
A Combined Molecular Docking/Dynamics Approach to Probe the Binding Mode of Cancer Drugs with Cytochrome P450 3A4

Abstract
:1. Introduction
2. Results and Discussion
2.1. Drug Interaction with CYP3A4


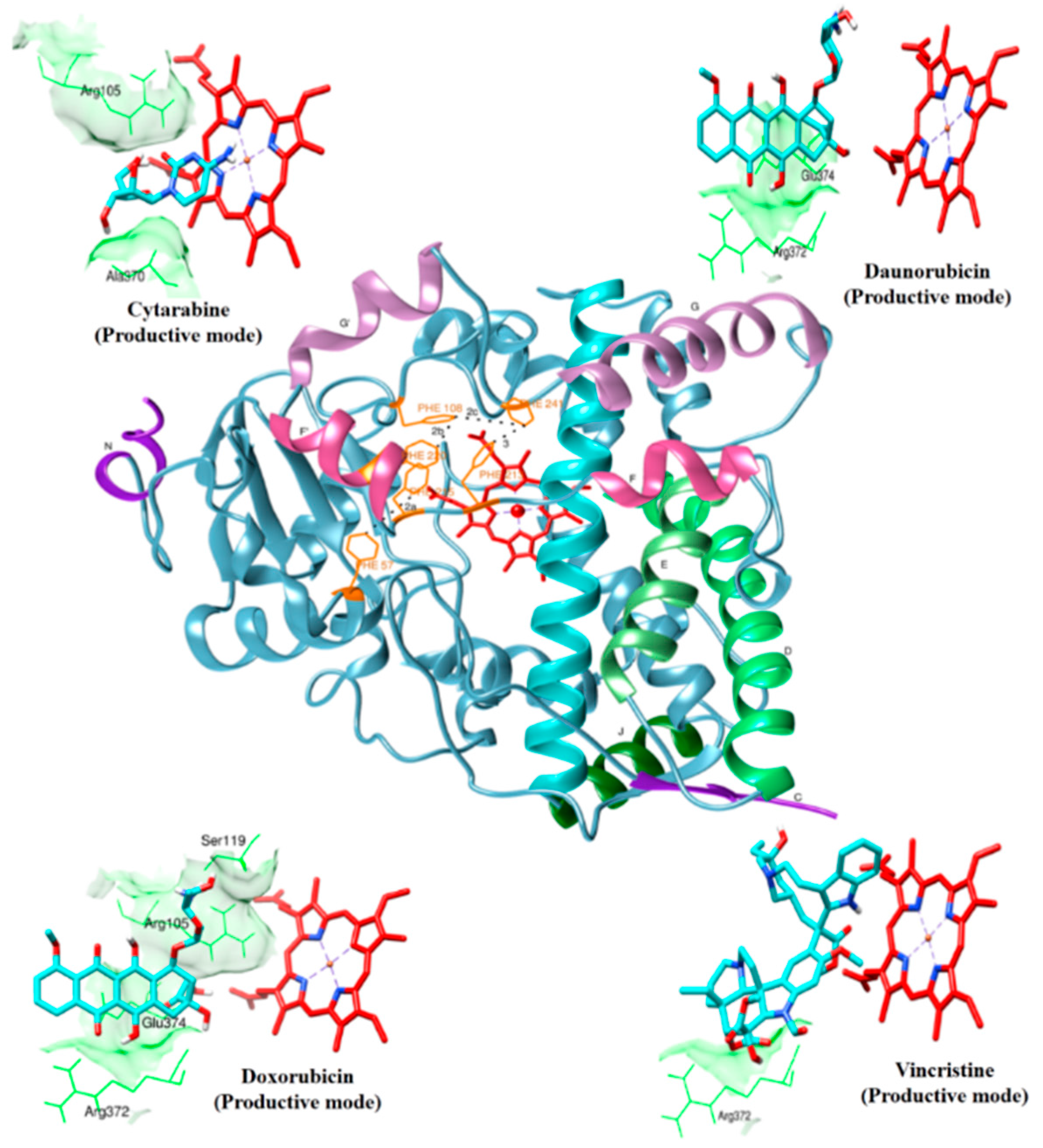{kind=link}
{kind=link}
{kind=link}
{kind=link}
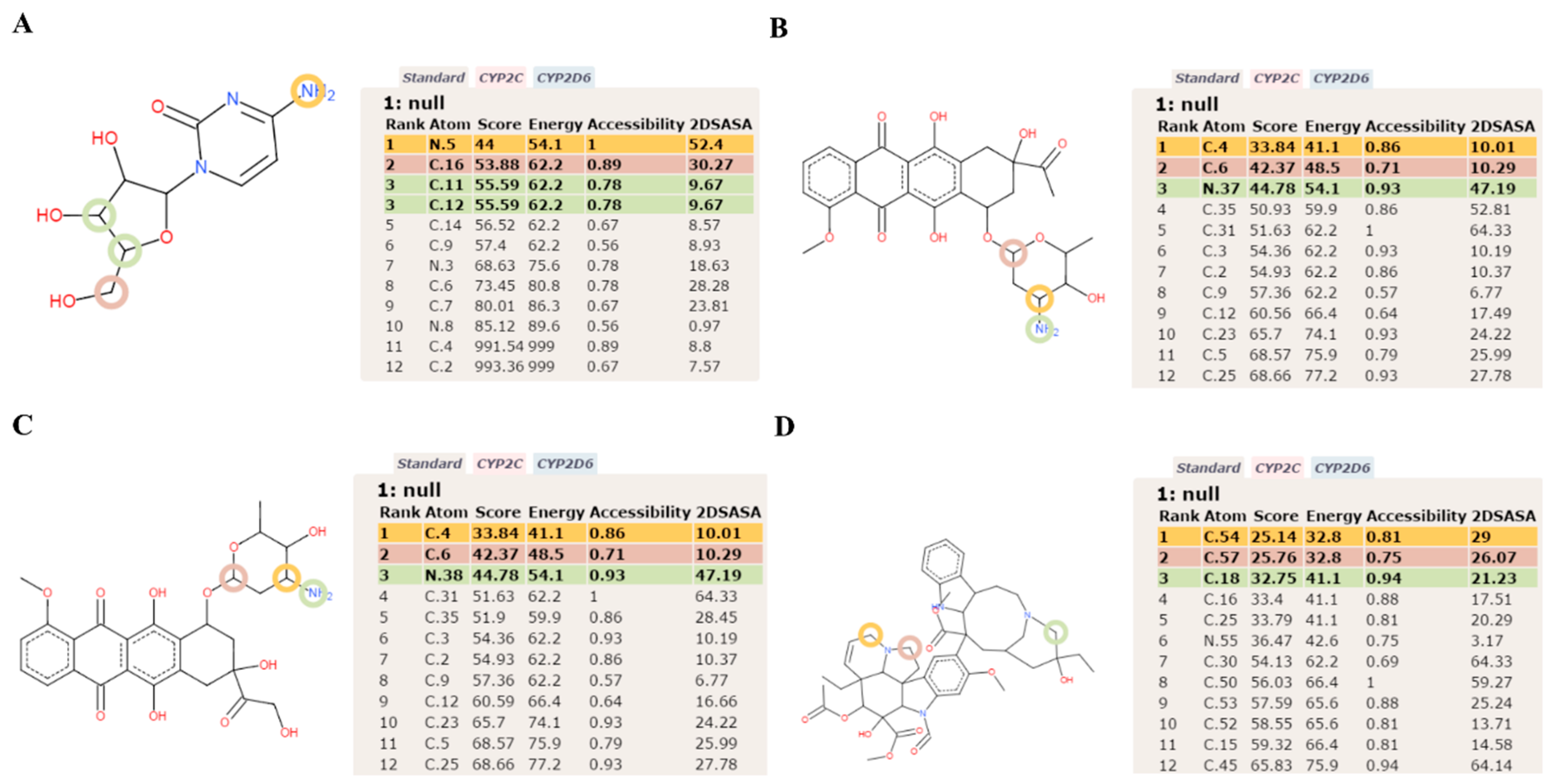{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Binding Pose | Conformational Clusters Out of 100 Runs | H-Bond Interactions (nm) | Binding Energy (kcal/mol) | Distance between Heme and Ligand (nm) | Van der Waals Interacting Residues |
|---|---|---|---|---|---|---|
| Cytarabine | Productive binding pose | (R105)N-H…O=0.211 | R105, R212, A305, T309, A370 | |||
| 16 | (A305)O-H…N=0.193 | −5.54 | 0.223 | |||
| (A370)O-H…O=0.194 | ||||||
| Non-productive binding pose | (R212)N-H…O=0.193 | R212, F215, S312, I369, A370, M371, L382, G481, L483 | ||||
| 20 | (L483)O-H…N=0.184 | −6.17 | 0.72 | |||
| (L483)N-H…N=0.220 | ||||||
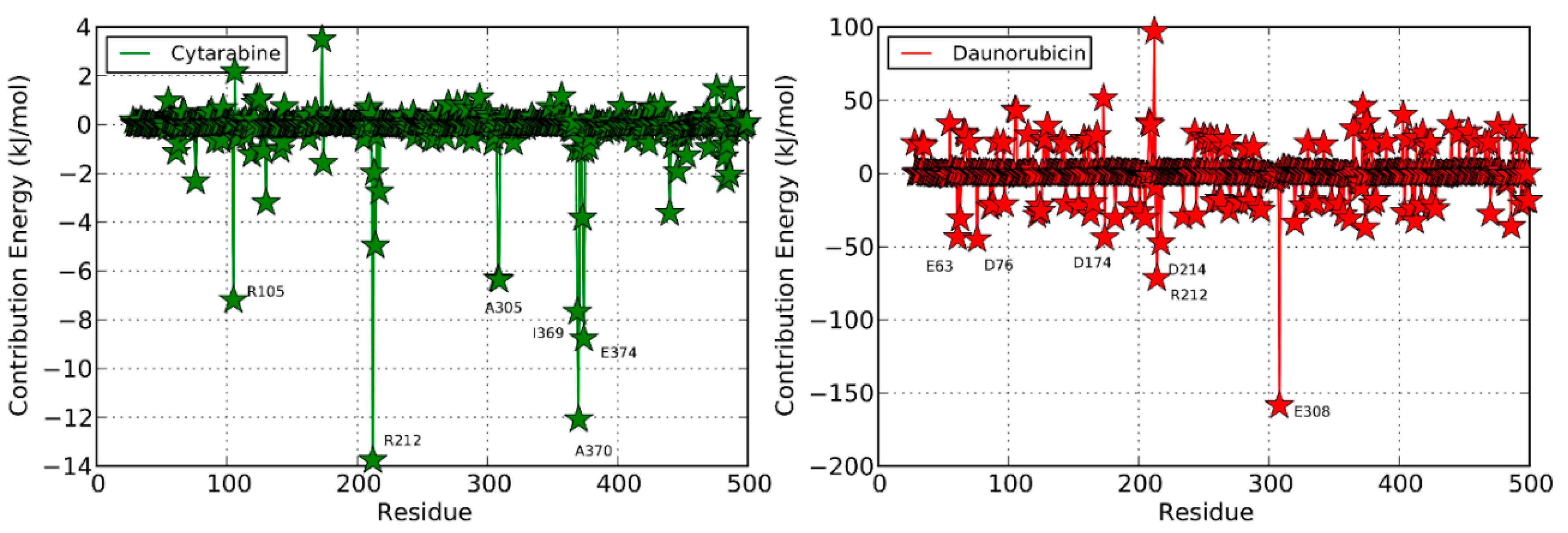
| Daunorubicin | Productive binding pose | (R372)N-H…O=0.215 | F57, R105, R106, F108, S119, F213, F304, R372, E374, heme | |||
| (E374)N-H…O=0.216 | −11.81 | 0.297 | ||||
| 68 | (E374)O…H-O=0.220 | |||||
| Non-productive binding pose I | (T224)O…H-N=0.108 | F108, F215, F304, S119, R105, F57, R372, E374, L373, A370, F304, heme | ||||
| (R372)N-H…O=0.194 | −11.69 | 0.483 | ||||
| 10 | (E374)O…H-O=0.177 | |||||
| Non-productive binding pose II | 4 | (N420)N-H…O=0.273 | − | 0.69 | Y99, W126, R128, R130, R375, P439, N441, I443, F435 | |
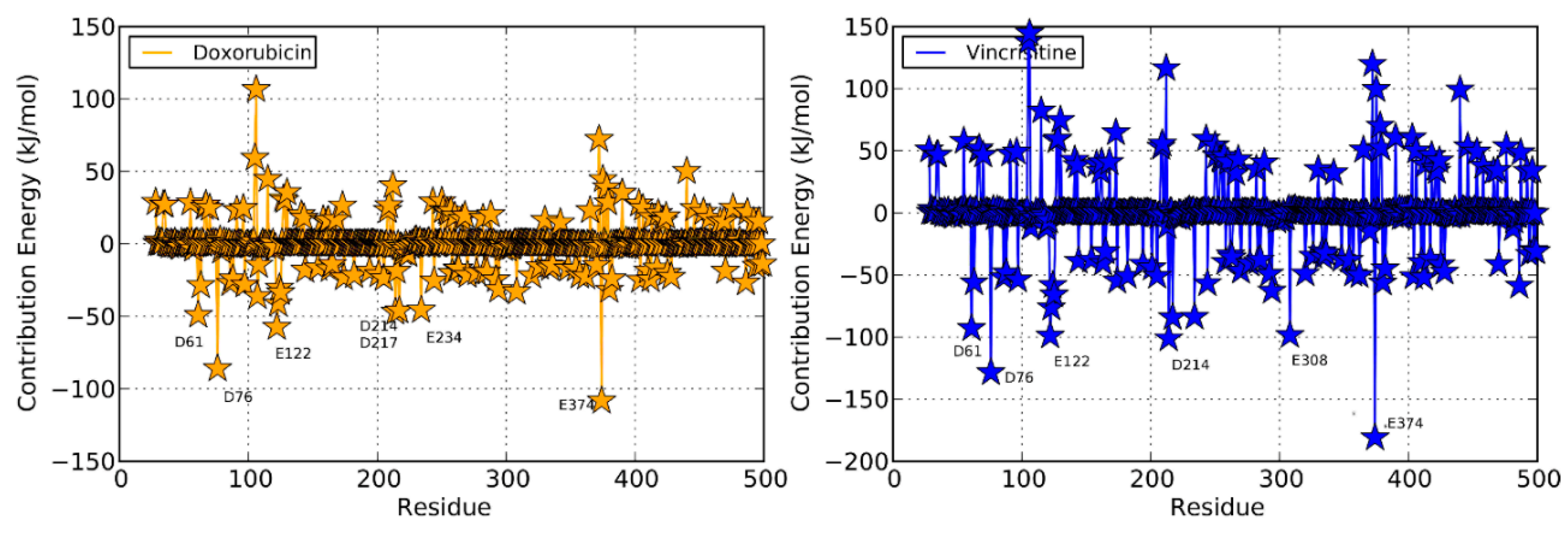
| Doxorubicin | Productive binding pose | (R105)N-H…O=0.225 | F57, D76, R105, R106, S119, F213, F215, F304, A370, R372, E374, R375, heme | |||
| (S119)O…O-H=0.222 | ||||||
| 69 | (R372)O…O-H=0.182 | −12.17 | 0.208 | |||
| (E374)N-H…O=0.132 | ||||||
| (Heme)O…H-O=0.190 | ||||||
| Non-productive binding pose | (E374)O…H-O=0.202 | R105, R106, F108, S119, R212, F215, I301, A305, A370, R372, E374, heme | ||||
| 15 | (E374)O…H-N=0.203 | −10.24 | 0.275 | |||
| Vincristine | Productive binding pose | (R372)N-H…O=0.222 | F57, R105, F108, S119, I120, R212, F304, A305, F213, F215, M371, R372, G481, L482, heme | |||
| 12 | −4.42 | 0.272 | ||||
| Non-productive binding pose | 2 | - | +53.43 | 0.845 | L351, D357, N361, K424, I427, P429, Y432, P434, F435, G436, S437, M445, R446, L449, K453 |
2.2. Cytarabine
2.2.1. Productive Binding Mode of Cytarabine



2.2.2. Non-Productive Binding Mode of Cytarabine Reoriented into Productive Binding Mode during MD Simulation
2.3. Daunorubicin
2.3.1. Productive Binding Mode of Daunorubicin


2.3.2. Non-Productive Binding Modes of Daunorubicin
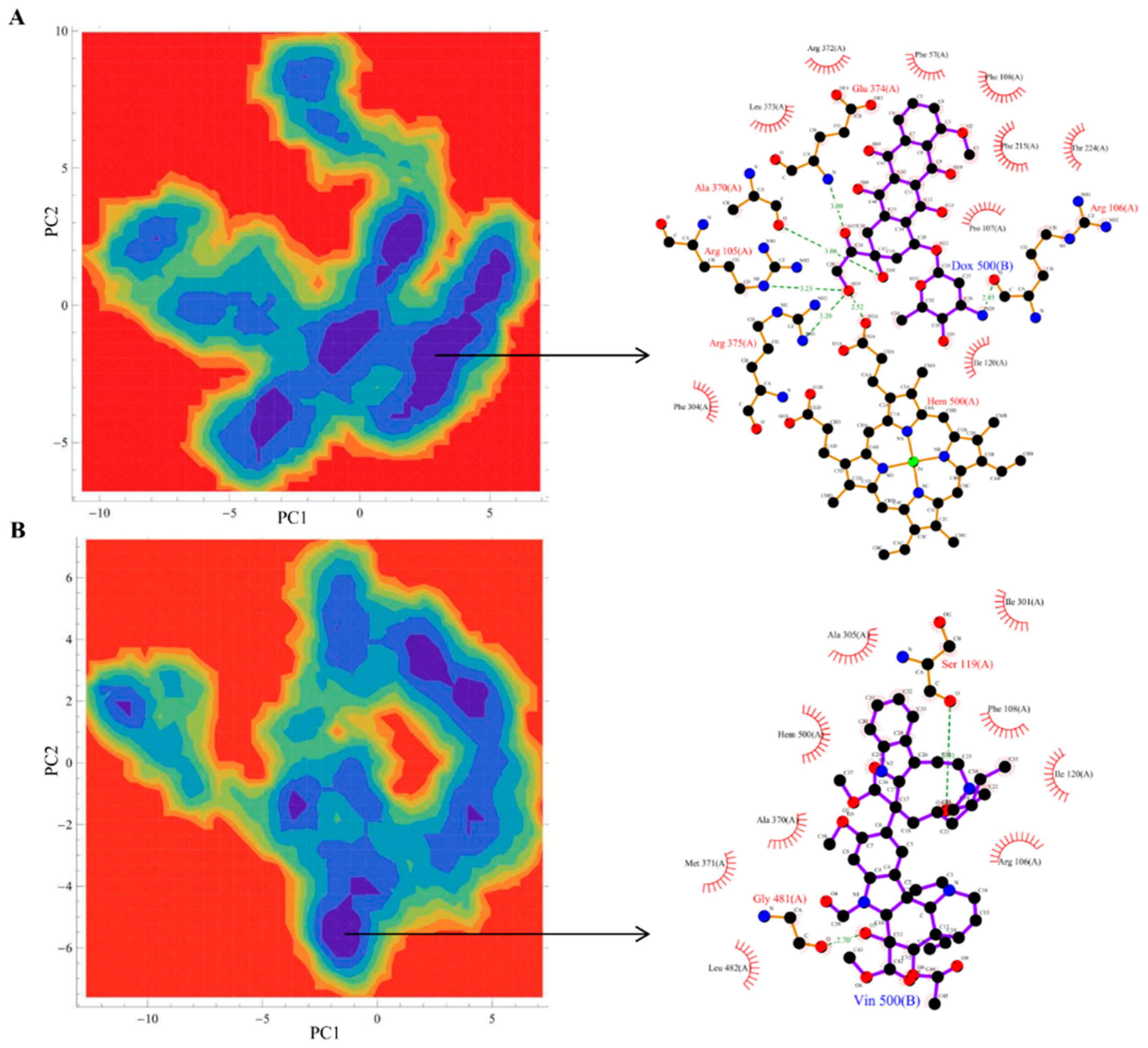
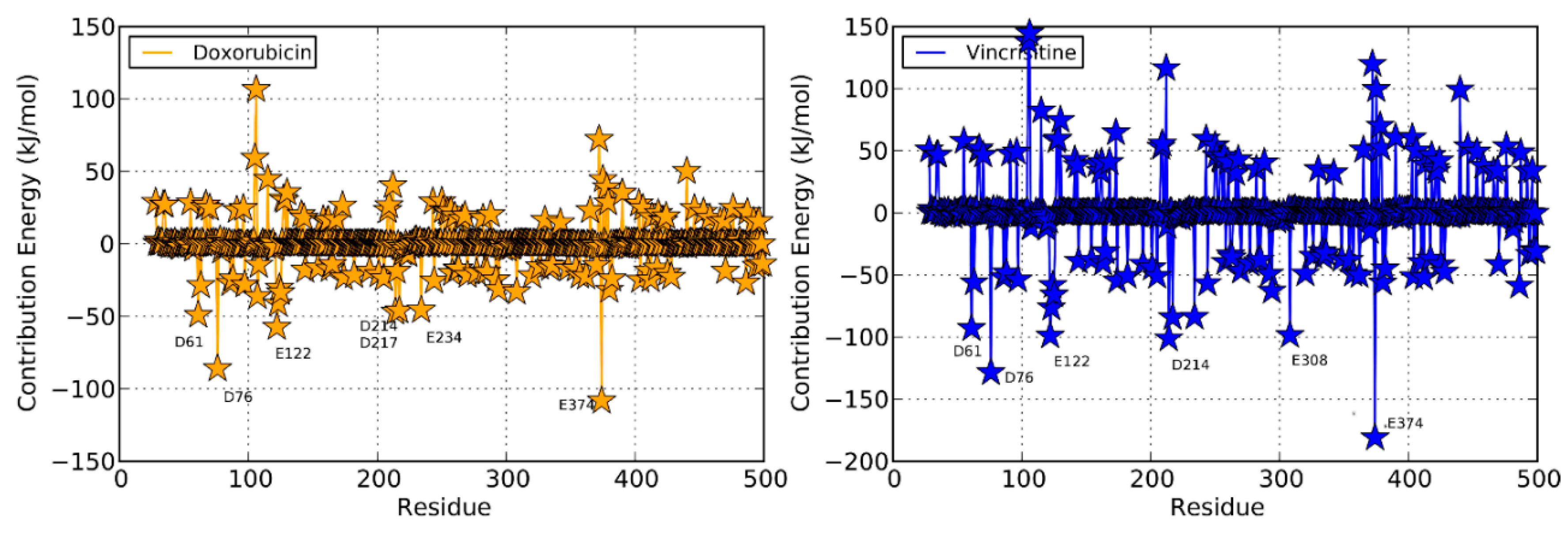
2.4. Doxorubicin
2.4.1. Productive Binding Mode of Doxorubicin

2.4.2. Non-Productive Binding Mode of Doxorubicin
2.5. Vincristine
2.6. Control Docking and Simulation
2.7. Discussion
3. Experimental Section
3.1. Docking
3.2. Molecular Dynamic Simulations
3.3. Heme Parameters
3.4. Principal Component Analysis and Free Energy Landscape Analysis
4. Conclusions
Supplementary Materials
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Nebert, D.W.; Russell, D.W. Clinical importance of the cytochromes P450. Lancet 2002, 360, 1155–1162. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome P-450 3A4: Regulation and role in drug metabolism. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 1–17. [Google Scholar] [CrossRef] [PubMed]
- De Groot, M.J. Designing better drugs: Predicting cytochrome P450 metabolism. Drug Discov. Today 2006, 11, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr. Drug Metab. 2008, 9, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Scripture, C.D.; Figg, W.D. Drug interactions in cancer therapy. Nat. Rev. Cancer 2006, 6, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Sevrioukova, I.F.; Poulos, T.L. Understanding the mechanism of cytochrome P450 3A4: Recent advances and remaining problems. Dalton Trans. 2013, 42, 3116–3126. [Google Scholar] [CrossRef] [PubMed]
- Bolwell, B.J.; Cassileth, P.A.; Gale, R.P. High dose cytarabine: A review. Leukemia 1988, 2, 253–260. [Google Scholar] [PubMed]
- Aubel-Sadron, G.; Londos-Gagliardi, D. Daunorubicin and doxorubicin, anthracycline antibiotics, a physicochemical and biological review. Biochimie 1984, 66, 333–352. [Google Scholar] [CrossRef]
- Keglevich, P.; Hazai, L.; Kalaus, G.; Szantay, C. Modifications on the basic skeletons of vinblastine and vincristine. Molecules 2012, 17, 5893–5914. [Google Scholar] [CrossRef] [PubMed]
- Dennison, J.B. Vincristine Metabolism and the Role of CYP3A5. Ph.D. Thesis, Indiana University, Indianapolis, IN, USA, 2007. [Google Scholar]
- Bello, M.; Mendieta-Wejebe, J.E.; Correa-Basurto, J. Structural and energetic analysis to provide insight residues of CYP2C9, 2C11 and 2E1 involved in valproic acid dehydrogenation selectivity. Biochem. Pharmacol. 2014, 90, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Cojocaru, V.; Wade, R.C. Conformational diversity and ligand tunnels of mammalian cytochrome P450s. Biotechnol. Appl. Biochem. 2013, 60, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Mannu, J.; Jenardhanan, P.; Mathur, P.P. A computational study of CYP3A4 mediated drug interaction profiles for anti-HIV drugs. J. Mol. Model. 2011, 17, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Cojocaru, V.; Winn, P.J.; Wade, R.C. Multiple, ligand-dependent routes from the active site of cytochrome P450 2C9. Curr. Drug Metab. 2012, 13, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Scott, D.O. Metabolism of 4-aminopiperidine drugs by cytochrome P450s: Molecular and quantum mechanical insights into drug design. ACS Med. Chem. Lett. 2011, 2, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Borhani, D.W.; Shaw, D.E. The future of molecular dynamics simulations in drug discovery. J. Comput. Aided Mol. Des. 2012, 26, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Dodson, G.G.; Lane, D.P.; Verma, C.S. Molecular simulations of protein dynamics: New windows on mechanisms in biology. EMBO Rep. 2008, 9, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Rydberg, P.; Rostkowski, M.; Gloriam, D.E.; Olsen, L. The contribution of atom accessibility to site of metabolism models for cytochromes P450. Mol. Pharm. 2013, 10, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Preissner, S.; Kroll, K.; Dunkel, M.; Senger, C.; Goldsobel, G.; Kuzman, D.; Guenther, S.; Winnenburg, R.; Schroeder, M.; Preissner, R. Supercyp: A comprehensive database on cytochrome P450 enzymes including a tool for analysis of cyp-drug interactions. Nucleic Acids Res. 2010, 38, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Davydov, D.R.; Rumfeldt, J.A.; Sineva, E.V.; Fernando, H.; Davydova, N.Y.; Halpert, J.R. Peripheral ligand-binding site in cytochrome P450 3A4 located with fluorescence resonance energy transfer (FRET). J. Biol. Chem. 2012, 287, 6797–6809. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.R.; Kim, S.Y.; Boysen, G.; Yun, C.H.; Miller, G.P. Contribution of three CYP3A isoforms to metabolism of R- and S-warfarin. Drug Metab. Lett. 2010, 4, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Sevrioukova, I.F.; Poulos, T.L. Structural and mechanistic insights into the interaction of cytochrome P4503A4 with bromoergocryptine, a type I ligand. J. Biol. Chem. 2012, 287, 3510–3517. [Google Scholar] [CrossRef] [PubMed]
- Hayes, C.; Ansbro, D.; Kontoyianni, M. Elucidating substrate promiscuity in the human cytochrome 3A4. J. Chem. Inf. Model 2014, 54, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Rydberg, P.; Olsen, L. Predicting drug metabolism by cytochrome P450 2C9: Comparison with the 2D6 and 3A4 isoforms. ChemMedChem 2012, 7, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Cojocaru, V.; Balali-Mood, K.; Sansom, M.S.; Wade, R.C. Structure and dynamics of the membrane-bound cytochrome P450 2C9. PLoS Comput. Biol. 2011, 7, e1002152. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Grinkova, Y.V.; Baylon, J.L.; Tajkhorshid, E.; Sligar, S.G. Mechanism of drug-drug interactions mediated by human cytochrome P450 CYP3A4 monomer. Biochemistry 2015, 54, 2227–2239. [Google Scholar] [CrossRef] [PubMed]
- Sevrioukova, I.F.; Poulos, T.L. Interaction of human cytochrome p4503a4 with ritonavir analogs. Arch. Biochem. Biophys. 2012, 520, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Lee, S.; Suh, J. Structural and dynamical basis of broad substrate specificity, catalytic mechanism, and inhibition of cytochrome P450 3A4. J. Am. Chem. Soc. 2005, 127, 13634–13642. [Google Scholar] [CrossRef] [PubMed]
- Shahrokh, K.; Cheatham, T.E., 3rd; Yost, G.S. Conformational dynamics of CYP3A4 demonstrate the important role of Arg212 coupled with the opening of ingress, egress and solvent channels to dehydrogenation of 4-hydroxy-tamoxifen. Biochim. Biophys. Acta 2012, 1820, 1605–1617. [Google Scholar] [CrossRef] [PubMed]
- Ekroos, M.; Sjogren, T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. USA 2006, 103, 13682–13687. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Cosme, J.; Vinkovic, D.M.; Ward, A.; Angove, H.C.; Day, P.J.; Vonrhein, C.; Tickle, I.J.; Jhoti, H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 2004, 305, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.K.; Wester, M.R.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. The structure of human microsomal cytochrome p450 3a4 determined by X-ray crystallography to 2.05-Å resolution. J. Biol. Chem. 2004, 279, 38091–38094. [Google Scholar] [CrossRef] [PubMed]
- Sevrioukova, I.F.; Poulos, T.L. Pyridine-substituted desoxyritonavir is a more potent inhibitor of cytochrome P450 3A4 than ritonavir. J. Med. Chem. 2013, 56, 3733–3741. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Available online: http://www.cancer.gov/cancertopics/druginfo/alphalist (accessed on 13 August 2015).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and autodocktools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. Gromacs 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. Charmm general force field: A force field for drug-like molecules compatible with the charmm all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [PubMed]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. Swissparam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Figshare. Available online: http://dx.doi.org/10.6084/m9.figshare.1289214 (accessed on 13 August 2015).
- Kumari, R.; Kumar, R.; Lynn, A. G_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Silva, D.A.; Yan, Y.; Huang, X. Force field development for cofactors in the photosystem II. J. Comput. Chem. 2012, 33, 1969–1980. [Google Scholar] [CrossRef] [PubMed]
- Figshare. Available online: http://dx.doi.org/10.6084/m9.figshare.1254117 (accessed on 13 August 2015).
- Amadei, A.; Linssen, A.B.; Berendsen, H.J. Essential dynamics of proteins. Proteins 1993, 17, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panneerselvam, S.; Yesudhas, D.; Durai, P.; Anwar, M.A.; Gosu, V.; Choi, S. A Combined Molecular Docking/Dynamics Approach to Probe the Binding Mode of Cancer Drugs with Cytochrome P450 3A4. Molecules 2015, 20, 14915-14935. https://doi.org/10.3390/molecules200814915
Panneerselvam S, Yesudhas D, Durai P, Anwar MA, Gosu V, Choi S. A Combined Molecular Docking/Dynamics Approach to Probe the Binding Mode of Cancer Drugs with Cytochrome P450 3A4. Molecules. 2015; 20(8):14915-14935. https://doi.org/10.3390/molecules200814915
Chicago/Turabian StylePanneerselvam, Suresh, Dhanusha Yesudhas, Prasannavenkatesh Durai, Muhammad Ayaz Anwar, Vijayakumar Gosu, and Sangdun Choi. 2015. "A Combined Molecular Docking/Dynamics Approach to Probe the Binding Mode of Cancer Drugs with Cytochrome P450 3A4" Molecules 20, no. 8: 14915-14935. https://doi.org/10.3390/molecules200814915