
Review of Platensimycin and Platencin: Inhibitors of β-Ketoacyl-acyl Carrier Protein (ACP) Synthase III (FabH)

Abstract
:1. Introduction

2. Isolation, Antibacterial Activities, and Biosynthesis
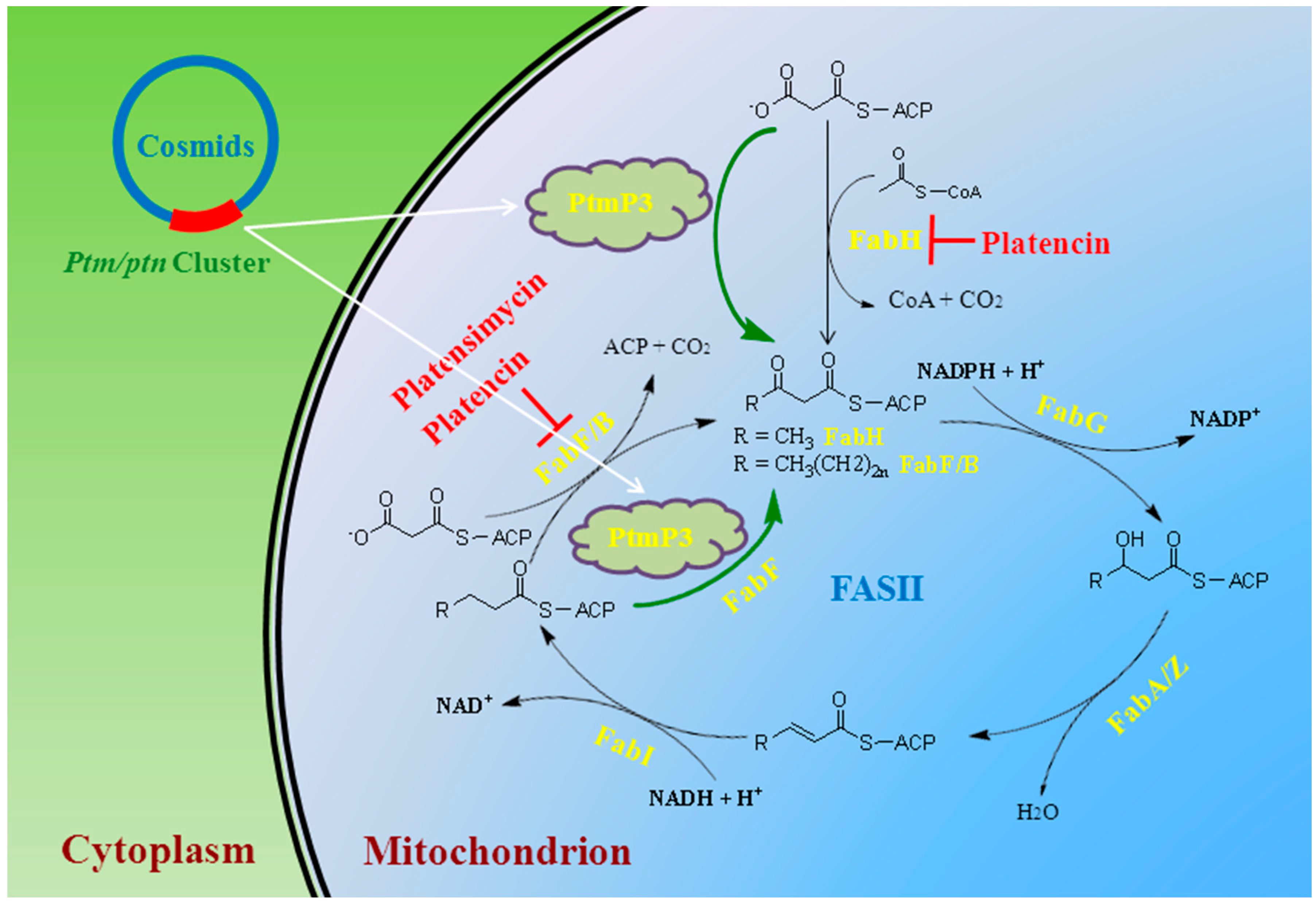
3. Antibacterial and Self-Resistant Mechanism

4. Recent Total Synthesis

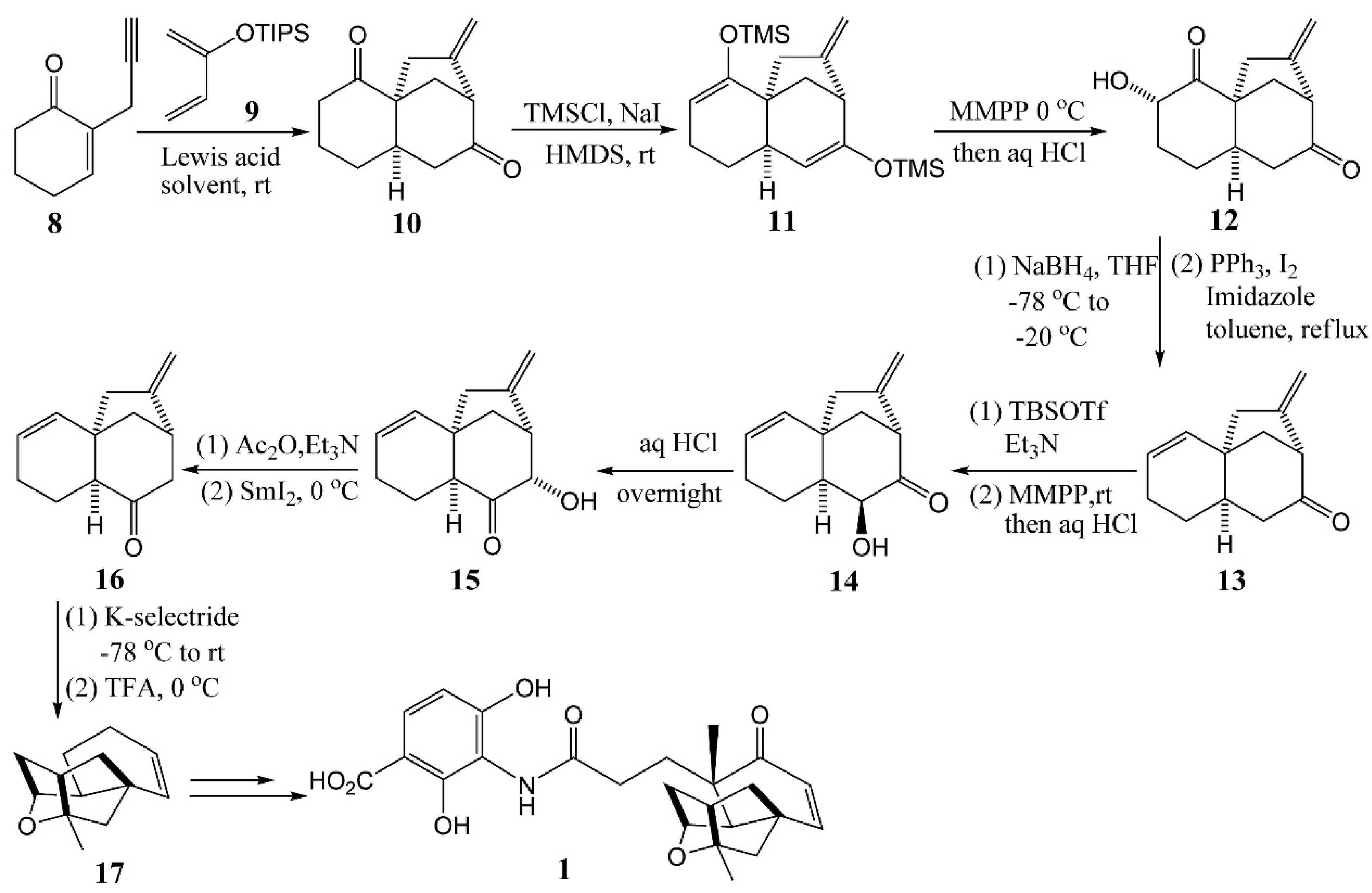
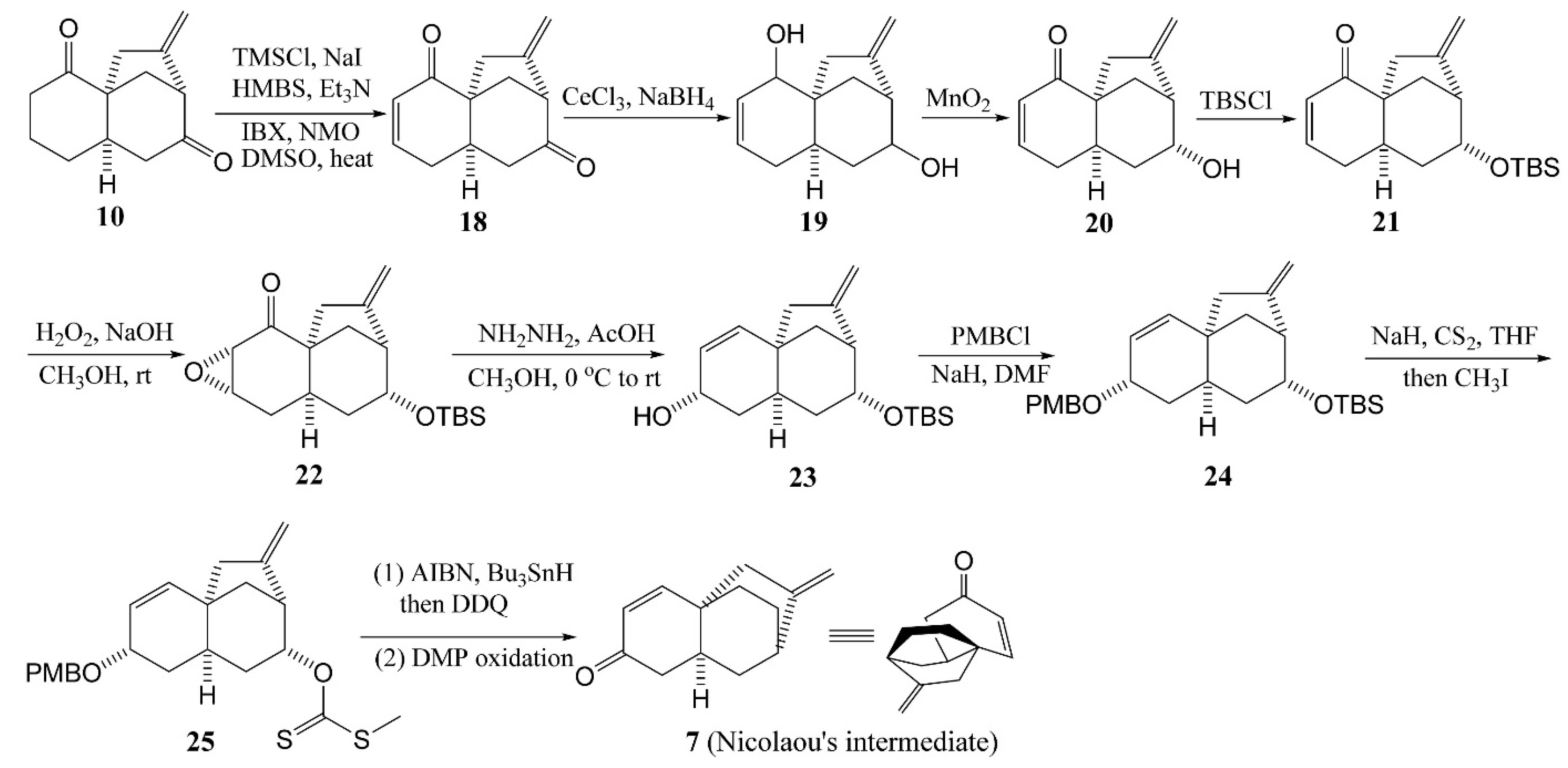
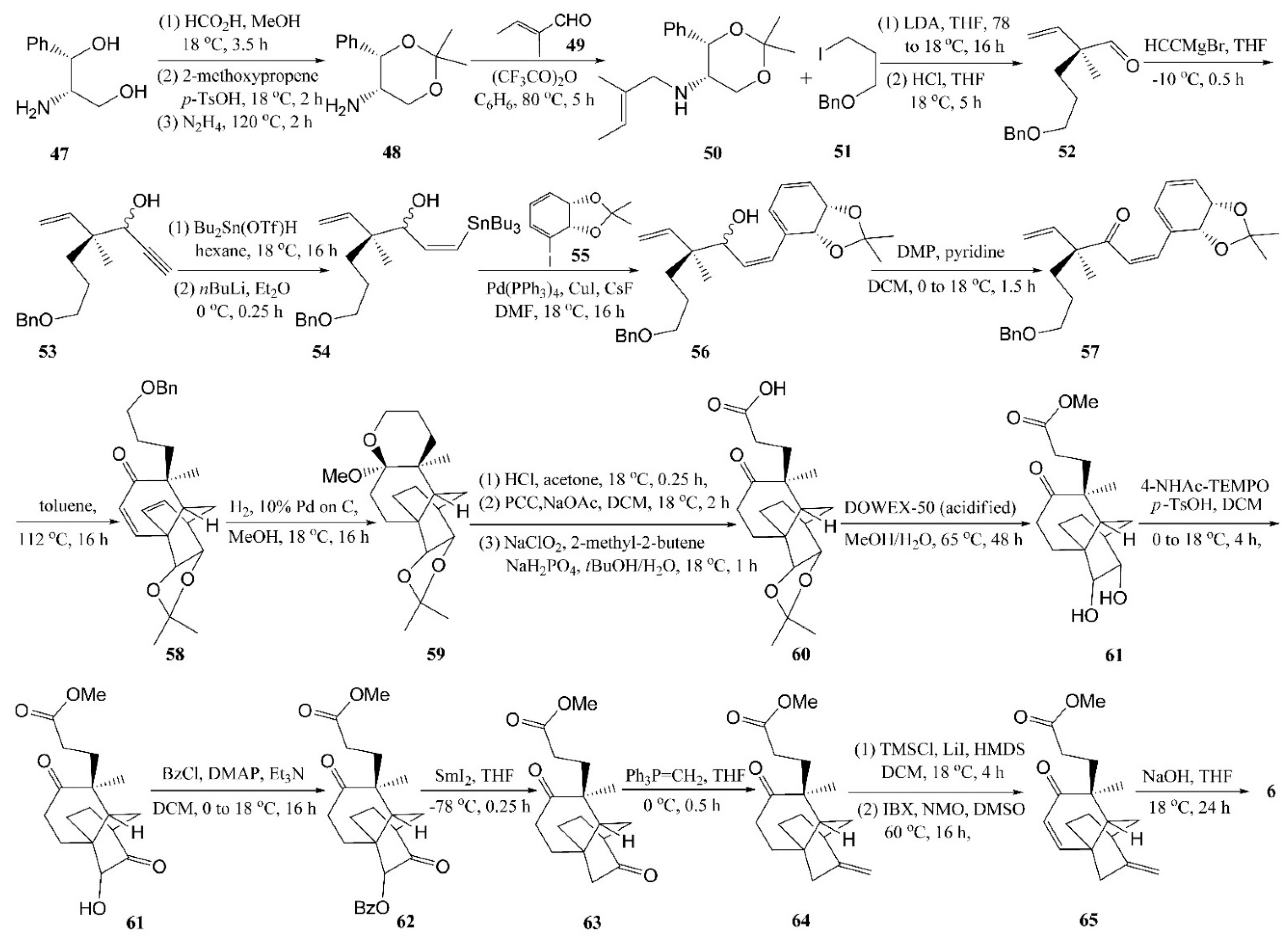
4.1. Zhu’s Total Syntheses of (±)-Platensimycin and Platencinusing a Cascade Cyclization Approach
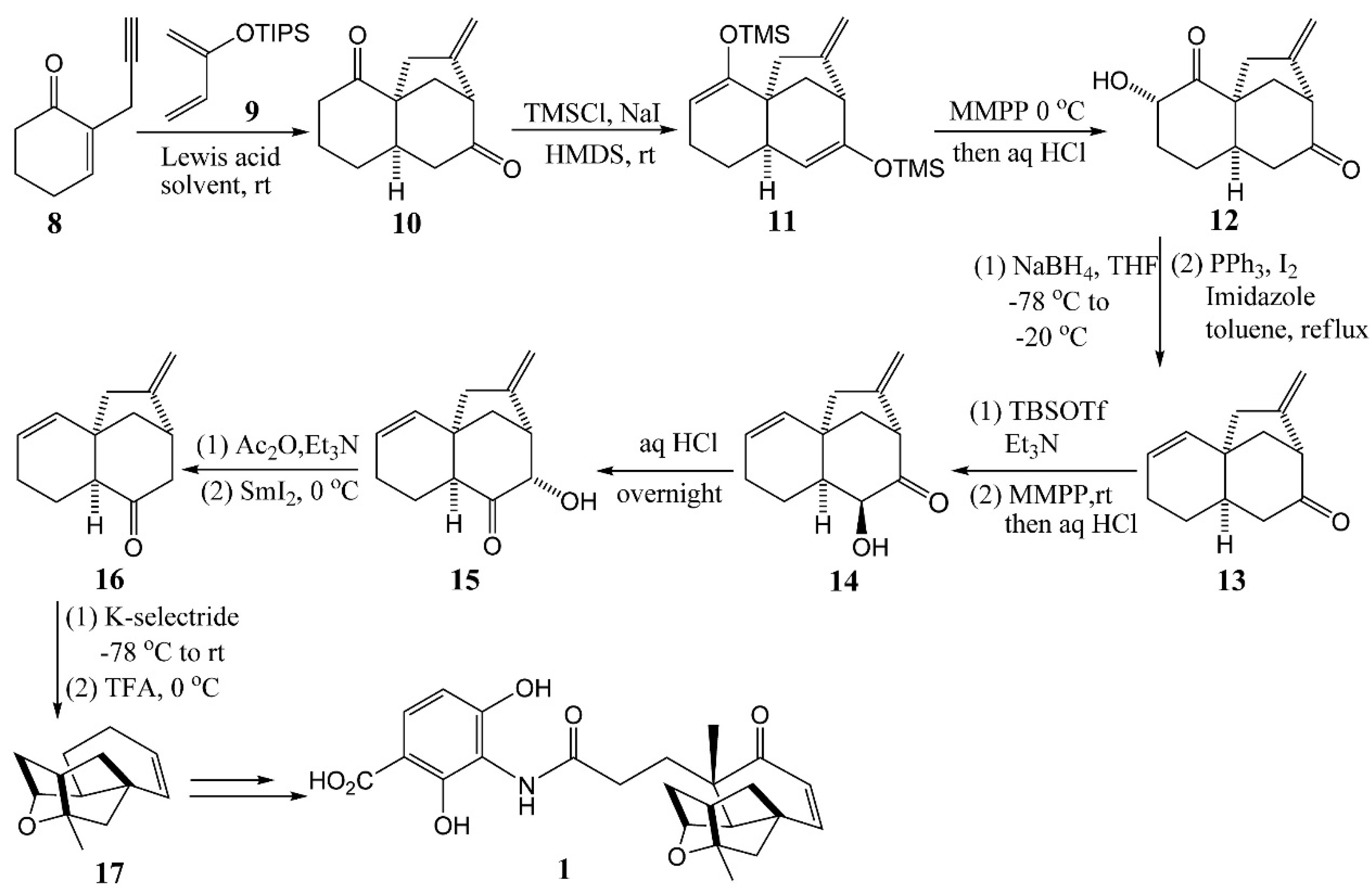

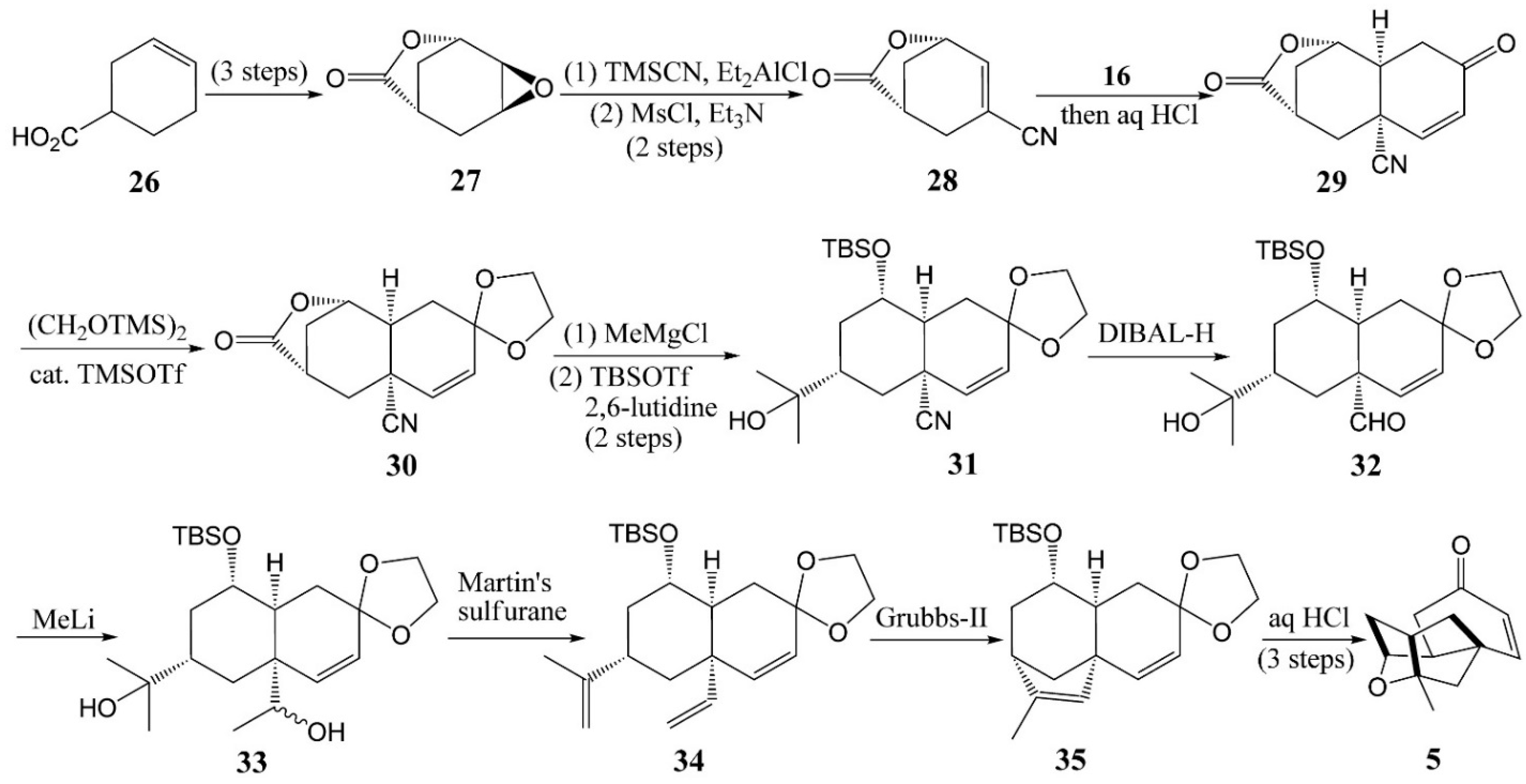
4.2. Horii’s Synthesis of Tetracyclic Cage 5

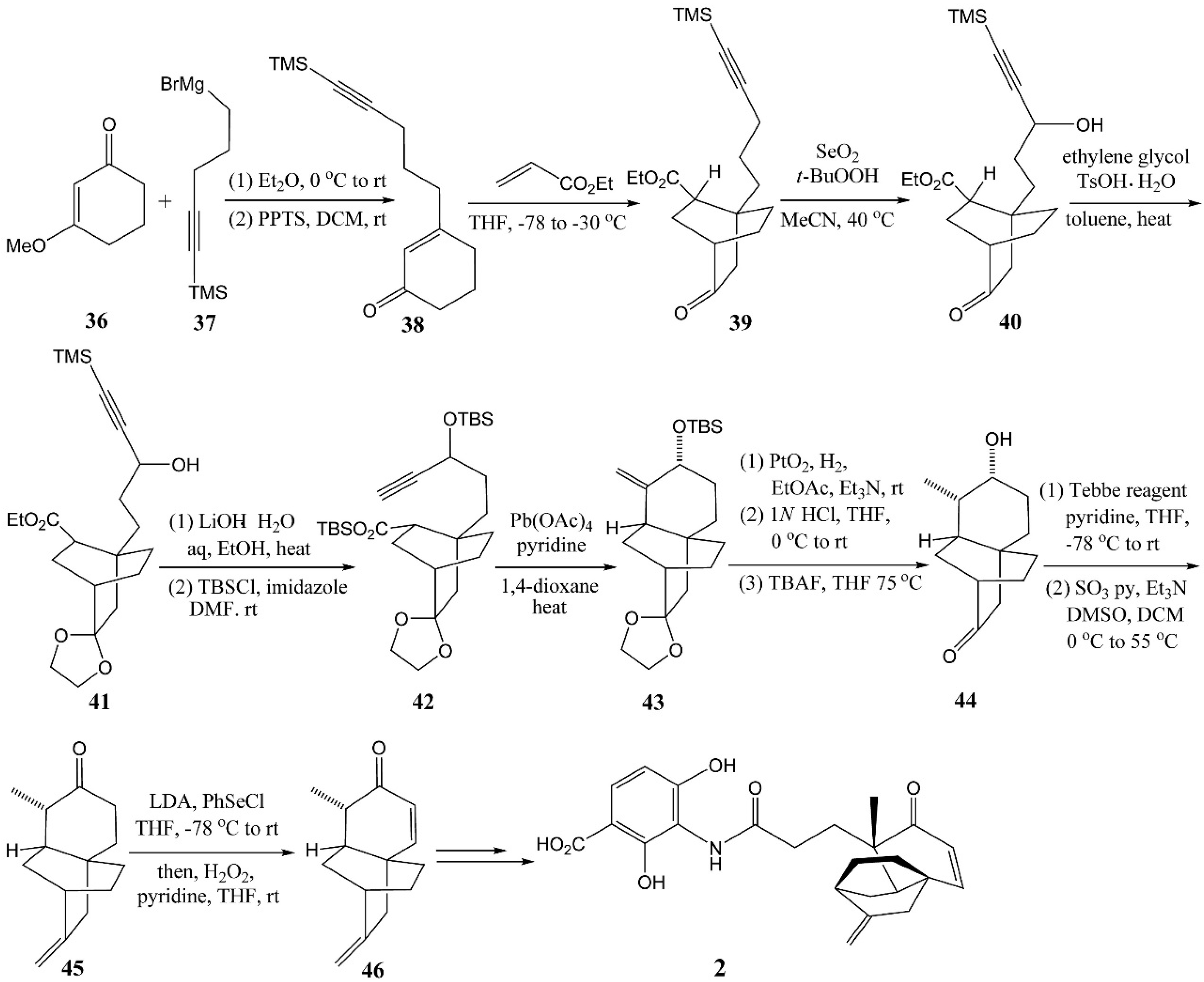
4.3. Gamal’s Synthesis of Platencin

4.4. Chang’s Synthesis of Platencin

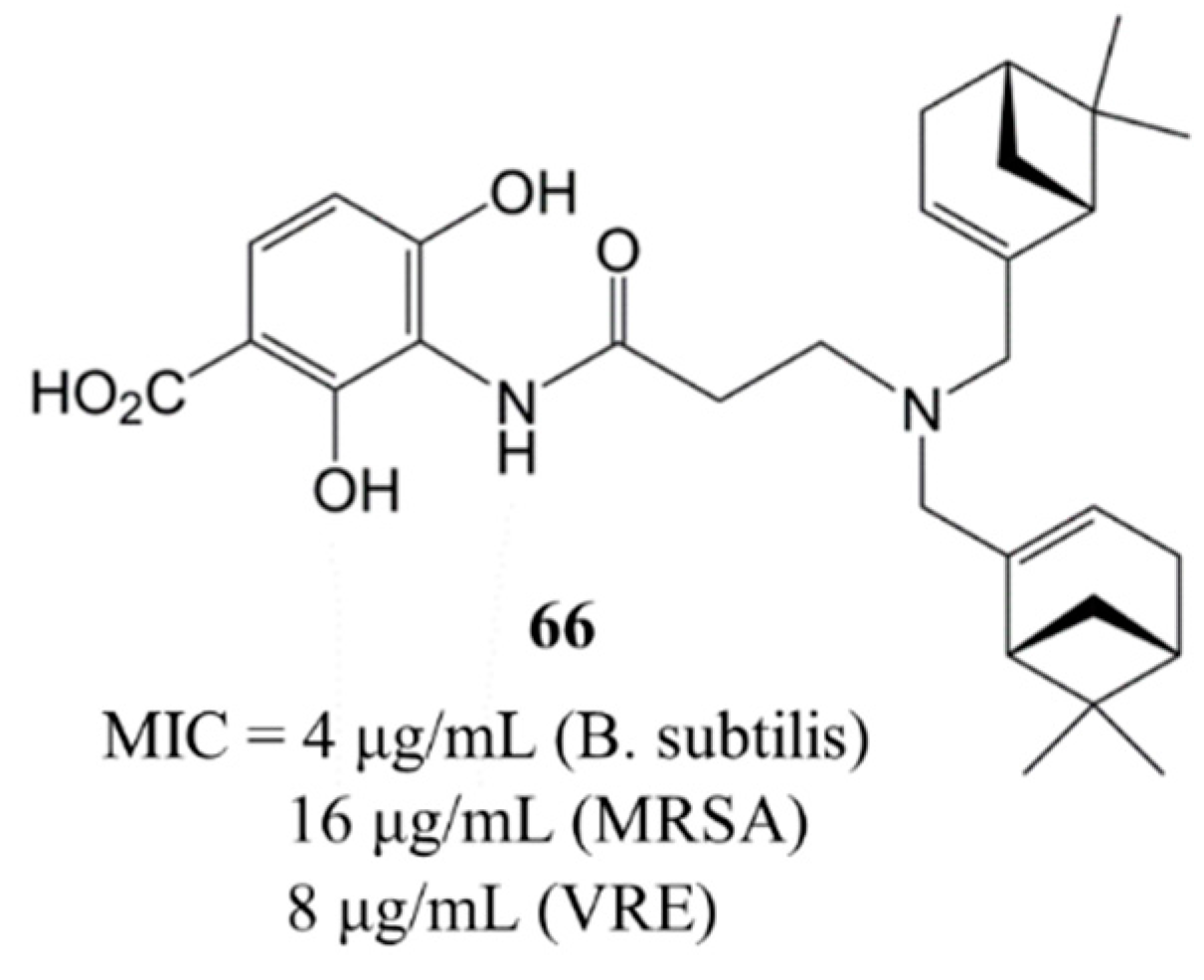
5. Recent Analogues and Their Antibacterial Activities



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | 67: R = Ph | 68: R = Trans H3C-CH=CH- | Tetracycline | Clotrimazol |
|---|---|---|---|---|
| Escherichia coli | 11 | 10 | 25 | 0 |
| Pseudomonas antimicrobia | 14 | 10 | 23 | 0 |
| Staphylococcus equorum | 0 | 11 | 23 | 9 |
| Streptococcus entericus | 16 | 14 | 12 | 11 |
| Candida glabrata | 20 | 17 | nt | 15 |
| Aspergillus niger | 0 | 6 | nt | 15 |
| Yarrowia lipolytica | 0 | 7 | nt | 20 |
| Hyphopichia burtonii | 10 | 15 | nt | 17 |
| Structure | R | Compounds | Kd/μM |
|---|---|---|---|
 | -SO2NHBOC | 69 | 50 ± 10 |
| -SO2NH2 | 70 | 100 ± 20 | |
| -CONHiPr | 71 | 110 ± 30 | |
| 3-pyridyl | 72 | 120 ± 20 | |
 | 73 | 650 ± 90 |
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviation
| FabF | Fatty acid acyl carrier protein synthases II |
| FabH | Fatty acid acyl carrier protein synthases III |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| VISA | Vancomycin-intermediate S. aureus |
| VRE | Vancomycin-resistant Enterococci |
| HPLC | High performance liquid chromatography |
| NMR | Nuclear magnetic resonance |
| E. coli | Escherichia coli |
| MIC | Minimal inhibitory concentration |
| FASII | Type II bacterial fatty acid synthesis |
| TMS | Tetramethylsilane |
| MMPP | Magnesium monoperoxyphthalate |
| TBS | t-Butyldimethylsilyl |
| PMB | p-Methoxybenzyl |
| THF | Tetrahydrofuran |
| RCM | Ring closing metathesis |
| IMDA | Intramolecular Diels-Alder |
| SAR | Structure activity relationship |
References
- Spicknall, I.H.; Foxman, B.; Marrs, C.F.; Eisenberg, J.N. A Modeling Framework for the Evolution and Spread of Antibiotic Resistance: Literature Review and Model Categorization. Am. J. Epidemiol. 2013, 178, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.G.; May-Dracka, T.L.; Gagnon, M.M.; Tommasi, R. Trends and exceptions of physical properties on antibacterial activity for gram-positive and gram-negative pathogens. J. Med. Chem. 2014, 57, 10144–10161. [Google Scholar] [CrossRef] [PubMed]
- Shang, R.F.; Wang, J.T.; Guo, W.Z.; Liang, J.P. Efficient antibacterial agents: A review of the synthesis, biological evaluation and mechanism of pleuromutilin derivatives. Curr. Top. Med. Chem. 2013, 13, 3013–3025. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kodali, S.; Lee, S.H.; Galgoci, A.; Painter, R.; Dorso, K.; Racine, F.; Motyl, M.; Hernandez, L.; Tinney, E.; et al. Discovery of platencin, a dual FabF and FabH inhibitor with in vivo antibiotic properties. Proc. Natl. Acad. Sci. USA. 2007, 104, 7612–7616. [Google Scholar] [CrossRef] [PubMed]
- Kirst, H.A. Developing new antibacterials through natural product research. Expert Opin. Drug Discv. 2013, 8, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Soisson, S.M.; Young, K.; Shoop, W.; Kodali, S.; Galgoci, A.; Painter, R.; Parthasarathy, G.; Tang, Y.; Cummings, R.; et al. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature 2006, 441, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Young, K.; Jayasuriya, H.; Ondeyka, J.G.; Herath, K.; Zhang, C.; Kodali, S.; Galgoci, A.; Painter, R.; Brown-Driver, V.; Yamamoto, R.; et al. Discovery of FabH/FabF Inhibitors from Natural Products. Antimicrob. Agents Chemother. 2006, 50, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.W.; Ondeyka, J.; Herath, K.; Jayasuriya, H.; Guan, Z.Q.; Zink, D.L.; Dietrich, L.; Burgess, B.; Ha, S.N.; Wang, J.; et al. Platensimycin and platencin congeners from Streptomyces platensis. J. Nat. Prod. 2011, 74, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Martens, E.; Demain, A.L. Platensimycin and platencin: Promising antibiotics for future application in human medicine. J. Antibiot. 2011, 64, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Peterson, R.M.; Huang, T.; Rudolf, J.D.; Smanski, M.J.; Shen, B. Mechanisms of self-resistance in the platensimycin- and platencin- producing Streptomyces platensis MA7327 and MA7339 strains. Chem. Bio. 2014, 21, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Jayasuriya, H.; Ondeyka, J.G.; Herath, K.B.; Zhang, C.; Zink, D.L.; Tsou, N.N.; Ball, R.G.; Basilio, A.; Genilloud, O.; et al. Isolation, Structure, and Absolute Stereochemistry of Platensimycin, A Broad Spectrum Antibiotic Discovered Using an Antisense Differential Sensitivity Strategy. J. Am. Chem. Soc. 2006, 128, 11916–11920. [Google Scholar] [CrossRef] [PubMed]
- Habich, D.; von Nussbaum, F. Platensimycin, a new antibiotic and “superbug challenger” from nature. Chem. Med. Chem. 2006, 1, 951–954. [Google Scholar] [CrossRef] [PubMed]
- Barton, S. New antibiotic on the horizon? Nat. Rev. Microbiol. 2006, 4. [Google Scholar] [CrossRef]
- Palanichamy, K.; Kaliappan, K.P. Discovery and syntheses of “superbug challengers”—platensimycin and platencin. Chem. Asian J. 2010, 5, 668–703. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Young, K. New antibiotic structures from fermentations. Expert Opin.Ther. Pat. 2010, 20, 1359–1371. [Google Scholar] [CrossRef] [PubMed]
- Jayasuriya, H.; Herath, K.B.; Zhang, C.; Zink, D.L.; Basilio, A.; Genilloud, O.; Diez, M.T.; Vicente, F.; Gonzalez, I.; Salazar, O.; et al. Isolation and Structure of Platencin: A FabH and FabF Dual Inhibitor with Potent Broad-Spectrum Antibiotic Activity. Angew. Chem. 2007, 46, 4768–4772. [Google Scholar] [CrossRef]
- Herath, K.B.; Attygalle, A.B.; Singh, S.B. Biosynthetic studies of platensimycin. J. Am. Chem. Soc. 2007, 129, 15422–15423. [Google Scholar] [CrossRef] [PubMed]
- Jayasuriya, H.; Herath, K.B.; Ondeyka, J.G.; Zink, D.L.; Burgess, B.; Wang, J.; Singh, S.B. Structure of homoplatensimide A: A potential key biosynthetic intermediate of platensimycin isolated from Streptomyces platensis. Tetrahedron Lett. 2008, 49, 3648–3651. [Google Scholar] [CrossRef]
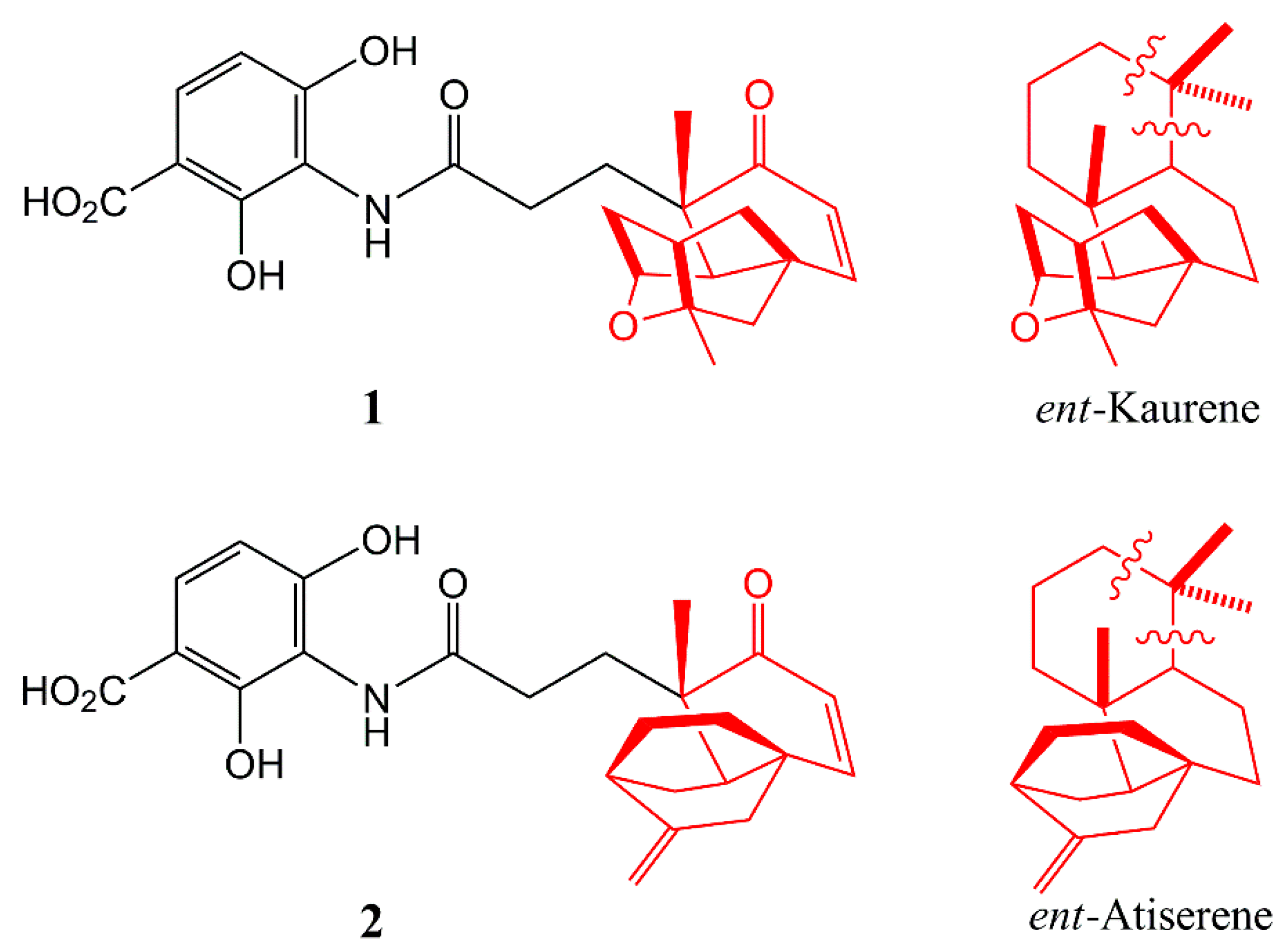
- Smanski, M.J.; Yu, Z.; Casper, J.; Lin, S.; Peterson, R.M.; Chen, Y.; Wendt-Pienkowski, E.; Rajski, S.R.; Shen, B. Dedicated ent-kaurene and ent-atiserene synthases for platensimycin and platencin biosynthesis. Proc. Natl. Acad. Sci. USA. 2011, 108, 13498–13503. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.J.; Warren, P.V.; Holmes, D.J.; Ji, Y.; Lonsdale, J.T. Bacterial fatty-acid biosynthesis: A genomics-driven target for antibacterial drug discovery. Drug Disc. Today 2001, 6, 537–544. [Google Scholar] [CrossRef]
- Hopwood, D.A. How do antibiotic-producing bacteria ensure their self-resistance before antibiotic biosynthesis incapacitates them? Mol. Microbiol. 2007, 63, 937–940. [Google Scholar] [CrossRef] [PubMed]
- McGrath, N.A.; Bartlett, E.S.; Sittihan, S.; Njardarson, J.T. A concise ring-expansion route to the compact core of platensimycin. Angew. Chem. Int. Ed. Engl. 2009, 48, 8543–8546. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M.; Hussain, H.; Ahmed, I.; van Ree, T.; Krohn, K. Platensimycin and its relatives: A recent story in the struggle to develop new naturally derived antibiotics. Nat. Prod. Rep. 2011, 28, 1534–1579. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y. From sigma- to pi- electrophilic Lewis acids. Application to selective organic transformations. J. Org. Chem. 2007, 72, 7817–7831. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.Z.; Han, Y.J.; Du, G.Y.; Lee, C.S. A bifunctional Lewis Acid induced cascade cyclization to the tricyclic core of ent-kaurenoids and its application to the formal synthesis of (±)-platensimycin. Org. Lett. 2013, 15, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Li, A.; Edmonds, D.J. Total synthesis of platensimycin. Angew. Chem. Int. Ed. Engl. 2006, 45, 7086–7090. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Chen, C.H.; Taylor, C.D.; Foxman, B.M.; Snider, B.B. Formal synthesis of (+/−)-platensimycin. Org. Lett. 2007, 9, 1825–1828. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.Z.; Zhou, C.S.; Yang, W.; He, S.Z.; Cheng, G.J.; Zhang, X.H.; Lee, C.S. Formal syntheses of (±)-platensimycin and (±)-platencin via a dual-mode Lewis acid induced cascade cyclization approach. J. Org. Chem. 2013, 78, 7912–7929. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Tria, G.S.; Edmonds, D.J. Total synthesis of platencin. Angew. Chem. Int. Ed. Engl. 2008, 47, 1780–1783. [Google Scholar] [CrossRef] [PubMed]
- Horii, S.; Torihata, M.; Nagasawa, T.; Kuwahara, S. Stereoselective approach to the racemic oxatetracyclic core of platensimycin. J. Org. Chem. 2013, 78, 2798–2801. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, G.A.; Saku, Y.; Aoyama, H.; Yoshimitsu, T. A new route to platencin via decarboxylative radical cyclization. Chem. Commun. 2014, 50, 15706–15709. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.L.; Schwartz, B.D.; Draffan, A.G.; Banwell, M.G.; Willis, A.C. A chemoenzymatic and fully stereo-controlled total synthesis of the antibacterial natural product (±)-platencin. Chem. Asian. J. 2015, 10, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Nordin, I.C.; Thomas, J.A. An improved synthesis of (4S,5S)-2,2-dimethyl-4-phenyl-1,3-dioxan-5-amine. Tetrahedron Lett. 1984, 25, 5723–5724. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Stepan, A.F.; Lister, T.; Li, A.; Montero, A.; Tria, G.S.; Turner, C.I.; Tang, Y.; Wang, J.; Denton, R.M.; et al. Design, synthesis, and biological evaluation of platensimycin analogues with varying degrees of molecular complexity. J. Am. Chem. Soc. 2008, 130, 13110–13119. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lee, V.; Sintim, H.O. Efforts towards the Identification of Simpler Platensimycin Analogues—The Total Synthesis of Oxazinidinyl Platensimycin. Chem. Eur. J. 2009, 15, 2747–2750. [Google Scholar] [CrossRef] [PubMed]
- Tiefenbacher, K.; Gollner, A.; Mulzer, J. Syntheses and antibacterial properties of iso-platencin, Cl-iso-platencin and Cl-platencin: Identification of a new lead structure. Chem. Eur. J. 2010, 16, 9616–9622. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.C.; Ding, F.X.; Singh, S.B.; Parthasarathy, G.; Soisson, S.M.; Ha, S.N.; Chen, X.; Kodali, S.; Wang, J.; Dorso, K.; et al. Synthesis and biological evaluation of platensimycin analogs. Bioorg. Med. Chem. Lett. 2009, 19, 1623–1627. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Sintim, H.O. Dialkylamino-2,4-dihydroxybenzoic acids as easily synthesized analogues of platensimycin and platencin with comparable antibacterial properties. Chem. Eur. J. 2011, 17, 3352–3357. [Google Scholar] [CrossRef] [PubMed]
- Plesch, E.; Bracher, F.; Krauss, J. Synthesis and Antimicrobial Evaluation of Novel Platensimycin Analogues. Arch. Pharm. 2012, 345, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.; Basak, R.; Kalverda, A.P.; Fishwick, C.W.; Turnbull, W.B.; Nelson, A. Discovery of novel FabF ligands inspired by platensimycin by integrating structure-based design with diversity-oriented synthetic accessibility. Org. Biomol. Chem. 2014, 12, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C.; Fasolini, M.; Flocco, M.; Knapp, S.; Pevarello, P.; Veronesi, M. NMR-Based screening with competition water-ligand observed via gradient spectroscopy experiments: Detection of high-affinity ligands. J. Med. Chem. 2002, 45, 2610–2614. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shang, R.; Liang, J.; Yi, Y.; Liu, Y.; Wang, J. Review of Platensimycin and Platencin: Inhibitors of β-Ketoacyl-acyl Carrier Protein (ACP) Synthase III (FabH). Molecules 2015, 20, 16127-16141. https://doi.org/10.3390/molecules200916127
Shang R, Liang J, Yi Y, Liu Y, Wang J. Review of Platensimycin and Platencin: Inhibitors of β-Ketoacyl-acyl Carrier Protein (ACP) Synthase III (FabH). Molecules. 2015; 20(9):16127-16141. https://doi.org/10.3390/molecules200916127
Chicago/Turabian StyleShang, Ruofeng, Jianping Liang, Yunpeng Yi, Yu Liu, and Jiatu Wang. 2015. "Review of Platensimycin and Platencin: Inhibitors of β-Ketoacyl-acyl Carrier Protein (ACP) Synthase III (FabH)" Molecules 20, no. 9: 16127-16141. https://doi.org/10.3390/molecules200916127