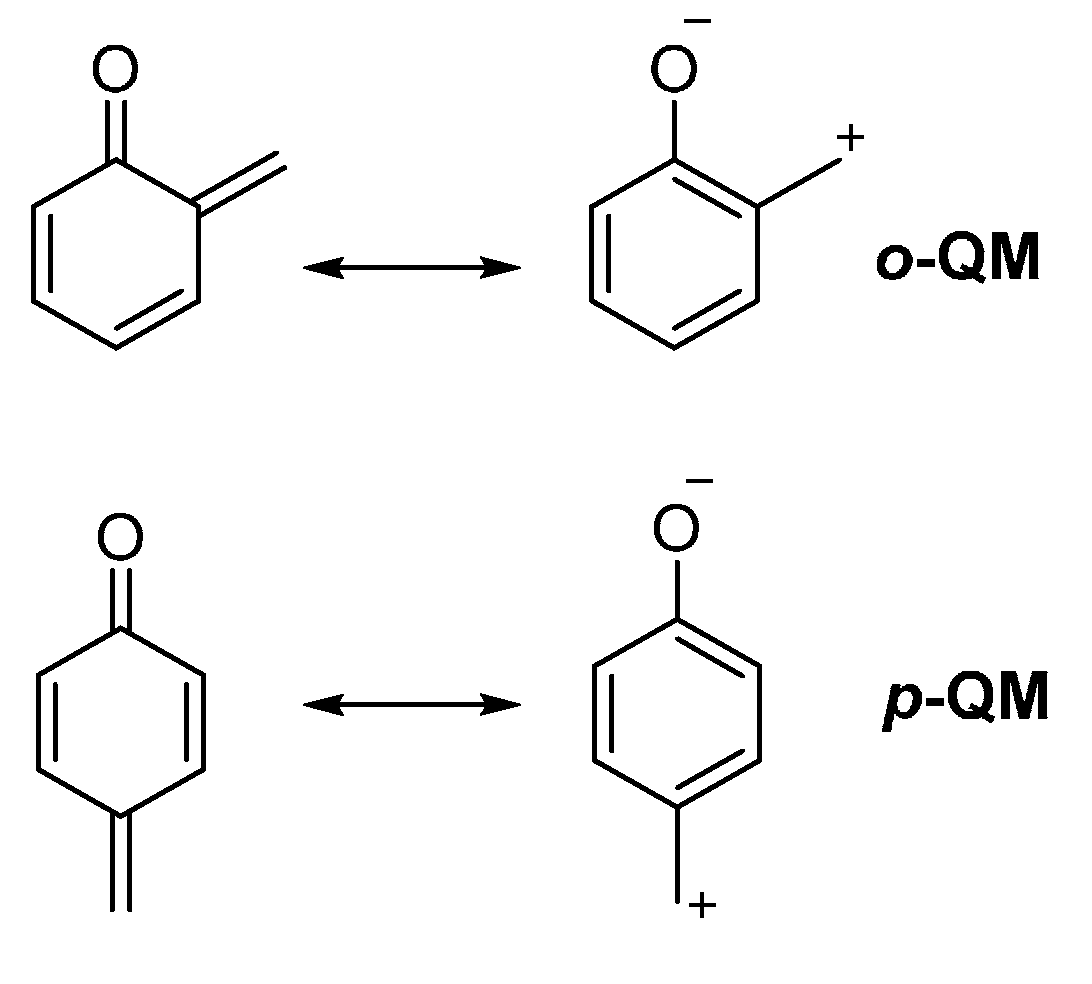
Although the generation of an
o-QM
in situ can in principle provide a more general approach, the disclosure of conditions compatible with both formation of a reactive
o-QM and a catalytic asymmetric reaction appeared to be a very challenging task, unmet until very recently. Furthermore, ease of formation, reactivity and stability of these intermediates are highly dependent on the substitution pattern of their triene portion, with electron donating substituents facilitating their formation and isolation but at the same decreasing reactivity [
19]. The most common method for the generation of
o-QMs is the elimination of a stable molecule (e.g., water) from the benzylic position of 2-substituted phenols. In fact, although several other methods for the generation of
o-QM are known (oxidations, olefinations,
etc.), such type of elimination has certainly been the most common platform to form
o-QMs
in situ for asymmetric organocatalytic reactions, as summarized in the next sections. The conditions under which the
o-QM is generated determine the type of catalysis that can be used in the asymmetric step, or, in a complementary perspective, the utilization of a type of catalysis mandates the conditions that can be used for the generation of the
o-QM. The combinations of both acidic and basic conditions with appropriate catalysts have been implemented, thus allowing the productive engagement of a broad range of substrates in organocatalytic enantioselective reactions with
o-QMs.
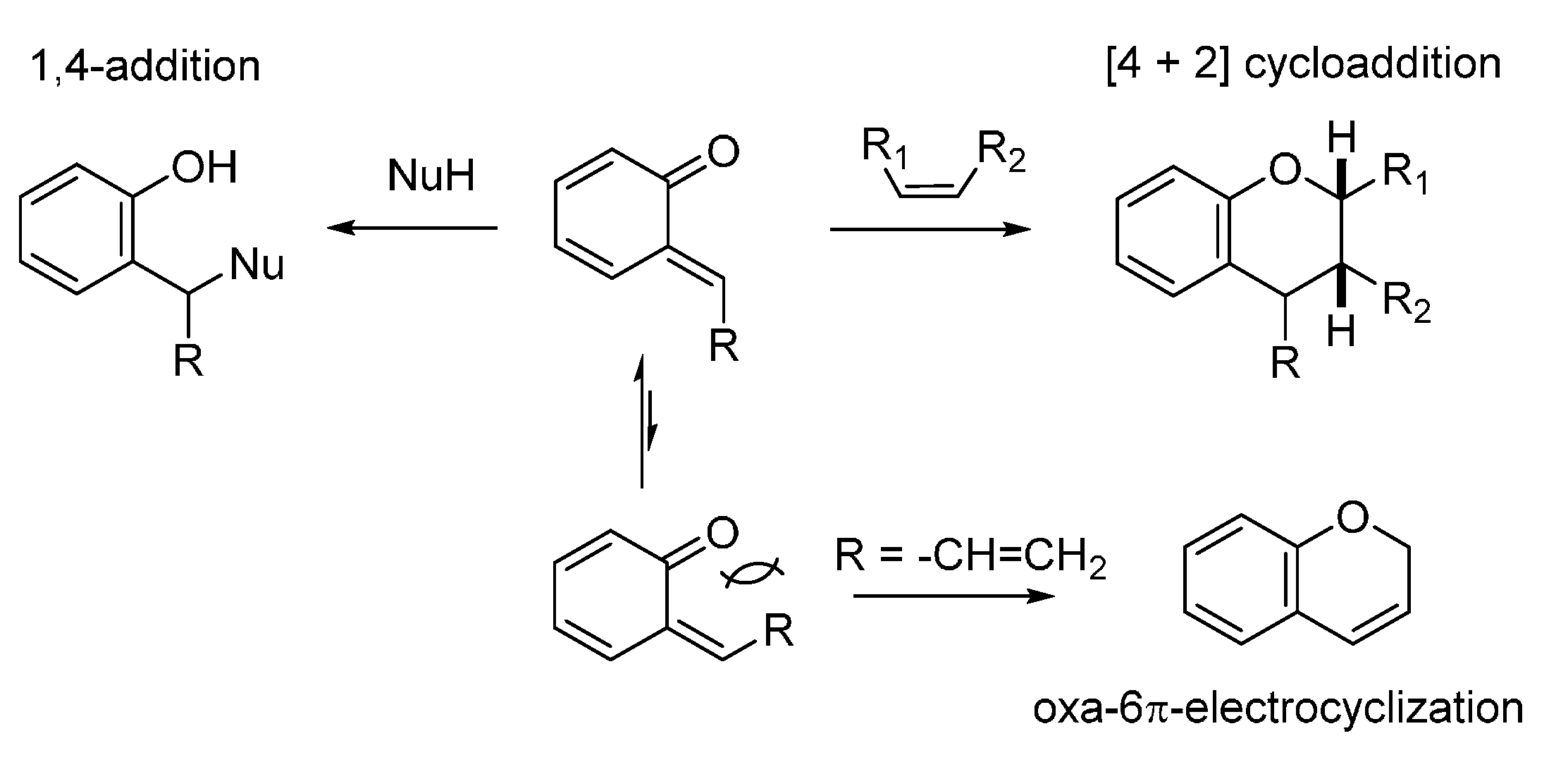
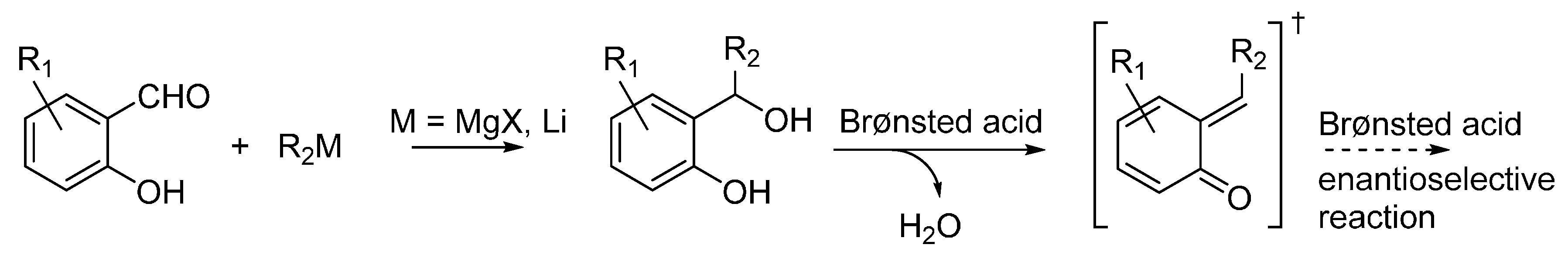
3.1. o-QMs Generated in Situ by Dehydration of Ortho-Hydroxybenzylic Alcohols under Brønsted Acid Conditions
The generation of
o-QM by dehydration of
ortho-hydroxybenzyl alcohols is perhaps the most common methodology exploited for the utilization of
o-QM in chemical biology, wherein QMs bearing a terminal methylene group have been the most investigated. The position of the equilibrium existing between the alcohol and the
o-QM, as well as the kinetics of
o-QM formation, has been shown to be highly dependent on the conditions (especially pH) [
20] and the substituents at the phenolic ring [
19]. A general synthetic route to
ortho-hydroxybenzylic alcohols is represented by the addition of organometallic reagents (Grignard and organolithium) to salicylaldehydes (
Scheme 13). Useful protocols can be found in the papers highlighted in this section, which show how these benzylic alcohols can be engaged in Brønsted acid catalyzed transformations, wherein the acid promotes both the formation of the
o-QM by dehydration and the ensuing enantioselective reaction.
Scheme 13.
Preparation of ortho-hydroxybenzylic alcohols and reactions under acidic conditions.
Scheme 13.
Preparation of ortho-hydroxybenzylic alcohols and reactions under acidic conditions.
Speculating on the analogy between
o-QMs and alkylideneindolenines, which can be formed by dehydration and activated upon the action of chiral phosphoric acid catalysts, Bach reported in 2011 that some
ortho-hydroxybenzylic alcohols react with indoles in the presence of chiral catalysts
7–
10, to give the expected adducts through the intermediacy of
o-QMs (
Scheme 14) [
21]. An electron donating substituent at the phenolic ring was found to be necessary for the reaction to proceed, presumably assisting formation of the intermediate. Whereas enantioselectivities were moderate at best, and no optimal catalyst giving uniformly good results with all substrates was found, this article represented the proof of concept of the viability of this approach, which other authors have later proven to be remarkably general. The intermediacy of an
o-QM, formed upon the action of the acidic chiral phosphoric acid
7–
10, was demonstrated by the following control experiments: (i) a free phenol moiety was found to be mandatory for the reaction to proceed enantioselectively; (ii) the substitution at the alcohol was not stereospecific (
i.e., racemic or enantiopure benzylalcohol substrates gave comparable enantiomeric enrichments in the products); and (iii) in reaction which were not run to completion, the remaining benzylalcohol starting material was optically active.
Scheme 14.
Addition of indoles to o-QMs generated from benzylic alcohols catalyzed by phosphoric acid catalysts 7–10.
Scheme 14.
Addition of indoles to o-QMs generated from benzylic alcohols catalyzed by phosphoric acid catalysts 7–10.
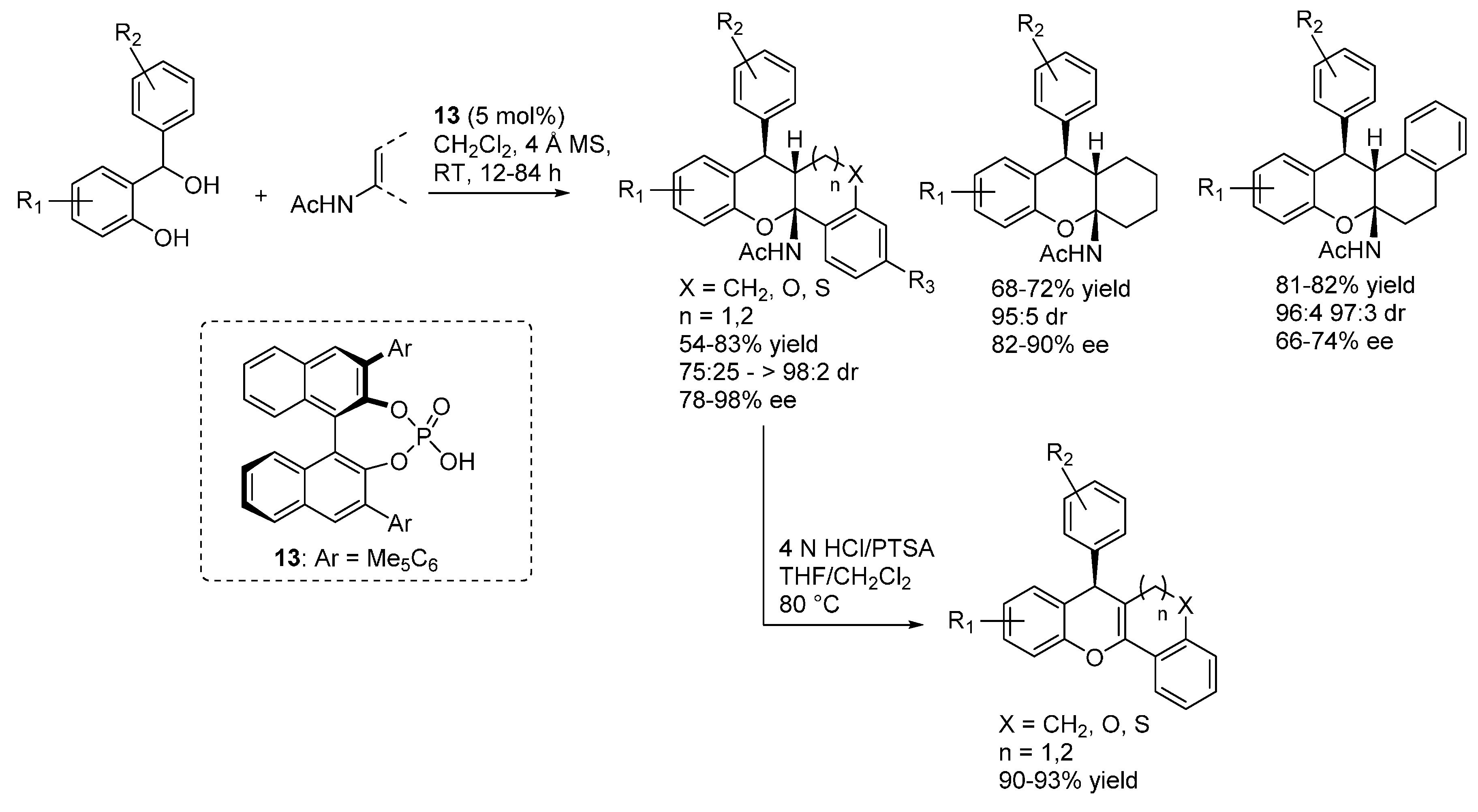
A catalytic enantioselective intramolecular cyclization involving dehydration of
ortho-hydroxybenzylic alcohols in the presence of acidic catalysts was then reported by Rueping in 2011 [
22]. However, the reaction was shown to proceed through a cationic intermediate, rather than through an
o-QM implying a 6π-electrocyclization. While in 2013 a non-enantioselective example of a hetero-Diels-Alder cycloaddition involving an
o-QM generated with this strategy was reported by Gharpure [
23], it was not until 2014 that highly enantioselective examples of the combination of dehydration of 2-hydroxy benzylic alcohols and ensuing asymmetric additions promoted by chiral acidic catalysts appeared in the literature. Two nearly simultaneous reports by Schneider and by Rueping showed the viability of this approach in the addition of 1,3-carbonyl compounds, which upon intramolecular hemiacetalization and dehydration render 4
H-chromenes or related polycyclic derivatives (
Scheme 15) [
24,
25]. In both cases, the reactions appeared to be limited to 2-hydroxy benzhydryl alcohols as starting materials. Taking also into account computational studies by Freccero showing that hydrogen bonding to the QMs carbonyl can enhance their reactivity [
26], and the commonly accepted mode of action of phosphoric acids, the catalysts were assumed to act in a bifunctional fashion. The acidic proton coordinates the carbonyl group of the
o-QM and the Lewis basic P=O moiety the enolic proton of the nucleophile in the reaction transition state.
Scheme 15.
Addition of 1,3-dicarbonyl compounds to o-QMs catalyzed by CPA. CPA = Chiral Phosphoric Acid.
Scheme 15.
Addition of 1,3-dicarbonyl compounds to o-QMs catalyzed by CPA. CPA = Chiral Phosphoric Acid.
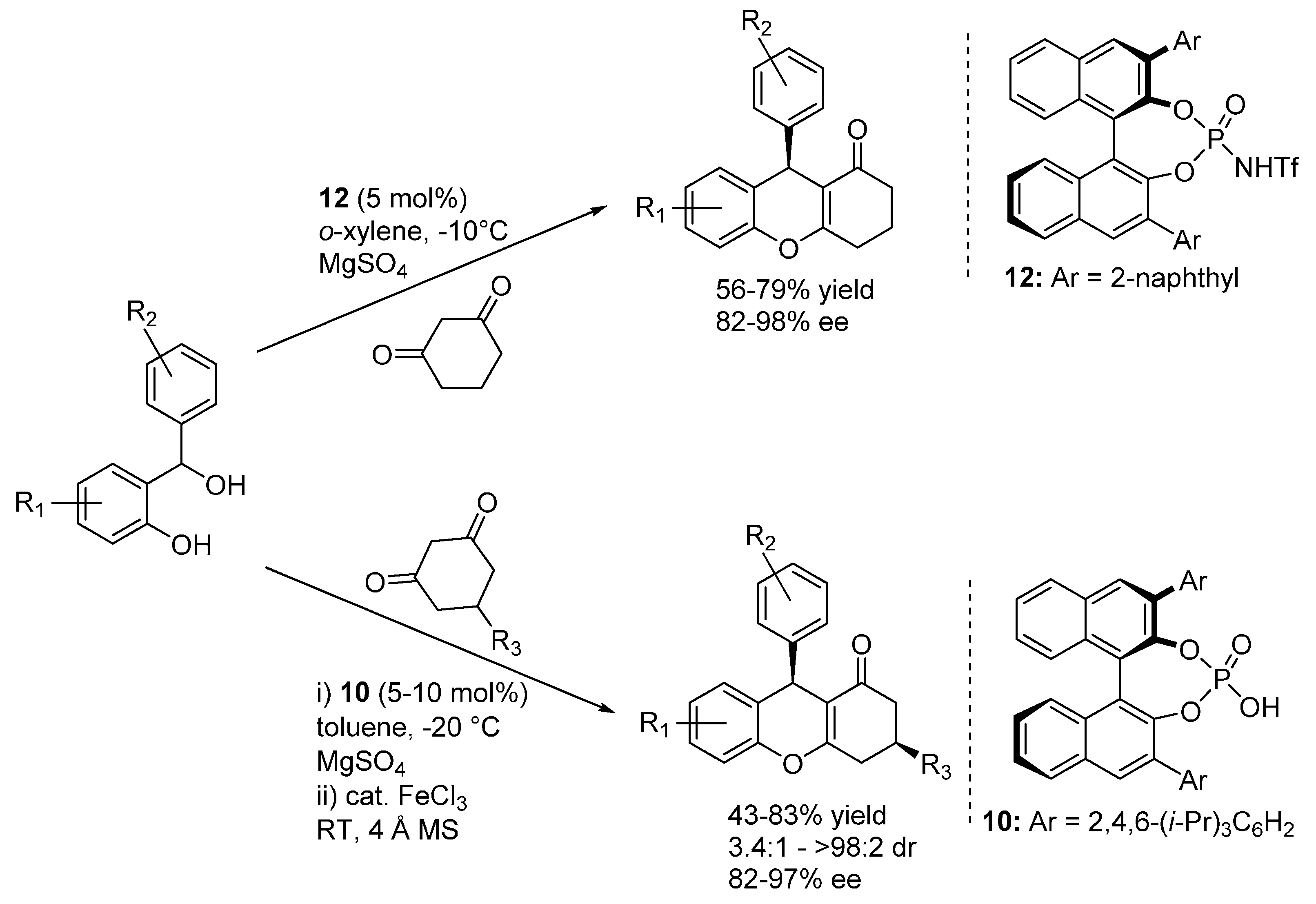
In more detail, Schneider reported the reaction with acetylacetone and cyclic 1,3-diketones featuring different ring sizes, and one example with a 3-ketoester, catalyzed by the BINOL derived phosphoric acid catalyst
11 (
Scheme 16) [
24]. A simple acidic treatment was performed to ensure full dehydration to the 4
H-chromene products, which were obtained with very good results irrespective of the nucleophile and benzhydrylic alcohol employed.
Scheme 16.
Addition of 1,3-dicarbonyl compounds to o-QMs catalyzed by phosphoric acid 11 developed by Schneider.
Scheme 16.
Addition of 1,3-dicarbonyl compounds to o-QMs catalyzed by phosphoric acid 11 developed by Schneider.
Rueping presented a similar catalytic reaction, using phosphorimide
12 or phosphoric acid
10 catalysts (
Scheme 17) [
25]; in the case of the more acidic phosphorimide
12, acidic treatment after the reaction was not required, the catalytic product cyclized spontaneously to the 4
H-chromene. While being limited to six membered diones, this report set great attention to a desymmetrization in the ensuing hemiacetalization and dehydration reactions, employing 5-substituted 1,3-cyclohexadiones as nucleophilic substrates and rendering tetrahydroxanthenes bearing two distant stereocenters with good diastereoselectivities.
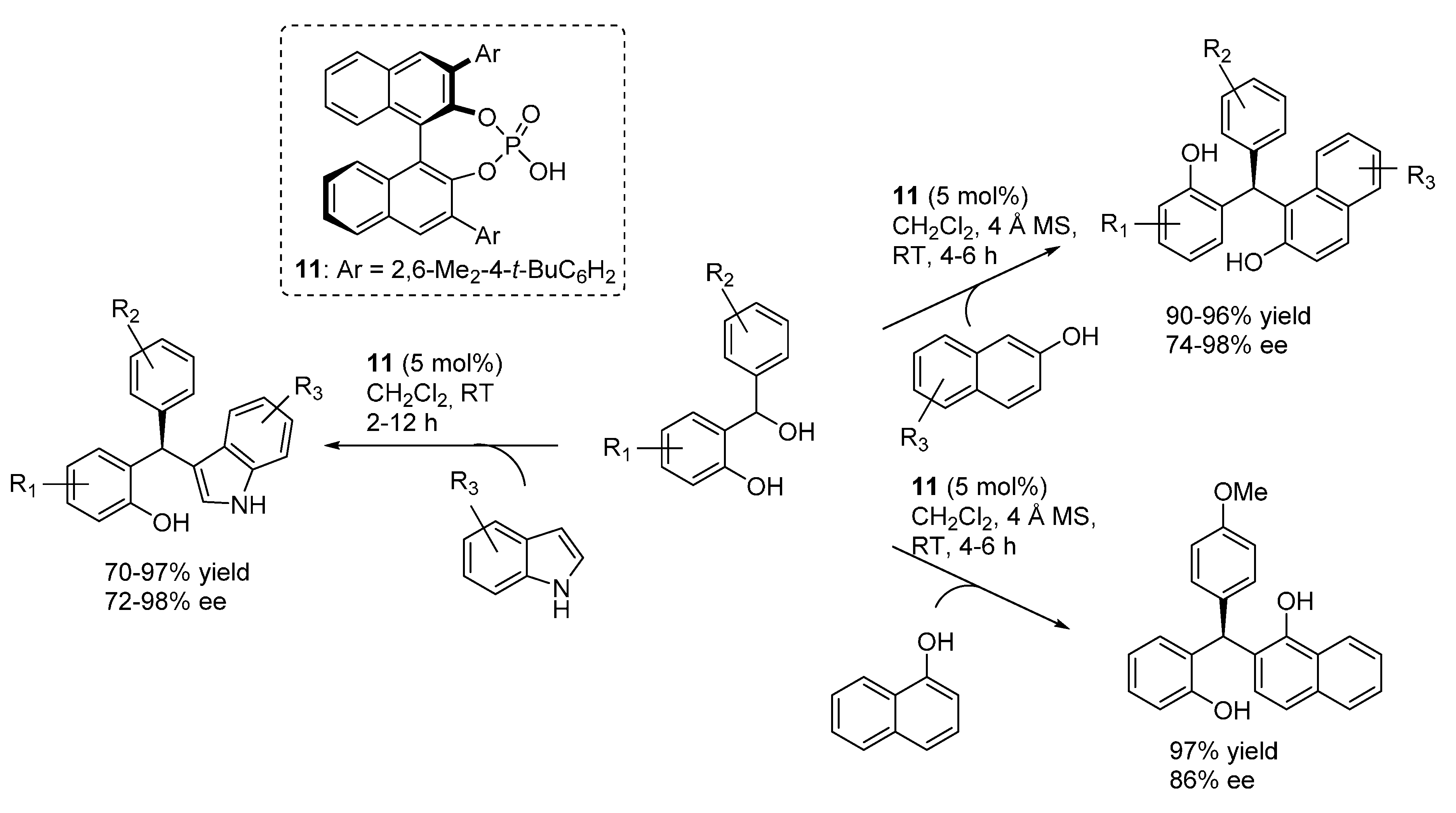
In a very short time, capitalizing on these disclosures, Schneider and co-workers extended this approach to other nucleophilic reaction partners. Reinvestigating reactions related to the low enantioselective examples reported by Bach (
Scheme 14), they first prepared diarylindolylmethanes and triarylmethanes through the addition of indoles and naphthols (
Scheme 18) [
27]. These transformations exhibited a remarkable scope in terms of nucleophile partners, whereas once again appeared limited to benzhydrylic alcohols as
o-QM precursors. Based on the low enantioselectivity obtained with an
N-Boc indole, a usual bifunctional mode of action involving simultaneous coordination of
o-QM and the indole or naphthol acidic proton, reminding the model depicted in
Scheme 15 for 1,3-dicarbonyls, was put forward. Remarkably, the same catalyst
11 that proved to be optimal in the addition of 1,3-dicarbonyl compounds was found to be the best performing in the terms of enantioselectivity also for these unrelated Friedel-Crafts type reactions.
Scheme 17.
Addition of 1,3-dicarbonyl compounds to o-QMs catalyzed by phosphorimide 12 or phosphoric acid 10 developed by Rueping.
Scheme 17.
Addition of 1,3-dicarbonyl compounds to o-QMs catalyzed by phosphorimide 12 or phosphoric acid 10 developed by Rueping.
Scheme 18.
Addition of indoles and naphthols to o-QMs catalyzed by phosphoric acid catalyst 11.
Scheme 18.
Addition of indoles and naphthols to o-QMs catalyzed by phosphoric acid catalyst 11.
A structurally distinct catalyst
13 was instead required for the catalytic asymmetric addition of enamides and enecarbamates, reported soon after by the same laboratory (
Scheme 19) [
28]. In this case, acetalization followed, resulting in a formal [4 + 2] cycloaddition. Acidic treatment on some of the adducts gave elimination of the amide, giving 4
H-chromenes. These enamides thus acted as masked ketones, and allowed to prepare 4
H-chromenes previously not accessible. In fact, whereas 1,3-dicarbonyls participate in the reaction thanks to their high enol content, direct utilization of mono-ketones was not possible.
Scheme 19.
Addition of enamides to o-QMs catalyzed by phosphoric acid 13.
Scheme 19.
Addition of enamides to o-QMs catalyzed by phosphoric acid 13.
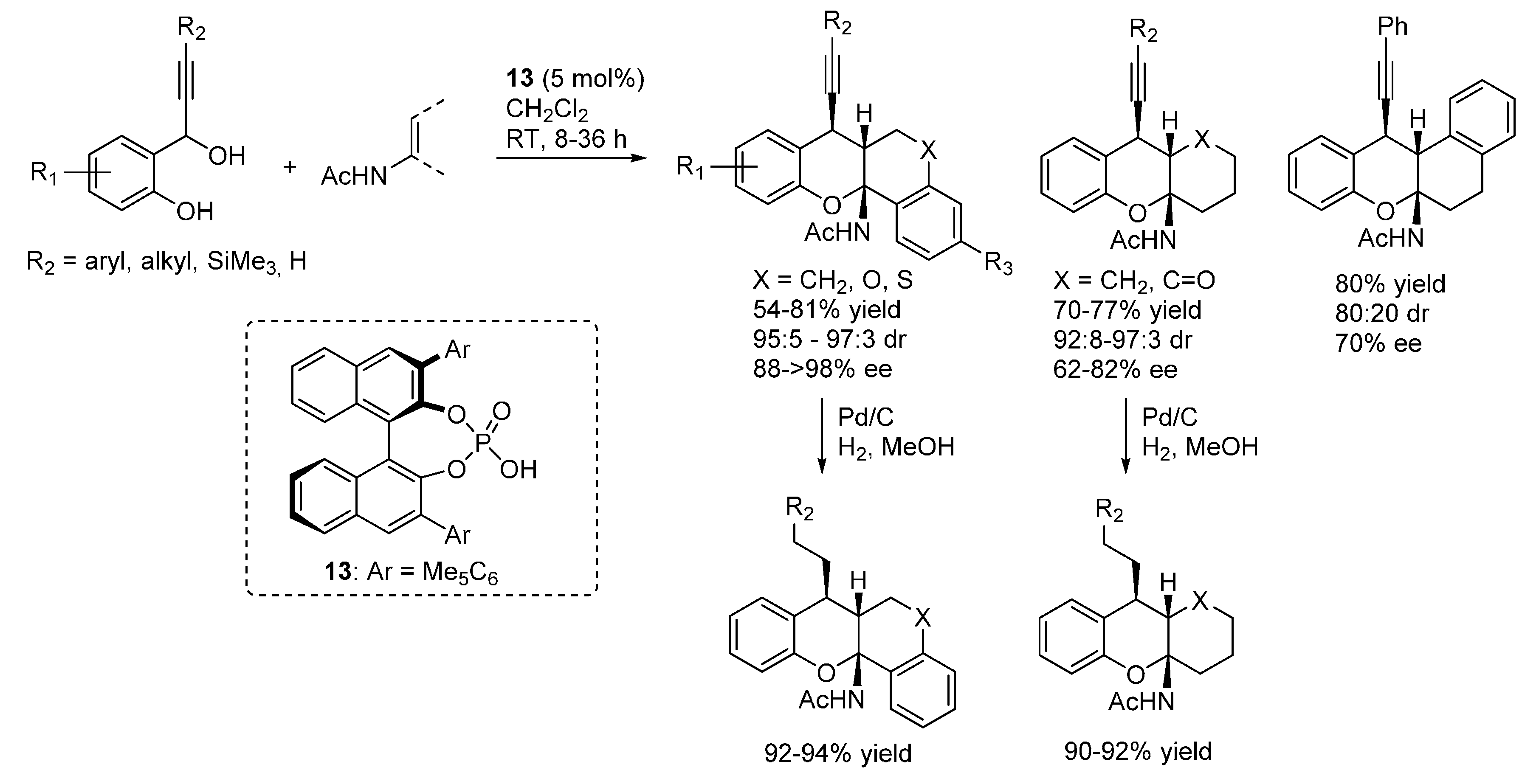
In a subsequent publication, the same authors reported the important extension of this reaction to propargylic alcohols as starting materials, thus demonstrating the possibility of using
o-QM intermediates different from β-aryl ones (
Scheme 20) [
29]. Furthermore, reduction of the triple bond ensured access 7-alkyl substituted xanthenes.
Scheme 20.
Addition of enamides to β-alkynyl substituted o-QMs catalyzed by phosphoric acid 13.
Scheme 20.
Addition of enamides to β-alkynyl substituted o-QMs catalyzed by phosphoric acid 13.
Another highly relevant extension of the
o-QM structures which can be engaged in this type of Brønsted acid catalyzed reactions was reported by Sun, who demonstrated that tertiary benzhydryl alcohols react with indoles in the presence of catalyst
14 delivering products bearing a challenging all-carbon quaternary stereocenter (
Scheme 21) [
30]. The reaction appeared limited to electron-rich phenols, and the intermediacy of an
o-QM intermediate, which structure was more thoroughly elucidated in a subsequent work (see
Section 3.3), was suggested by several control experiments. Since
N-methyl indole furnished exclusively a styrene elimination product, in this case the usual double activation furnished by the acid through coordination at the indole N-H was also invoked. Curiously, the same styrene side-product was also observed when some non-optimal phosphoric acid catalysts were applied to the reaction. Considering the results reported in
Section 3.3, it is not clear whether this styrene is an intermediate of the reaction or a dead-end side-product.
Scheme 21.
Addition of indoles o-QMs delivering products with quaternary stereocenters.
Scheme 21.
Addition of indoles o-QMs delivering products with quaternary stereocenters.
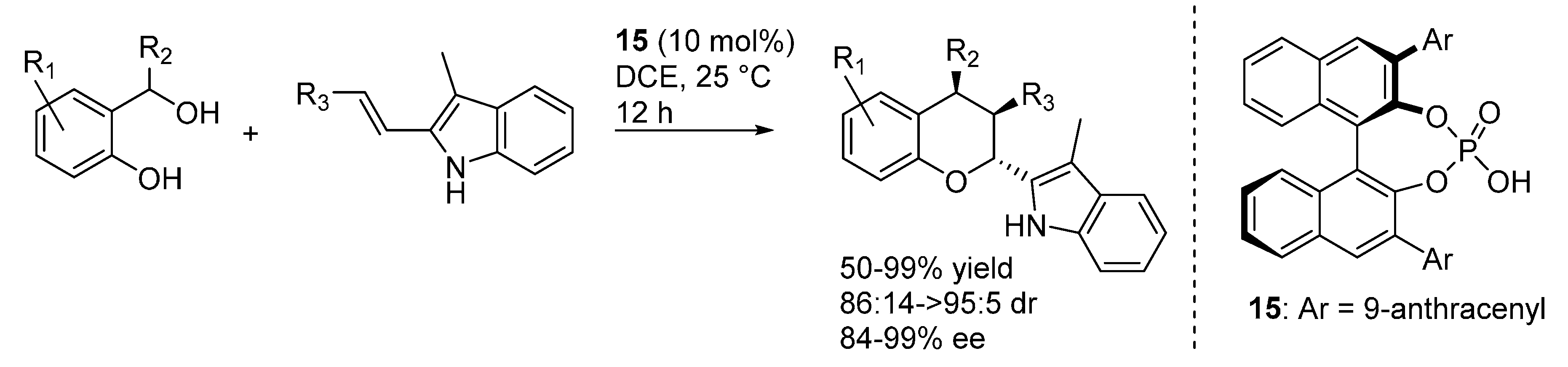
The coordination of these phosphoric acid catalysts to the indole N-H was exploited in a different reaction by Shi, who employed the activated olefin of 2-vinyl-3-methyl substituted indoles in a [4 + 2] inverse electron demand oxa-Diels-Alder reaction with
o-QMs catalyzed by
15 (
Scheme 22) [
31]. A substituent at the 3-position of the indole was found to be mandatory for the reaction to proceed through the cycloaddition pathway, instead of the simple addition of the indole (see e.g.,
Scheme 14 and
Scheme 18). Furthermore,
Z-vinylindoles were found to isomerize to their more stable (and more reactive towards the cycloaddition)
E-isomers under the reaction conditions, thus allowing using
E/
Z mixtures in the reactions, which were assumed to occur through a concerted pathway. Remarkably and in contrast with most of the examples highlighted so far, the reaction worked well with substrates bearing not only aryl but also alkyl substituents at the benzylic position (
i.e., R
2 in
Scheme 22).
Scheme 22.
Inverse electron demand [4 + 2] oxa-Diels-Alder of o-QMs with 2-vinylindoles catalyzed by phosphoric acid 15.
Scheme 22.
Inverse electron demand [4 + 2] oxa-Diels-Alder of o-QMs with 2-vinylindoles catalyzed by phosphoric acid 15.
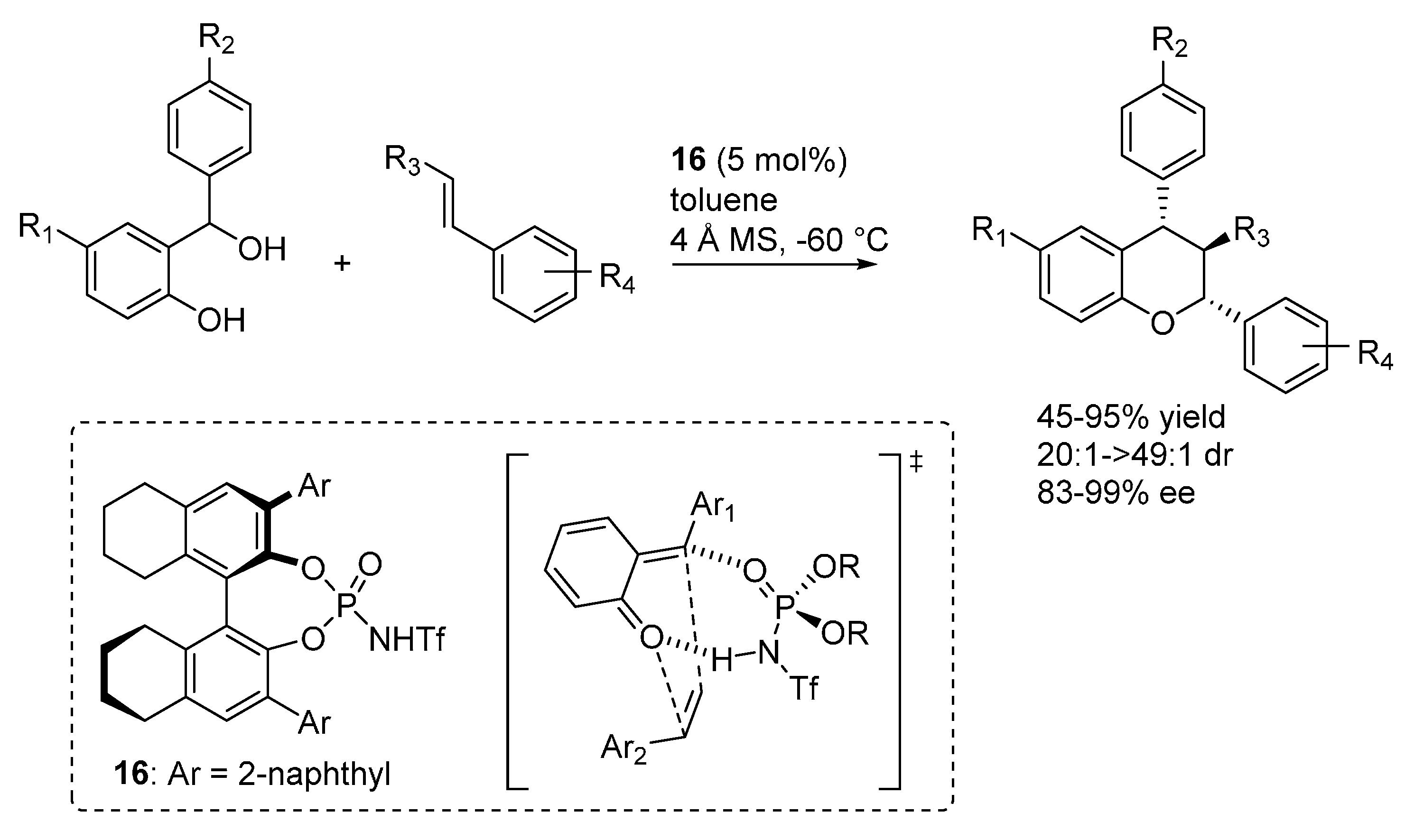
A different hetero-Diels-Alder reaction of
o-QMs involving styrenes as dienes was reported by Rueping (
Scheme 23) [
32]. This reaction presents some very challenging aspects, related to the low nucleophlicity of styrenes, to the tendency of these olefins to undergo polymerization under acidic reaction conditions, and especially to their lack of an anchor for catalyst coordination. In fact, the reaction represents one of the rare examples wherein nucleophile coordination does not seem to be possible, in contrast with the usual bifunctional activation mode expressed by phosphoric acid and related catalysts, also seen in the previous examples highlighted in this section. To account for the excellent enantioselectivites observed when the phosphorimide catalyst
16 was employed, an open transition state model involving bicoordination of the phosphorimide to the
o-QM was proposed. The formation of such a complex, wherein the phosphorimide coordinates the
o-QM oxygen with its acidic proton and the electropositive exocyclic olefin carbon with its Lewis basic oxygen, was also supported by NMR experiments.
Scheme 23.
Inverse electron demand [4 + 2] oxa-Diels-Alder reaction of o-QMs with styrenes catalyzed by phosphorimide 16.
Scheme 23.
Inverse electron demand [4 + 2] oxa-Diels-Alder reaction of o-QMs with styrenes catalyzed by phosphorimide 16.
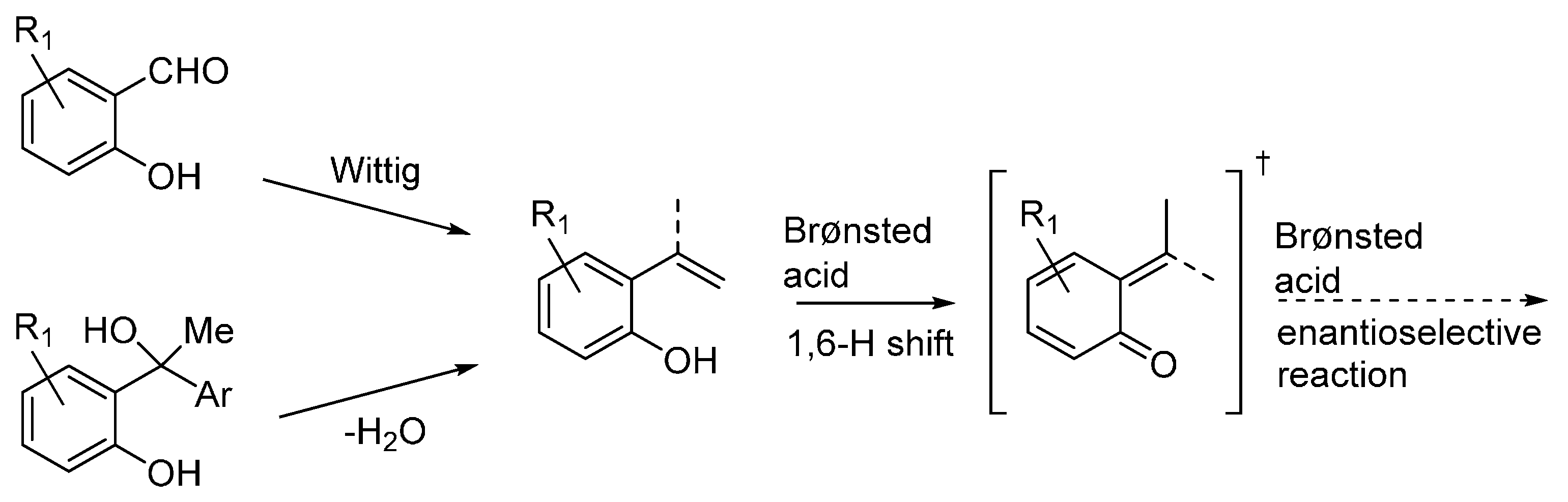
3.3. o-QMs Generated in Situ by 1,6-H Shift of Ortho-Hydroxystyrenes under Brønsted Acid Conditions
The possibility of using
ortho-hydroxystyrenes as
o-QMs precursors in organocatalytic asymmetric reactions with nucleophiles has been reported by three laboratories almost simultaneously in 2015 [
33,
34,
35]. All three reports dealt with chiral phosphoric acid catalysts, providing protocols alternative to the synthetic platform developed by Sigman giving
o-QM through palladium-hydride initiated H-shifts from these styrenes [
11]. The
ortho-hydroxystyrenes substrates can be prepared by Wittig olefination of the corresponding salicylaldehydes, or by dehydration of suitable tertiary alcohols (
Scheme 26). The phosphoric acid catalysts are then able to promote a 1,6-H shift by protonating the electron-rich olefin while abstracting the phenolic proton (see discussion below).
Scheme 26.
Preparation of ortho-hydroxystyrenes and reactions under acidic conditions.
Scheme 26.
Preparation of ortho-hydroxystyrenes and reactions under acidic conditions.
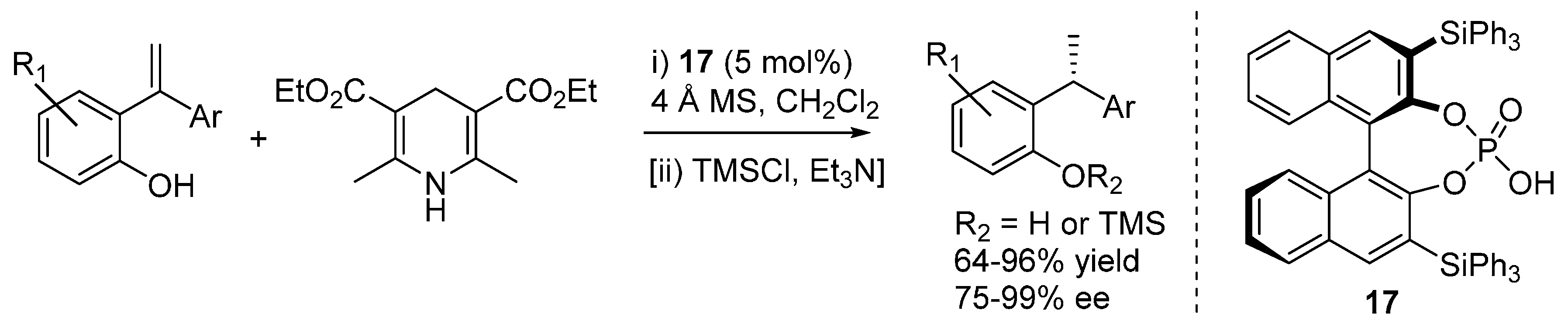
Sun
et al. used α,α-disubstituted styrenes and described the transfer hydrogenation reaction with Hantzsch esters as hydride donors, delivering unsymmetrically substituted 1,1-diarylethanes with very good results under the action of catalyst
17 (
Scheme 27) [
33]. Six randomly selected compounds were tested for cytotoxicity against human cancer cell lines, and one of them displayed remarkable activity. In some cases, it was found to be convenient to isolate the products as the corresponding trimethylsilyl ethers.
Scheme 27.
Transfer hydrogenation of 2-hydroxystyrenes with Hantzsch ester catalyzed by phosphoric acid 17.
Scheme 27.
Transfer hydrogenation of 2-hydroxystyrenes with Hantzsch ester catalyzed by phosphoric acid 17.
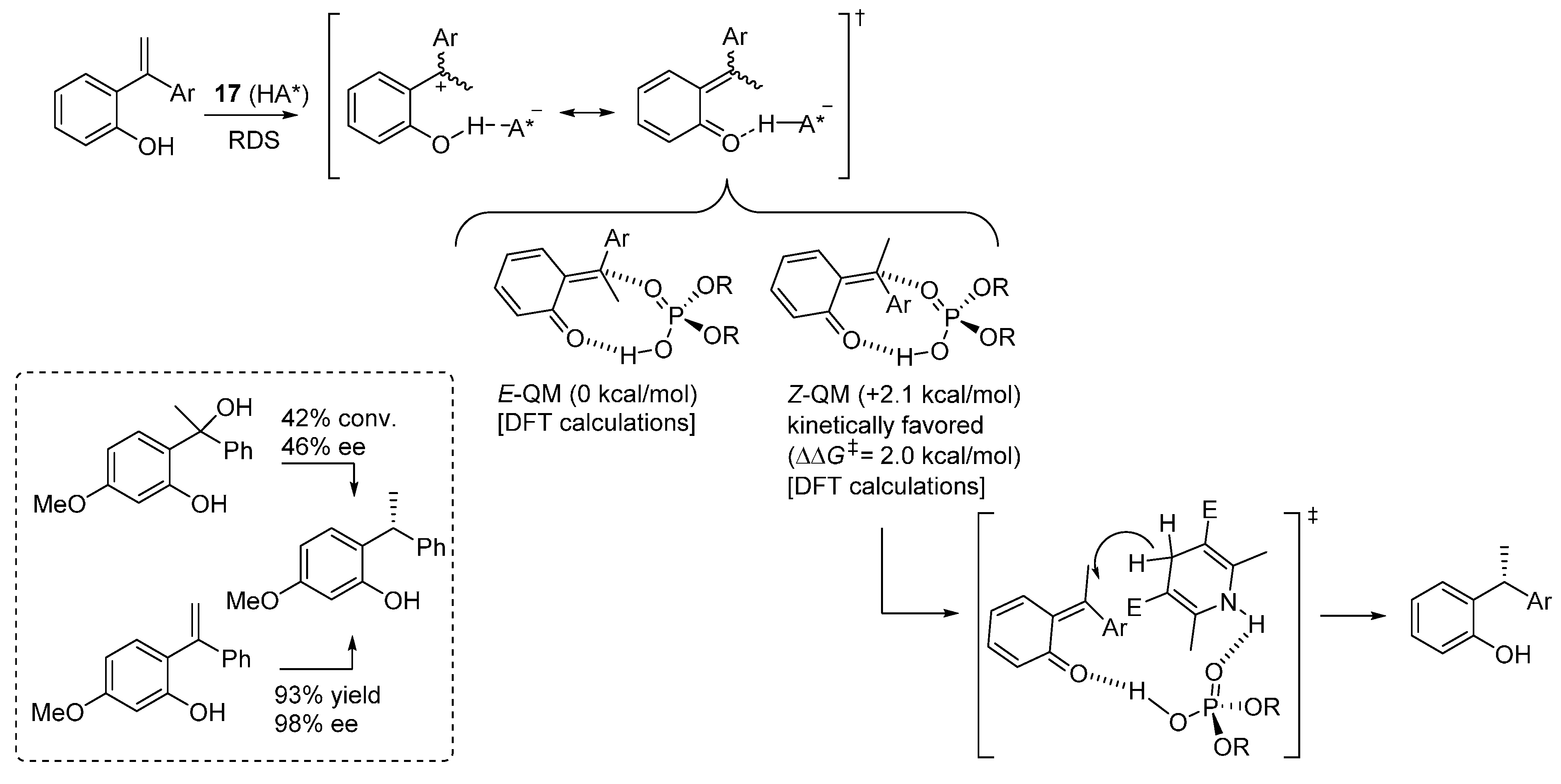
Substantiated by several control experiments, the proposed reaction pathway involves interaction of the electron rich styrene olefin with the acidic catalyst, resulting in a species which can be represented by two limiting resonance structures, a zwitterionic and a neutral (
o-QM-like) one (
Scheme 28). DFT (B3LYP-D3) calculations pinpointed that the neutral structure, bicoordinated to the catalyst, is a more accurate representation of the electronic distribution of this intermediate, and that formation of the less stable
Z-QM is kinetically favored over its
E-counterpart. Kinetic experiments showed that the reaction is zeroth order in Hantzsch ester. Therefore,
o-QM generation is the rate determining step, and the reaction is likely to occur via the addition of the Hantzsch ester to the less stable
Z-QM intermediate, through a stereodetermining transition state involving the usual bifunctional action of the catalyst, which coordinates the
o-QM with its acidic proton and the Hantzsch ester NH with the phosphoryl oxygen. An interesting control experiment, wherein a benzylic alcohol as
o-QM precursor gave dramatically reduced results compared to the
ortho-hydroxystyrene, demonstrated the importance of the strategy employed for the
o-QM generation, especially for the control of its geometry and thus ensuing stereoselectivity in the following addition step.
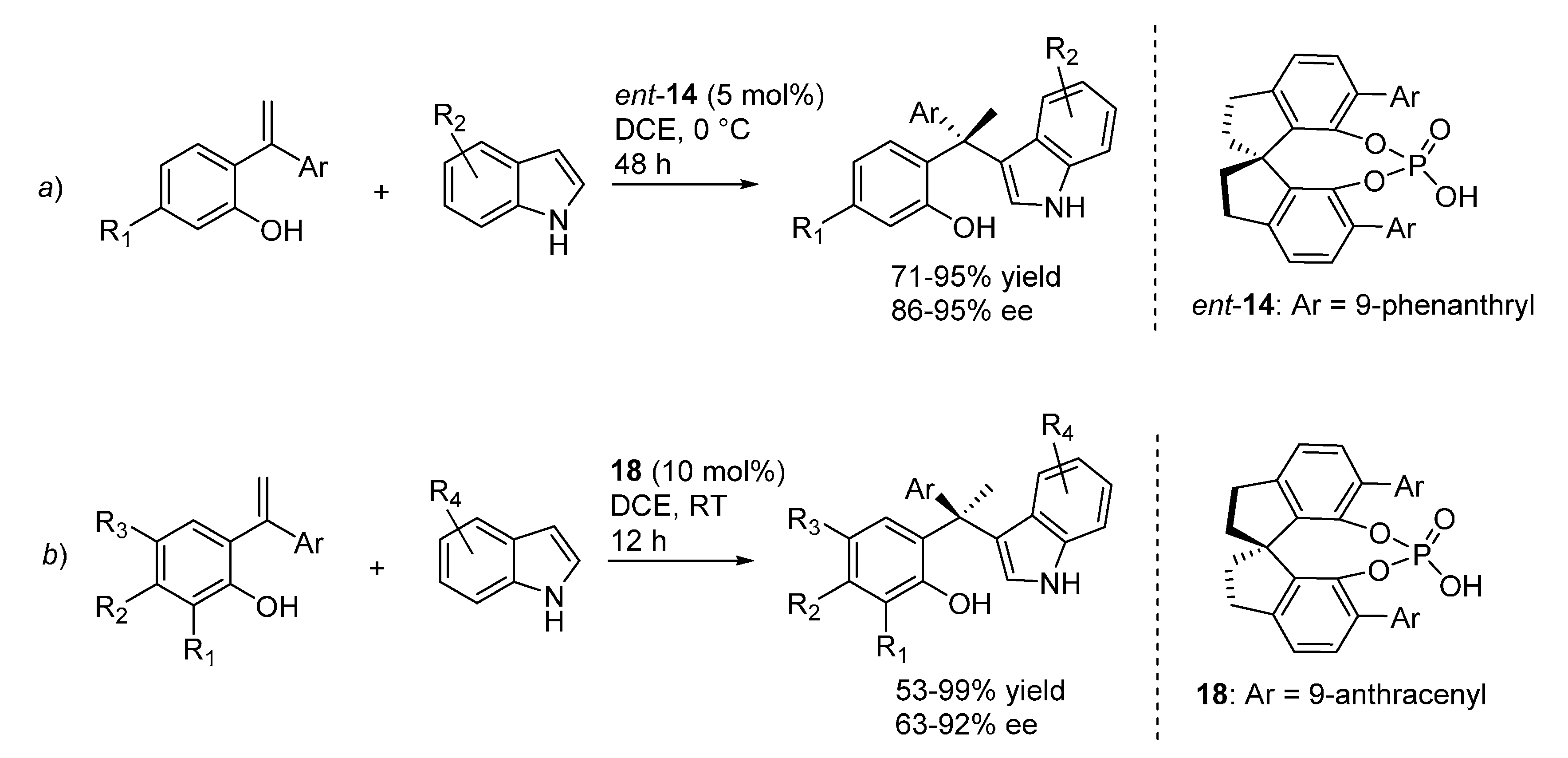
Despite these latter considerations, the same catalyst
14 previously applied by the same laboratory to the addition of indoles to
o-QMs generated from benzylic alcohols (
Scheme 21), proved to also be an excellent promoter when the
o-QMs were generated from
ortho-hydroxystyrenes (
Scheme 29a), indicating perhaps a change in the RDS between the transfer hydrogenation and the Friedel-Crafts reactions. Some of the previously highlighted limitations of the Friedel-Crafts reaction (the requirement of an ether substituent at the phenol ring to stabilize the
o-QM and assist its formation) were somehow alleviated with this new protocol, which, however, still necessitated the presence of an electron releasing group in at least one of the two aryl substituents of the styrenes. This latter limitation is also present in a similar protocol for the same transformation, reported almost simultaneously by Wu
et al., which is based on a closely related catalyst
18 and includes a more thorough study of the reaction scope (
Scheme 29b) [
34].
Scheme 28.
Proposed reaction pathway and control experiment with the benzylic alcohol in the transfer hydrogenation reaction of 2-hydroxystyrenes with Hantzsch esters.
Scheme 28.
Proposed reaction pathway and control experiment with the benzylic alcohol in the transfer hydrogenation reaction of 2-hydroxystyrenes with Hantzsch esters.
Scheme 29.
Catalytic asymmetric additions of indoles to
o-QMs generated
in situ from
ortho-hydroxystyrenes reported by Sun
et al. (
a, [
33]) and by Wu
et al. (
b, [
34]).
Scheme 29.
Catalytic asymmetric additions of indoles to
o-QMs generated
in situ from
ortho-hydroxystyrenes reported by Sun
et al. (
a, [
33]) and by Wu
et al. (
b, [
34]).
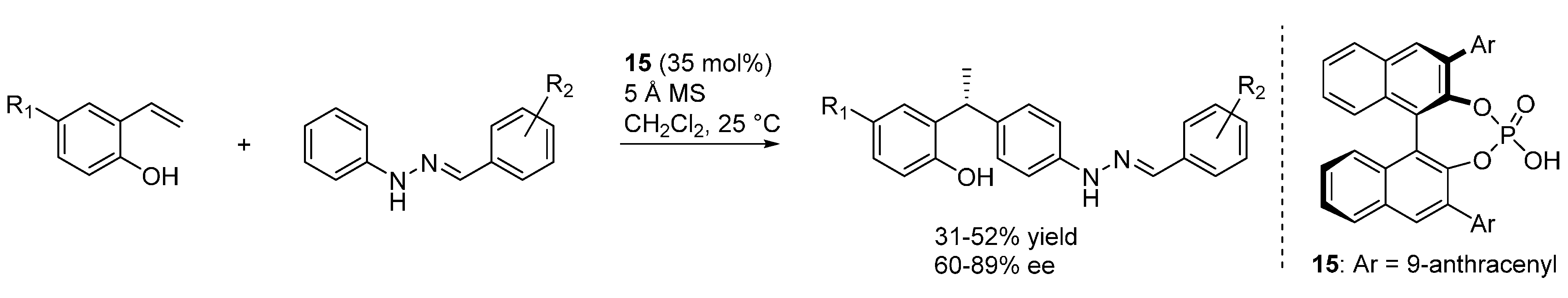
A distinct transformation based on the generation of
o-QM intermediates from
ortho-hydroxystyrenes was reported by Shi [
35]. In contrast with the above examples involving α,α-disubstituted
ortho-hydroxystyrenes, in this case simpler α-monosubstituted substrates were used for the reaction, a hydroarylation of the
o-QM promoted by the chiral Brønsted acid catalyst
15 at high loadings (
Scheme 30). A hydrazone served as an activating unit, rendering aryl rings sufficiently nucleophilic at their
para-position to undergo the asymmetric additions, although the products were obtained with moderate results. A transition state involving two catalyst units, one activating the
o-QM and the other coordinating the remote hydrazone was proposed.
Scheme 30.
Catalytic asymmetric additions of hydrazone activated aryls to o-QMs generated in situ from ortho-hydroxystyrenes catalyzed by phosphoric acid 15.
Scheme 30.
Catalytic asymmetric additions of hydrazone activated aryls to o-QMs generated in situ from ortho-hydroxystyrenes catalyzed by phosphoric acid 15.
Furthermore, as we recently reviewed [
36], the intermediacy of
o- and
p-QMs has been invoked in some Brønsted acid catalyzed Povarov and related cycloaddition reactions, wherein addition of hydroxystyrenes to acid activated electrophiles brings about the formation of a QM undergoing an intramolecular conjugate addition, resulting in formal cycloadditions between the activated styrene olefin and various dienes.
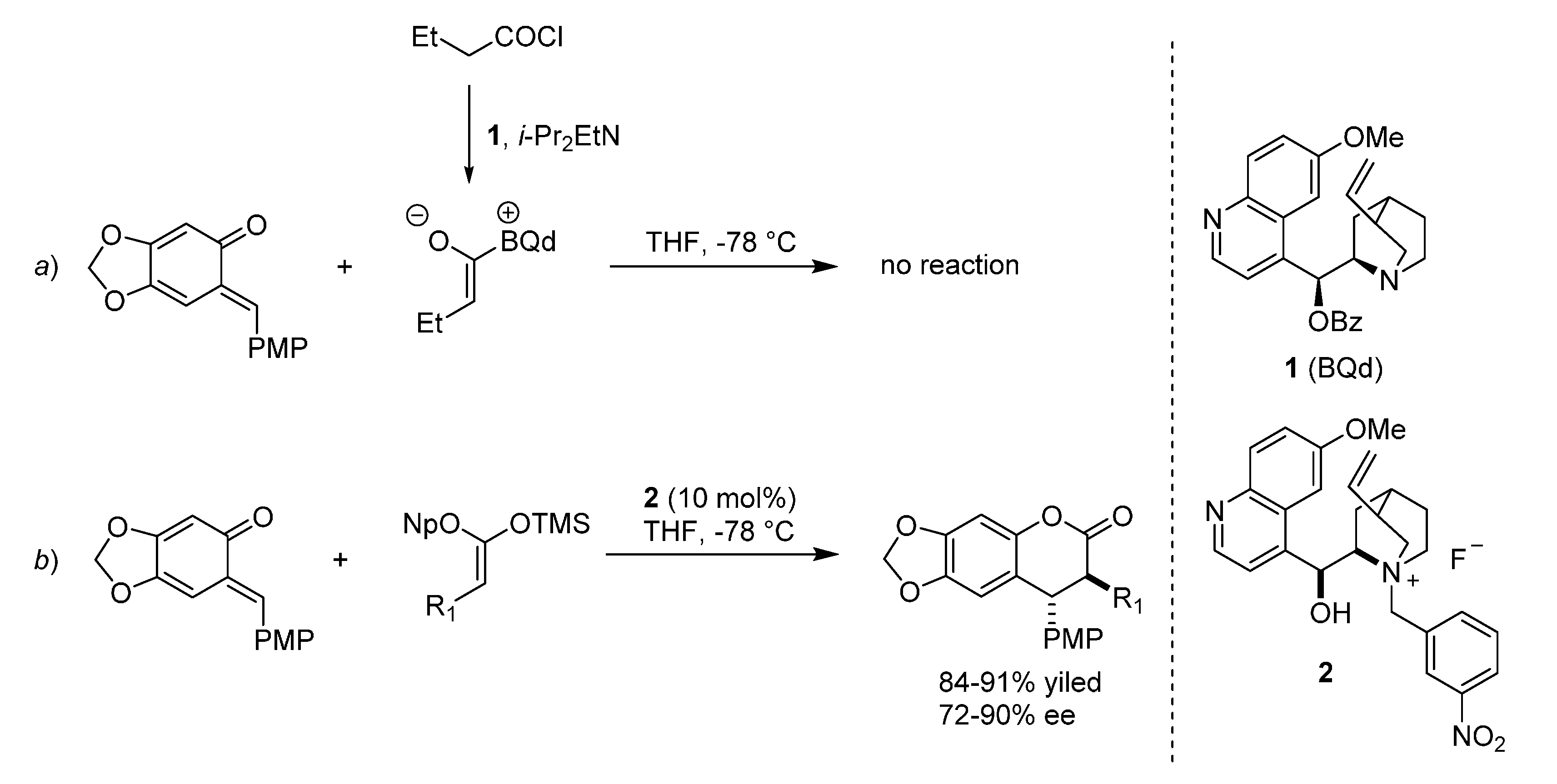
3.4. o-QMs Generated in Situ by Desilylation—Halide Elimination from Ortho-Silyloxy Benzylic Halides under Lewis Basic Conditions
O-Silyl protected phenols bearing a leaving group at the benzylic position, such as an halide, can be employed for the generation of
o-QMs
in situ under the promotion of Lewis bases (typically fluorides) (
Scheme 31). These substrates were introduced to overcome the poor stability of the corresponding free phenols, and to guarantee a ionic control over the
o-QM generation [
37]. Their preparation entails silyl protection of the phenol followed by either radical halogenation [
38] or hydroxide substitution [
39], depending on the phenolic structure employed. This method of
o-QM formation has been successfully exploited in asymmetric organocatalysis by Scheidt, by flanking the stoichiometric fluoride Lewis base, used to generate the
o-QM, with chiral Lewis basic catalysts able to combine selectively with the nucleophilic component:
N-heterocyclic carbenes (NHCs) [
40,
41].
Scheme 31.
Preparation of O-silyl phenols bearing a leaving group at the benzylic position, and ensuing organocatalytic reaction.
Scheme 31.
Preparation of O-silyl phenols bearing a leaving group at the benzylic position, and ensuing organocatalytic reaction.
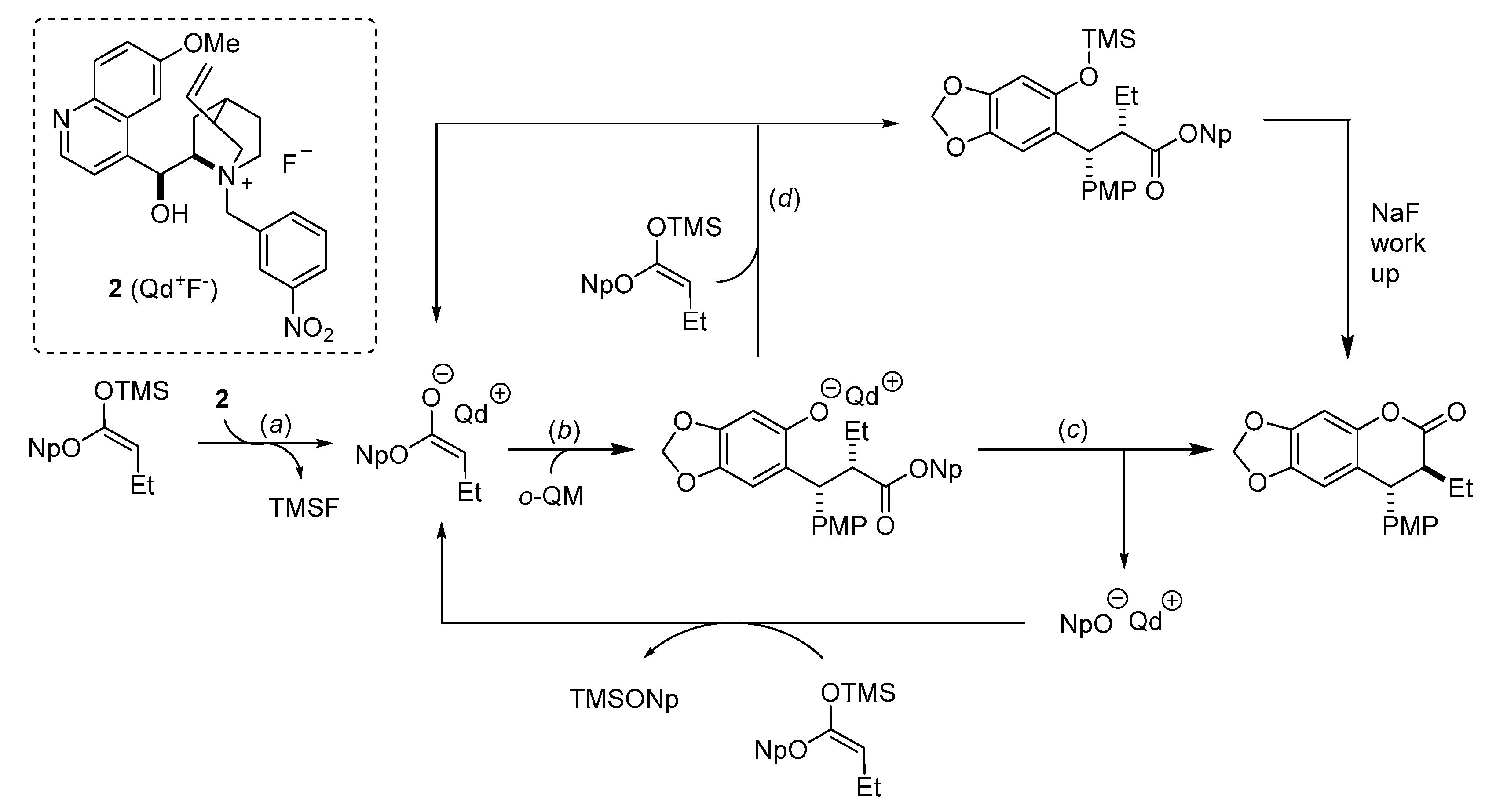
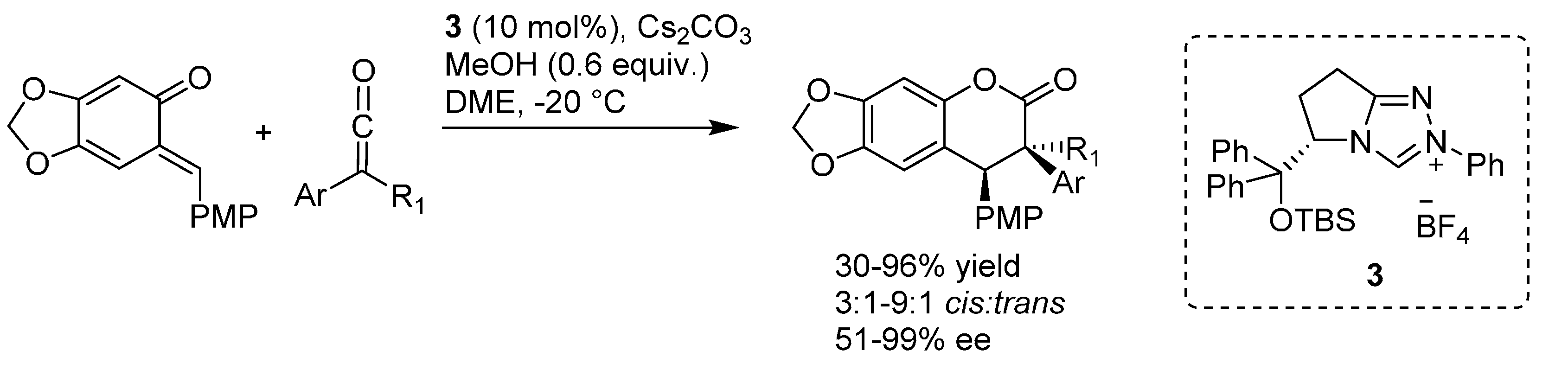
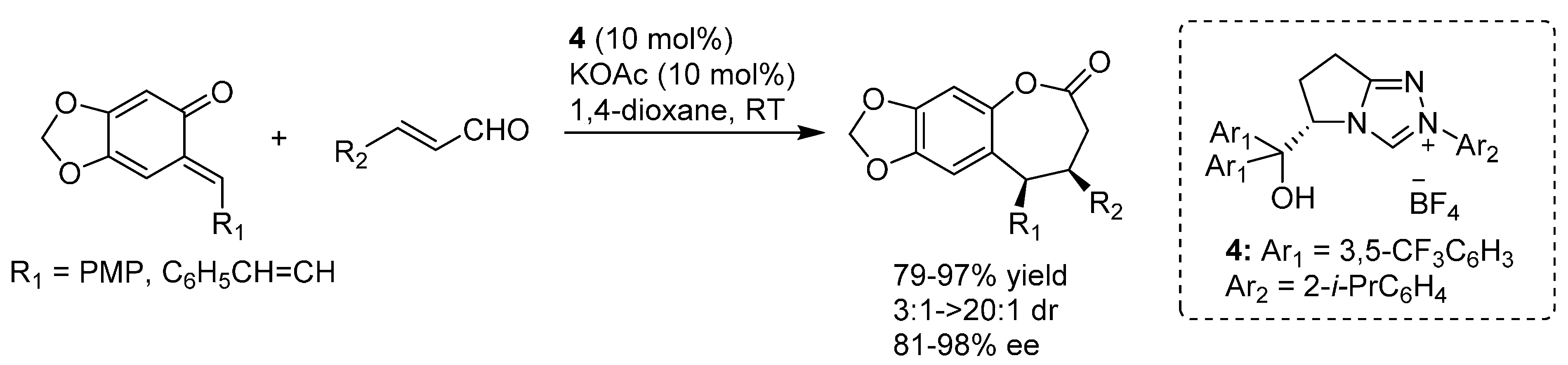
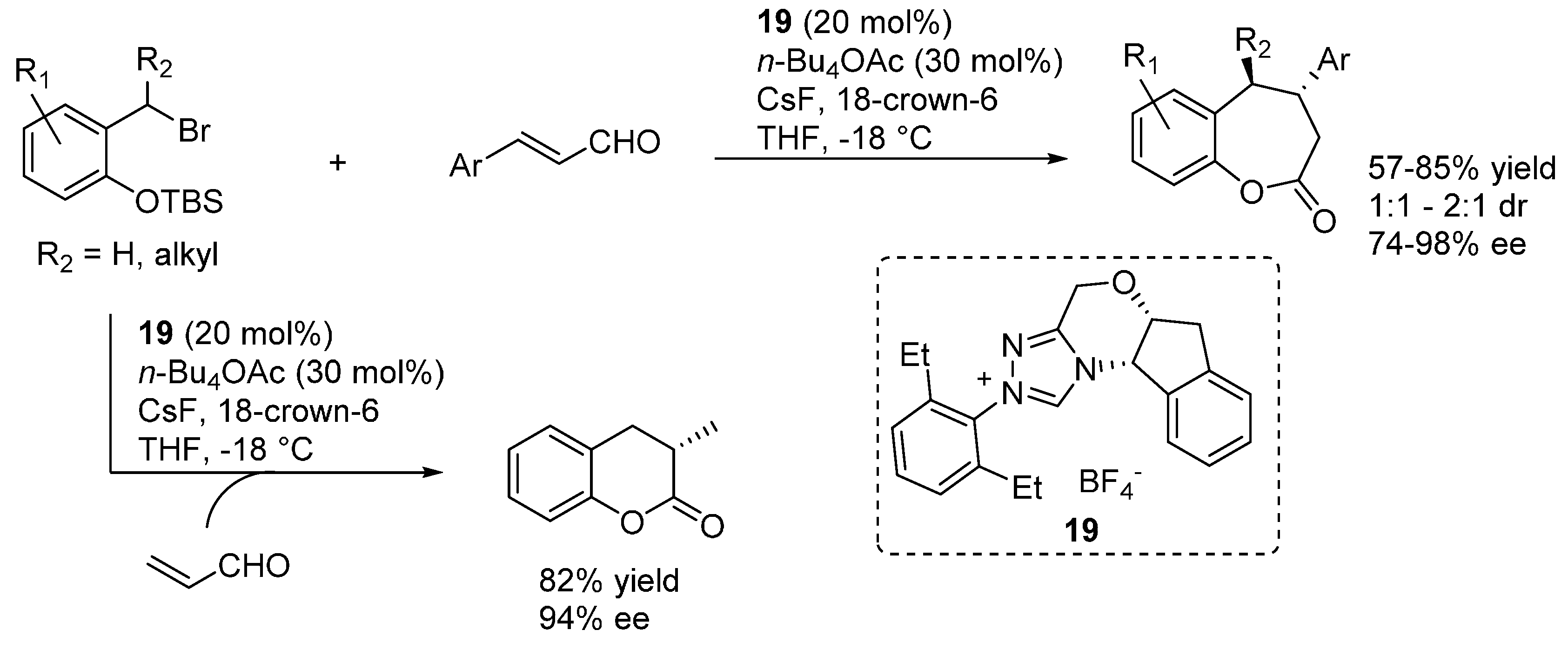
In more detail, two formal cycloaddition reactions were developed exploiting this strategy, where a substantial optimization process was required to find conditions suitable for combining the desilylation-elimination step with the NHC catalyzed reactions. The first report described a formal [4 + 3] cycloaddition with cinnamaldehydes, and employed cesium fluoride/18-crown-6 as fluoride source, with tetra-
n-butylammonium acetate as a mild Brønsted base to generate the NHC catalyst from pre-catalyst
19 (
Scheme 32) [
40]. While chloride and bromide could be equally used as leaving groups, the choice of the silyl substituents proved to be crucial, with robust moieties such as TIPS and TBS allowing a controlled generation of the
o-QM and giving much better results than more labile groups such as TES. Remarkably, also β-unsubstituted, and thus highly unstable,
o-QMs could participate in the reaction. Whereas cinnamaldehydes followed the [4 + 3] cycloaddition pathway, aliphatic enals or acrolein delivered a [4 + 2] cycloaddition product.
Scheme 32.
Formal [4 + 3] cycloaddition reaction of cinnamaldehydes with o-QMs catalyzed by NHC generated from precursor 19.
Scheme 32.
Formal [4 + 3] cycloaddition reaction of cinnamaldehydes with o-QMs catalyzed by NHC generated from precursor 19.
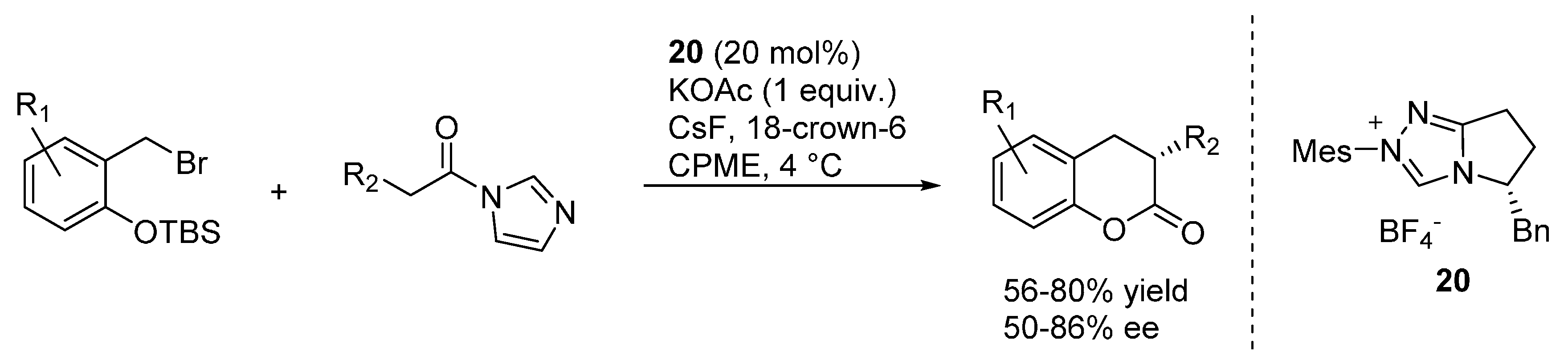
A related [4 + 2] cycloaddition reaction was indeed the subject of the subsequent publication, wherein
N-acylimidazoles were applied as carbonyl donors in combination with pre-catalyst
20 (
Scheme 33) [
41]. To avoid racemization of the obtained 3,4-dihydrocoumarins, it turned out to be necessary to change the weak Brønsted base used for catalyst generation. Eventually, by swapping the previously employed tetra-
n-butyl ammonium acetate with its potassium salt, it turned out to be possible to obtain adducts with moderate to good enantioselectivities. These transformations well complement the related NHC-catalyzed transformations reported by Ye and highlighted in
Section 2, wherein pre-formed
o-QMs were employed.
Scheme 33.
[4 + 2] cycloaddition reaction of N-acylimidazoles with o-QMs catalyzed by NHC generated from precursor 20.
Scheme 33.
[4 + 2] cycloaddition reaction of N-acylimidazoles with o-QMs catalyzed by NHC generated from precursor 20.
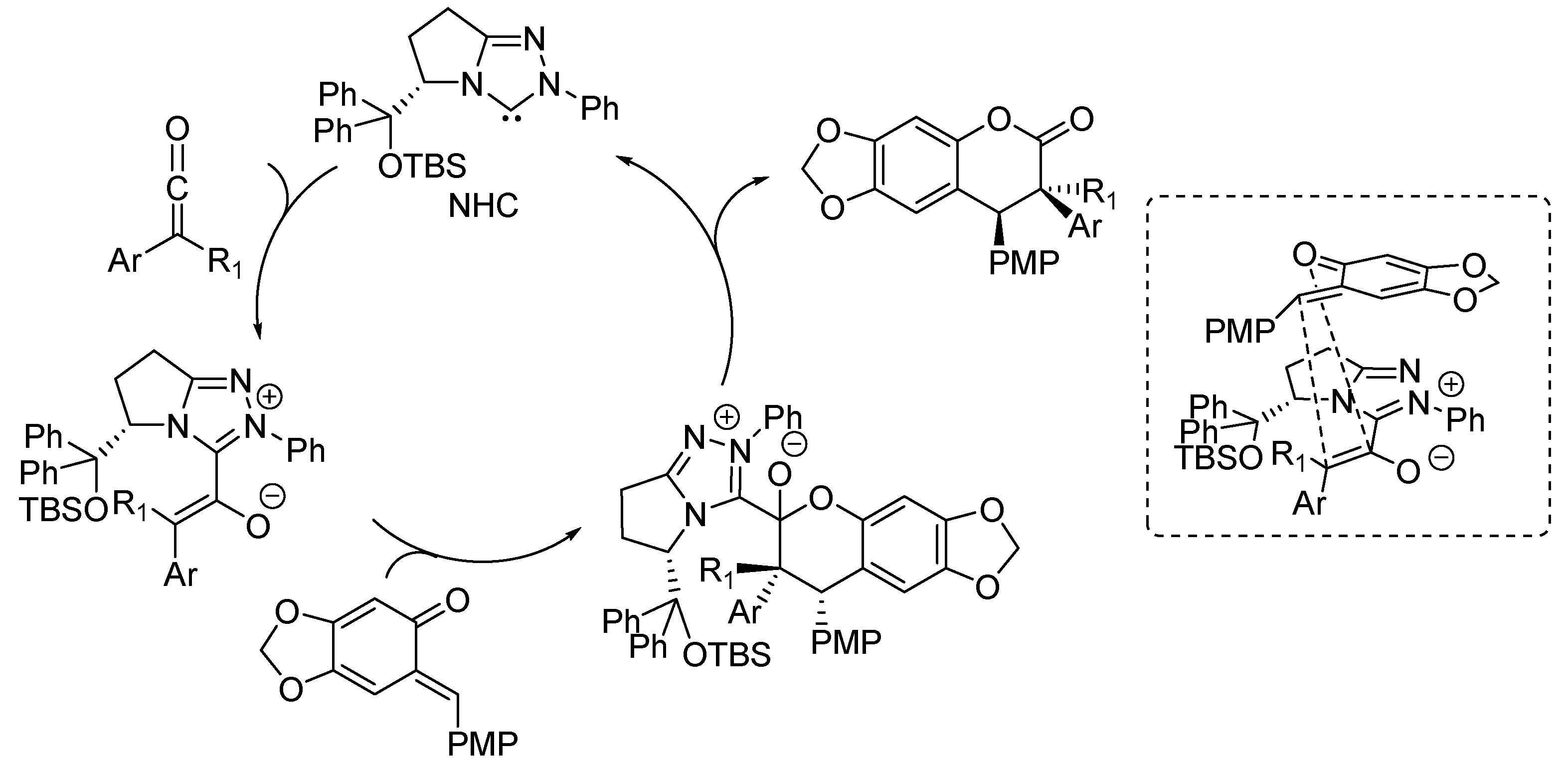
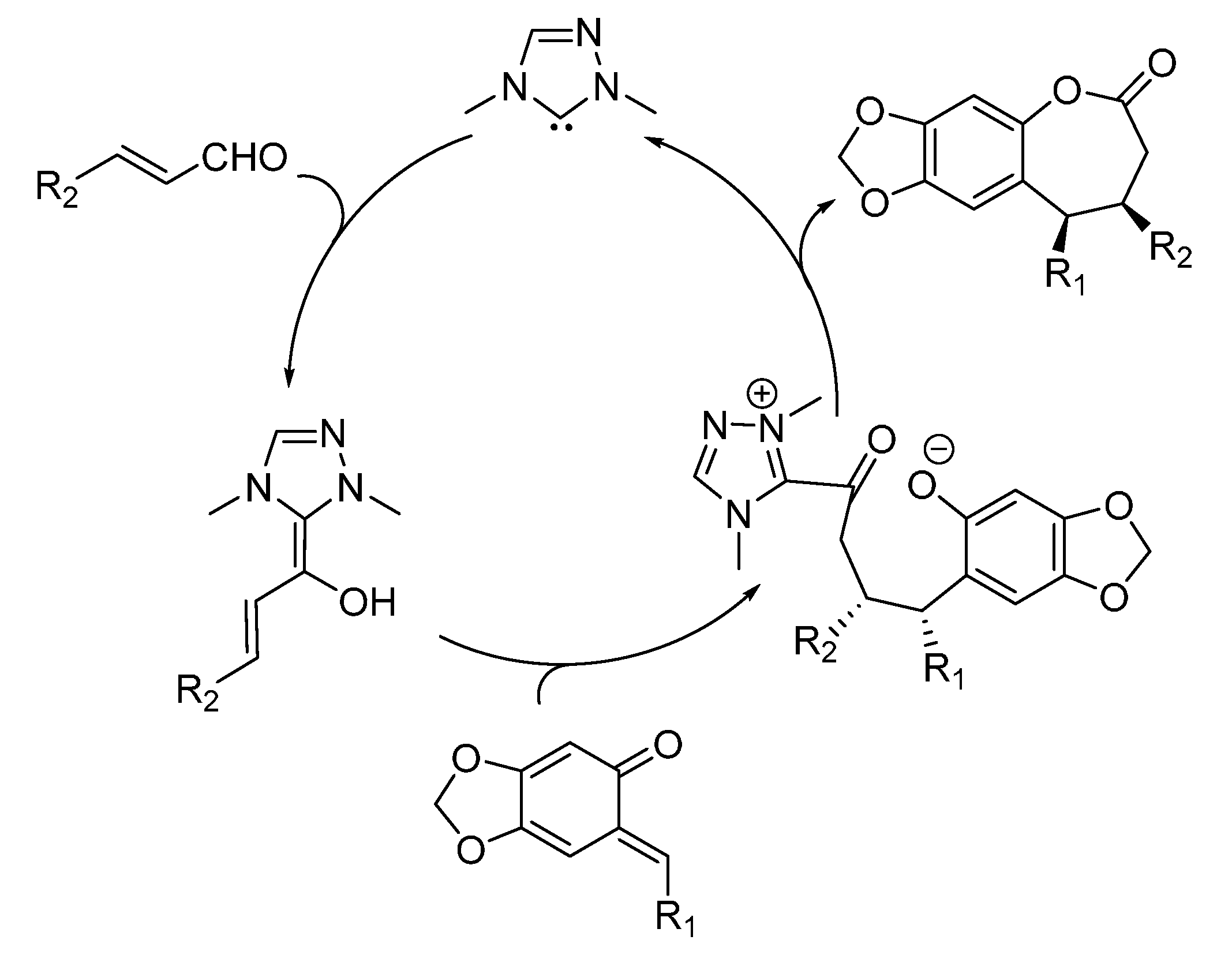
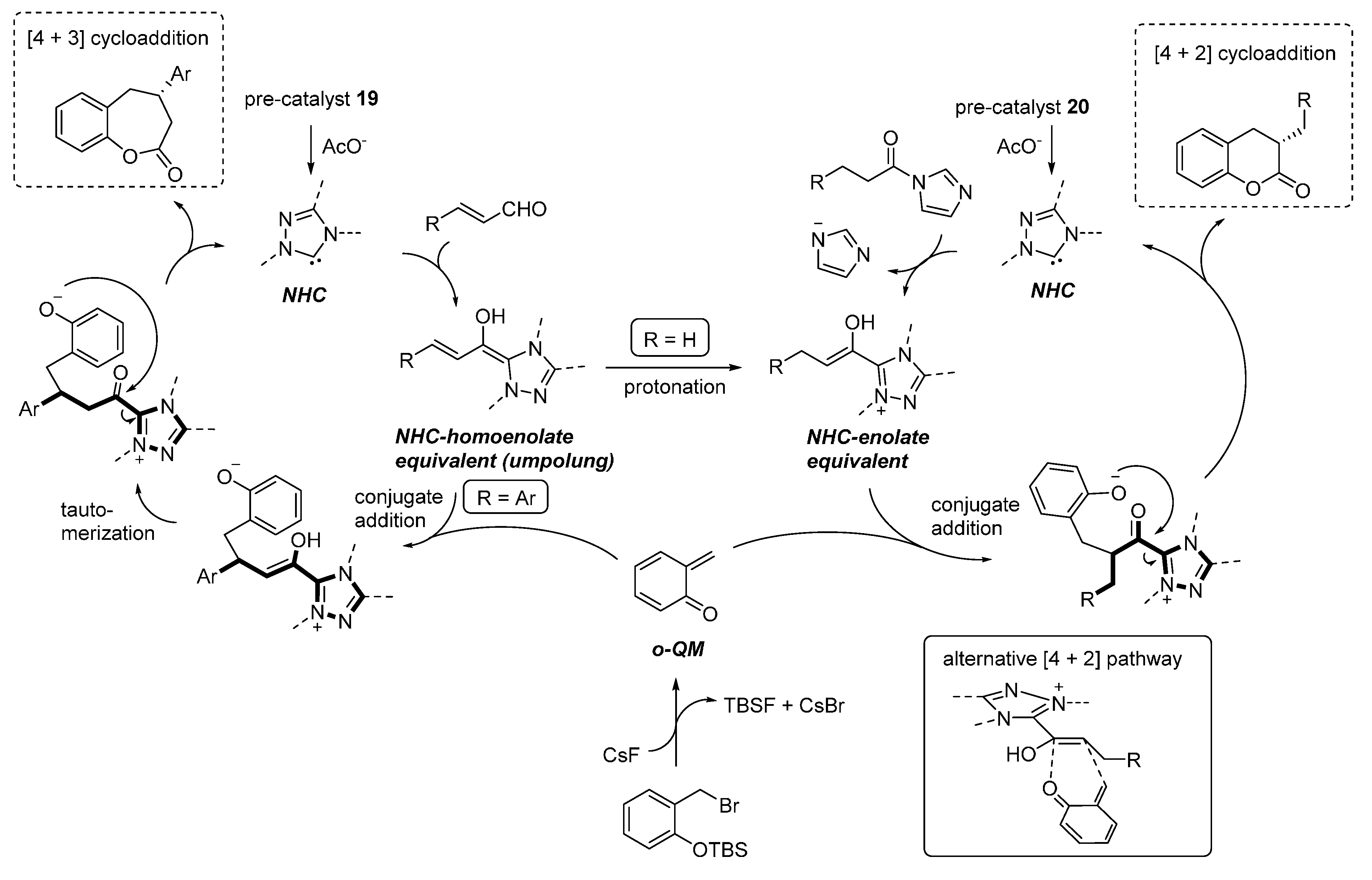
The [4 + 3] and [4 + 2] cycloadditions were proposed to follow related reaction pathways, involving the generation of the reactive
o-QM by the action of fluoride (
Scheme 34). In the case of enals reaction partners, their simultaneous combination with the NHC catalyst, obtained by deprotonation of precatalyst
19, brings about an
umpolung of these substrates by forming an NHC homoenolate equivalent. Here, the fate and the reactivity of this species depends on the enal involved. The NHC-homoenolates derived from cinnamaldehydes add to the
o-QM in a conjugate fashion. Upon tautomerization, the phenoxide displaces the NHC catalyst ensuring completion of the catalytic cycle and formation of a formal [4 + 3] cycloaddition product. Conversely, the NHC-homoenolate derived from acrolein undergoes protonation at the terminal position faster than the conjugate addition, giving an NHC enolate equivalent and ultimately resulting in a [4 + 2] cycloaddition. Related NHC-enolates are produced directly from
N-acylimidazoles and the NHC catalyst derived from
20. As an alternative to the sequential conjugate addition phenoxide displacement pathway, a concerted [4 + 2] cycloaddition between the
o-QM and the NHC enolate was also considered to be possible.
Scheme 34.
Reaction pathways followed by the cycloaddition reactions catalyzed by NHCs.
Scheme 34.
Reaction pathways followed by the cycloaddition reactions catalyzed by NHCs.
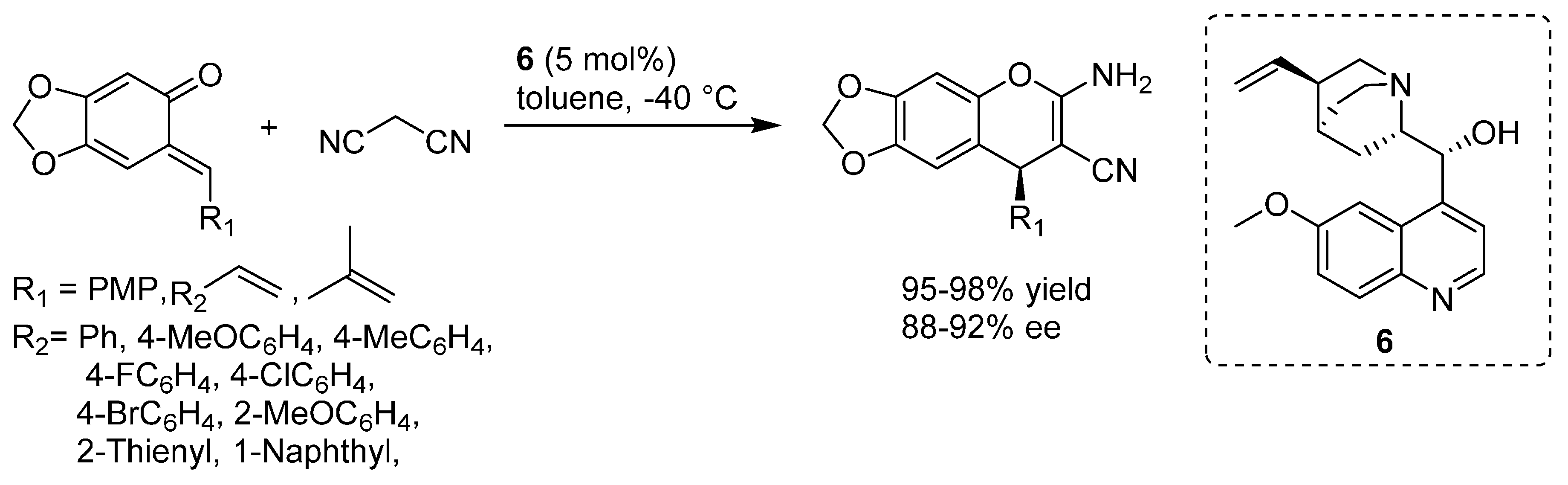
3.6. o-QMs Generated in Situ by Sulfinic Acid Elimination from 2-Sulfonylalkyl Phenols under Brønsted Basic Conditions
A straightforward approach to
o-QMs generation is the base induced elimination of a leaving group from the benzylic position of suitably functionalized phenols. However, the aforementioned poor stability of
ortho-hydroxybenzylic halides prevented this approach from being fully adopted and exploited, with silyl protection at the phenolic oxygen, to be removed under Lewis basic conditions (see
Section 3.4), being the most pursued alternative strategy. This stability issue was circumvented only very recently, by applying a leaving group different from a halide, namely an arylsulfonyl moiety. Inspired by the strategic employment of arylsulfonyl moieties to temporarily trap relatively unstable intermediates such as alkylideneindolenines [
44] and
N-carbamoyl imines [
45], Zhou reported in 2013 the straightforward preparation of 2-arylsulfonylalkyl phenols from the corresponding alcohols and their employment in a synthesis of 2,3-benzofurans, wherein
o-QMs intermediates were generated from these sulfonyl species under mild Brønsted basic reaction conditions (
Scheme 36) [
46]. The same strategy could also be applied to the synthetically appealing additions of cyanide [
47] and ammonia [
48].
Scheme 36.
Preparation of 2-sulfonylalkyl phenols and their use as o-QM precursors.
Scheme 36.
Preparation of 2-sulfonylalkyl phenols and their use as o-QM precursors.
These reports paved the way to the application of chiral Brønsted basic catalyst for activation of
o-QMs reaction partners. Despite its considerable synthetic utility, employment of chiral Brønsted bases results unfeasible with the other methodologies for
o-QMs generation described in
Section 3.1,
Section 3.2,
Section 3.3,
Section 3.4 and
Section 3.5. Whereas a low, enantioselective example restricted to one substrate was first reported by Zhou in the frame of the addition of ammonia [
48], two papers, appearing almost simultaneously, demonstrated that high enantioselectivity can be achieved in this approach, and highlighted relevant challenges.
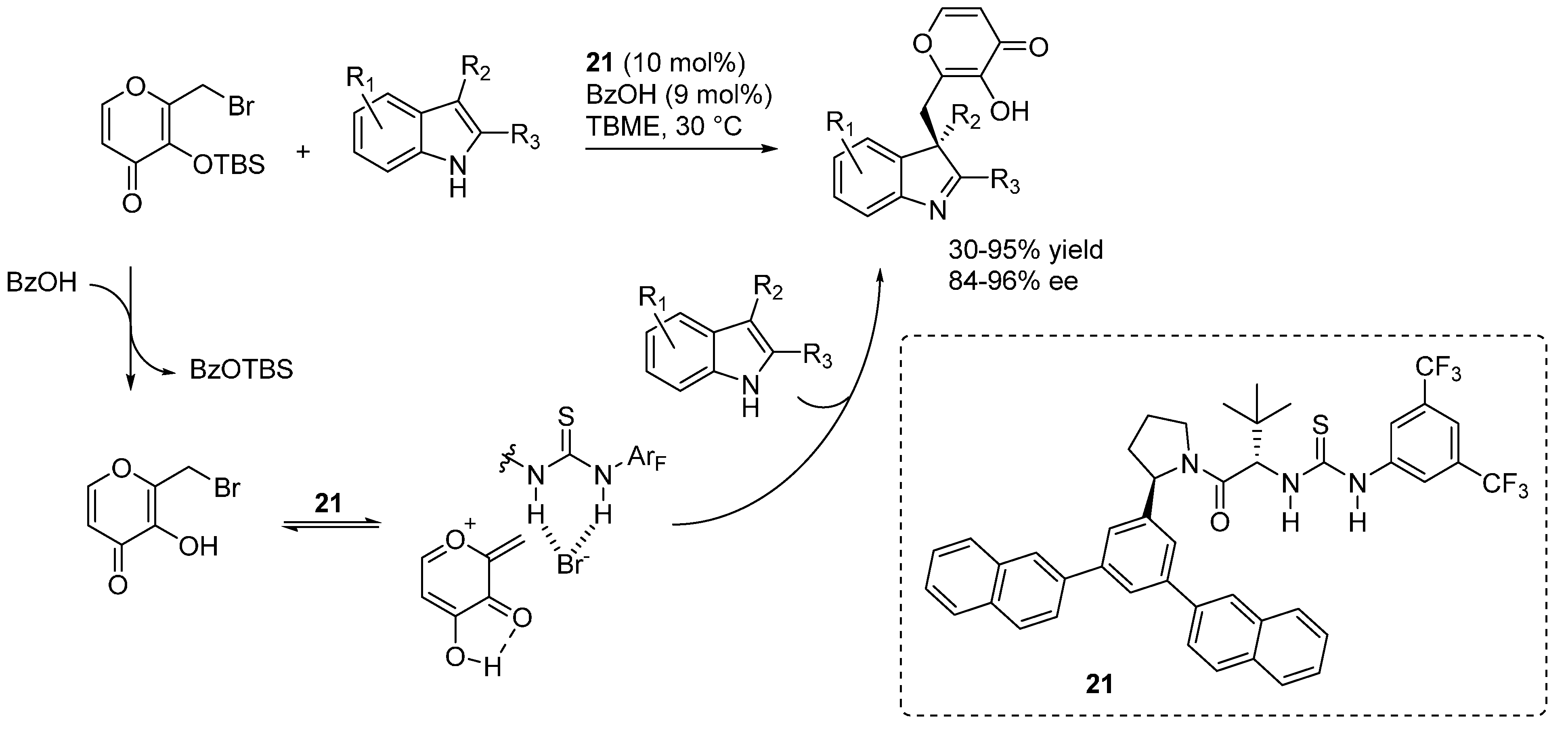
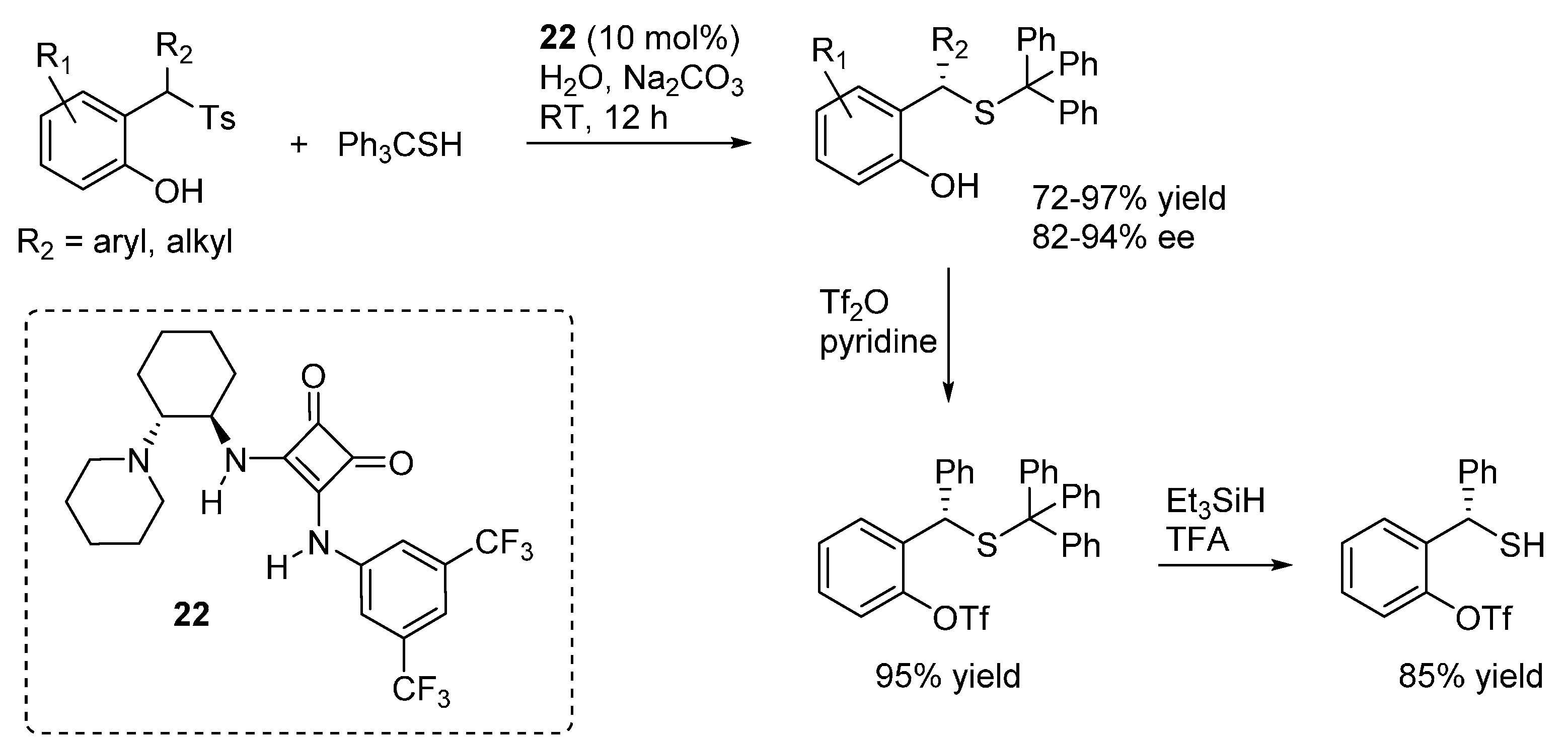
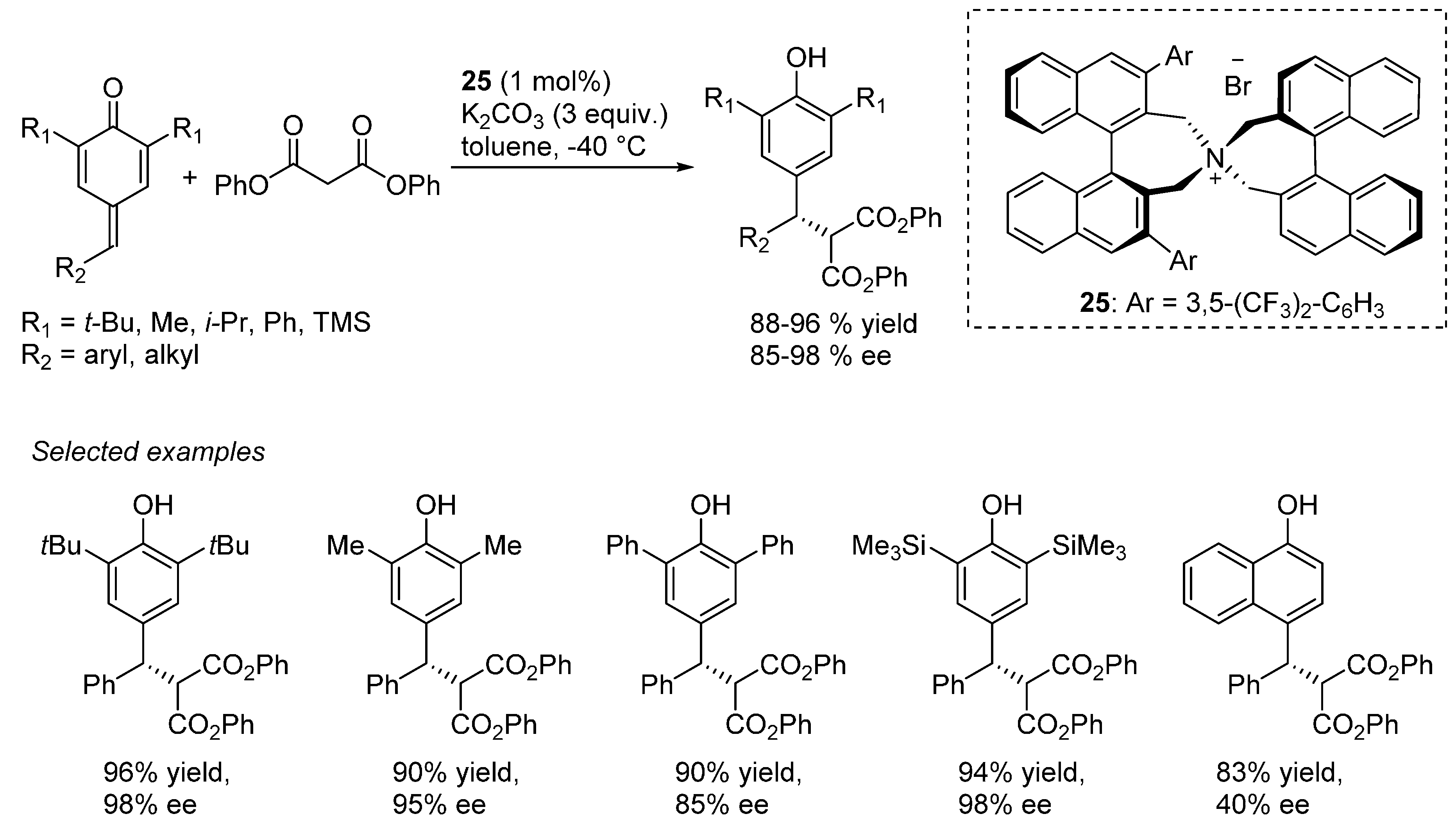
Liu and Li reported the addition of tritylthiol to
o-QMs generated from 2-tosylalkyl phenols (
Scheme 37), promoted by the bifunctional organocatalyst
22 and proceeding in the absence of organic solvents (just a small amount of dichloromethane was added to dissolve the substrates) [
49]. Aqueous sodium carbonate was used as the stoichiometric inorganic base to neutralize the sulfinic acid formed in the reaction. It was suggested that spatial separation between the stoichiometric inorganic base and the chiral organic base
22 was the key in achieving excellent enantioselectivities. Remarkably, the reaction could be applied successfully not only to β-aryl
o-QMs but also to their less stable β-alkyl counterparts, whereas limitations appeared with more stabilized
o-QMs (
i.e., R
1 = electron releasing substitutent in
Scheme 37). Furthermore, deprotection of the thiols was demonstrated to be feasible after phenol triflation, thus giving an entry to otherwise difficult to access benzyl thiols in highly enantioenriched form.
Scheme 37.
Catalytic asymmetric additions of tritylthiol to o-QMs catalyzed by 22.
Scheme 37.
Catalytic asymmetric additions of tritylthiol to o-QMs catalyzed by 22.
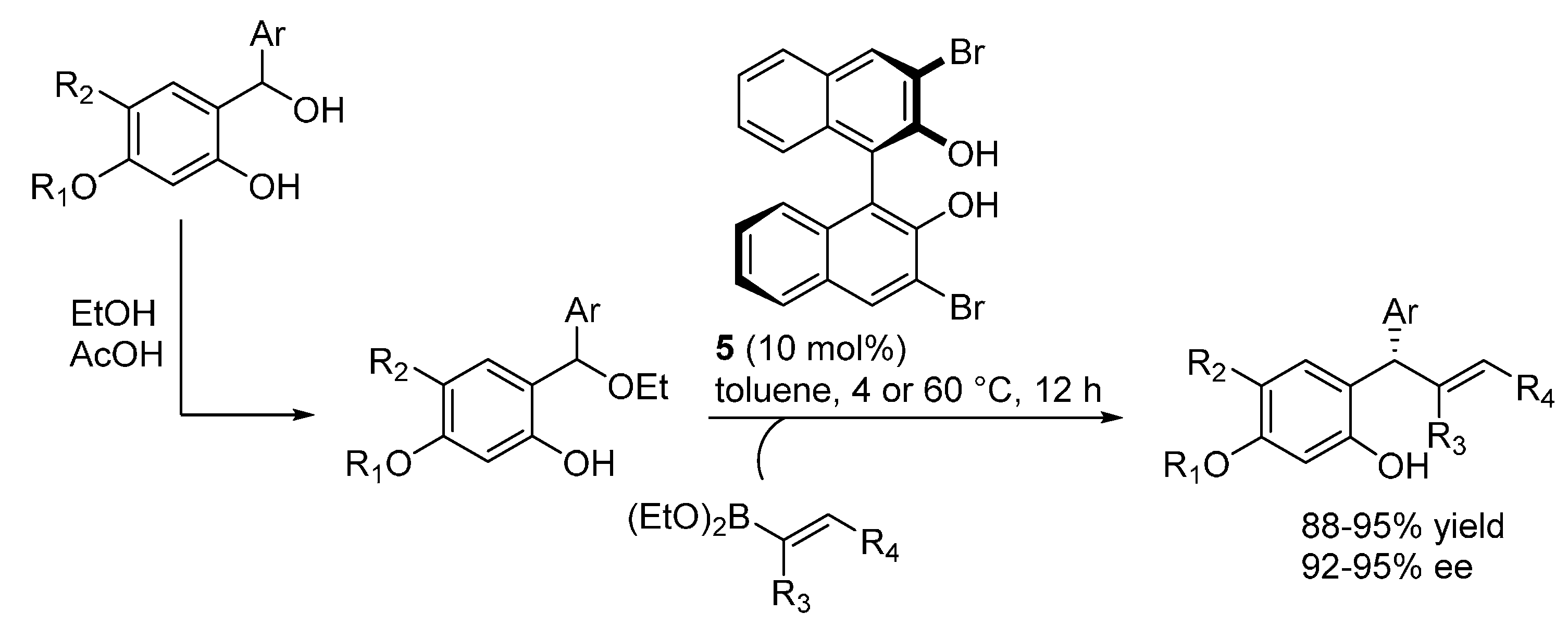
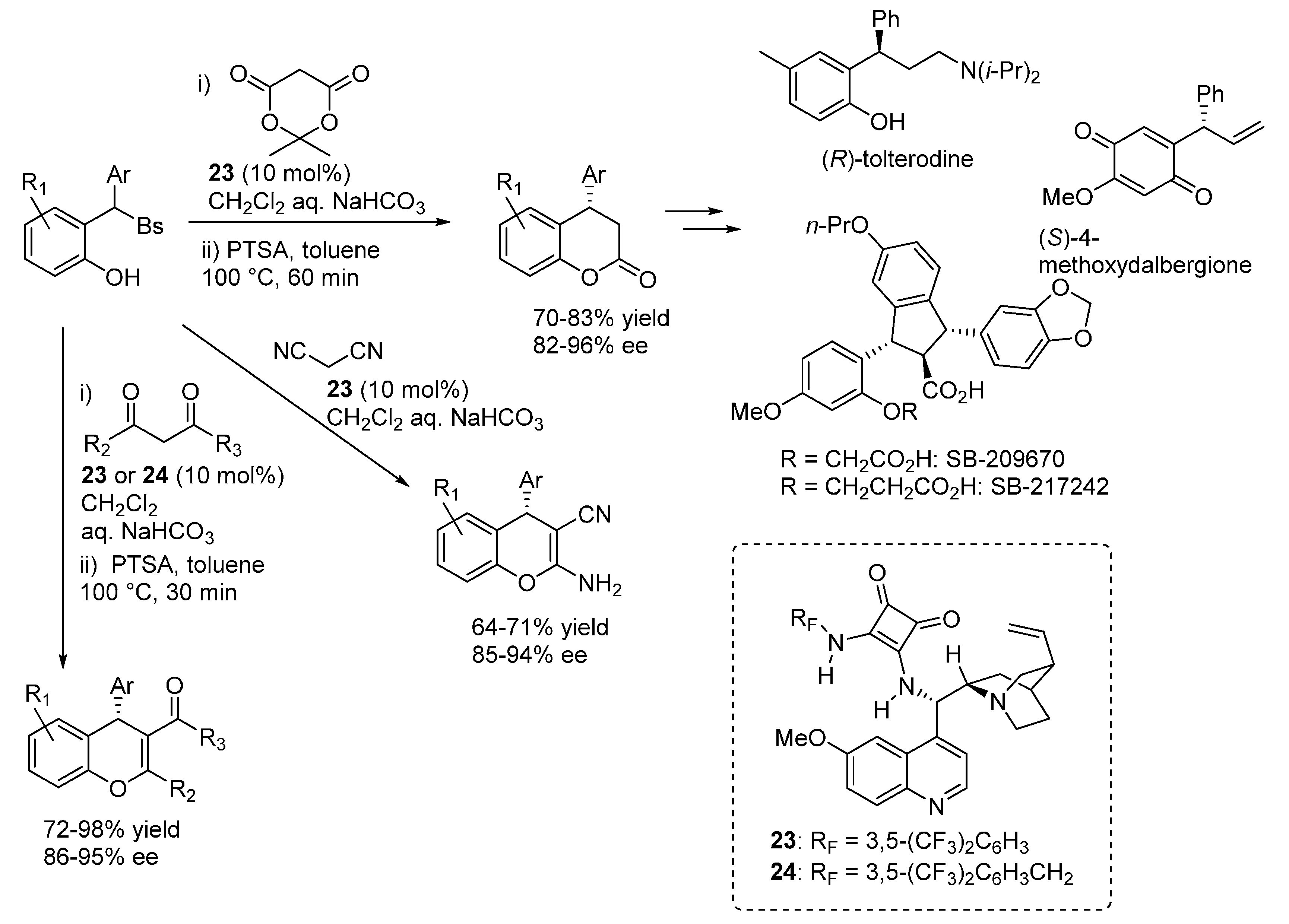
Nearly at the same time, our laboratory reported on the addition of various 1,3-dicarbonyl compounds (Meldrum’s acid, malononitrile, 1,3-diketones and 3-ketoesters) to
o-QMs generated
in situ from 2-sulfonylalkyl phenols, proceeding under the combined action of a stoichiometric inorganic base (aq. NaHCO
3, used in large excess) and the bifunctional catalysts
23 and
24 (
Scheme 38) [
50]. Key to success was the discovery that a subtle tuning of the leaving group properties of the sulfonyl moiety, using a less electron rich phenylsulfone instead of the usual
p-tolyl one, had a profound impact on the reaction outcome. Cyclizations, sometimes ensured by dehydrative acidic treatment after the catalytic step, followed the conjugate additions, delivering a range of 3,4-dihydrocoumarins and 4
H-chromenes with a fully complementary scope with respect to the catalytic additions of 1,3-diketones highlighted in
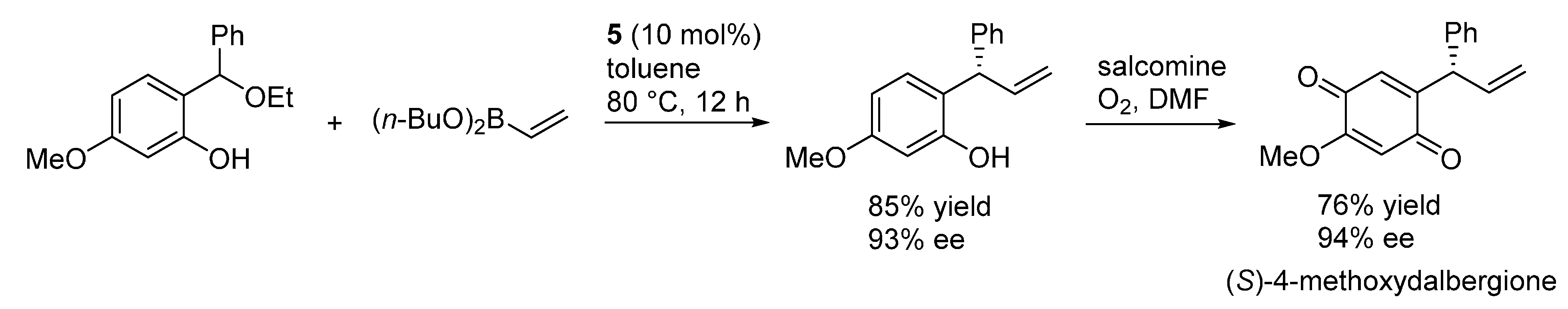
Section 3.1 and proceeding under acidic conditions. Some of the 3,4-dihydrocoumarin adducts obtained from the reactions with Meldrum’s acid are known intermediates for the synthesis of tolterodine, the active pharmaceutical ingredient of the antimuscarinic drug Detrol, and the endothelin antagonists SB-209670 and SB-217242, whereas another compound was converted in a synthetic precursor of the natural compound (
S)-4-methoxydalbergione. Both electron releasing and withdrawing substituents could be installed at the phenol ring, however the reaction appeared limited to β-aryl
o-QMs.
Scheme 38.
Catalytic asymmetric addition of 1,3-dicarbonyl compounds to o-QMs catalyzed by 23 and 24.
Scheme 38.
Catalytic asymmetric addition of 1,3-dicarbonyl compounds to o-QMs catalyzed by 23 and 24.
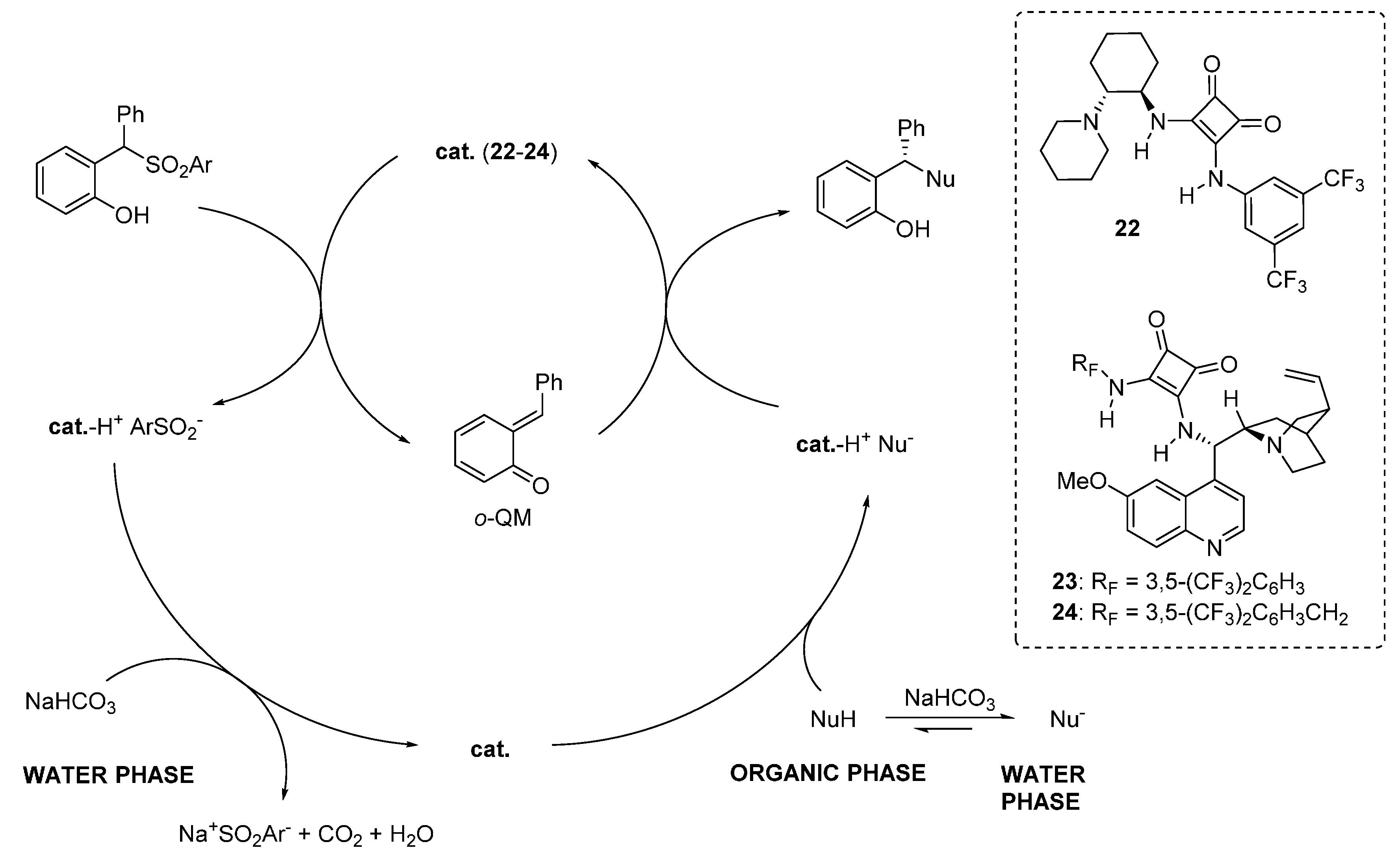
A nearly identical reaction pathway was proposed in the two papers, wherein the chiral organic base is responsible not only for the asymmetric addition, but also for the generation of the
o-QM by deprotonating the phenol (
Scheme 39). Thus, the role of the inorganic base is to regenerate the catalyst in its active form (the free amine), allowing the reaction to proceed. Whereas catalyst regeneration is the rate determining step of the overall catalytic cycle, it was determined that at least in some cases a large part of the nucleophile reaction partners are in the aqueous phase, due to their considerable acidity. Despite this unfavorable partition, both reactions appeared highly efficient indicating perhaps the requirement of a substantial amount of free catalyst (
i.e., not complexed with the pro-nucleophiles) in the organic phase for
o-QM generation.
Scheme 39.
Proposed reaction pathway for catalytic asymmetric additions to o-QMs generated in situ from 2-sulfonylalkyl phenols.
Scheme 39.
Proposed reaction pathway for catalytic asymmetric additions to o-QMs generated in situ from 2-sulfonylalkyl phenols.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










































