
Studies on Cytotoxic Activity against HepG-2 Cells of Naphthoquinones from Green Walnut Husks of Juglans mandshurica Maxim

Abstract
: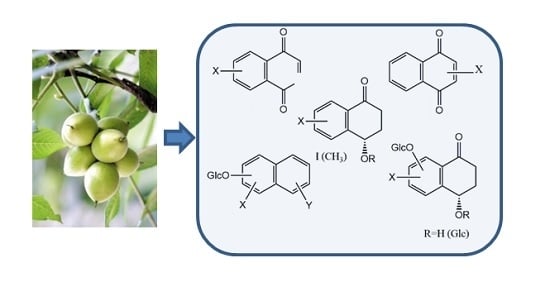
1. Introduction
2. Results and Discussion
2.1. Isolation and Characterization of Compounds 18, 25–27



{kind=link}
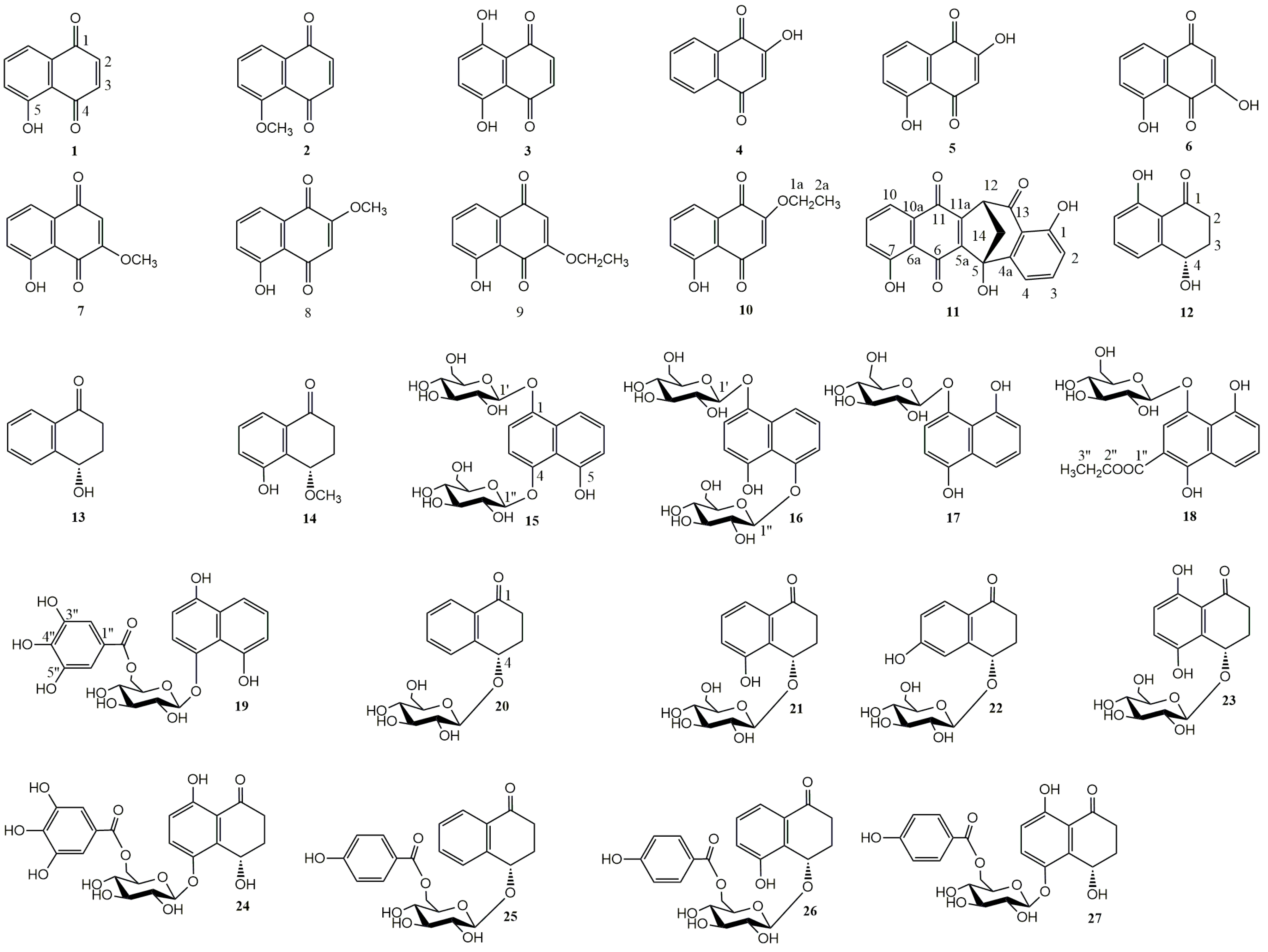{kind=link}
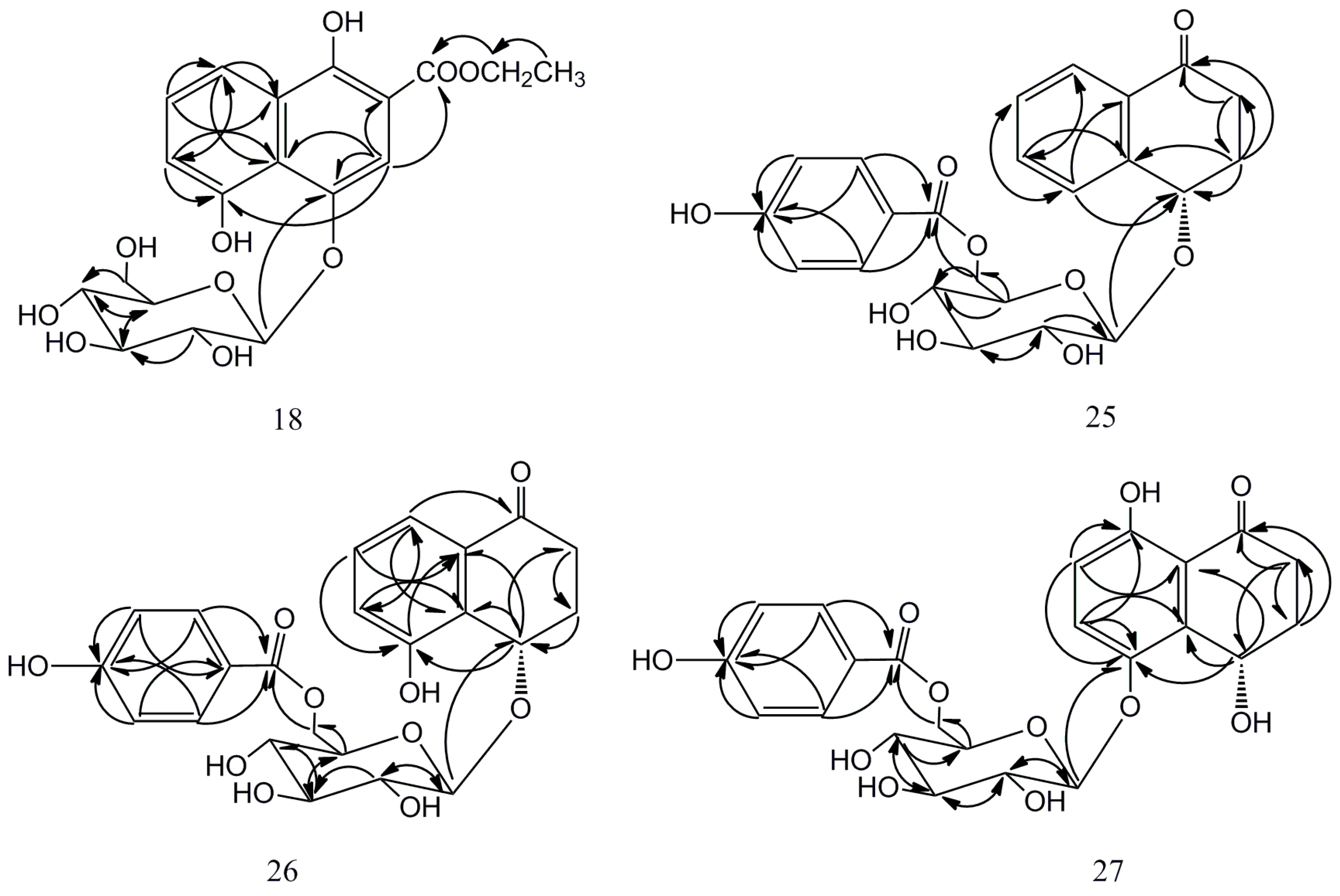{kind=link}
| No. | 18 | 25 | 26 | 27 | ||||
|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | — | 148.0 | — | 200.0 | — | 200.9 | — | 206.4 |
| 2 | 7.72, s | 109.9 | Hax: 2.87, ddd (4.5, 8.9, 17.5) | 35.5 | Hax: 3.03, ddd (5.0, 13.4, 17.0) | 33.9 | Hax: 3.01, ddd (5.9,12.9, 17.6) | 33.5 |
| Heq: 2.41, ddd (4.5, 6.5, 17.5) | Heq: 2.37, dt (3.6, 17.0) | Heq: 2.44, dt (3.6, 17.6) | ||||||
| 3 | — | 105.8 | Hax: 2.34, dddd (2.2, 4.5, 8.9, 13.4) | 31.5 | Hax: 2.48, dddd (1.3, 3.2, 4.7, 12.6) | 30.0 | 2.16, m | 30.3 |
| Heq: 2.28, dddd (3.8, 4.5, 6.5, 13.4) | Heq: 2.10, tt (4.2, 12.6) | |||||||
| 4 | — | 155.1 | 4.97, dd (3.6, 6.5) | 75.9 | 5.37, t (2.9) | 69.9 | 5.32, t (3.1) | 61.3 |
| 5 | 6.99, dd (1.0, 7.8) | 116.0 | 7.65, brd (7.6) | 130.0 | — | 157.0 | — | 148.3 |
| 6 | 7.40, t (7.8) | 128.6 | 7.52, dt (1.2, 7.6) | 134.8 | 7.08, dd (0.8, 8.0) | 122.3 | 7.40, d (9.1) | 128.9 |
| 7 | 7.86, dd (1.0, 7.8) | 116.1 | 7.43, dt (1.2, 7.6) | 129.4 | 7.27, t (8.0) | 130.6 | 6.67, d (9.1) | 118.9 |
| 8 | — | 158.0 | 7.93, dd (1.2, 7.6) | 127.9 | 7.46, dd (0.8, 8.0) | 118.9 | — | 159.3 |
| 9 | — | 120.2 | — | 132.9 | — | 134.4 | — | 116.2 |
| 10 | — | 131.1 | — | 143.9 | — | 129.6 | — | 135.5 |
| 1′ | 4.99, d (7.6) | 105.5 | 4.42, d (7.6) | 103.7 | 4.60, d (7.8) | 103.8 | 4.81, d (7.5) | 104.4 |
| 2′ | 3.50, m | 78.9 | 3.34, m | 75.2 | 3.2, m | 75.3 | 3.53, dd (8.8, 16.5) | 75.2 |
| 3′ | 3.53, m | 75.0 | 3.36, dd (2.5, 7.0) | 78.1 | 3.38, m | 78.0 | 3.49, t (8.6) | 78.0 |
| 4′ | 3.43, m | 71.3 | 3.36, dd (2.5, 7.0) | 72.2 | 3.38, m | 72.1 | 3.43, dd (10.6, 16.2) | 72.0 |
| 5′ | 3.48, m | 78.2 | 3.62, m | 75.5 | 3.65, m | 75.7 | 3.68, dt (2.2, 8.4) | 75.8 |
| 6′a | 3.96, dd (2.1, 12.0) | 62.5 | 4.66, dd (2.2, 11.8) | 65.0 | 4.64, dd (2.2, 11.8) | 65.0 | 4.61, dd (2.2, 11.8) | 64.8 |
| 6′b | 3.77, dd (5.5, 12.0) | 4.47, dd (7.2, 11.8) | 4.43, dd (6.8, 11.8) | 4.40, dd (7.4, 11.8) | ||||
| 1′′ | — | 171.8 | — | 122.2 | — | 121.9 | — | 122.1 |
| 2′′ | 4.44, dq (3.6, 17.7) | 62.8 | 7.95, d (8.8) | 132.9 | 7.95, dt (2.6, 8.8) | 133.0 | 7.82, dt (2.7, 8.8) | 132.9 |
| 3′′ | 1.43, t (7.2) | 14.5 | 6.84, d (8.8) | 116.3 | 6.83, dt (2.6, 8.8) | 116.2 | 6.81, dt (2.7, 8.8) | 116.2 |
| 4′′ | — | — | — | 163.7 | — | 163.6 | — | 163.7 |
| 5′′ | — | — | 6.84, d (8.8) | 116.3 | 6.83, dt (2.6, 8.8) | 116.2 | 6.81, dt (2.7, 8.8) | 116.2 |
| 6′′ | — | — | 7.95, d (8.8) | 132.9 | 7.95, dt (2.6, 8.8) | 133.0 | 7.82, dt (2.7, 8.8) | 132.9 |
| 7′′ | — | — | — | 167.9 | — | 168.1 | — | 167.8 |
2.2. Cytotoxic Activity
| Compd. | Structural Features | IC50 (μM) a | SD b | Compd. | Structural Features | IC50 (μM) a | SD b |
|---|---|---|---|---|---|---|---|
| 1 |  | 8.14 | 1.95 | 15 |  | NA | 3.21 |
| 2 | 68.72 | 1.50 | 16 | NA | - | ||
| 3 | 16.11 | 3.54 | 17 | 83.32 | 4.54 | ||
| 4 | 18.83 | 2.98 | 18 | NA | - | ||
| 5 | 15.37 | 1.63 | 19 | 78.61 | 2.38 | ||
| 6 |  | 7.33 | 0.52 | 20 |  | NA | - |
| 7 | 43.54 | 0.15 | 21 | NA | - | ||
| 8 | 22.38 | 0.66 | 22 | NA | - | ||
| 9 | 30.42 | 2.48 | 23 | NA | - | ||
| 10 | 32.51 | 0.46 | 24 | NA | - | ||
| 11 | 34.80 | 0.33 | 25 | NA | - | ||
| 12 |  | 56.87 | 4.27 | 26 | NA | - | |
| 13 | 67.95 | 3.22 | 27 | NA | - | ||
| 14 | 88.23 | 1.90 | PC c | metal complex | 4.51 | 0.38 |
3. Experimental Section
3.1. General Information
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Spectral Data
3.5. Acid Hydrolysis and Sugar Analysis
3.6. Cytotoxicity Assays
3.6.1. Cell Culture
3.6.2. Measurement of Cell Proliferation by MTT Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lu, S.; Tian, J.; Sun, W.; Meng, J.; Wang, X.; Fu, X.; Wang, A.; Lai, D.; Liu, Y.; Zhou, L. Bis-naphtho-gamma-pyrones from fungi and their bioactivities. Molecules 2014, 19, 7169–7188. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.L.; Zhang, C.H.; Luo, J.; Jin, M.; Zheng, M.S.; Cui, J.M.; Son, J.K.; Li, G. Chemical constituents from the leaves of Juglans mandshurica. Arch. Pharm. Res. 2015, 38, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Zhang, Y.W.; Hua, Y.; Bao, Y.L.; Wu, Y.; Sun, L.G.; Yu, C.L.; Huang, Y.X.; Wang, E.B.; Jiang, H.Y.; et al. Three new compounds from the stem bark of Juglans mandshurica. J. Asian Nat. Prod. Res. 2014, 16, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lee, K.S.; Son, J.K.; Je, G.H.; Lee, J.S.; Lee, C.H.; Cheng, C.J. Cytotoxic Compounds from the Roots of Juglans mandshurica. J. Nat. Prod. 1998, 61, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Bai, W.N.; Liao, W.J.; Zhang, D.Y. Nuclear and chloroplast DNA phylogeography reveal two refuge areas with asymmetrical gene flow in a temperate walnut tree from East Asia. New Phytol. 2010, 188, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Cui, Y.Q.; Zhu, J.Y.; Li, H.Z.; Mao, J.W.; Jin, X.B.; Wang, X.S.; Du, Y.F.; Lu, J.Z. The anti-tumor effect and biological activities of the extract JMM6 from the stem-barks of the Chinese Juglans mandshurica Maxim on human hepatoma cell line bel-7402. Afr. J. Tradit. Complement. Altern. Med. 2013, 10, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Park, G.; Oh, M.S. Inhibitory effects of Juglans mandshurica leaf on allergic dermatitis-like skin lesions-induced by 2, 4-dinitrochlorobenzene in mice. Exp. Toxicol. Pathol. 2014, 66, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Park, G.H.; Jang, D.S.; Oh, M.S. Juglans mandshurica leaf extract protects skin fibroblasts from damage by regulating the oxidative defense system. Biochem. Biophys. Res. Commun. 2012, 421, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Negi, A.S.; Luqman, S.; Srivastava, S.; Krishna, V.; Gupta, N.; Darokar, M.P. Antiproliferative and antioxidant activities of Juglans regia fruit extracts. Pharm. Biol. 2011, 49, 669–673. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy Doherty, M.; Rodgers, A.; Cohen, G.M. Mechanisms of toxicity of 2-and 5-hydroxy-1,4-naphthoquinone; absence of a role for redox cycling in the toxicity of 2-hydroxy-1,4-naphthoquinone to isolated hepatocytes. J. Appl. Toxicol. 1987, 7, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.L.; Yu, X.F.; Qu, S.C.; Sui, D.Y. Juglone, isolated from Juglans mandshurica Maxim, induces apoptosis via down-regulation of AR expression in human prostate cancer LNCaP cells. Bioorg. Med. Chem. Lett. 2013, 23, 3631–3634. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Matsuoka, E.; Kasahara, T.; Kikuchi, M. Studies on the constituents of Juglans Species. I. structural determination of (4S)- and (4R)-4-hydroxy-α-tetralone derivatives from the fruit of Juglans mandshurica Maxim var. sieboldiana MAKINO. Chem. Pharm. Bull. 2005, 53, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Pi, X.M.; Yu, C.Y. A new naphthalenone isolated from the green walnut husks of Juglans mandshurica Maxim. Nat. Prod. Res. 2015, 29, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sun, J.X.; Yu, H.Y.; Chen, Z.Y.; Zhao, X.Y.; Ruan, H.L. Diarylheptanoids from the root bark of Juglans cathayensis. Chin. Chem. Lett. 2013, 24, 521–523. [Google Scholar] [CrossRef]
- Yang, H.J.; Cho, H.J.; Sim, S.H.; Chung, Y.K.; Kim, D.D.; Sung, S.H.; Kim, J.; Kim, Y.C. Cytotoxic terpenoids from Juglans sinensis leaves and twigs. Bioorg. Med. Chem. Lett. 2012, 22, 2079–2083. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Yogiashi, Y.; Matsuda, S.; Suzuki, A.; Kikuchi, M. A new phenolic glycoside syringate from the bark of Juglans mandshurica Maxim var. sieboldiana MAKINO. J. Nat. Med. 2009, 63, 220–222. [Google Scholar] [CrossRef] [PubMed]
- Wellington, K.W. Understanding cancer and the anticancer activities of naphthoquinones—A review. RSC Adv. 2015, 5, 20309–20338. [Google Scholar] [CrossRef]
- Liu, L.J.; Li, W.; Kazuo, K.; Zhang, S.J.; Tamotsu, N. New α-Tetralonyl Glucosides from the Fruit of Juglans mandshurica. Chem. Pharm. Bull. 2004, 52, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Min, B.S.; Nakamura, N.; Miyashiro, H.; Kim, Y.H.; Hattori, M. Inhibition of human immunodeficiency virus type 1 reverse transcriptase and ribonuclease H activities by constituents of Juglans mandshurica. Chem. Pharm. Bull. 2000, 48, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Noureini, S.K.; Wink, M. Antiproliferative effect of the isoquinoline alkaloid papaverine in hepatocarcinoma HepG-2 cells—Inhibition of telomerase and induction of senescence. Molecules 2014, 19, 11846–11849. [Google Scholar] [CrossRef] [PubMed]
- Noureini, S.K.; Wink, M. Dose-dependent cytotoxic effects of boldine in HepG-2 cells- telomerase inhibition and apoptosis induction. Molecules 2015, 20, 3730–3743. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.Y.; Zhang, X.Q.; Li, X.; Zeng, F.B.; Ruan, H.L. 2-methoxyjuglone induces apoptosis in HepG-2 human hepatocellular carcinoma cells and exhibits in vivo antitumor activity in a H22 mouse hepatocellular carcinoma model. J. Nat. Prod. 2013, 76, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Raquel, C.M.; Ana, J.A.; Maria, T.M.; José, D.B.M.F.; Danilo, D.R.; Eulogio, L.M.; Marília, O.F.G.; Bento, E.S.; Ana, P.N.N.A.; Cláudia, P.; et al. Cytotoxic activity of naphthoquinones with special emphasis on juglone and its 5-O-methyl derivative. Chem. Biol. Interact. 2010, 184, 439–448. [Google Scholar]
- Liu, L.J.; Qi, F.Q.; Gong, X.F. Studies on the cytotoxicity of naphthoquinone derivatives from the fresh rejvenated fruits of Juglans mandshurica. Chin. JMAP 2010, 27, 574–577. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Carmichael, J.; DeGraff, W.G.; Gazdar, A.F.; Minna, J.D.; Mitchell, J.B. Evaluation of a tetrazolium-based semiautomated colorimetric assay: Assessment of chemosensitivity testing. Cancer Res. 1987, 47, 936–942. [Google Scholar] [PubMed]
- Sample Availability: Samples of the compounds 1–27 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Yang, B.; Jiang, Y.; Liu, Z.; Liu, Y.; Wang, X.; Kuang, H. Studies on Cytotoxic Activity against HepG-2 Cells of Naphthoquinones from Green Walnut Husks of Juglans mandshurica Maxim. Molecules 2015, 20, 15572-15588. https://doi.org/10.3390/molecules200915572
Zhou Y, Yang B, Jiang Y, Liu Z, Liu Y, Wang X, Kuang H. Studies on Cytotoxic Activity against HepG-2 Cells of Naphthoquinones from Green Walnut Husks of Juglans mandshurica Maxim. Molecules. 2015; 20(9):15572-15588. https://doi.org/10.3390/molecules200915572
Chicago/Turabian StyleZhou, Yuanyuan, Bingyou Yang, Yanqiu Jiang, Zhaoxi Liu, Yuxin Liu, Xiaoli Wang, and Haixue Kuang. 2015. "Studies on Cytotoxic Activity against HepG-2 Cells of Naphthoquinones from Green Walnut Husks of Juglans mandshurica Maxim" Molecules 20, no. 9: 15572-15588. https://doi.org/10.3390/molecules200915572