
The Potential Use of Natural and Structural Analogues of Antimicrobial Peptides in the Fight against Neglected Tropical Diseases


Abstract
:1. Introduction
2. Antimicrobial Peptides
2.1. Classification of AMPs
2.1.1. Classification of AMPs According to Net Charge
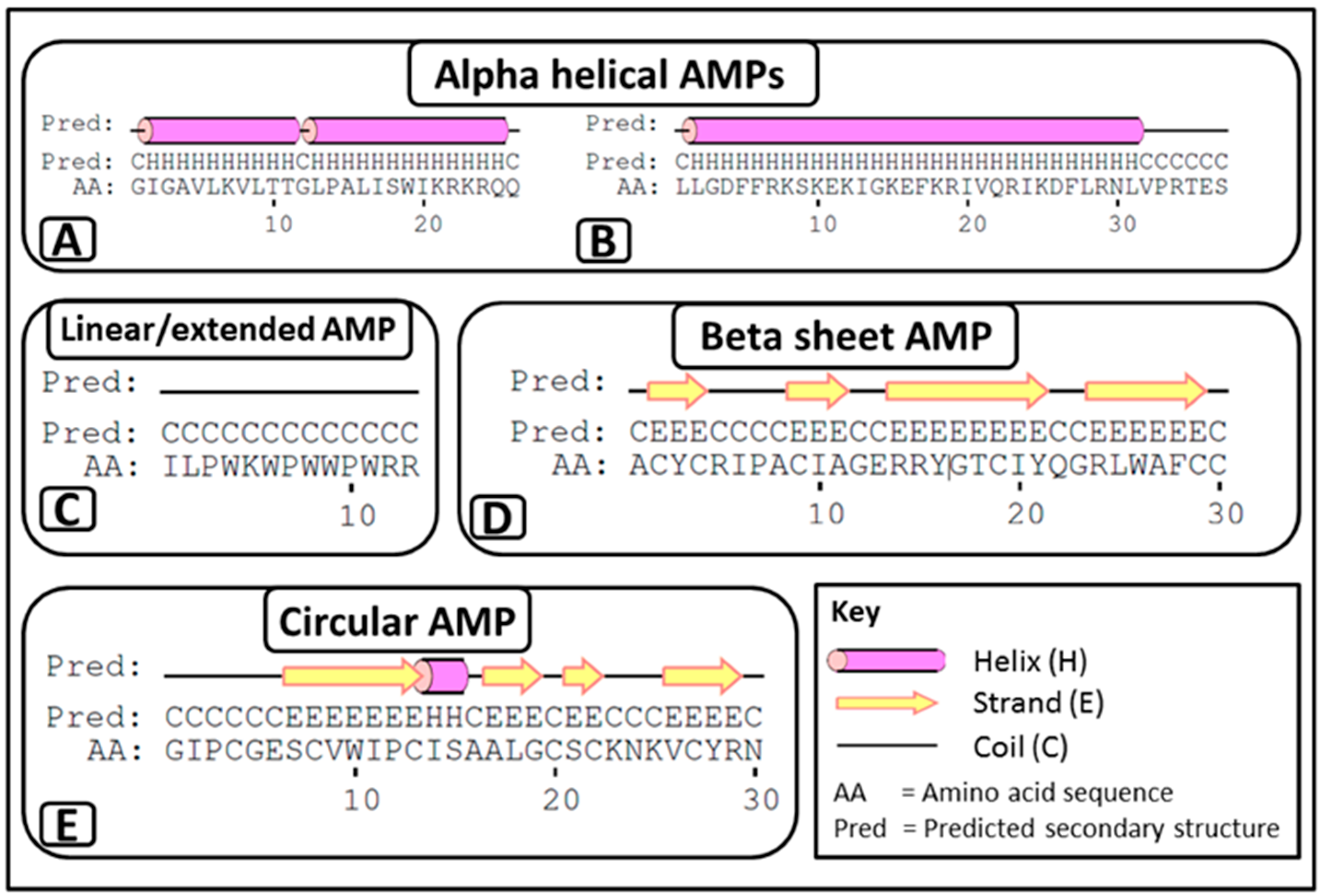
2.1.2. Classification According to Secondary Structure
- i
- α-helical AMPs: These peptides are unstructured linear peptides free of cysteine residues that fold into α-helixes upon contact with membranes. They consist of approximately 50% hydrophobic residues, favouring an amphiphilic conformation upon interaction with membranes, which enables them to permeabilise microbial membranes. Some of these peptides are not strictly α-helical and may possess an internal kink and/or a flexible unstructured segment at the N- and/or C-terminus. For example, melittin (APD ID: 00146) (Figure 1A) from bee venom and the human cathelicidin LL-37 (APD ID: AP00310) (Figure 1B).
- ii
- Linear/extended AMPs: These peptides are linear without cysteine residues and contain a high proportion of proline, arginine, glycine, tryptophan and histidine. Some of these AMPs form extended coils. Examples include indolicidin (APD ID: AP00150) (Figure 1C) from bovine leukocytes.
- iii
- β-sheet AMPs: These peptides contain six to eight cysteine residues, forming two or more disulphide bonds, resulting in a stabilized β-sheet structure. For example α and β defensins such as human neutrophil peptide-1 (HNP-1, APD ID: AP00176) (Figure 1D) from mammals.

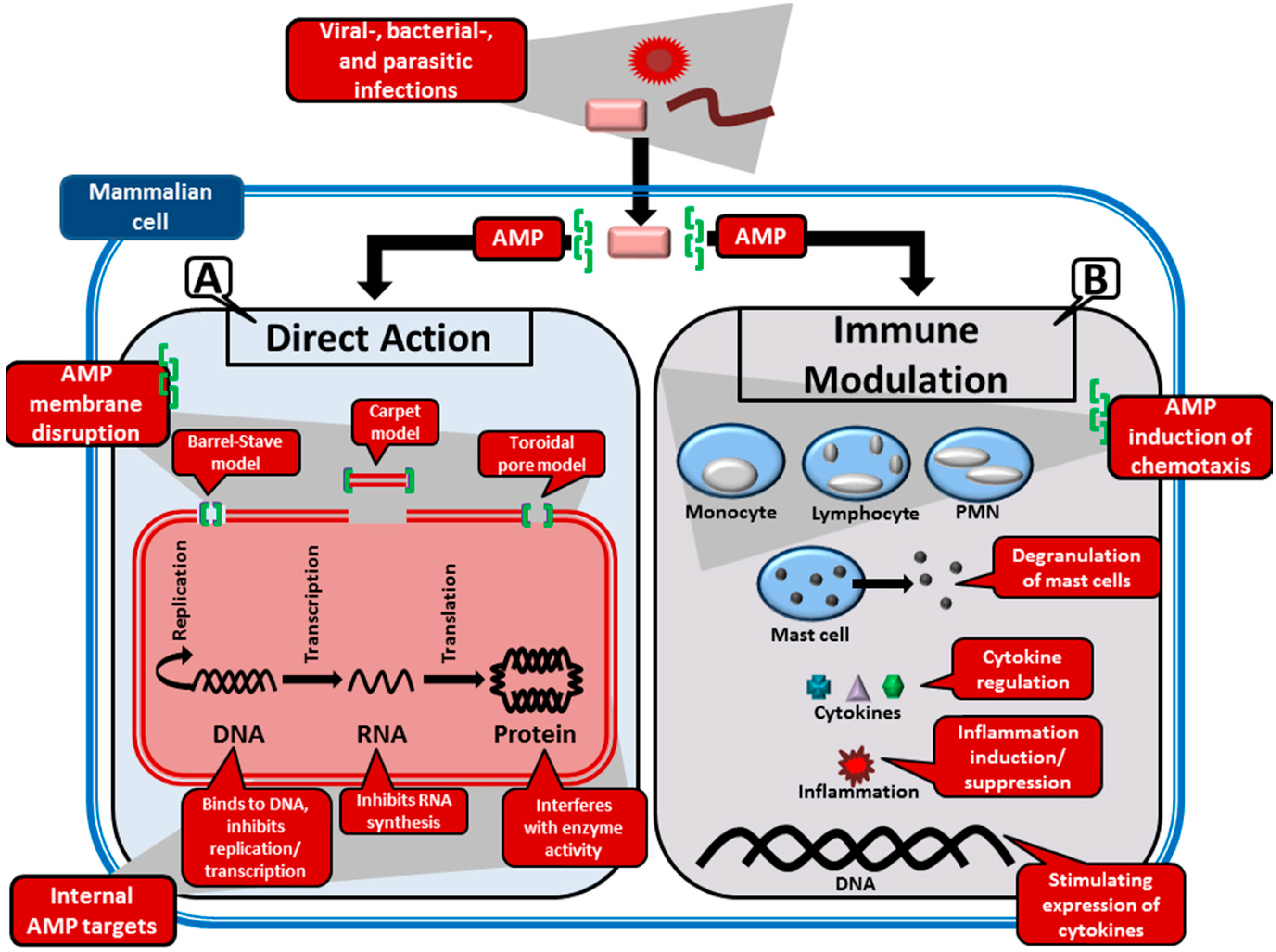
2.2. Target Organisms and Mode of Action of AMPs

{kind=link}
{kind=link}
| AMP | APD ID | Source | Sequence | Structure |
|---|---|---|---|---|
| Magainin 2 | AP00144 | African clawed frog (Xenopus laevis) | GIGKFLHSAKKFGKAFVGEIMNS | α-helix |
| Melittin | AP00146 | Honey bee (Apis mellifera) (also A. florea, A. cerana) | GIGAVLKVLTTGLPALISWIKRKRQQ | α-helix |
| Dermaseptin-S1 | AP00157 | Sauvages leaf frog (Phyllomedusa sauvagii) | ALWKTMLKKLGTMALHAGKAALGAAADTISQGTQ | α-helix |
| HNP-1 | AP00176 | Human (Homo sapiens) | ACYCRIPACIAGERRYGTCIYQGRLWAFCC | β-sheet |
| LL-37 | AP00310 | Human (Homo sapiens) Chimpanzee (Pan troglodytes) | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | α-helix |
| BMAP-27 | AP00366 | Domestic cattle (Bos taurus) | GRFKRFRKKFKKLFKKLSPVIPLLHLG | α-helix |
2.2.1. Perturbation of the Microbial Membranes by AMPs

- i
- Barrel-stave model: This model is used to describe the formation of pores in the lipid membrane. This is achieved through the amphipathic peptides that insert in a perpendicular orientation to the membrane and align so that the hydrophilic side chains point inward and form the transmembrane pore, while the hydrophobic side chains face outward and interact with the lipid bilayer [10,60]. This was the first model to be proposed as the mechanism of peptide induced pores [61]. AMPs that this model typically applies to are those that promote loss of electrochemical potential at low, well-defined peptide:phospholipid ratios [54]. The only AMP for which there is clear evidence of the barrel-stave mechanism is the non-ribosomally synthesised alamethicin (APD ID: AP02197), produced by the fungi Trichoderma viride [62].
- ii
- Toroidal model: In this model, pores, consisting of peptides intercalated with lipids, are formed in the lipid membrane [63]. Amphipathic peptides accumulate parallel to the membrane surface, where their partial insertion causes the outer (but not the inner) leaflet to expand. The peptides re-orientate perpendicularly to the bilayer once a critical threshold is reached, thereby relieving tension. The bilayer undergoes a positive curvature and its thickness is modified, which creates transient pores consisting of both peptide and lipid molecules [54].
- iii
- Carpet model: Partial membrane solubilisation in a detergent-like manner occurs with this model [54]. The amphipathic peptides accumulate parallel to the membrane via electrostatic interaction with anionic phospholipids—covering the membrane in a carpet-like fashion. When a threshold concentration is reached, detergent-like activity causes the formation of micelles and membrane pores [10].
2.2.2. Interaction of AMPs with Internal Targets
2.2.3. Modulation of the Immune System by AMPs
2.3. Therapeutic Potential and Obstacles Associated with AMPs
3. Neglected Tropical Diseases
3.1. Neglected Bacterial Infections
3.1.1. AMPs against Mycobacterium Infections and Hansen’s Disease
“And if the priest sees that the scab has indeed spread on the skin, then the priest shall pronounce him unclean. It is leprosy.”Leviticus 13:8
3.1.2. AMPs against Trachoma
3.2. Neglected Protozoan Infections
3.2.1. AMPs against Chagas Disease
3.2.2. AMP against Human African Trypanosomiasis
3.2.3. AMPs against Leishmaniosis
3.2.4. AMPs against Malaria
3.3. Neglected Helminth-Related Infections
3.3.1. Possible AMPs against Taeniasis and Cysticercosis
3.3.2. AMPs from Onchocerciasis
3.4. Antiviral Peptides and Their Potential Use to Treat Viral Diseases and Possible Application to Viral Neglected Tropical Diseases
3.4.1. AMPs against Dengue Viral Disease
3.4.2. AMPs against Rabies
4. Conclusions
| Type | Infection (Causative Agent) | AMP | Source | Notes | Ref |
|---|---|---|---|---|---|
| Bacterial | Leprosy (Mycobacterium leprae) | Human β-defensin 3 | Homo sapiens (Human) | Up-regulated during a leprosy type 1 infections | [106] |
| LL-37 | Homo sapiens (Human) Pan troglodytes (Chimpanzee) | M. leprae inhibits CAMP—the gene encoding for LL-37 | [116] | ||
| Hepcidin | Homo sapiens (Human) | Involved in the degradation of ferroportin in multibacillary lesions | [118] | ||
| Trachoma (Chlamydia trachomatis) | SMAP-29 | Ovis aries (Sheep) | Reduced inclusion number at concentration of 10 µg/mL. Compromised integrity of extracellular elementary body | [130] | |
| Melittin | Apis mellifera (Honey bee) | Direct cytotoxic effect on C. trachomatis. Hindering normal process of intracellular development by lowering the transdermal potential and disrupting the adhesion of the bacteria to the cell | [133] | ||
| Cyto-insectotoxin 1a | Lachesana tarabaevi (Central Asian ant spider) | Inhibit C. trachomatis infection at an early stage, with higher efficacy than melittin and negligible effect on cell viability | [134] | ||
| Parasites | Chagas disease (Trypanosoma cruzi) | Melittin | A. mellifera (Honey bee) | Induced morphological alterations in the different developmental forms of the parasite, which could be characterized as apoptosis and autophagy. | [145] |
| Dermasiptin-01 | Phyllomedusa oreades (Tree frog) | Lytic activity | [149] | ||
| Parasites | Chagas disease (Trypanosoma cruzi) | Tachyplesin I | Tachypleus tridentatus (Horseshoe crab) | Able to kill T. cruzi epimastigote and promastigotes at micromolar concentrations. | [151] |
| Human African Trypanosomiasis (Trypanosoma brucei) | Temporin-SHd | Pelophylax saharica (Sahara frog) | Growth inhibitory activity against T. brucei and T. cruzi | [155] | |
| BMAP-27 | Bos Taurus (Cattle) | Inhibited both life cycles of both T. brucei at low micromolar concentrations | [157] | ||
| Leishmaniasis (L. donovani, L. major, L. tropica, L. Mexicana and L. panamensis) | Temporin A and B | Rana temporaria (European common frog) | Active against insect and mammalian stages at 15-25µM concentrations. Rapid disruption of plasma membrane potential and decrease in intracellular ATP levels | [160] | |
| Dermaseptin S4 | Phyllomedusa sauvagii, (South America Sauvages leaf frog) | Potent lysis for L. major promastigotes. | [51] | ||
| Dermasiptin-01 | Phyllomedusa oreades (tree frog) | Active against L. infantum promastigotes by membrane damage and flagella alterations | [164] | ||
| Gomesin | Acanthoscurria gomesiana (Tarantula spider) | Decreased the viability of L. amazonensis promastigotes with little activity against human erythrocytes | [166] | ||
| Indolicidin | Bovine leukocytes | Membrane effects and able to induce autophagy in L. donovani | [167] | ||
| Parasites | Leishmaniasis (L. donovani, L. major, L. tropica, L. Mexicana and L. panamensis) | Thionins | Triticum aestivum (Wheat) | HNP1–3 can bind to Onchocerca volvulus surfaces and is known to mediate the macrophage response against other micro-organisms by the release of IFN-γ and TNF-α | [200] |
| Helminths | Onchocerciasis (River blindness) (Onchocerca volvulus) | HNP1-3 | Homo sapiens (Human) | Proved effective against L. donovani promastigotes in the low micromolar range, membrane permeability | [168] |
| Galectin Peroxidoxin-2 | Onchocerca ochengi (Black fly) | Excretory/secretory products of Onchocerca volvulus with significant antibacterial activity against E. coli | [11] | ||
| ALT-1 (all peptides are AMP precursors) | Onchocerca volvulus (Roundworm) | The peptides Galectin, Peroxidoxin-2 and ALT-1 are all AMP precursors | |||
| Parasites | Dengue viral disease (family Flaviviridae) | Latarcin | Lachesana tarabaevi (Ant spider) | Able to inhibit the replication of dengue viruses in in vitro cultured cells | [232,232] |
| Rabies (Lyssavirus) | C2, C6, C8 and P16 | Synthetic | Synthetic AMPs based on naturally according peptides C2, C6, C8 and P16 peptides exhibited strong rabies virus inhibitory properties | [252] |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- WHO. Neglected Tropical Diseases. Available online: http://www.who.int/neglected_diseases/diseases/en/ (accessed on 18 August 2015).
- Hotez, P.J.; Molyneuz, D.H.; Fenwick, A.; Ottesen, E.; Ehrlich Sachs, S.; Sachs, J.D. Incorporating a rapid-impact package for Neglected tropical diseases with programs for HIV/AIDS, tuberculosis, maleria. PLoS Med. 2006, 3. [Google Scholar] [CrossRef] [PubMed]
- Feasey, N.; Wansbrough-Jones, M.; Mabey, D.C.; Solomon, A.W. Neglected tropical diseases. Br. Med. Bull. 2010, 93, 179–200. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.L. Antimicrobial peptides stage a comeback. Nat. Biotechnol. 2013, 31, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Lehrer, R. Cationic peptides: A new source of antibiotics. Trends Biotechnol. 1998, 16, 82–88. [Google Scholar] [CrossRef]
- Wagner, D.; Young, L.S. Nontuberculous mycobacterial infections: A clinical review. Infection 2004, 32, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Gordon, Y.J.; Romanowski, E.G.; McDermott, A.M. A review of antimicrobial peptides and their therapeutic potential as anti-infective drugs. Curr. Res. 2005, 30, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.; Salin, V.; Williams, G.W. Nisin and the Market for Commercial Bacteriocins. TAMRC Consumer and Product Research Report No. CP-01-05. 2005. http://wwww.ageconsearch.umn.edu/bitstream/90779/2/CP%2001%2005%20Nisin%20Report.pdf (accessed on 18 August 2015).
- Jenssen, H.; Hamill, P.; Hancock, R.E. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Eberle, R.; Brattig, N.W.; Trusch, M.; Schlüter, H.; Achukwi, M.D.; Eisenbarth, A.; Renz, A.; Liebau, E.; Perbandt, M.; Betzel, C. Isolation, identification and functional profile of excretory-secretory peptides from Onchocerca ochengi. Acta Tropica 2015, 142, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. The Antimicrobial Peptide Database. Available online: http://wwww.aps.unmc.edu/AP/main.php (accesed on 22 June 2015).
- Wang, G.; Li, X.; Wang, Z. APD2: The updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 2009, 37, D933–D937. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, G. APD: The Antimicrobial Peptide Database. Nucleic Acids Res. 2004, 32, D590–D592. [Google Scholar] [CrossRef] [PubMed]
- Papagianni, M. Ribosomally synthesized peptides with antimicrobial properties: Biosynthesis, structure, function, and applications. Biotechnol. Adv. 2003, 21, 465–499. [Google Scholar] [CrossRef]
- Felnagle, E.A.; Jackson, E.E.; Chan, Y.A.; Podevels, A.M.; Berti, A.D.; McMahon, M.D.; Thomas, M.G. Nonribosomal peptide synthetases involved in the production of medically relevant natural products. Mol. Pharm. 2008, 5, 191–211. [Google Scholar] [CrossRef] [PubMed]
- Niggemann, J.; Bozko, P.; Bruns, N.; Wodtke, A.; Gieseler, M.T.; Thomas, K.; Jahns, C.; Nimtz, M.; Reupke, I.; Bruser, T.; et al. Baceridin, a cyclic hexapeptide from an epiphytic Bacillus strain, inhibits the proteasome. Chem. Biol. Chem. 2014, 15, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Mishra, B.; Lau, K.; Lushnikova, T.; Golla, R.; Wang, X. Antimicrobial peptides in 2014. Pharmaceuticals 2015, 8, 123–150. [Google Scholar] [CrossRef] [PubMed]
- Midorikawa, K.; Ouhara, K.; Komatsuzawa, H.; Kawai, T.; Yamada, S.; Fujiwara, T.; Yamazaki, K.; Sayama, K.; Taubman, M.A.; Kurihara, H.; et al. Staphylococcus aureus susceptibility to innate antimicrobial peptides, beta-defensins and CAP18, expressed by human keratinocytes. Infect. Immun. 2003, 71, 3730–3739. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Diamond, G. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol. 2000, 8, 402–410. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.; Thomsen, A.R. Sensing of RNA viruses: A review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 2012, 86, 2900–2910. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Fornek, J.; Crochet, N.; Bajwa, G.; Perwitasari, O.; Martinez-Sobrido, L.; Akira, S.; Gill, M.A.; Garcia-Sastre, A.; Katze, M.G.; et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 2008, 82, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Cotter, P.D.; Hill, C.; Ross, R.P. Bacteriocins: Developing innate immunity for food. Nat. Rev. Microbiol. 2005, 3, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta 2009, 1788, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.; Shukla, S.K.; Prakash, O.; Zhang, G. Structural determinants of host defense peptides for antimicrobial activity and target cell selectivity. Biochimie 2010, 92, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Harris, F.; Dennison, S.R.; Phoenix, D.A. Anionic antimicrobial peptides from eukaryotic organisms. Curr. Protein Pept. Sci. 2009, 10, 585–606. [Google Scholar] [CrossRef] [PubMed]
- Laverty, G.; Gorman, S.P.; Gilmore, B.F. The potential of antimicrobial peptides as biocides. Int. J. Mol. Sci. 2011, 12, 6566–6596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brogden, K.A.; Ackermann, M.; McCray, P.B., Jr.; Tack, B.F. Antimicrobial peptides in animals and their role in host defences. Int. J. Antimicrob. Agents 2003, 22, 465–478. [Google Scholar] [CrossRef]
- Huang, Y.; Huang, J.; Chen, Y. Alpha-helical cationic antimicrobial peptides: Relationships of structure and function. Protein Cell 2010, 1, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Boman, H.G. Gene-encoded peptide antibiotics and the concept of innate immunity: An update review. Scand. J. Immunol. 1998, 48, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Buchan, D.W.A.; Minneci, F.; Nugent, T.C.O.; Bryson, K.; Jones, D.T. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 2013, 41, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Vale, N.; Aguiar, L.; Gomes, P. Antimicrobial peptides: A new class of antimalarial drugs? Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, K.; Suketa, Y.; Hayashi, K.; Suzuki, T. Chemical structure of circulin A. Experientia 1965, 21, 307–308. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Daly, N.L.; Bond, T.; Waine, C. Plant cyclotides: A unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif. J. Mol. Biol. 1999, 294, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I.; Cole, A.M.; Selsted, M.E. Theta-Defensins: Cyclic peptides with endless potential. J. Biol. Chem. 2012, 287, 27014–27019. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, A.; Molhoek, E.M.; Bikker, F.J.; Yu, P.L.; Veldhuizen, E.J.; Haagsman, H.P. Avian cathelicidins: Paradigms for the development of anti-infectives. Vet. Microbiol. 2011, 153, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Zhou, C.; Zhang, M.; Han, Z.; Shao, Y.; Liu, S. Functional analysis and induction of four novel goose (Anser cygnoides) avian beta-defensins in response to salmonella enteritidis infection. Comp. Immunol. Microbiol. Infecti. Dis. 2012, 35, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Scocchi, M.; Pallavicini, A.; Salgaro, R.; Bociek, K.; Gennaro, R. The salmonid cathelicidins: A gene family with highly varied C-terminal antimicrobial domains. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2009, 152, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Casadei, E.; Wang, T.; Zou, J.; Gonzalez Vecino, J.L.; Wadsworth, S.; Secombes, C.J. Characterization of three novel beta-defensin antimicrobial peptides in rainbow trout (Oncorhynchus mykiss). Mol. Immunol. 2009, 46, 3358–3366. [Google Scholar] [CrossRef] [PubMed]
- Alibardi, L. Immunocytochemical detection of beta-defensins and cathelicidins in the secretory granules of the tongue in the lizard Anolis carolinensis. Acta Histochem. 2015, 117, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Gan, T.X.; Liu, X.D.; Jin, Y.; Lee, W.H.; Shen, J.H.; Zhang, Y. Identification and characterization of novel reptile cathelicidins from elapid snakes. Peptides 2008, 29, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.F.; Lee, T.R.; Tsai, P.H.; Hsu, M.P.; Chen, C.S.; Lyu, P.C. Structure-based protein engineering for alpha-amylase inhibitory activity of plant defensin. Proteins 2007, 68, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Cornet, B.; Bonmatin, J.M.; Hetru, C.; Hoffmann, J.A.; Ptak, M.; Vovelle, F. Refined three-dimensional solution structure of insect defensin A. Structure 1995, 3, 435–448. [Google Scholar] [CrossRef]
- Ramanathan, B.; Davis, E.G.; Ross, C.R.; Blecha, F. Cathelicidins: Microbicidal activity, mechanisms of action, and roles in innate immunity. Microbes Infect. 2002, 4, 361–372. [Google Scholar] [CrossRef]
- Ganz, T. Defensins: Antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 2003, 3, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Carmona, M.; Hubert, P.; Delvenne, P.; Herfs, M. Defensins: “Simple” antimicrobial peptides or broad-spectrum molecules? Cytokine Growth Factor Rev. 2015, 26, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, D.; Landuyt, B.; Luyten, W.; Schoofs, L. A comprehensive summary of LL-37, the factotum human cathelicidin peptide. Cellular Immunol. 2012, 280, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, M.; Gennaro, R.; Romeo, D. Cathelicidins: A novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett. 1995, 374, 1–5. [Google Scholar] [CrossRef]
- Feder, R.; Dagan, A.; Mor, A. Structure-activity relationship study of antimicrobial dermaseptin S4 showing the consequences of peptide oligomerization on selective cytotoxicity. J. Biol. Chem. 2000, 275, 4230–4238. [Google Scholar] [CrossRef] [PubMed]
- Unger, T.; Oren, Z.; Shai, Y. The effect of cyclization of magainin 2 and melittin analogues on structure, function, and model membrane interactions: Implication to their mode of action. Biochemistry 2001, 40, 6388–6397. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; Pulido, D.; Rivas, L.; Andreu, D. Antimicrobial peptide action on parasites. Curr. Drug Targets 2012, 13, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Lee, D.G. Fungicidal effect of antimicrobial peptide arenicin-1. Biochim. Biophys. Acta 2009, 1788, 1790–1796. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.E.; O’Brien, L.M.; Thwaite, J.E.; Fox, M.A.; Atkins, H.; Ulaeto, D.O. A carpet-based mechanism for direct antimicrobial peptide activity against vaccinia virus membranes. Peptides 2010, 31, 1966–1972. [Google Scholar] [CrossRef] [PubMed]
- Klotman, M.E.; Chang, T.L. Defensins in innate antiviral immunity. Nat. Rev. Immunol. 2006, 6, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.Q.; Mukhopadhyay, K.; Yeaman, M.R.; Adler-Moore, J.; Bayer, A.S. Functional interrelationships between cell membrane and cell wall in antimicrobial peptide-mediated killing of Staphylococcus aureus. Antimicrob. Agents Chemother. 2005, 49, 3114–3121. [Google Scholar] [CrossRef] [PubMed]
- Gee, M.L.; Burton, M.; Grevis-James, A.; Hossain, M.A.; McArthur, S.; Palombo, E.A.; Wade, J.D.; Clayton, A.H. Imaging the action of antimicrobial peptides on living bacterial cells. Sci. Rep. 2013, 3, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.P.; Hancock, R.E. The relationship between peptide structure and antibacterial activity. Peptides 2003, 24, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Baumann, G.; Mueller, P. A molecular model of membrane excitability. J. Supramol. Struct. 1974, 2, 538–557. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Harroun, T.A.; Weiss, T.M.; Ding, L.; Huang, H.W. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys. J. 2001, 81, 1475–1485. [Google Scholar] [CrossRef]
- Carnicelli, V.; Lizzi, A.R.; Ponzi, A.; Amicosante, G.; Bozzi, A.; di Giulio, A. Interaction between antimicrobial peptides (AMPs) and their primary target, the biomembranes. In Microbial Pathogens and Strategies for Combating Them: Science, Technology and Education; Méndez-Vilas, A., Ed.; Formatex Research Center: Badajoz, Spain, 2013; Volume 2, pp. 1123–1134. [Google Scholar]
- Matsuzaki, K.; Sugishita, K.; Harada, M.; Fujii, N.; Miyajima, K. Interactions of an antimicrobial peptide, magainin 2, with outer and inner membranes of Gram-negative bacteria. Biochim. Biophys. Acta 1997, 1327, 119–130. [Google Scholar] [CrossRef]
- Pandey, B.K.; Srivastava, S.; Singh, M.; Ghosh, J.K. Inducing toxicity by introducing a leucine-zipper-like motif in frog antimicrobial peptide, magainin 2. Biochem. J. 2011, 436, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.P.; Bayer, A.S.; Yeaman, M.R. Diversity in antistaphylococcal mechanisms among membrane-targeting antimicrobial peptides. Infect. Immun. 2001, 69, 4916–4922. [Google Scholar] [CrossRef] [PubMed]
- Podda, E.; Benincasa, M.; Pacor, S.; Micali, F.; Mattiuzzo, M.; Gennaro, R.; Scocchi, M. Dual mode of action of Bac7, a proline-rich antibacterial peptide. Biochim. Biophys. Acta 2006, 1760, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, P. Multifunctional host defense peptides: Intracellular-targeting antimicrobial peptides. FEBS J. 2009, 276, 6483–6496. [Google Scholar] [CrossRef] [PubMed]
- Otvos, L., Jr. Antibacterial peptides and proteins with multiple cellular targets. J. Pept. Sci. An Off. Publ. Europ. Pept. Soc. 2005, 11, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Bowdish, D.M.; Davidson, D.J.; Hancock, R.E. A re-evaluation of the role of host defence peptides in mammalian immunity. Curr. Protein Pept. Sci. 2005, 6, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Ding, J.; Li, H.; Li, L.; Zhao, R.; Shen, Z.; Fan, X.; Xi, T. Effects of cations and pH on antimicrobial activity of thanatin and s-thanatin against Escherichia coli ATCC25922 and B. subtilis ATCC 21332. Curr. Microbiol. 2008, 57, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.; Gudmundsson, G.H.; Rottenberg, M.E.; Berndt, K.D.; Agerberth, B. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J. Biol. Chem. 1998, 273, 3718–2374. [Google Scholar] [CrossRef] [PubMed]
- Niyonsaba, F.; Hirata, M.; Ogawa, H.; Nagaoka, I. Epithelial cell-derived antibacterial peptides human beta-defensins and cathelicidin: Multifunctional activities on mast cells. Curr. Drug Targets Inflamm. Allergy 2003, 2, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, J.J.; Biragyn, A.; Kwak, L.W.; Yang, D. Roles of antimicrobial peptides such as defensins in innate and adaptive immunity. Annals Rheum. Dis. 2003, 62, ii17–ii21. [Google Scholar] [CrossRef]
- Scott, M.G.; Davidson, D.J.; Gold, M.R.; Bowdish, D.; Hancock, R.E. The human antimicrobial peptide LL-37 is a multifunctional modulator of innate immune responses. J. Immunol. 2002, 169, 3883–3891. [Google Scholar] [CrossRef] [PubMed]
- Tjabringa, G.S.; Aarbiou, J.; Ninaber, D.K.; Drijfhout, J.W.; Sorensen, O.E.; Borregaard, N.; Rabe, K.F.; Hiemstra, P.S. The antimicrobial peptide LL-37 activates innate immunity at the airway epithelial surface by transactivation of the epidermal growth factor receptor. J. Immunol. 2003, 171, 6690–6696. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Biragyn, A.; Hoover, D.M.; Lubkowski, J.; Oppenheim, J.J. Multiple roles of antimicrobial defensins, cathelicidins, and eosinophil-derived neurotoxin in host defense. Annu. Rev. Immunol. 2004, 22, 181–215. [Google Scholar] [CrossRef] [PubMed]
- Gough, M.; Hancock, R.E.; Kelly, N.M. Antiendotoxin activity of cationic peptide antimicrobial agents. Infect. Immun. 1996, 64, 4922–4927. [Google Scholar] [PubMed]
- Bucki, R.; Pastore, J.J.; Randhawa, P.; Vegners, R.; Weiner, D.J.; Janmey, P.A. Antibacterial activities of rhodamine B-conjugated gelsolin-derived peptides compared to those of the antimicrobial peptides cathelicidin LL37, magainin II, and melittin. Antimicrob. Agents Chemother. 2004, 48, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Marr, A.K.; Gooderham, W.J.; Hancock, R.E. Antibacterial peptides for therapeutic use: Obstacles and realistic outlook. Curr. Opin. Pharmacol. 2006, 6, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Chen, S.; Li, S.; Yan, H.; Fan, Y.; Mi, H. Deletion of two C-terminal Gln residues of 12-26-residue fragment of melittin improves its antimicrobial activity. Peptides 2005, 26, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Blondelle, S.E.; Houghten, R.A. Hemolytic and antimicrobial activities of the twenty-four individual omission analogues of melittin. Biochemistry 1991, 30, 4671–4678. [Google Scholar] [CrossRef] [PubMed]
- Asthana, N.; Yadav, S.P.; Ghosh, J.K. Dissection of antibacterial and toxic activity of melittin: A leucine zipper motif plays a crucial role in determining its hemolytic activity but not antibacterial activity. J. Biol. Chem. 2004, 279, 55042–55050. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Wang, Q.; Zheng, Z.; Zhao, L.; Shang, Y.; Wei, X.; Liao, X.; Zhang, R. Design, characterization and expression of a novel hybrid peptides melittin (1-13)-LL37 (17-30). Mol. Biol. Rep. 2014, 41, 4163–4169. [Google Scholar] [CrossRef] [PubMed]
- Brogden, N.K.; Brogden, K.A. Will new generations of modified antimicrobial peptides improve their potential as pharmaceuticals? Int. J. Antimicrob. Agents 2011, 38, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Finlay, B.B.; Hancock, R.E. Can innate immunity be enhanced to treat microbial infections? Nat. Rev. Microbiol. 2004, 2, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Marais, E.; Hamman, J.; Plessis, L.; Lemmer, R.; Steenekamp, J. Eudragit(R) L100/N-trimethylchitosan chloride microspheres for oral insulin delivery. Molecules 2013, 18, 6734–6747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarmento, B.; Ribeiro, A.; Veiga, F.; Ferreira, D.; Neufeld, R. Oral bioavailability of insulin contained in polysaccharide nanoparticles. Biomacromolecules 2007, 8, 3054–3060. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Jin, H.; Qian, Y.; Qi, S.; Luo, H.; Luo, Q.; Zhang, Z. Hybrid melittin cytolytic Peptide-driven ultrasmall lipid nanoparticles block melanoma growth in vivo. ACS Nano 2013, 7, 5791–5800. [Google Scholar] [CrossRef] [PubMed]
- Piras, A.M.; Maisetta, G.; Sandreschi, S.; Gazzarri, M.; Bartoli, C.; Grassi, L.; Esin, S.; Chiellini, F.; Batoni, G. Chitosan nanoparticles loaded with the antimicrobial peptide temporin B exert a long-term antibacterial activity in vitro against clinical isolates of Staphylococcus epidermidis. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Mataraci, E.; Dosler, S. In vitro activities of antibiotics and antimicrobial cationic peptides alone and in combination against methicillin-resistant Staphylococcus aureus biofilms. Antimicrob. Agents Chemother. 2012, 56, 6366–6371. [Google Scholar] [CrossRef] [PubMed]
- Engels, D.; Savioli, L. Reconsidering the underestimated burden caused by Neglected tropical diseases. Trends Parasitol. 2006, 22, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, M.; Malhotra, I.; Mungai, P.L.; Wamachi, A.N.; Kioko, J.M.; Ouma, J.H.; Muchiri, E.; King, C.L. The effects of maternal helminth and malaria infections on mother-to-child HIV transmission. Aids 2005, 19, 1849–1855. [Google Scholar] [CrossRef] [PubMed]
- Hotez, P.J.; Molyneux, D.H.; Fenwick, A.; Kumaresan, J.; Sachs, S.E.; Sachs, J.D.; Savioli, L. Control of Neglected tropical diseases. N. Engl. J. Med. 2007, 357, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Hotez, P.J.; Fenwick, A.; Savioli, L.; Molyneux, D.H. Rescuing the bottom billion through control of Neglected tropical diseases. Lancet 2009, 373, 1570–1575. [Google Scholar] [CrossRef]
- Norris, J.; Adelman, C.; Spantchak, Y.; Marano, K. Social and Economic Impact Review on Neglected Tropical Diseases; Hudson Institute’s Center for Science in Public Policy: Washington, DC, USA, 2012; pp. 1–26. [Google Scholar]
- WHO. Antimicrobial Resistance: Fact sheet N° 194. Available online: http://www.who.int/mediacentre/factsheets/fs194/en/ (accessed on 18 August 2015).
- Zucca, M.; Savoia, D. The post-antibiotic era: Promising developments in the therapy of infectious diseases. Int. J. Biomed. Sci.: IJBS 2010, 6, 77–86. [Google Scholar] [PubMed]
- Barrow, W.W. Processing of mycobacterial lipids and effects on host responsiveness. Front. Biosci. A J. Virtual Lib. 1997, 2, d387–d400. [Google Scholar]
- Barrow, W.W. Treatment of mycobacterial infections. Rev. Scientifique Tech. 2001, 20, 55–70. [Google Scholar]
- Mendez-Samperio, P. The human cathelicidin hCAP18/LL-37: A multifunctional peptide involved in mycobacterial infections. Peptides 2010, 31, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.; Cho, Y.; Dinh, N.N.; Waring, A.J.; Lehrer, R.I. Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob. Agents Chemother. 1998, 42, 2206–2214. [Google Scholar] [PubMed]
- Gilliet, M.; Lande, R. Antimicrobial peptides and self-DNA in autoimmune skin inflammation. Curr. Opin. Immunol. 2008, 20, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Monot, M.; Honore, N.; Garnier, T.; Araoz, R.; Coppee, J.Y.; Lacroix, C.; Sow, S.; Spencer, J.S.; Truman, R.W.; Williams, D.L.; et al. On the origin of leprosy. Science 2005, 308, 1040–1042. [Google Scholar] [CrossRef] [PubMed]
- WHO Leprosy Fact Sheet N° 101. Available online: http://www.who.int/mediacentre/factsheets/fs101/en/ (accessed on 18 August 2015).
- Cogen, A.L.; Walker, S.L.; Roberts, C.H.; Hagge, D.A.; Neupane, K.D.; Khadge, S.; Lockwood, D.N. Human beta-defensin 3 is up-regulated in cutaneous leprosy type 1 reactions. PLoS Negl. Trop. Dis. 2012, 6, e1869. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.H.; Parker, D.L.; Robson, M. Leprosy: Steps along the journey of eradication. Public Health Rep. 2008, 123, 198–205. [Google Scholar] [PubMed]
- Cole, S.T.; Eiglmeier, K.; Parkhill, J.; James, K.D.; Thomson, N.R.; Wheeler, P.R.; Honore, N.; Garnier, T.; Churcher, C.; Harris, D.; et al. Massive gene decay in the leprosy bacillus. Nature 2001, 409, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., 3rd; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Irgens, L.M. The discovery of the leprosy bacillus. Tidsskr. Norske Laegeforening Tidsskr. Prakt. Med. Ny Raekke 2002, 122, 708–709. [Google Scholar]
- De Messias-Reason, I.; Kremsner, P.G.; Kun, J.F. Functional haplotypes that produce normal ficolin-2 levels protect against clinical leprosy. J. Infect. Dis. 2009, 199, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Rees, R.J.; Pearson, J.M.; Waters, M.F. Experimental and clinical studies on rifampicin in treatment of leprosy. Br. Med. J. 1970, 1, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, R.O.; de Souza Salles, J.; Sarno, E.N.; Sampaio, E.P. Mycobacterium leprae-host-cell interactions and genetic determinants in leprosy: An overview. Future Microbiol. 2011, 6, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Bochud, P.Y.; Hawn, T.R.; Siddiqui, M.R.; Saunderson, P.; Britton, S.; Abraham, I.; Argaw, A.T.; Janer, M.; Zhao, L.P.; Kaplan, G.; et al. Toll-like receptor 2 (TLR2) polymorphisms are associated with reversal reaction in leprosy. J. Infect. Dis. 2008, 197, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.T.; Wheelwright, M.; Teles, R.; Komisopoulou, E.; Edfeldt, K.; Ferguson, B.; Mehta, M.D.; Vazirnia, A.; Rea, T.H.; Sarno, E.N.; et al. MicroRNA-21 targets the vitamin D-dependent antimicrobial pathway in leprosy. Nat. Med. 2012, 18, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.E.; Wessling-Resnick, M. Iron metabolism and the innate immune response to infection. Microbes Infect. 2012, 14, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rojas, A.; Rodriguez-Beltran, J.; Couce, A.; Blazquez, J. Antibiotics and antibiotic resistance: A bitter fight against evolution. Int. J. Med. Microbiol. 2013, 303, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Bechtle, M.; Chen, S.; Efferth, T. Neglected diseases caused by bacterial infections. Curr. Med. Chem. 2010, 17, 42–60. [Google Scholar] [CrossRef] [PubMed]
- WHO. Trachoma: Fact Sheet N° 382. Available online: http://www.who.int/mediacentre/factsheets/fs382/en/ (accessed on 18 August 2015).
- Courtright, P.; West, S.K. Contribution of sex-linked biology and gender roles to disparities with trachoma. Emerg. Infect. Dis. 2004, 10, 2012–2016. [Google Scholar] [CrossRef] [PubMed]
- Solomon, A.W.; Peeling, R.W.; Foster, A.; Mabey, D.C. Diagnosis and assessment of trachoma. Clin. Microbiol.Rev. 2004, 17, 982–1011. [Google Scholar] [CrossRef] [PubMed]
- Mabey, D.C.; Solomon, A.W.; Foster, A. Trachoma. Lancet 2003, 362, 223–239. [Google Scholar] [CrossRef]
- Burton, M.J. Trachoma: An overview. Br. Med. Bull. 2007, 84, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Potroz, M.G.; Cho, N.J. Natural products for the treatment of trachoma and Chlamydia trachomatis. Molecules 2015, 20, 4180–4203. [Google Scholar] [CrossRef] [PubMed]
- WHO. Report of the 2nd Global Scientific Meeting on Trachoma. http://www.who.int/blindness/2nd%20GLOBAL%20SCIENTIFIC%20MEETING.pdf (accessed on 18 August 2015).
- Solomon, A.W.; Holland, M.J.; Alexander, N.D.; Massae, P.A.; Aguirre, A.; Natividad-Sancho, A.; Molina, S.; Safari, S.; Shao, J.F.; Courtright, P.; et al. Mass treatment with single-dose azithromycin for trachoma. N. Engl. J. Med. 2004, 351, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.R.; Solomon, A.W. Antibiotics for Trachoma. Cochrane Database Syst. Rev. 2011. [Google Scholar] [CrossRef]
- Donati, M.; di Leo, K.; Benincasa, M.; Cavrini, F.; Accardo, S.; Moroni, A.; Gennaro, R.; Cevenini, R. Activity of cathelicidin peptides against Chlamydia spp. Antimicrob. Agents Chemother. 2005, 49, 1201–1202. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.M.; Liu, C.Q. Cathelicidin peptide SMAP-29: Comprehensive review of its properties and potential as a novel class of antibiotics. Drug Dev. Res. 2009, 70, 481–498. [Google Scholar] [CrossRef]
- Huang, G.T.; Zhang, H.B.; Kim, D.; Liu, L.; Ganz, T. A model for antimicrobial gene therapy: Demonstration of human beta-defensin 2 antimicrobial activities in vivo. Hum. Gene Ther. 2002, 13, 2017–2025. [Google Scholar] [CrossRef] [PubMed]
- Lazarev, V.N.; Parfenova, T.M.; Gularyan, S.K.; Misyurina, O.Y.; Akopian, T.A.; Govorun, V.M. Induced expression of melittin, an antimicrobial peptide, inhibits infection by Chlamydia trachomatis and Mycoplasma hominis in a HeLa cell line. Int. J. Antimicrob. Agents 2002, 19, 133–137. [Google Scholar] [CrossRef]
- Lazarev, V.N.; Shkarupeta, M.M.; Polina, N.F.; Kostrjukova, E.S.; Vassilevski, A.A.; Kozlov, S.A.; Grishin, E.V.; Govorun, V.M. Antimicrobial peptide from spider venom inhibits Chlamydia trachomatis infection at an early stage. Arch. Microbiol. 2013, 195, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Lazarev, V.N.; Shkarupeta, M.M.; Titova, G.A.; Kostrjukova, E.S.; Akopian, T.A.; Govorun, V.M. Effect of induced expression of an antimicrobial peptide melittin on Chlamydia trachomatis and Mycoplasma hominis infections in vivo. Biochem. Biophys. Res. Commun. 2005, 338, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Cox, F.E.G. Parasitic protozoa. In Modern Parasitology: A Textbook of Parasitology, 2nd ed.; Cox, F.E.G., Ed.; Blackwell Science Ltd: Oxford, UK, 1996; p. 276. [Google Scholar]
- Spicer, J.W. Clinical Bacteriology, Mycology and Parasitology; Harcourt Publishers Ltd: London, UK, 2000; p. 221. [Google Scholar]
- Andrews, K.T.; Fisher, G.; Skinner-Adams, T.S. Drug repurposing and human parasitic protozoan diseases. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 95–111. [Google Scholar] [CrossRef] [PubMed]
- McGwire, B.S.; Kulkarni, M.M. Interactions of antimicrobial peptides with Leishmania and trypanosomes and their functional role in host parasitism. Exp. Parasitol. 2010, 126, 397–405. [Google Scholar] [CrossRef] [PubMed]
- WHO. Chagas Disease (American Trypanosomiasis). Fact Sheet No 340 March 2015. http://www.who.int/chagas/en/ (accessed on 18 August 2015).
- Bern, C.; Montgomery, S.P.; Herwaldt, B.L.; Rassi, A., Jr.; Marin-Neto, J.A.; Dantas, R.O.; Maguire, J.H.; Acquatella, H.; Morillo, C.; Kirchhoff, L.V.; et al. Evaluation and treatment of chagas disease in the United States: A systematic review. JAMA 2007, 298, 2171–2181. [Google Scholar] [CrossRef] [PubMed]
- Soy, D.; Aldasoro, E.; Guerrero, L.; Posada, E.; Serret, N.; Mejia, T.; Urbina, J.A.; Gascon, J. Population pharmacokinetics of benznidazole in adult patients with chagas disease. Antimicrob. Agents Chemother. 2015, 59, 3342–3349. [Google Scholar] [CrossRef] [PubMed]
- Urbina, J.A. Recent clinical trials for the etiological treatment of chronic chagas disease: Advances, challenges and perspectives. J. Eukaryot. Microbiol. 2015, 62, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Adade, C.M.; Chagas, G.S.; Souto-Padron, T. Apis mellifera venom induces different cell death pathways in Trypanosoma cruzi. Parasitology 2012, 139, 1444–1461. [Google Scholar] [CrossRef] [PubMed]
- Adade, C.M.; Oliveira, I.R.; Pais, J.A.; Souto-Padron, T. Melittin peptide kills Trypanosoma cruzi parasites by inducing different cell death pathways. Toxicon 2013, 69, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Fieck, A.; Hurwitz, I.; Kang, A.S.; Durvasula, R. Trypanosoma cruzi: Synergistic cytotoxicity of multiple amphipathic anti-microbial peptides to T. cruzi and potential bacterial hosts. Exp. Parasitol. 2010, 125, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.M.; Chen, H.C.; Zierdt, C.H. Magainin analogs effective against pathogenic protozoa. Antimicrob. Agents Chemother. 1990, 34, 1824–1826. [Google Scholar] [CrossRef] [PubMed]
- Jaynes, J.M.; Burton, C.A.; Barr, S.B.; Jeffers, G.W.; Julian, G.R.; White, K.L.; Enright, F.M.; Klei, T.R.; Laine, R.A. In vitro cytocidal effect of novel lytic peptides on Plasmodium falciparum and Trypanosoma cruzi. FASEB J. 1988, 2, 2878–2883. [Google Scholar] [PubMed]
- Brand, G.D.; Leite, J.R.; Silva, L.P.; Albuquerque, S.; Prates, M.V.; Azevedo, R.B.; Carregaro, V.; Silva, J.S.; Sa, V.C.; Brandao, R.A.; et al. Dermaseptins from Phyllomedusa oreades and Phyllomedusa distincta. Anti-Trypanosoma cruzi activity without cytotoxicity to mammalian cells. J. Biol. Chem. 2002, 277, 49332–49340. [Google Scholar] [CrossRef] [PubMed]
- Pinto, E.G.; Pimenta, D.C.; Antoniazzi, M.M.; Jared, C.; Tempone, A.G. Antimicrobial peptides isolated from Phyllomedusa nordestina (Amphibia) alter the permeability of plasma membrane of Leishmania and Trypanosoma cruzi. Exp. Parasitol. 2013, 135, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Lofgren, S.E.; Miletti, L.C.; Steindel, M.; Bachere, E.; Barracco, M.A. Trypanocidal and leishmanicidal activities of different antimicrobial peptides (AMPs) isolated from aquatic animals. Exp. Parasitol. 2008, 118, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.P.; Boykin, D.W.; Brun, R.; Tidwell, R.R. Human African trypanosomiasis: Pharmacololgical re-engagement with a neglected disease. Br. J. Pharmacol. 2007, 152, 1155–1171. [Google Scholar] [CrossRef] [PubMed]
- WHO. Trypanosomiasis, Human African (Sleeping Sickness). Fact Sheet No. 259 May 2015. http://www.who.int/mediacentre/factsheets/fs259/en/ (accessed on 18 August 2015).
- Brun, R.; Blum, J.; Chappuis, F.; Burri, C. Human African trypanosomiasis. Lancet 2010, 375, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Abbassi, F.; Raja, Z.; Oury, B.; Gazanion, E.; Piesse, C.; Sereno, D.; Nicolas, P.; Foulon, T.; Ladram, A. Antibacterial and leishmanicidal activities of temporin-SHd, a 17-residue long membrane-damaging peptide. Biochimie 2013, 95, 388–399. [Google Scholar] [CrossRef] [PubMed]
- McGwire, B.S.; Olson, C.L.; Tack, B.F.; Engman, D.M. Killing of African trypanosomes by antimicrobial peptides. J. Infect. Dis. 2003, 188, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Haines, L.R.; Thomas, J.M.; Jackson, A.M.; Eyford, B.A.; Razavi, M.; Watson, C.N.; Gowen, B.; Hancock, R.E.; Pearson, T.W. Killing of trypanosomatid parasites by a modified bovine host defense peptide, BMAP-18. PLoS Negl. Trop. Dis. 2009, 3, e373. [Google Scholar] [CrossRef] [PubMed]
- WHO. Leishmaniasis Fact Sheet No. 375. February 2015. http://www.who.int/mediacentre/factsheets/fs375/en/ (accessed on 18 August 2015).
- De Menezes, J.P.; Guedes, C.E.; Petersen, A.L.; Fraga, D.B.; Veras, P.S. Advances in Development of New Treatment for Leishmaniasis. Biomed. Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.L.; Saugar, J.M.; Dellisanti, M.; Barra, D.; Simmaco, M.; Rivas, L. Temporins, small antimicrobial peptides with leishmanicidal activity. J. Biol. Chem. 2005, 280, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Chadbourne, F.L.; Raleigh, C.; Ali, H.Z.; Denny, P.W.; Cobb, S.L. Studies on the antileishmanial properties of the antimicrobial peptides temporin A, B and 1Sa. J. Pept. Sci. An Off. Publ. Eur. Pept. Soc. 2011, 17, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Abbassi, F.; Oury, B.; Blasco, T.; Sereno, D.; Bolbach, G.; Nicolas, P.; Hani, K.; Amiche, M.; Ladram, A. Isolation, characterization and molecular cloning of new temporins from the skin of the North African ranid Pelophylax saharica. Peptides 2008, 29, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Eggimann, G.A.; Sweeney, K.; Bolt, H.L.; Rozatian, N.; Cobb, S.L.; Denny, P.W. The role of phosphoglycans in the susceptibility of Leishmania mexicana to the temporin family of anti-microbial peptides. Molecules 2015, 20, 2775–2785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, P.; Bittencourt, C.R.; Costa Silva, V.; Veras, L.M.; Costa, C.H.; Feio, M.J.; Leite, J.R. Anti-leishmanial activity of the antimicrobial peptide DRS 01 observed in Leishmania infantum (syn. Leishmania chagasi) cells. Nanomedicine 2014, 10, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Brand, G.D.; Leite, J.R.; de Sa Mandel, S.M.; Mesquita, D.A.; Silva, L.P.; Prates, M.V.; Barbosa, E.A.; Vinecky, F.; Martins, G.R.; Galasso, J.H.; et al. Novel dermaseptins from Phyllomedusa hypochondrialis (Amphibia). Biochem. Biophys. Res. Commun. 2006, 347, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.I., Jr.; Daffre, S.; Bulet, P. Isolation and characterization of gomesin, an 18-residue cysteine-rich defense peptide from the spider Acanthoscurria gomesiana hemocytes with sequence similarities to horseshoe crab antimicrobial peptides of the tachyplesin family. J. Biol. Chem. 2000, 275, 33464–33470. [Google Scholar] [CrossRef] [PubMed]
- Bera, A.; Singh, S.; Nagaraj, R.; Vaidya, T. Induction of autophagic cell death in Leishmania donovani by antimicrobial peptides. Mol. Biochem. Parasitol. 2003, 127, 23–35. [Google Scholar] [CrossRef]
- Berrocal-Lobo, M.; Molina, A.; Rodriguez-Palenzuela, P.; Garcia-Olmedo, F.; Rivas, L. Leishmania donovani: Thionins, plant antimicrobial peptides with leishmanicidal activity. Exp. Parasitol. 2009, 122, 247–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Cordero, J.J.; Lozano, J.M.; Cortes, J.; Delgado, G. Leishmanicidal activity of synthetic antimicrobial peptides in an infection model with human dendritic cells. Peptides 2011, 32, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, M.M.; McMaster, W.R.; Kamysz, E.; Kamysz, W.; Engman, D.M.; McGwire, B.S. The major surface-metalloprotease of the parasitic protozoan, Leishmania, protects against antimicrobial peptide-induced apoptotic killing. Mol. Microbiol. 2006, 62, 1484–1497. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, M.M.; McMaster, W.R.; Kamysz, W.; McGwire, B.S. Antimicrobial peptide-induced apoptotic death of leishmania results from calcium-dependent, caspase-independent mitochondrial toxicity. J. Biol. Chem. 2009, 284, 15496–15504. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, M.M.; Karafova, A.; Kamysz, W.; McGwire, B.S. Design of protease-resistant pexiganan enhances antileishmanial activity. Parasitol. Res. 2014, 113, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- WHO. Malaria. Fact Sheet No. 94 April 2015. http://www.who.int/mediacentre/factsheets/fs094/en/ (accessed on 18 August 2015).
- WHO. Guidelines for the Treatment of Malaria. http://apps.who.int/iris/bitstream/10665/162441/1/9789241549127_eng.pdf (accessed on 18 August 2015).
- Du Plessis, L.H.; van Niekerk, A.C.; Maritz, M.M.; Kotze, A.F. In vitro activity of Pheroid vesicles containing antibiotics against Plasmodium falciparum. J. Antibiot. 2012, 65, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Krugliak, M.; Feder, R.; Zolotarev, V.Y.; Gaidukov, L.; Dagan, A.; Ginsburg, H.; Mor, A. Antimalarial activities of dermaseptin S4 derivatives. Antimicrob. Agents Chemother. 2000, 44, 2442–2451. [Google Scholar] [CrossRef] [PubMed]
- Carter, V.; Underhill, A.; Baber, I.; Sylla, L.; Baby, M.; Larget-Thiery, I.; Zettor, A.; Bourgouin, C.; Langel, U.; Faye, I.; et al. Killer bee molecules: Antimicrobial peptides as effector molecules to target sporogonic stages of Plasmodium. PLoS Pathog. 2013, 9, e1003790. [Google Scholar] [CrossRef] [PubMed]
- Moreira, C.K.; Rodrigues, F.G.; Ghosh, A.; Varotti Fde, P.; Miranda, A.; Daffre, S.; Jacobs-Lorena, M.; Moreira, L.A. Effect of the antimicrobial peptide gomesin against different life stages of Plasmodium spp. Exp. Parasitol. 2007, 116, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Parker, G.A.; Ball, M.A.; Chubb, J.C. Why do larval helminths avoid the gut of intermediate hosts? J. Theor. Biol. 2009, 260, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Halton, D.W. Microscopy and the helminth parasite. Micron 2004, 35, 361–390. [Google Scholar] [CrossRef] [PubMed]
- Hotez, P.J.; Brindley, P.J.; Bethony, J.M.; King, C.H.; Pearce, E.J.; Jacobson, J. Helminth infections: The great Neglected tropical diseases. J. Clin. Investig. 2008, 118, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.; Bundy, D.A.; Savioli, L. Helminthic infections. Br. Med. J. 2003, 327, 431–433. [Google Scholar] [CrossRef] [PubMed]
- Crompton, D.W.T. How Much Human Helminthiasis Is There in the World? J. Parasitol. 1999, 85, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, L.S.; Latham, M.C.; Ottesen, E.A. Malnutrition and parasitic helminth infections. Parasitology 2000, 121, S23–S38. [Google Scholar] [CrossRef] [PubMed]
- WHO. World Heatlh Report 1999; World Health Organization: Geneva, Switzerland, 1999. [Google Scholar]
- Anthony, R.M.; Rutitzky, L.I.; Urban, J.F., Jr.; Stadecker, M.J.; Gause, W.C. Protective immune mechanisms in helminth infection. Nat. Rev. Immunol. 2007, 7, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Ito, A.; Chen, X.; Long, C.; Okamoto, M.; Raoul, F.; Giraudoux, P.; Yanagida, T.; Nakao, M.; Sako, Y.; et al. Usefulness of pumpkin seeds combined with areca nut extract in community-based treatment of human taeniasis in northwest Sichuan Province, China. Acta Trop. 2012, 124, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Flisser, A.; Avila, G.; Maravilla, P.; Mendlovic, F.; Leon-Cabrera, S.; Cruz-Rivera, M.; Garza, A.; Gomez, B.; Aguilar, L.; Teran, N.; et al. Taenia solium: Current understanding of laboratory animal models of taeniosis. Parasitology 2010, 137, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Bustos, J.A.; Rodriguez, S.; Jimenez, J.A.; Moyano, L.M.; Castillo, Y.; Ayvar, V.; Allan, J.C.; Craig, P.S.; Gonzalez, A.E.; Gilman, R.H.; et al. Detection of Taenia solium Taeniasis Coproantigen Is an Early Indicator of Treatment Failure for Taeniasis. Clin. Vaccine Immunol. 2012, 19, 570–573. [Google Scholar] [CrossRef] [PubMed]
- WHO Taeniasis/ cysticercosis: Fact Sheet N° 376. Available online: http://www.who.int/mediacentre/factsheets/fs376/en/# (accessed on 23 June 2015).
- García, H.H.; Gonzalez, A.E.; Evans, C.A.W.; Gilman, R.H.; for the Cysticercosis Working Group in Peru. Taenia solium cysticercosis. Lancet 2003, 362, 547–556. [Google Scholar] [CrossRef]
- Kościuczuk, E.M.; Lisowski, P.; Jarczak, J.; Strzałkowska, N.; Jóźwik, A.; Horbańczuk, J.; Krzyżewski, J.; Zwierzchowski, L.; Bagnicka, E. Cathelicidins: Family of antimicrobial peptides. A review. Mol. Biol. Rep. 2012, 39, 10957–10970. [Google Scholar] [CrossRef] [PubMed]
- Landa, A.; Jiménez, L.; Willms, K.; Jiménez-García, L.F.; Lara-Martínez, R.; Robert, L.; Cirioni, O.; Barańska–Rybak, W.; Kamysz, W. Antimicrobial peptides (Temporin A and Iseganan IB-367): Effect on the cysticerci of Taenia crassiceps. Mol. Biochem. Parasitol. 2009, 164, 126–130. [Google Scholar] [CrossRef] [PubMed]
- WHO Onchocerciasis: Fact Sheet N° 374. Available online: http://www.who.int/mediacentre/factsheets/fs374/en/# (accessed on 23 June 2015).
- CDC Parasites—Onchocerciasis (also known as River Blindness). Available online: http://www.cdc.gov/parasites/onchocerciasis/ (accessed on 23 June 2015).
- Noma, M.; Nwoke, B.E.; Nutall, I.; Tambala, P.A.; Enyong, P.; Namsenmo, A.; Remme, J.; Amazigo, U.V.; Kale, O.O.; Seketeli, A. Rapid epidemiological mapping of onchocerciasis (REMO): Its application by the African Programme for Onchocerciasis Control (APOC). Ann. Trop. Med. Parasitol. 2002, 96, S29–S39. [Google Scholar] [CrossRef] [PubMed]
- Tekle, A.H.; Elhassan, E.; Isiyaku, S.; Amazigo, U.V.; Bush, S.; Noma, M.; Cousens, S.; Abiose, A.; Remme, J. H. Impact of long-term treatment of onchocerciasis with ivermectin in Kaduna State, Nigeria: First evidence of the potential for elimination in the operational area of the African Programme for Onchocerciasis Control. Parasites Vectors 2012, 5. [Google Scholar] [CrossRef] [PubMed]
- Marti, T.; Erttmann, K.D.; Gallin, M.Y. Host-Parasite Interaction in Human Onchocerciasis: Identification and Sequence Analysis of a Novel Human Calgranulin. Biochem. Biophys. Res. Commun. 1996, 221, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Selsted, M.E.; Szklarek, D.; Harwig, S.S.; Daher, K.; Bainton, D.F.; Lehrer, R.I. Defensins. Natural peptide antibiotics of human neutrophils. J. Clin. Investig. 1985, 76, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Kai-Larsen, Y.; Frithiof, R.; Sorensen, O.E.; Kenne, E.; Scharffetter-Kochanek, K.; Eriksson, E.E.; Herwald, H.; Agerberth, B.; Lindbom, L. Neutrophil primary granule proteins HBP and HNP1-3 boost bacterial phagocytosis by human and murine macrophages. J. Clin. Investig. 2008, 118, 3491–3502. [Google Scholar] [CrossRef] [PubMed]
- Mookherjee, N.; Hancock, R.E. Cationic host defence peptides: Innate immune regulatory peptides as a novel approach for treating infections. Cell. Mol. Life Sci. 2007, 64, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Gwyer Findlay, E.; Currie, S.M.; Davidson, D.J. Cationic host defence peptides: Potential as antiviral therapeutics. BioDrugs 2013, 27, 479–493. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Antiviral drugs: Current state of the art. J. Clin. Virol. 2001, 22, 73–89. [Google Scholar] [CrossRef]
- Albiol Matanic, V.C.; Castilla, V. Antiviral activity of antimicrobial cationic peptides against Junin virus and herpes simplex virus. Int. J. Antimicrob. Agents 2004, 23, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Orsi, N. The antimicrobial activity of lactoferrin: Current status and perspectives. Biometals 2004, 17, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Finnegan, C.M.; Kish-Catalone, T.; Blumenthal, R.; Garzino-Demo, P.; la Terra Maggiore, G.M.; Berrone, S.; Kleinman, C.; Wu, Z.; Abdelwahab, S.; et al. Human beta-defensins suppress human immunodeficiency virus infection: Potential role in mucosal protection. J. Virol. 2005, 79, 14318–14329. [Google Scholar] [CrossRef] [PubMed]
- van der Strate, B.W.; Beljaars, L.; Molema, G.; Harmsen, M.C.; Meijer, D.K. Antiviral activities of lactoferrin. Antivir. Res. 2001, 52, 225–239. [Google Scholar] [CrossRef]
- Yasin, B.; Pang, M.; Turner, J.S.; Cho, Y.; Dinh, N.N.; Waring, A.J.; Lehrer, R.I.; Wagar, E.A. Evaluation of the inactivation of infectious Herpes simplex virus by host-defense peptides. Eur. J. Clin. Microbiol. Infect. Dis. 2000, 19, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Daher, K.A.; Selsted, M.E.; Lehrer, R.I. Direct inactivation of viruses by human granulocyte defensins. J. Virol. 1986, 60, 1068–1074. [Google Scholar] [PubMed]
- Duits, L.A.; Nibbering, P.H.; van Strijen, E.; Vos, J.B.; Mannesse-Lazeroms, S.P.; van Sterkenburg, M.A.; Hiemstra, P.S. Rhinovirus increases human beta-defensin-2 and -3 mRNA expression in cultured bronchial epithelial cells. FEMS Immunol. Med. Microbiol. 2003, 38, 59–64. [Google Scholar] [CrossRef]
- Buck, C.B.; Day, P.M.; Thompson, C.D.; Lubkowski, J.; Lu, W.; Lowy, D.R.; Schiller, J.T. Human alpha-defensins block papillomavirus infection. Proc. Natl. Acad. Sci. USA 2006, 103, 1516–1521. [Google Scholar] [CrossRef] [PubMed]
- Carriel-Gomes, M.C.; Kratz, J.M.; Barracco, M.A.; Bachere, E.; Barardi, C.R.; Simoes, C.M. In vitro antiviral activity of antimicrobial peptides against herpes simplex virus 1, adenovirus, and rotavirus. Mem. Inst. Oswaldo Cruz 2007, 102, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Edinger, A.L.; Thompson, C.B. Death by design: Apoptosis, necrosis and autophagy. Curr. Opin. Cell Biol. 2004, 16, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Marie, I.J.; Durbin, J.E. Induction and function of type I and III interferon in response to viral infection. Curr. Opin. Cell Biol. 2011, 1, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Stetson, D.B.; Medzhitov, R. Type I interferons in host defense. Immunity 2006, 25, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Randall, R.E.; Goodbourn, S. Interferons and viruses: An interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008, 89. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, M.; Garcia-Sastre, A.; Ruchala, P.; Lehrer, R.I.; Chang, T.; Klotman, M.E. Alpha-Defensin inhibits influenza virus replication by cell-mediated mechanism(s). J. Infect. Dis. 2007, 196, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Koczulla, A.R.; Bals, R. Antimicrobial peptides: Current status and therapeutic potential. Drugs 2003, 63, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Beatty, M.E.; Stone, A.; Fitzsimons, D.W.; Hanna, J.N.; Lam, S.K.; Vong, S.; Guzman, M.G.; Mendez-Galvan, J.F.; Halstead, S.B.; Letson, G.W.; et al. Americas Dengue Prevention Boards Surveillance Working, G., Best practices in dengue surveillance: A report from the Asia-Pacific and Americas Dengue Prevention Boards. PLoS Negl.Trop. Dis. 2010, 4, e890. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.B. Dengue virus-mosquito interactions. Ann. Rev. Entomol. 2008, 53, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.B. Pathogenesis of dengue: Challenges to molecular biology. Science 1988, 239, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Normile, D. Tropical medicine. Surprising new dengue virus throws a spanner in disease control efforts. Science 2013, 342. [Google Scholar] [CrossRef] [PubMed]
- Vasilakis, N.; Holmes, E.C.; Fokam, E.B.; Faye, O.; Diallo, M.; Sall, A.A.; Weaver, S.C. Evolutionary processes among sylvatic dengue type 2 viruses. J. Virol. 2007, 81, 9591–9595. [Google Scholar] [CrossRef] [PubMed]
- Rodenhuis-Zybert, I.A.; Wilschut, J.; Smit, J.M. Dengue virus life cycle: Viral and host factors modulating infectivity. Cell. Mol. Life Sci. 2010, 67, 2773–2786. [Google Scholar] [CrossRef] [PubMed]
- Steinhauer, D.A.; Domingo, E.; Holland, J.J. Lack of evidence for proofreading mechanisms associated with an RNA virus polymerase. Gene 1992, 122, 281–288. [Google Scholar] [CrossRef]
- Wittke, V.; Robb, T.E.; Thu, H.M.; Nisalak, A.; Nimmannitya, S.; Kalayanrooj, S.; Vaughn, D.W.; Endy, T.P.; Holmes, E.C.; Aaskov, J.G. Extinction and rapid emergence of strains of dengue 3 virus during an interepidemic period. Virology 2002, 301, 148–156. [Google Scholar] [CrossRef] [PubMed]
- CDC. Dengue and the Aedes aegypti Mosquito. http://www.cdc.gov/dengue/resources/30Jan2012/aegyptifactsheet.pdf (accessed on 21 August 2015).
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Shepard, D.S.; Undurraga, E.A.; Halasa, Y.A. Economic and disease burden of dengue in Southeast Asia. PLoS Negl.Trop. Dis. 2013, 7, e2055. [Google Scholar] [CrossRef] [PubMed]
- Rothman, A.L. Immunity to dengue virus: A tale of original antigenic sin and tropical cytokine storms. Nat. Rev. Immunol. 2011, 11, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.A.; Vassilevski, A.A.; Feofanov, A.V.; Surovoy, A.Y.; Karpunin, D.V.; Grishin, E.V. Latarcins, antimicrobial and cytolytic peptides from the venom of the spider Lachesana tarabaevi (Zodariidae) that exemplify biomolecular diversity. J. Biol. Chem. 2006, 281, 20983–20992. [Google Scholar] [CrossRef] [PubMed]
- Rothan, H.A.; Bahrani, H.; Rahman, N.A.; Yusof, R. Identification of natural antimicrobial agents to treat dengue infection: In vitro analysis of latarcin peptide activity against dengue virus. BMC Microbiol. 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M. Molecular biology of flaviviruses. Adv. Virus Res. 2003, 59, 23–61. [Google Scholar] [PubMed]
- Perera, R.; Kuhn, R.J. Structural proteomics of dengue virus. Curr. Opin. Microbiol. 2008, 11, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Noble, C.G.; Seh, C.C.; Chao, A.T.; Shi, P.Y. Ligand-bound structures of the dengue virus protease reveal the active conformation. J. Virol. 2012, 86, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Alhoot, M.A.; Rathinam, A.K.; Wang, S.M.; Manikam, R.; Sekaran, S.D. Inhibition of dengue virus entry into target cells using synthetic antiviral peptides. Int. J. Med. Sci. 2013, 10, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Maguire, T.; Marks, R.M. Demonstration of binding of dengue virus envelope protein to target cells. J. Virol. 1996, 70, 8765–8772. [Google Scholar] [PubMed]
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S.C. Structure of the dengue virus envelope protein after membrane fusion. Nature 2004, 427, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Wallace, R.M.; Gilbert, A.; Slate, D.; Chipman, R.; Singh, A.; Cassie, W.; Blanton, J.D. Right place, wrong species: A 20-year review of rabies virus cross species transmission among terrestrial mammals in the United States. PLoS ONE 2014, 9, e107539. [Google Scholar] [CrossRef] [PubMed]
- Cleaveland, S.; Fevre, E.M.; Kaare, M.; Coleman, P.G. Estimating human rabies mortality in the United Republic of Tanzania from dog bite injuries. Bull. World Health Organ. 2002, 80, 304–310. [Google Scholar] [PubMed]
- Schnell, M.J.; McGettigan, J.P.; Wirblich, C.; Papaneri, A. The cell biology of rabies virus: Using stealth to reach the brain. Nat. Rev. Microbiol. 2010, 8, 51–61. [Google Scholar] [CrossRef] [PubMed]
- WHO. Rabies Fact Sheet N° 99. Available online: http://www.who.int/mediacentre/factsheets/fs099/en/ (accessed on 18 August 2015).
- Lyles, D.S.; Rupprecht, C.E. Rhabdoviridae. Fields’ Virology; Fields, B.N., Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Roizman, B., Straus, S.E., Eds.; Williams and Wilkins: Philadelphia, PA, USA, 2007; Volume 4, pp. 1364–1408. [Google Scholar]
- Brzozka, K.; Finke, S.; Conzelmann, K.K. Identification of the rabies virus alpha/beta interferon antagonist: Phosphoprotein P interferes with phosphorylation of interferon regulatory factor 3. J. Virol. 2005, 79, 7673–7681. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.M.; Strick, P.L. Rabies as a transneuronal tracer of circuits in the central nervous system. J. Neurosci. Methods 2000, 103, 63–71. [Google Scholar] [CrossRef]
- Warrell, D.A. The clinical picture of rabies in man. Trans. R. Soc. Trop. Med. Hyg. 1976, 70, 188–195. [Google Scholar] [CrossRef]
- Manning, S.E.; Rupprecht, C.E.; Fishbein, D.; Hanlon, C.A.; Lumlertdacha, B.; Guerra, M.; Meltzer, M.I.; Dhankhar, P.; Vaidya, S.A.; Jenkins, S.R.; et al. Advisory Committee on Immunization Practices Centers for Disease, C.; Prevention, Human rabies prevention—United States, 2008: Recommendations of the Advisory Committee on Immunization Practices. MMWR Recomm. Rep. 2008, 57, 1–28. [Google Scholar] [PubMed]
- Cenna, J.; Tan, G.S.; Papaneri, A.B.; Dietzschold, B.; Schnell, M.J.; McGettigan, J.P. Immune modulating effect by a phosphoprotein-deleted rabies virus vaccine vector expressing two copies of the rabies virus glycoprotein gene. Vaccine 2008, 26, 6405–6414. [Google Scholar] [CrossRef] [PubMed]
- Lodmell, D.L.; Parnell, M.J.; Bailey, J.R.; Ewalt, L.C.; Hanlon, C.A. Rabies DNA vaccination of non-human primates: Post-exposure studies using gene gun methodology that accelerates induction of neutralizing antibody and enhances neutralizing antibody titers. Vaccine 2002, 20, 2221–2228. [Google Scholar] [CrossRef]
- Dodet, B.; Africa Rabies Expert, B.; Adjogoua, E.V.; Aguemon, A.R.; Amadou, O.H.; Atipo, A.L.; Baba, B.A.; Ada, S.B.; Boumandouki, P.; Bourhy, H.; et al. Fighting rabies in Africa: The Africa Rabies Expert Bureau (AfroREB). Vaccine 2008, 26, 6295–6298. [Google Scholar] [CrossRef] [PubMed]
- Real, E.; Rain, J.C.; Battaglia, V.; Jallet, C.; Perrin, P.; Tordo, N.; Chrisment, P.; D’Alayer, J.; Legrain, P.; Jacob, Y. Antiviral drug discovery strategy using combinatorial libraries of structurally constrained peptides. J. Virol. 2004, 78, 7410–7417. [Google Scholar] [CrossRef] [PubMed]
- A Database of Antiviral Peptides. Available online: http://crdd.osdd.net/servers/avpdb/browse.php?by=Rabies%20virus&TYPE=Virus (accessed on 21 August 2015).
- Jacob, Y.; Real, E.; Tordo, N. Functional interaction map of lyssavirus phosphoprotein: Identification of the minimal transcription domains. J. Virol. 2001, 75, 9613–9622. [Google Scholar] [CrossRef] [PubMed]
- Wojczyk, B.S.; Stwora-Wojczyk, M.; Shakin-Eshleman, S.; Wunner, W.H.; Spitalnik, S.L. The role of site-specific N-glycosylation in secretion of soluble forms of rabies virus glycoprotein. Glycobiology 1998, 8, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Saint-Jean, S.; De las Heras, A.; Carrillo, W.; Recio, I.; Ortiz-Delgado, J.B.; Ramos, M.; Gomez-Ruiz, J.A.; Sarasquete, C.; Perez-Prieto, S.I. Antiviral activity of casein and alphas2 casein hydrolysates against the infectious haematopoietic necrosis virus, a rhabdovirus from salmonid fish. J. Fish Dis. 2013, 36, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Falco, A.; Mas, V.; Tafalla, C.; Perez, L.; Coll, J.M.; Estepa, A. Dual antiviral activity of human alpha-defensin-1 against viral haemorrhagic septicaemia rhabdovirus (VHSV): Inactivation of virus particles and induction of a type I interferon-related response. Antivir. Res. 2007, 76, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.P.; Yoshimatsu, K.; Suzuki, T.; Kato, T.; Park, E.Y. Expression and purification of cyto-insectotoxin (Cit1a) using silkworm larvae targeting for an antimicrobial therapeutic agent. Appl. Microbiol. Biotechnol. 2014, 98, 6973–6982. [Google Scholar] [CrossRef] [PubMed]
- Bommarius, B.; Jenssen, H.; Elliott, M.; Kindrachuk, J.; Pasupuleti, M.; Gieren, H.; Jaeger, K.E.; Hancock, R.E.; Kalman, D. Cost-effective expression and purification of antimicrobial and host defense peptides in Escherichia coli. Peptides 2010, 31, 1957–1965. [Google Scholar] [CrossRef] [PubMed]
- Li, Y. Recombinant production of antimicrobial peptides in Escherichia coli: A review. Protein Expr. Purif. 2011, 80, 260–267. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewies, A.; Wentzel, J.F.; Jacobs, G.; Du Plessis, L.H. The Potential Use of Natural and Structural Analogues of Antimicrobial Peptides in the Fight against Neglected Tropical Diseases. Molecules 2015, 20, 15392-15433. https://doi.org/10.3390/molecules200815392
Lewies A, Wentzel JF, Jacobs G, Du Plessis LH. The Potential Use of Natural and Structural Analogues of Antimicrobial Peptides in the Fight against Neglected Tropical Diseases. Molecules. 2015; 20(8):15392-15433. https://doi.org/10.3390/molecules200815392
Chicago/Turabian StyleLewies, Angélique, Johannes Frederik Wentzel, Garmi Jacobs, and Lissinda Hester Du Plessis. 2015. "The Potential Use of Natural and Structural Analogues of Antimicrobial Peptides in the Fight against Neglected Tropical Diseases" Molecules 20, no. 8: 15392-15433. https://doi.org/10.3390/molecules200815392