
Salix daphnoides: A Screening for Oligomeric and Polymeric Proanthocyanidins

Abstract
:1. Introduction
2. Results and Discussion
2.1. Screening for Oligomeric Proanthocyanidins

2.2. Characterization and Comparison of Fractions Enriched with Oligomeric or Polymeric Proanthocyanidins

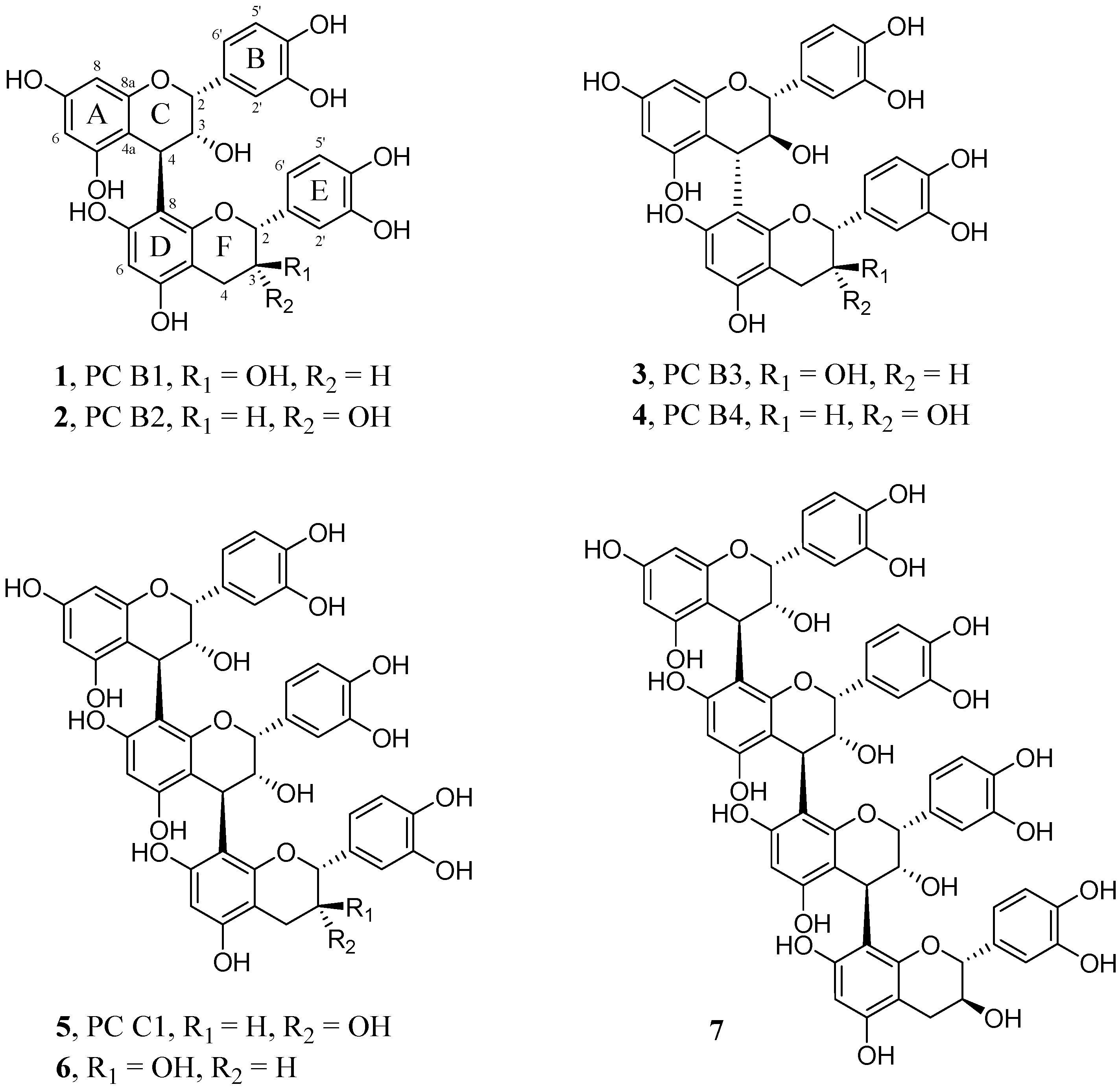{kind=link}
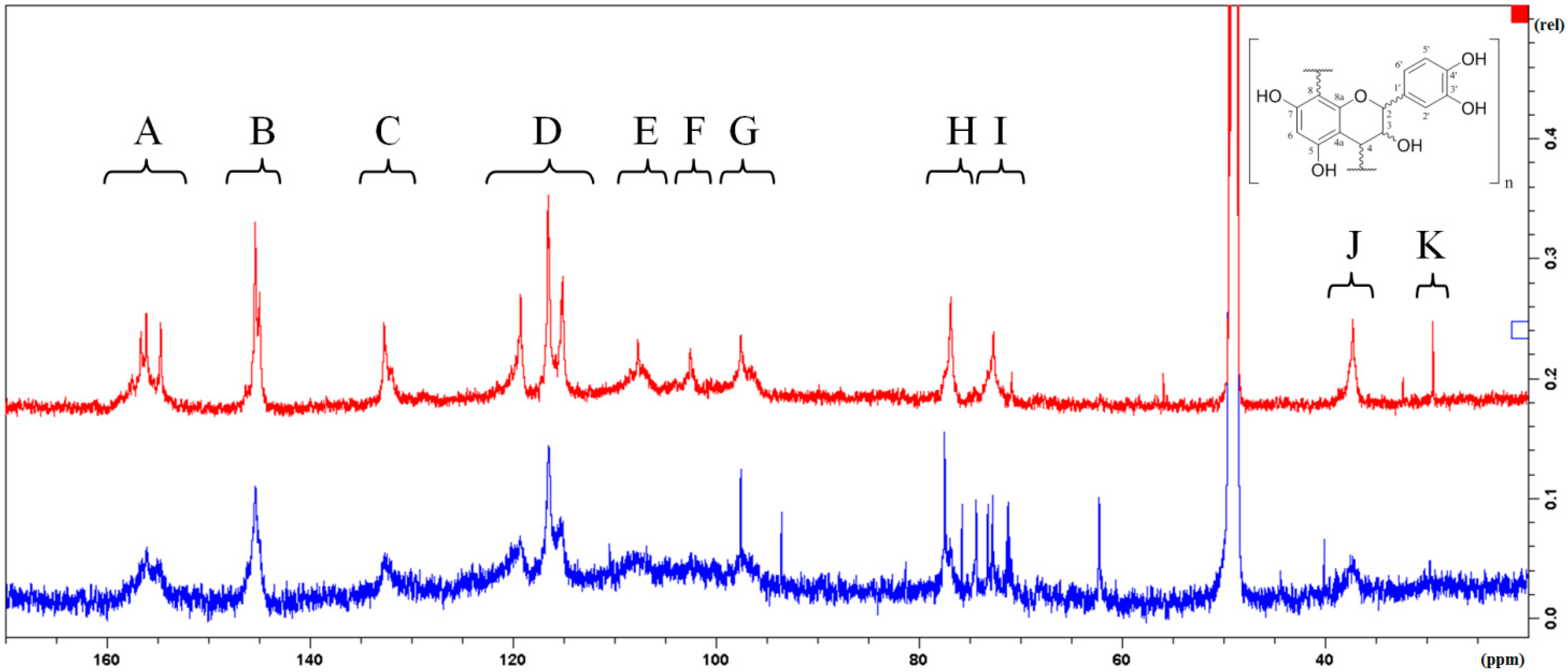{kind=link}
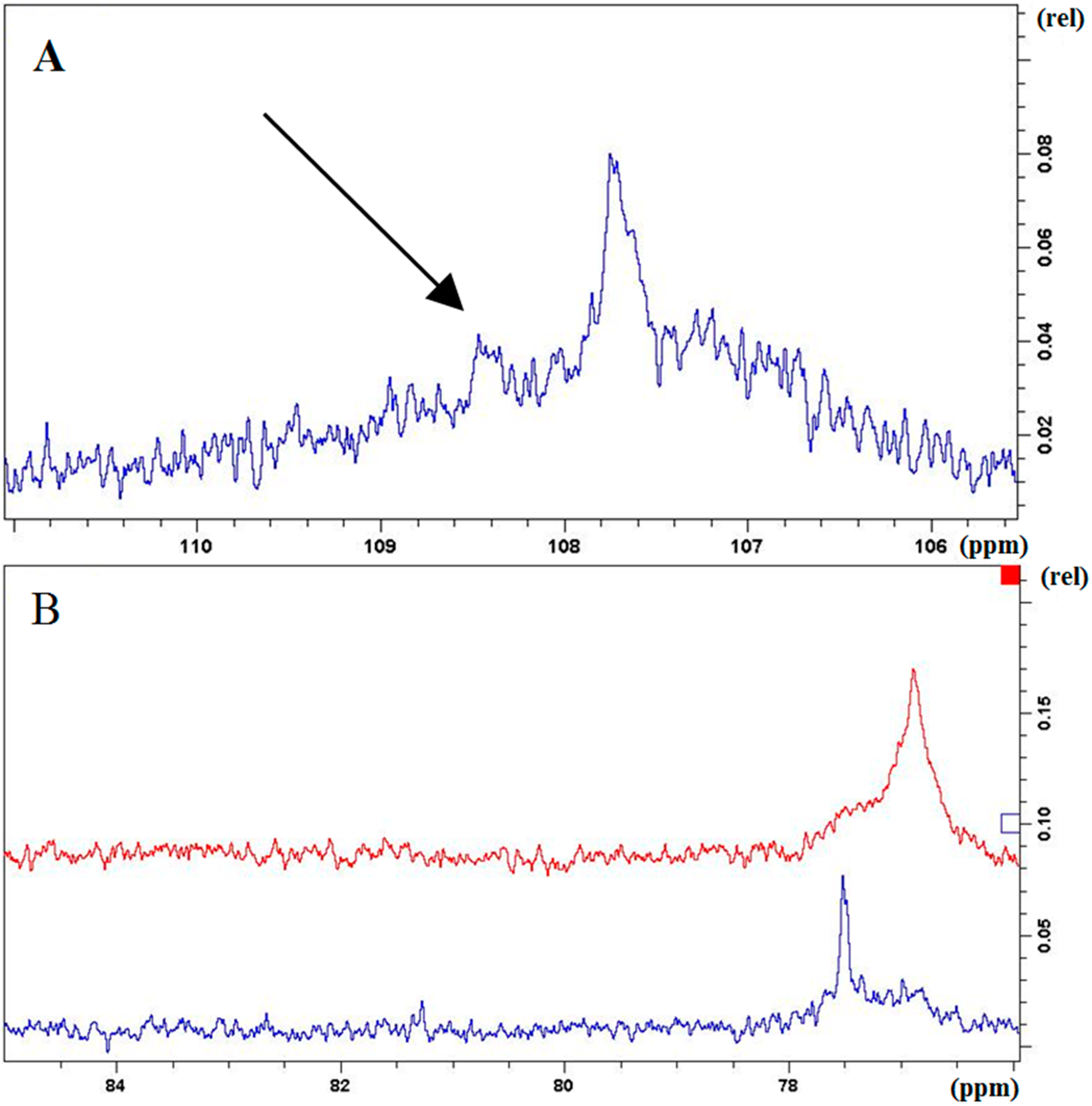{kind=link}
| Fraction | Delphinidin (RT = 5.91 min) | Cyanidin (RT = 10.07 min) | Pelargonidin (RT = 18.28 min) |
|---|---|---|---|
| “regular” PAs | 4.3% | 94.3% | 1.4% |
| “unusual” PAs | 0.8% | 97.8% | 1.4% |
| Peak | RT (min) (LC-HRMS) | RT (min) Anal. HPLC | m/z [M − H]− | Chemical Formula (calcd [M − H]−) |
|---|---|---|---|---|
| 1 | 20.97 | 22.82 | 289.0725 | C15H14O6 (289.0718) |
| 2 | 23.25 | 25.14 | 289.0723 | C15H14O6 (289.0718) |
| 3 ** | 35.06 | 36.92 | 699.1544 | C37H32O12S (699.1542) |
| 4 * | 36.60 | 38.40 | 427.0862 | C22H20O7S (427.0857) |
| 5 | 37.24 | 39.08 | 699.1548 | C37H32O12S (699.1542) |
| 6 | 39.08 | 40.81 | 699.1545 | C37H32O12S (699.1542) |
| 7 ** | 39.33 | 41.71 | 699.1540 | C37H32O12S (699.1542) |
| 8 * | 40.50 | 42.19 | 699.1543 | C37H32O12S (699.1542) |
| 9 | 41.39 | 43.12 | 411.0908 | C22H20O6S (411.0908) |
| 10 | 44.45 | 46.12 | 395.0963 | C22H20O5S (395.0959) |


3. Experimental Section
3.1. Reagents and Standards
3.2. Plant Material
3.3. Extraction Procedure
3.4. Sephadex® LH-20
3.5. Centrifugal Partition Chromatography (CPC)
3.6. MCI-Gel®
3.7. Preparative HPLC
3.8. Quantification of Tannins
3.9. Quantification of Total Amount of Procyanidins and Qualitative Evaluation of Monomeric Constitution
3.10. Estimation of the Degree of Polymerization (mDP) via Thiolysis
3.11. Phloroglucinol Degradation
3.12. Peracetylation
3.13. NMR Spectroscopy
3.14. Mass Spectrometry
3.15. Circular Dichroism
3.16. Polarimetry
3.17. Isolated Compounds
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Weidenrinde: Salicis cortex. In Europäisches Arzneibuch 8. Ausgabe, 2. Nachtrag, 8th ed.; Deutscher Apotheker Verlag: Stuttgart, Germany, 2015; Volume 2, pp. 2135–2136.
- Salicis cortex. In E/S/C/O/P Monographs: The Scientific Foundation for Herbal Medicinal Products, 2nd ed.; Thieme: Exeter, UK; Stuttgart, Germany; New York, NY, USA, 2003; pp. 445–451.
- Meier, B.; Meier-Liebi, M. Salix. In Hagers Enzyklopädie der Arzneistoffe und Drogen, 6th ed.; Blaschek, W., Hager, H., Eds.; Wissenschaftliche Verlagsgesellschaft: Stuttgart, Germany, 2007; Volume 14, pp. 81–115. [Google Scholar]
- Knuth, S.; Abdelsalam, R.M.; Khayyal, M.T.; Schweda, F.; Heilmann, J.; Kees, M.G.; Mair, G.; Kees, F.; Jürgenliemk, G. Catechol conjugates are in vivo metabolites of Salicis cortex. Planta Med. 2013, 79, 1489–1494. [Google Scholar] [CrossRef] [PubMed]
- Nahrstedt, A.; Schmidt, M.; Jäggi, R.; Metz, J.; Khayyal, M.T. Willow bark extract: the contribution of polyphenols to the overall effect. Wien. Med. Wochenschr. 2007, 157, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Butterweck, V.; Liefländer-Wulf, U.; Winterhoff, H.; Nahrstedt, A. Plasma levels of hypericin in presence of procyanidin B2 and hyperoside: A pharmacokinetic study in rats. Planta Med. 2003, 69, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Jürgenliemk, G.; Nahrstedt, A. Dissolution, solubility and cooperativity of phenolic compounds from Hypericum perforatum L. in aqueous systems. Die Pharm. 2003, 58, 200–203. [Google Scholar]
- Pobłocka-Olech, L.; Krauze-Baranowska, M. SPE-HPTLC of procyanidins from the barks of different species and clones of Salix. J. Pharm. Biomed. Anal. 2008, 48, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Shoji, T.; Mutsuga, M.; Nakamura, T.; Kanda, T.; Akiyama, H.; Goda, Y. Isolation and structural elucidation of some procyanidins from apple by low-temperature nuclear magnetic resonance. J. Agric. Food Chem. 2003, 51, 3806–3813. [Google Scholar] [CrossRef] [PubMed]
- Kolodziej, H. Synthesis and characterization of procyanidin dimers as their peracetates and octamethyl ether diacetates. Phytochemistry 1986, 25, 1209–1215. [Google Scholar] [CrossRef]
- Saito, A.; Nakajima, N.; Tanaka, A.; Ubukata, M. Synthetic studies of proanthocyanidins. Part 2: Stereoselective gram-scale synthesis of procyanidin-B3. Tetrahedron 2002, 58, 7829–7837. [Google Scholar] [CrossRef]
- Saito, A.; Nakajima, N.; Tanaka, A.; Ubukata, M. Synthetic studies of proanthocyanidins. Part 4. The synthesis of procyanidin B1 and B4: TMSOTf-catalyzed cyclization of catechin and epicatechin condensation. Heterocycles 2003, 61, 287. [Google Scholar] [CrossRef]
- Mohri, Y.; Sagehashi, M.; Yamada, T.; Hattori, Y.; Morimura, K.; Kamo, T.; Hirota, M.; Makabe, H. An efficient synthesis of procyanidins. Rare earth metal Lewis acid catalyzed equimolar condensation of catechin and epicatechin. Tetrahedron Lett. 2007, 48, 5891–5894. [Google Scholar] [CrossRef]
- Thompson, R.S.; Jacques, D.; Haslam, E.; Tanner, R.J.N. Plant proanthocyanidins. Part I. Introduction; the isolation, structure, and distribution in nature of plant procyanidins. J. Chem. Soc. Perkin Trans. 1 1972, 1387–1399. [Google Scholar] [CrossRef]
- Barrett, M.W.; Klyne, W.; Scopes, P.M.; Fletcher, A.C.; Porter, L.J.; Haslam, E. Plant proanthocyanidins. Part 6. Chiroptical studies. Part 95. Circular dichroism of procyanidins. J. Chem. Soc. Perkin Trans. 1 1979, 2375–2377. [Google Scholar] [CrossRef]
- Botha, J.J.; Ferreira, D.; Roux, D.G. Condensed tannins. Circular dichroism method of assessing the absolute configuration at C-4 of 4-arylflavan-3-ols, and stereochemistry of their formation from flavan-3,4-diols. J. Chem. Soc. Chem. Commun. 1978, 698–700. [Google Scholar] [CrossRef]
- Botha, J.J.; Young, D.A.; Ferreira, D.; Roux, D.G. Synthesis of condensed tannins. Part 1. Stereoselective and stereospecific syntheses of optically pure 4-arylflavan-3-ols, and assessment of their absolute stereochemistry at C-4 by means of circular dichroism. J. Chem. Soc. Perkin Trans. 1 1981, 1213–1219. [Google Scholar] [CrossRef]
- Saito, A.; Tanaka, A.; Ubukata, M.; Nakajima, N. Efficient stereoselective synthesis of proanthocyanidin trimers with TMSOTf-catalyzed intermolecular condensation. Synlett 2004, 1069–1073. [Google Scholar] [CrossRef]
- Esatbeyoglu, T.; Jaschok-Kentner, B.; Wray, V.; Winterhalter, P. Structure elucidation of procyanidin oligomers by low-temperature 1H-NMR spectroscopy. J. Agric. Food Chem. 2011, 59, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Mizushina, Y.; Tanaka, A.; Nakajima, N. Versatile synthesis of epicatechin series procyanidin oligomers, and their antioxidant and DNA polymerase inhibitory activity. Tetrahedron 2009, 65, 7422–7428. [Google Scholar] [CrossRef]
- Czochanska, Z.; Foo, L.Y.; Newman, R.H.; Porter, L.J. Polymeric proanthocyanidins. Stereochemistry, structural units, and molecular weight. J. Chem. Soc. Perkin Trans. 1 1980, 2278–2286. [Google Scholar] [CrossRef]
- 2.8.14: Gerbstoffe in pflanzlichen Drogen. In Europäisches Arzneibuch 8. Ausgabe, 2. Nachtrag, 8th ed.; Deutscher Apotheker Verlag: Stuttgart, Germany, 2015; Volume 1, p. 383.
- Glasl, H. Zur Photometrie in der Drogenstandardiserung. DAZ 1983, 123, 1979–1987. [Google Scholar]
- Weißdornfrüchte: Crataegi fructus. In Europäisches Arzneibuch 7. Ausgabe, 7th ed.; Deutscher Apotheker Verlag: Stuttgart, Germany, 2011; Volume 1, pp. 1933–1934.
- Zhang, Z.; Kou, X.; Fugal, K.; McLaughlin, J. Comparison of HPLC methods for determination of anthocyanins and anthocyanidins in bilberry extracts. J. Agric. Food Chem. 2004, 52, 688–691. [Google Scholar] [CrossRef] [PubMed]
- Dauer, A.; Rimpler, H.; Hensel, A. Polymeric Proanthocyanidins from the bark of Hamamelis virginiana. Planta Med. 2003, 69, 89–91. [Google Scholar] [CrossRef] [PubMed]
- Matthews, S.; Mila, I.; Scalbert, A.; Pollet, B.; Lapierre, C.; Hervé du Penhoat, C.L.M.; Rolando, C.; Donelly, D.M.X. Method for estimation of proanthocyanidins based on their acid depolymerization in the presence of nucleophiles. J. Agric. Food Chem. 1997, 45, 1195–1201. [Google Scholar] [CrossRef]
- Eberhardt, T.L.; Young, R.A. Conifer seed cone proanthocyanidin polymers: Characterization by 13C-NMR spectroscopy and determination of antifungal activities. J. Agric. Food Chem. 1994, 42, 1704–1708. [Google Scholar] [CrossRef]
- Newman, R.H.; Porter, L.J.; Foo, L.Y.; Johns, S.R.; Willing, R.I. High-resolution 13C-NMR studies of proanthocyanidin polymers (condensed tannins). Magn. Reson. Chem. 1987, 25, 118–124. [Google Scholar] [CrossRef]
- Jürgenliemk, G.; Petereit, F.; Nahrstedt, A. Flavan-3-ols and procyanidins from the bark of Salix purpurea L. Pharmazie 2007, 2007, 231–234. [Google Scholar]
- Porter, L.J. Condensed Tannins. In Natural Products of Woody Plants: Chemicals Extraneous to the Lignocellulosic Cell Wall; Rowe, J.W., Ed.; Springer Verlag: Berlin, Germany, 1989; Volume 1, pp. 651–690. [Google Scholar]
- Bicker, J.; Petereit, F.; Hensel, A. Proanthocyanidins and a phloroglucinol derivative from Rumex acetosa L. Fitoterapia 2009, 80, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Foo, L.Y.; Porter, L.J. Prodelphinidin polymers: Definition of structural units. J. Chem. Soc. Perkin Trans. 1 1978, 1186–1190. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiesneth, S.; Petereit, F.; Jürgenliemk, G. Salix daphnoides: A Screening for Oligomeric and Polymeric Proanthocyanidins. Molecules 2015, 20, 13764-13779. https://doi.org/10.3390/molecules200813764
Wiesneth S, Petereit F, Jürgenliemk G. Salix daphnoides: A Screening for Oligomeric and Polymeric Proanthocyanidins. Molecules. 2015; 20(8):13764-13779. https://doi.org/10.3390/molecules200813764
Chicago/Turabian StyleWiesneth, Stefan, Frank Petereit, and Guido Jürgenliemk. 2015. "Salix daphnoides: A Screening for Oligomeric and Polymeric Proanthocyanidins" Molecules 20, no. 8: 13764-13779. https://doi.org/10.3390/molecules200813764