
Heterocyclic Anticancer Compounds: Recent Advances and the Paradigm Shift towards the Use of Nanomedicine’s Tool Box

 ,
,  ,
,
Abstract
:1. Introduction

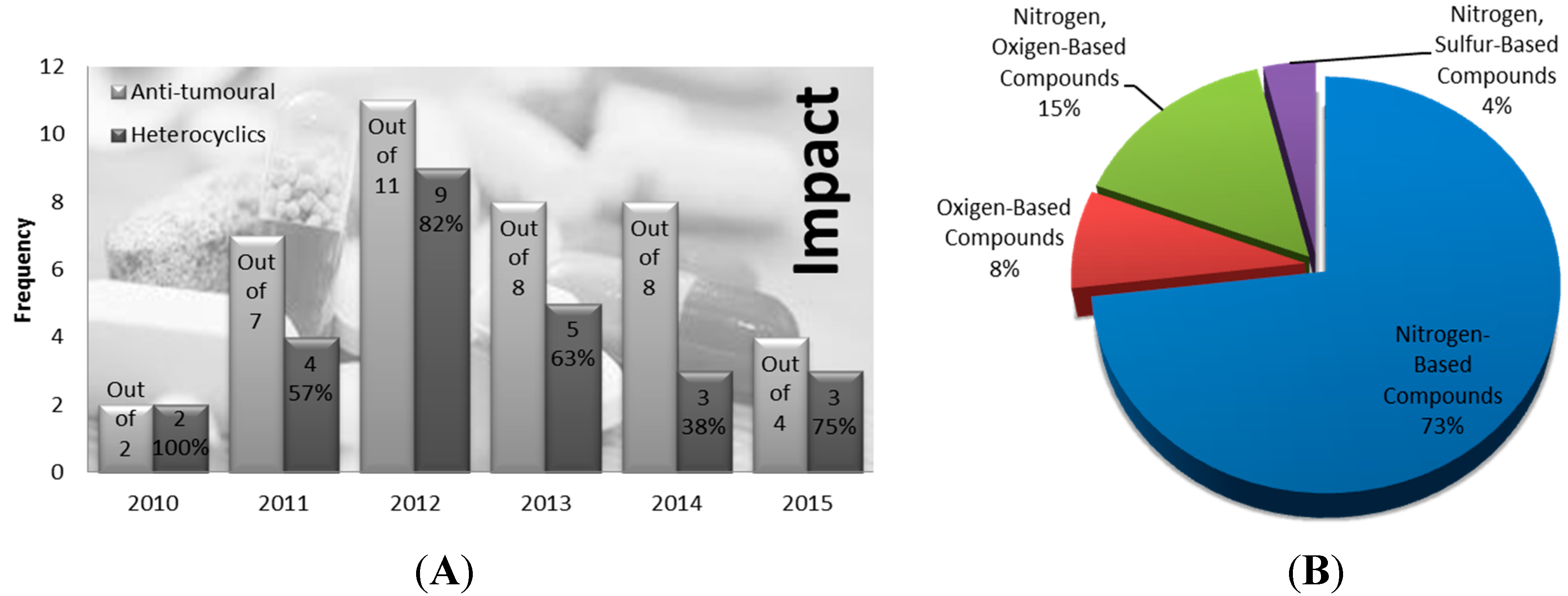
1.1. Heterocycles’ Clinical Relevance in Cancer Therapy
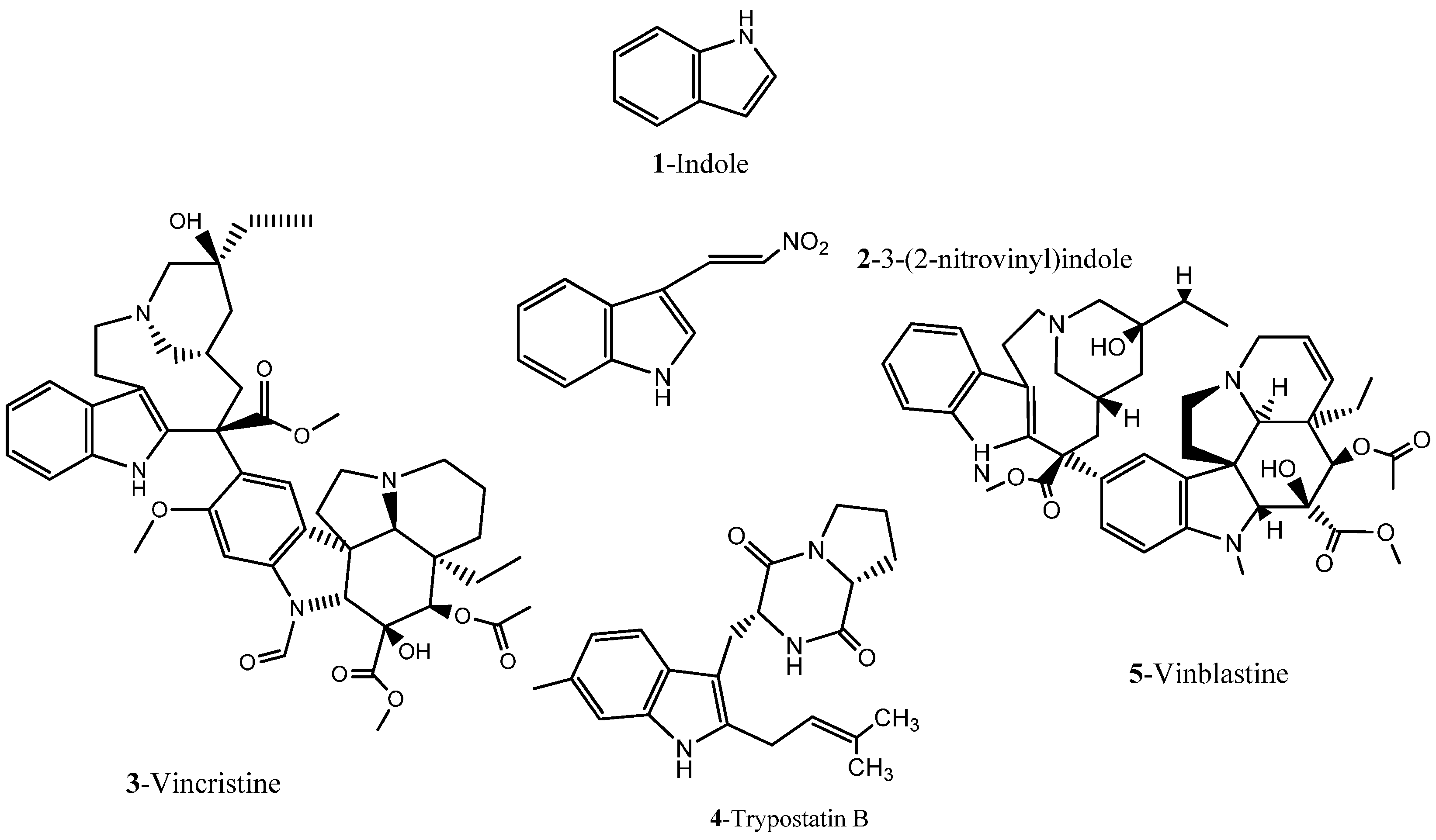
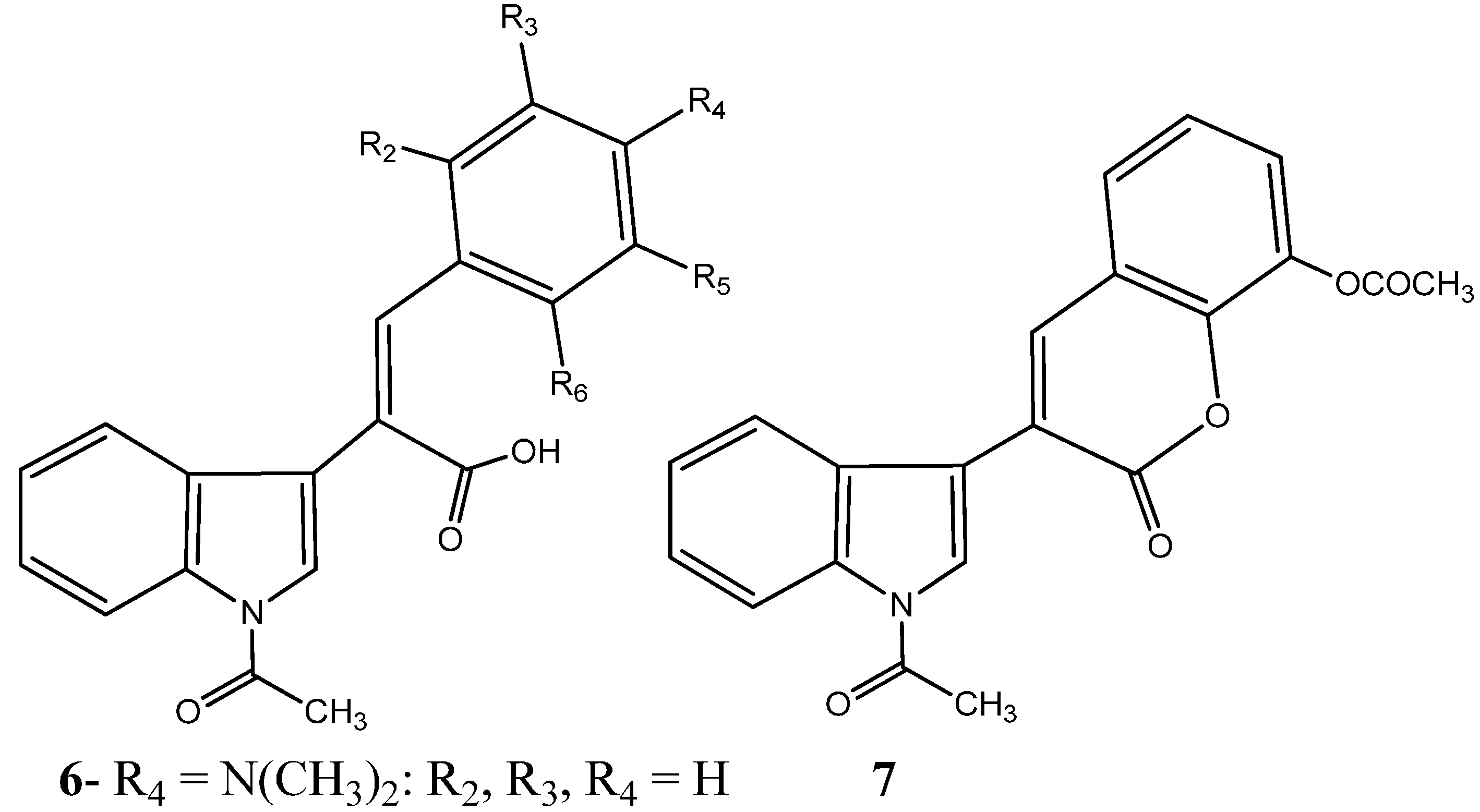
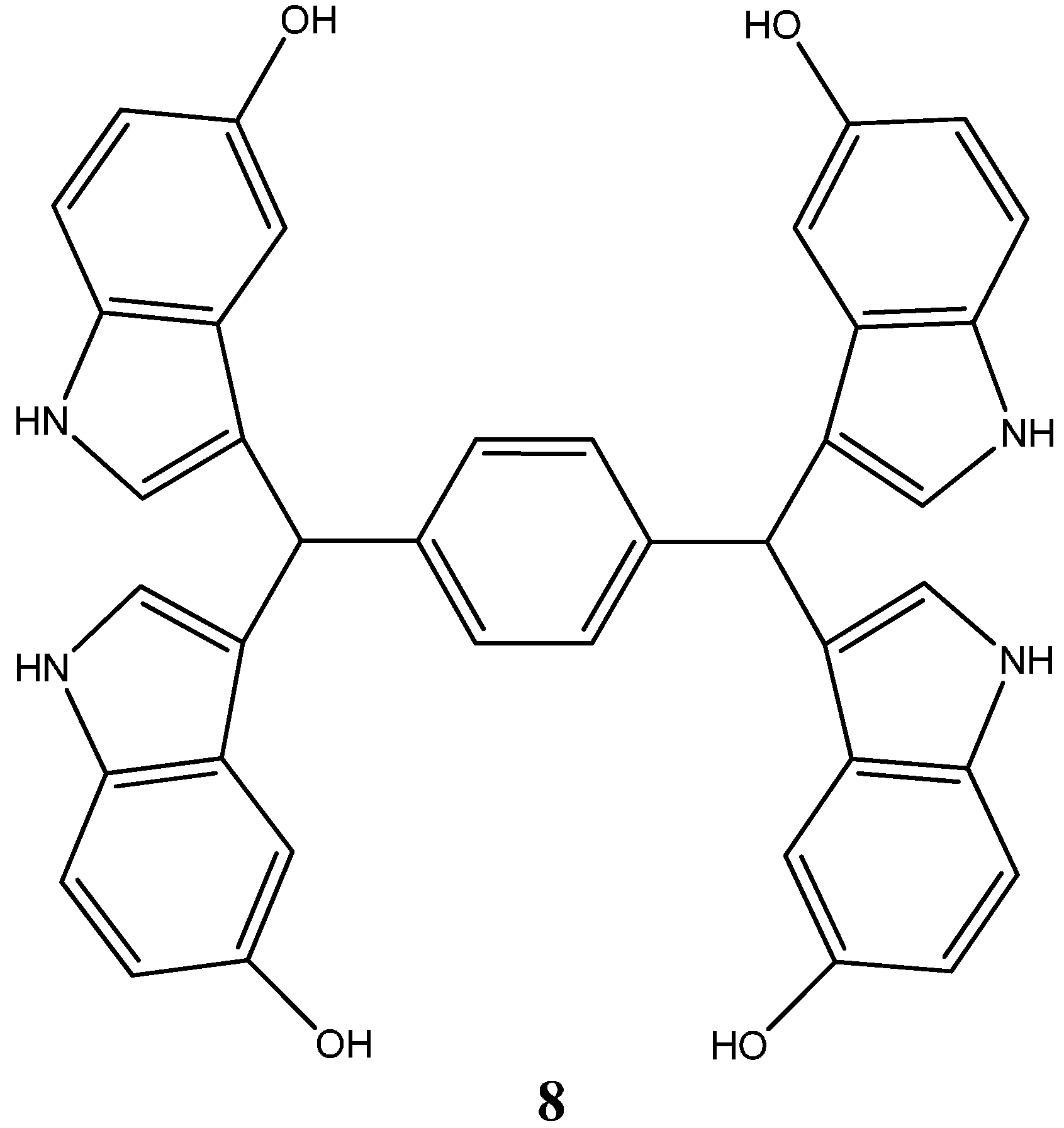
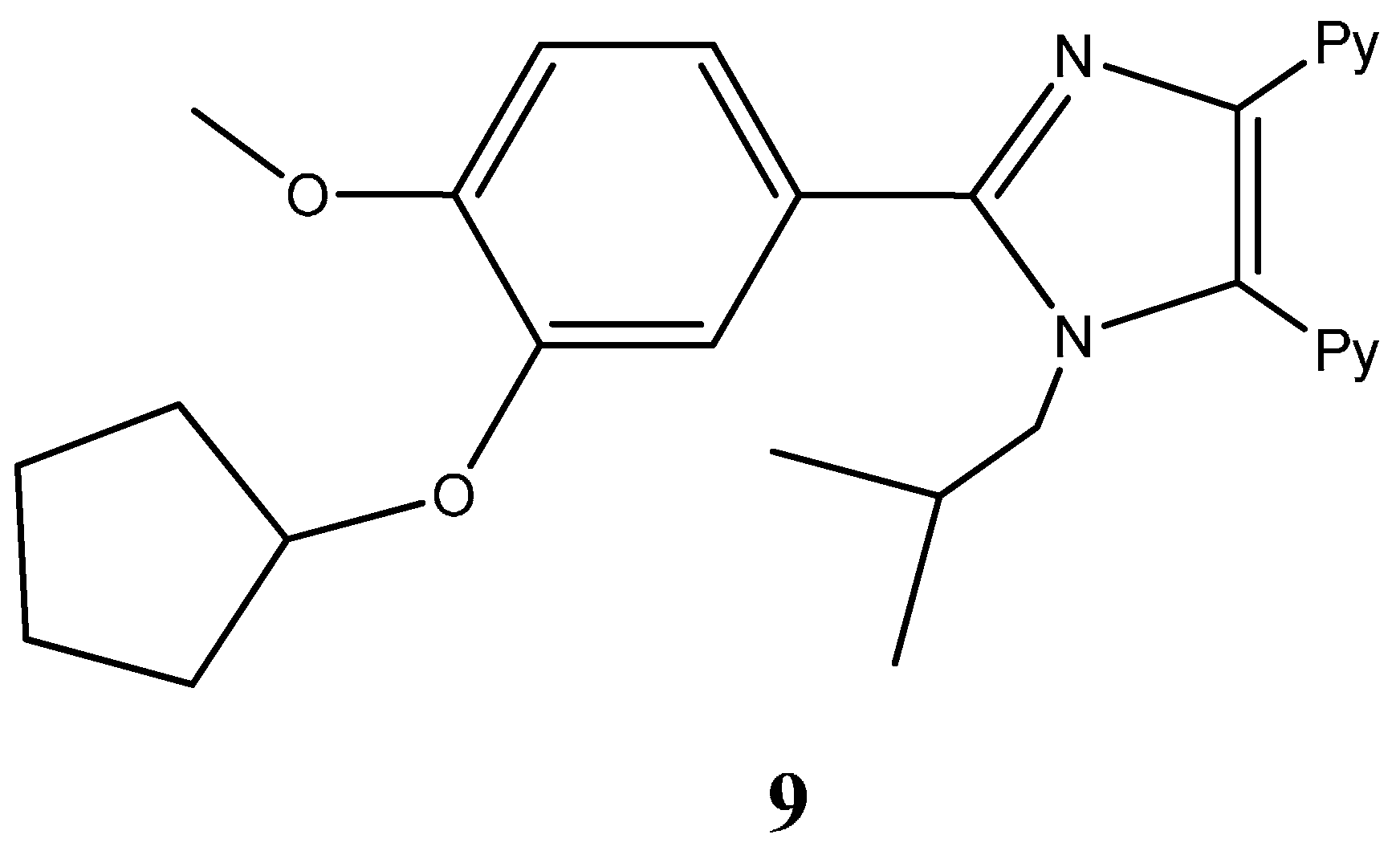
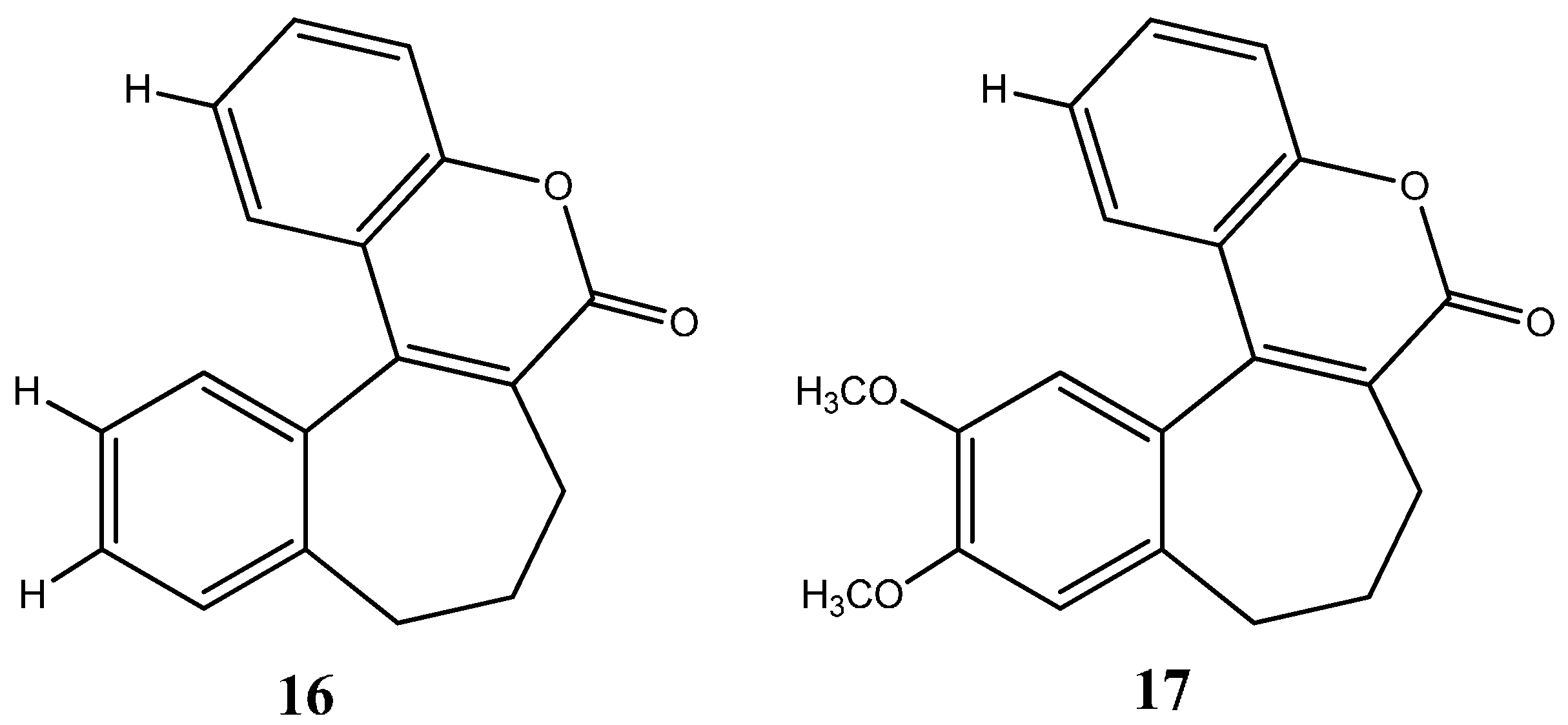
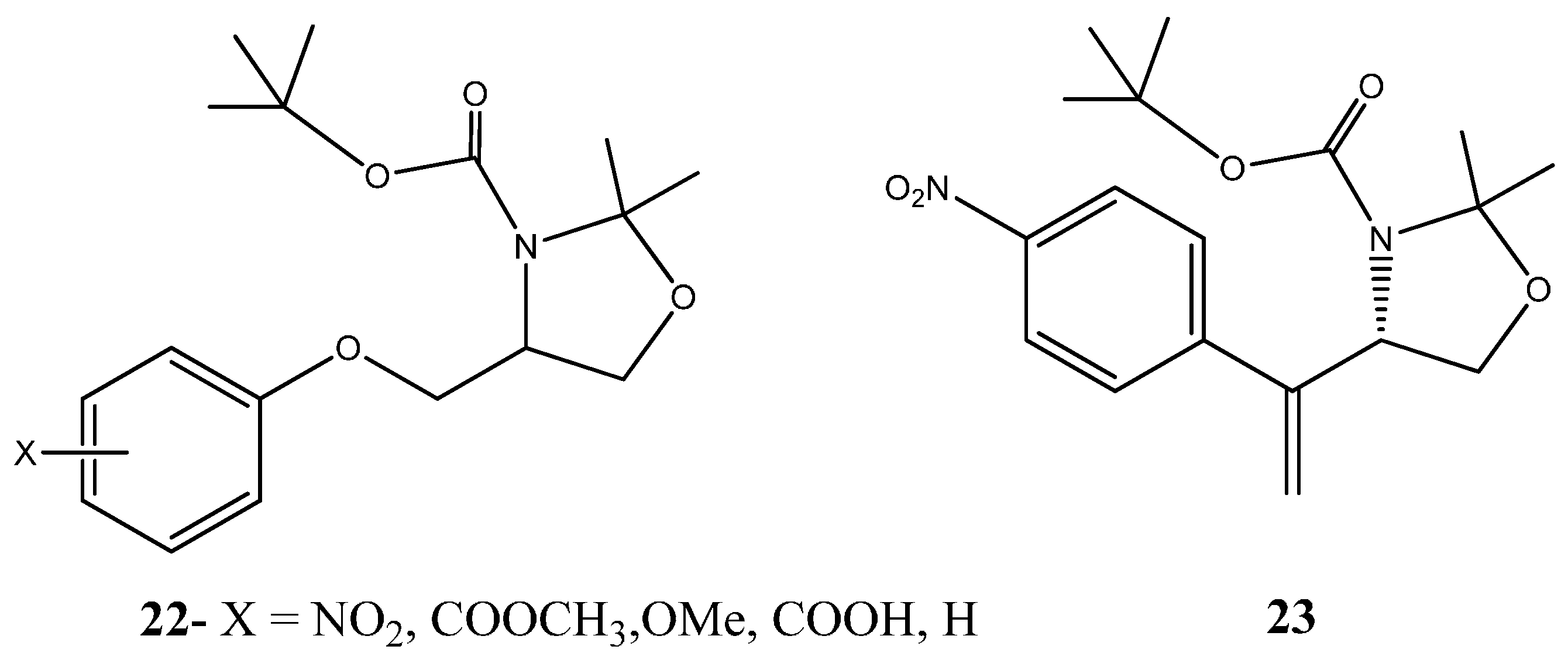
1.1.1. Nitrogen-Based Heterocycle







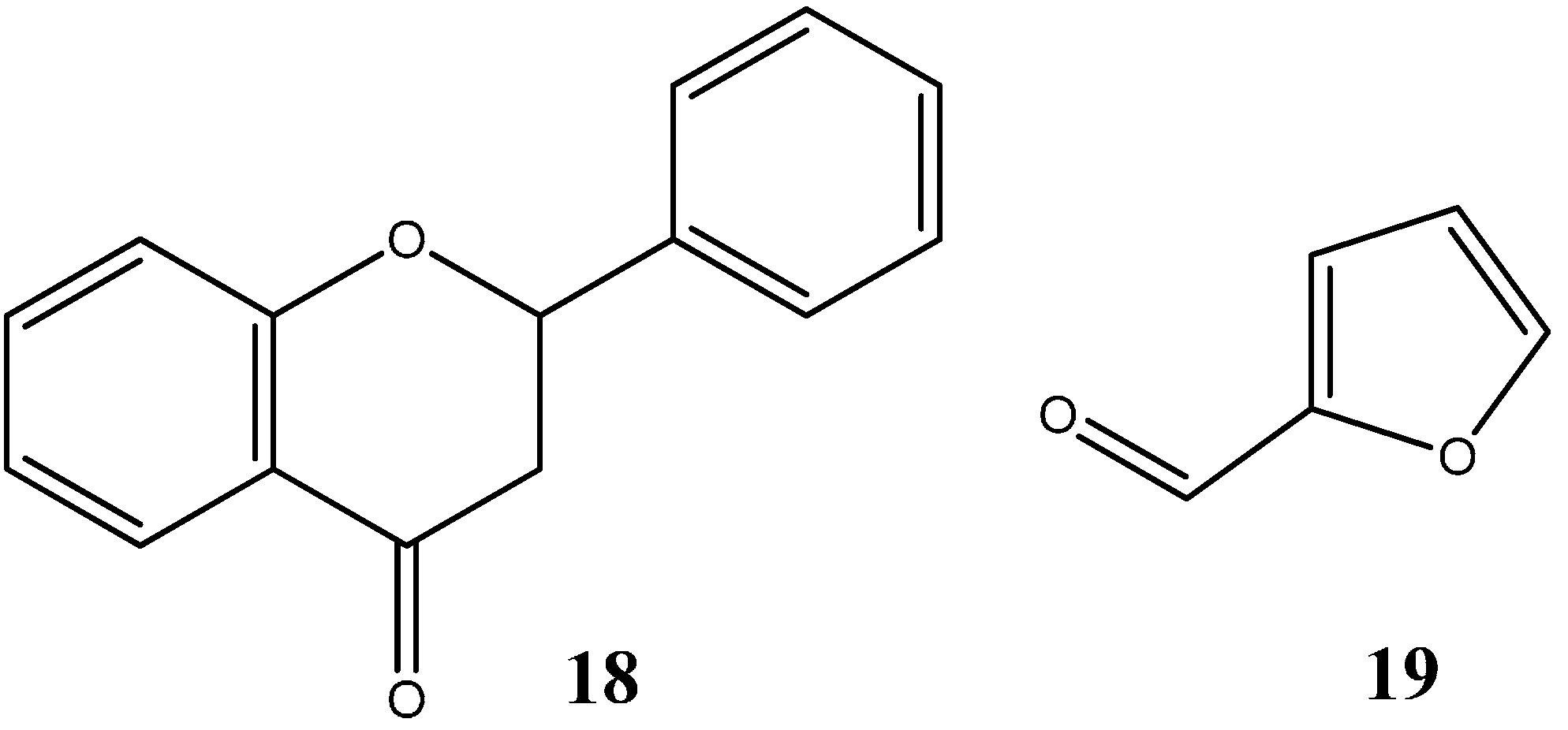
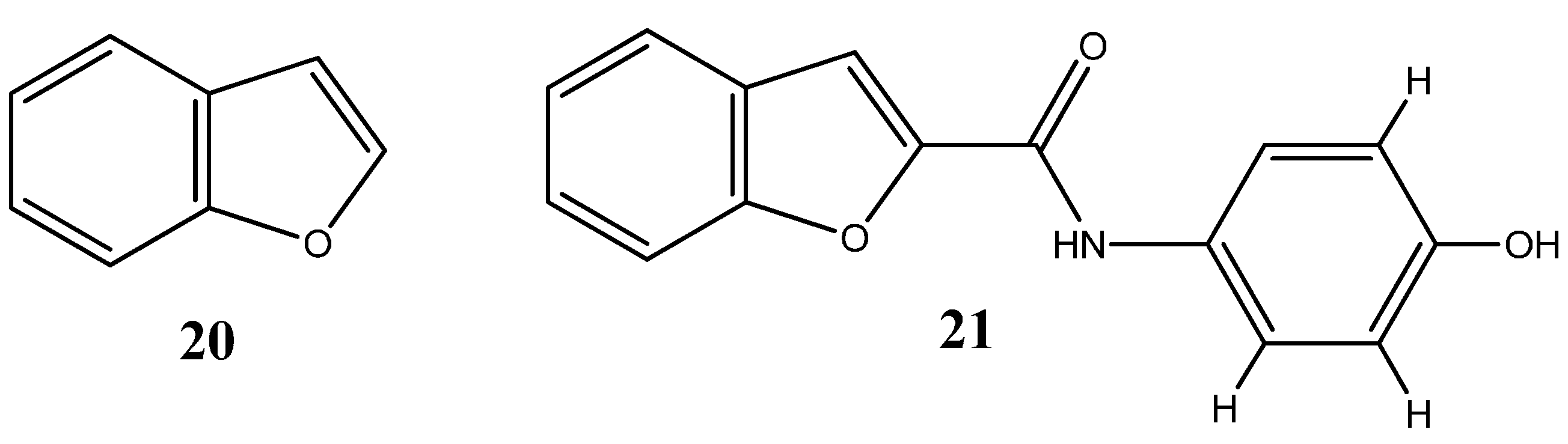
1.1.2. Oxygen-Based Heterocycles





1.1.3. Sulfur-Based Heterocycles




1.2. Approved Molecular Entities

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
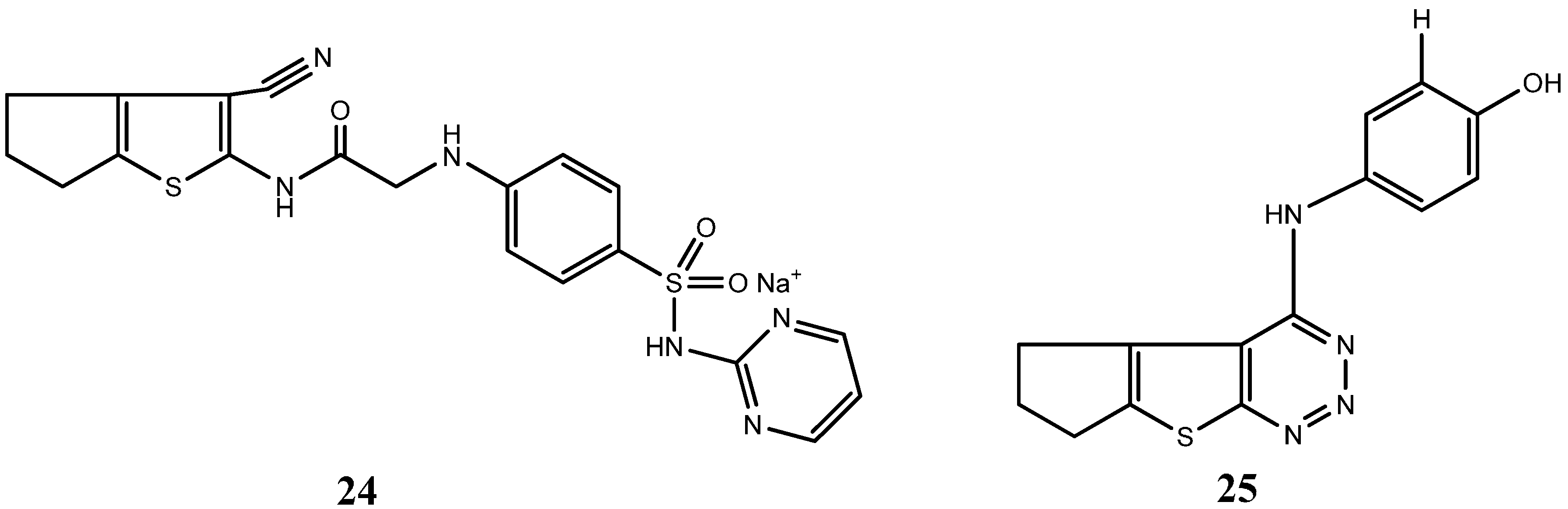{kind=link}
{kind=link}
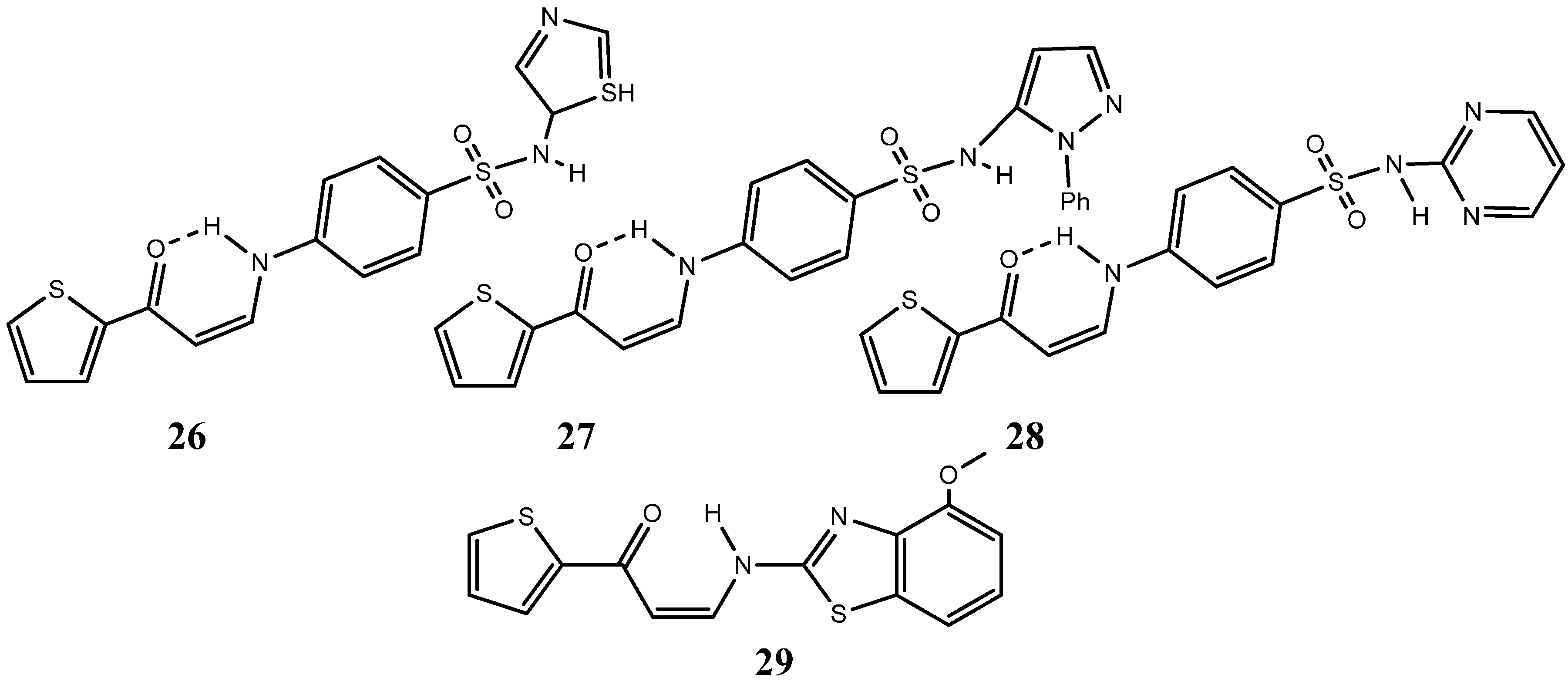
{kind=link}
{kind=link}
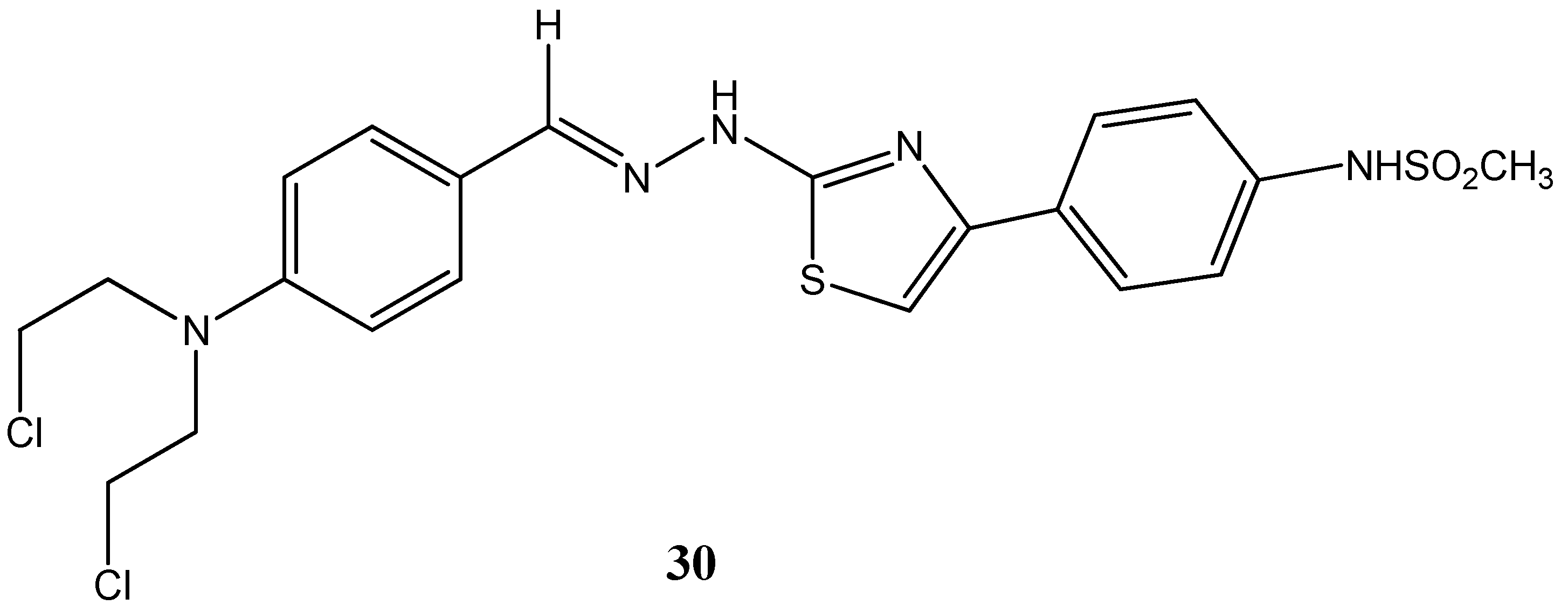
{kind=link}
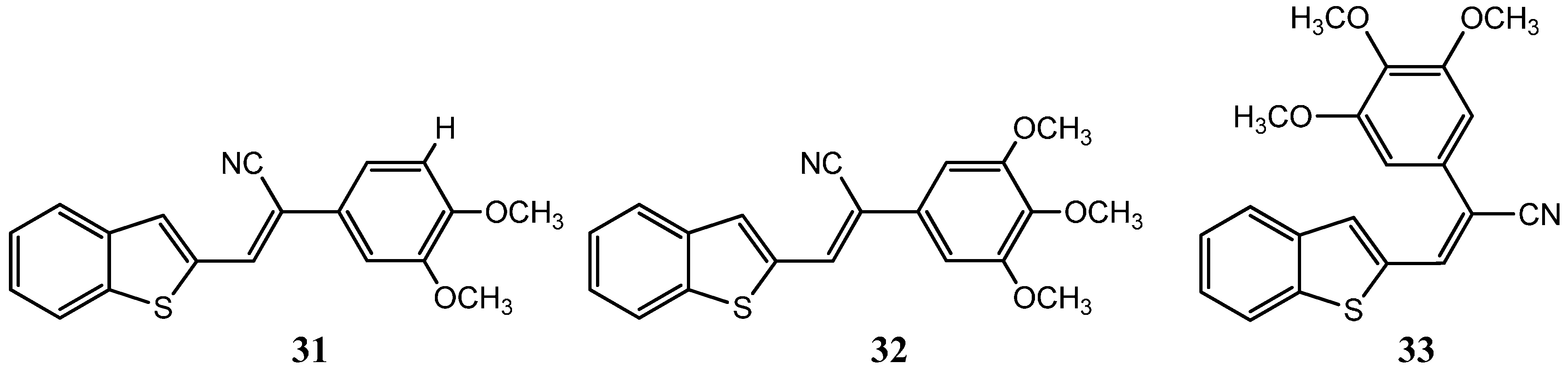
{kind=link}
{kind=link}
| Drug Name (Company) | Chemical Structure | Bioactive Compound | Therapeutic Indication | Approval Date |
|---|---|---|---|---|
| Approved Nitrogen-Based Heterocycle Drugs | ||||
| Xalkori® (Pfizer) |  | Crizotinib | Late-stage Non-small-cell lung carcinoma (NSCLC) | 2011 |
| Zelboraf® (Hoffmann-La Roche) |  | Vemurafenib | Metastatic or unresectable melanoma | 2011 |
| Zytiga® (Centocor Ortho Biotech) |  | Abiraterone acetate | Metastatic castration-resistant prostate cancer | 2011 |
| Caprelsa® (AstraZeneca) |  | Vandetanib | Metastatic medullary thyroid cancer | 2011 |
| Iclusig® (ARIAD Pharmaceuticals) |  | Ponatinib | Chronic myeloid leukemia/lymphoblastic leukemia | 2012 |
| Cometriq® (Exelixis) |  | Cabozantinib | Metastasized medullary thyroid cancer | 2012 |
| Stivarga® (Bayer HealthCare) |  | Regorafenib | Metastatic colorectal cancer | 2012 |
| Bosulif® (Pfizer) |  | Bosutinib | Chronic myelogenous leukemia | 2012 |
| Xtandi® (Astellas Pharma) |  | Enzalutamide | Metastatic castration-resistant prostate cancer | 2012 |
| Erivedge® (Genentech) |  | Vismodegib | Basal cell carcinoma | 2012 |
| Inlyta® (Pfizer) |  | Axitinib | Renal cell carcinoma | 2012 |
| Imbruvica® (Pharmacyclics/Janssen Biotec) |  | Ibrutinib | Mantle cell lymphoma | 2013 |
| Pomalyst® (Celgene) |  | Pomalidomide | Multiple myeloma | 2013 |
| Lynparza® (AstraZeneca) |  | Olaparib | Advanced ovarian cancer | 2014 |
| Zydelig® (Pharmacyclics/Janssen Biotec) |  | Idelalisib | Chronic lymphocytic leukemia | 2014 |
| Zycadia® (Novartis) |  | Ceritinib | Metastatic NSCLC | 2014 |
| Farydak® (Novartis) |  | Panobinostat | Multiple myeloma | 2015 |
| Lenvima® (Eisai) |  | Lenvatinib | Progressive and differentiated thyroid cancer | 2015 |
| Ibrance® (Pfizer) |  | Palbociclib | Metastatic breast cancer | 2015 |
| Approved Oxygen-Based Heterocycle Drugs | ||||
| Jevtana® (Sanofi-aventis) |  | Cabazitaxel | Metastatic prostate cancer | 2010 |
| Halaven® (Eisai) |  | Eribulin mesylate | Metastatic breast cancer | 2010 |
| Approved Nitrogen, Oxygen-Based Heterocycle Drugs | ||||
| Synribo® (Frazer) |  | Omacetaxine mepesuccinate | Chronic myelogenous leukemia | 2012 |
| Kyprolis® (Onyx) |  | Carfilzomib | Multiple myeloma | 2012 |
| Gilotrif® (Boehringer Ingelheim) |  | Afatinib | Metastatic NSCLC (EGFR mutations) | 2013 |
| Mekinist® (GlaxoSmithKline) |  | Trametinib | Tumors that express the BRAF V600E or V600K gene mutations | 2013 |
| Approved Nitrogen, Sulfur-Based Heterocycle Drugs | ||||
| Tafinlar® (GlaxoSmithKline) |  | Dabrafenib | Melanoma that express the BRAF V600E gene mutation | 2013 |

1.3. Drug Design and Structure–Activity Relationship

2. Nanomedicine for Heterocyclic Compound Vectorization in Cancer
2.1. Nanoparticle Diversity
2.1.1. Liposomes
2.1.2. Polymeric Nanocarriers
| Name | Formulation | Target Ligand | Bioactive Compound | Indication | Status | Ref. |
|---|---|---|---|---|---|---|
| DaunoXome® | Non-PEGylated liposomes | None | Daunorubicin | Kaposi’s sarcoma | Approved | [77,80,88] |
| Myocet® | Non-PEGylated liposomes | None | Doxorubicin | Breast cancer | Approved | [77,80,88] |
| Depocyt® | Non-PEGylated liposomes | None | Cytarabine | Leukemia; Glioblastoma | Approved | [77,80,88] |
| Doxil®/Caelyx® | PEGylated liposomes | None | Doxorubicin | Breast cancer; ovarian cancer; multiple myeloma; Kaposi’s sarcoma | Approved | [77,80,88] |
| Thermodox® | PEGylated liposomes | None | Doxorubicin | Liver cancer; breast cancer | Phase III | [88] |
| NK105 | PEG-poly(aspartic acid) | None | Paclitaxel | Breast cancer | Phase III | [88] |
| Opaxio™ | PGA-paclitaxel | None | Paclitaxel | Lung cancer; ovarian cancer | Phase III | [77,88] |
| NC-6004 | PEG-poly(glutamic acid) | None | Cisplatin | Pancreatic cancer | Phase II/III | [77,88] |
| Abraxane® | Albumin-based | None | Paclitaxel | Breast cancer | Approved | [77,88] |
| Paclical® | Micellar retinoid-derived | None | Paclitaxel | Ovarian cancer | Phase III | [77,88] |
| Oncaspar® | PEG-l-asparaginase | None | Asparagine specific enzyme | Acute lymphoblastic leukemia | Approved | [77,88] |
| Lipo-Dox | PEGylated liposomes | None | Doxorubicin | Kaposi’s sarcoma; breast cancer and ovarian cancer | Approved | [77,80] |
| Marqibo | Non-PEGylated liposomes | None | Vincristine | Acute lymphoblastic leukemia | Approved | [77,80] |
| CPX-351 | Non-PEGylated liposomes | None | Cytarabine:daunorubicin | Acute myeloid leukemia | Phase II/III | [77,80] |
| MM-398 | Non-PEGylated liposomes | None | CPT-11 | Gastric and pancreatic cancer | Phase III | [77,80] |
| Lipoplatin | Non-PEGylated liposomes | None | Cisplatin | Non-small cell lung cancer | Phase III | [77,80] |
| ThermoDox | Non-PEGylated liposomes | None | Thermosensitive doxorubicin | Primary hepatocellular carcinoma | Phase III | [77,80] |
| Stimuvax | Non-PEGylated liposomes | None | Anti-MUC1 cancer vaccine | Non-small cell lung cancer | Phase III | [77,80] |
| Mylotarg® | Antibody drug conjugate (Gemtuzumab ozogamicin) | CD33 | Calicheamicin | Acute myeloid leukemia | Approved | [77] |
| Adcetris® | Antibody drug conjugate (Brentuximab vedotin) | CD30 | MMAE | Non-Hodgkin lymphoma | Approved | [77] |
| Kadcyla® | Antibody drug conjugate (Trastuzumab emtansine) | HER2 | DM1 | Breast cancer | Approved | [77] |
| CMC-544 | Antibody drug conjugate (Inotuzumab ozogamicin) | CD22 | Calicheamicin | Acute lymphoblastic leukemia | Phase III | [89] |
| CMA-676 | Antibody drug conjugate (Gemtuzumab ozogamicin) | CD33 | Calicheamicin | Aute Myeloid Leukemia | Phase III | [89] |
| Genexol-PM® (IG-001) | PEGylated liposomes | None | Paclitaxel | Breast Cancer; Lung Cancer | Approved | [77] |
| Mepact® | Non-PEGylated liposomes | None | Mifamurtide | Osteosarcoma | Approved | [77] |
| Zinostatin stimalamer® | Polymer protein conjugate | None | Styrene maleic anhydride neocarzinostatin (SMANCS) | Liver cancer, renal cancer | Approved | [77] |
| NKTR-102 (Etirinotecan pegol) | PEG drug conjugate | None | Irinotecan | Breast cancer; Ovarian Cancer; Colorectal Cancer | Phase III | [77] |
| Taxoprexin | Docosahexaenoic acid drug conjugate | None | Paclitaxel | Melanoma; Liver cancer; Adenocarcinoma; Kidney Cancer; Non-small-cell lung cancer | Phase II/III | [77] |
| Lipusu | Non-PEGylated liposomes | None | Paclitaxel | Solid tumors; Metastatic Breast Cancer | Phase IV | [77] |
2.1.3. Albumin Bound Nanoparticles
2.1.4. Metallic Nanoparticles
2.1.5. Drug Conjugates
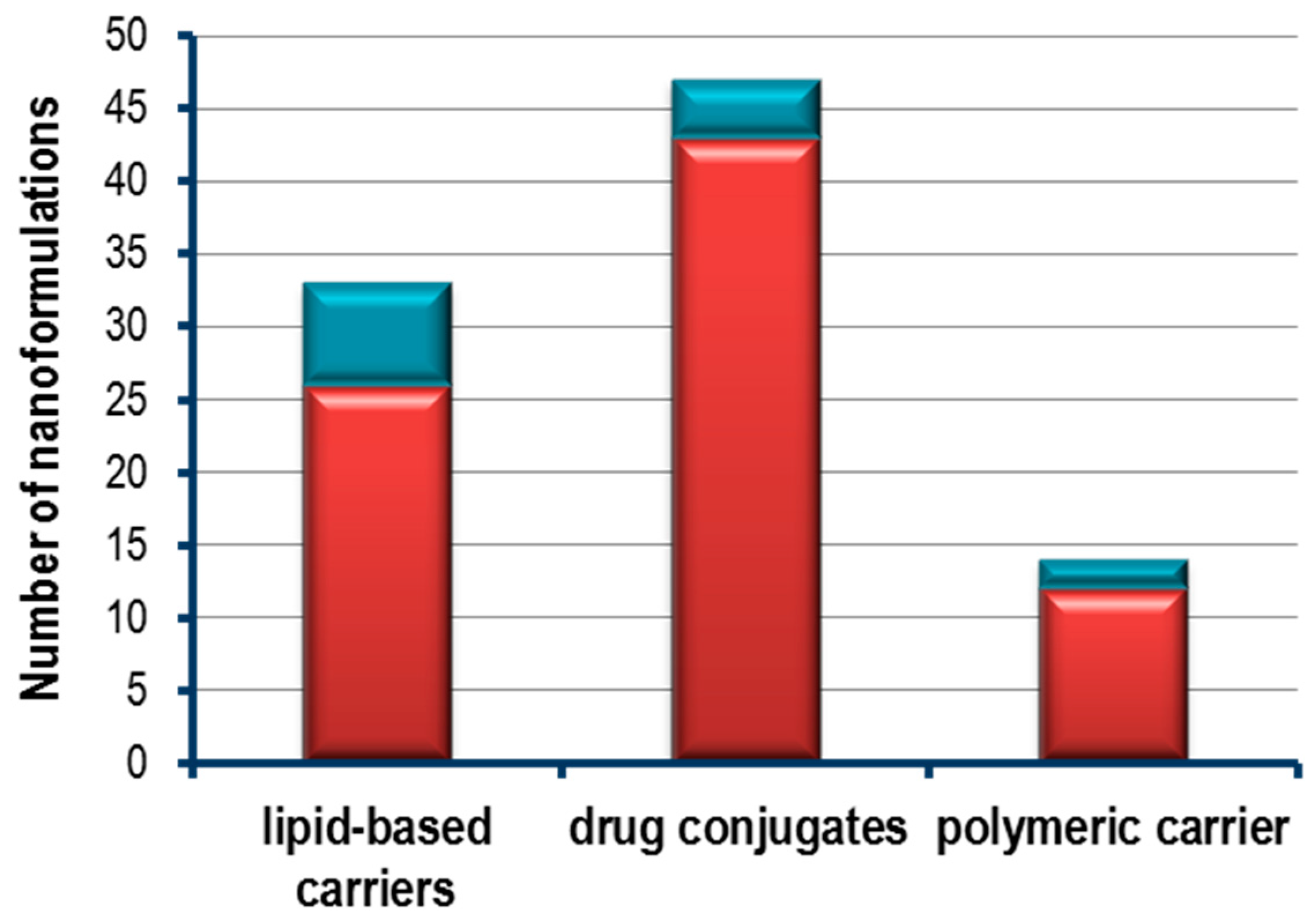
2.2. From Bench to Bedside

2.3. Fundamental Aspects of Nanoformulation Design and in Vivo Interactions
2.3.1. Nanomaterial–Cell Interactions and Cell Uptake
2.3.2. Nanoparticle Size and Shape
2.3.3. Natural Barriers
2.3.4. Drug Release Rate
2.4. Challenges Still to Overcome
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- IUPAC Gold Book—Heterocyclic Compounds. Available online: http://goldbook.iupac.org/H02798.html (accessed on 26 May 2015).
- Gomtsyan, A. Heterocycles in drugs and drug discovery. Chem. Heterocycl. Compd. 2012, 48, 7–10. [Google Scholar] [CrossRef]
- Dua, R.; Shrivastava, S.; Sonwane, S.K.; Srivastava, S.K. Pharmacological Significance of Synthetic Heterocycles Scaffold : A Review. Adv. Biol. Res. (Rennes). 2011, 5, 120–144. [Google Scholar]
- Eicher, T.; Hauptmann, S.; Speicher, A. (Eds.) The Structure of Heterocyclic Compounds. In The Chemistry of Heterocycles: Structure, Reactions, Synthesis, and Applications, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2012; pp. 1–4.
- Broughton, H.B.; Watson, I.A. Selection of heterocycles for drug design. J. Mol. Graph. Model. 2004, 23, 51–58. [Google Scholar] [CrossRef] [PubMed]
- El-salam, N.M.A.; Mostafa, M.S.; Ahmed, G.A.; Alothman, O.Y. Synthesis and Antimicrobial Activities of Some New Heterocyclic Compounds Based on 6-Chloropyridazine-3 (2H) -thione. J. Chem. 2013, 2013, 1–8. [Google Scholar] [CrossRef]
- Azab, M.E.; Youssef, M.M.; El-Bordany, E.A. Synthesis and antibacterial evaluation of novel heterocyclic compounds containing a sulfonamido moiety. Molecules 2013, 18, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.S.; Sakr, S.I.; El-Senousy, W.M.; Madkour, H.M.F. Synthesis, antibacterial, and antiviral evaluation of new heterocycles containing the pyridine moiety. Arch. Pharm. (Weinheim). 2013, 346, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Sun, Z.; Cao, Y.; Wang, R.; Cai, T.; Chu, W.; Hu, W.; Yang, Y. Design, Synthesis, and Structure—Activity Relationship Studies of Novel Fused Heterocycles-Linked Triazoles with Good Activity and Water Solubility. J. Med. Chem. 2014, 57, 3687–3706. [Google Scholar] [CrossRef] [PubMed]
- El-Sawy, E.R.; Ebaid, M.S.; Abo-Salem, H.M.; Al-Sehemi, A.G.; Mandour, A.H. Synthesis, anti-inflammatory, analgesic and anticonvulsant activities of some new 4,6-dimethoxy-5-(heterocycles)benzofuran starting from naturally occurring visnagin. Arab. J. Chem. 2013, 7, 914–923. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, K.; Tan, N.Y.; Qiu, R.H.; Liu, W.; Luo, N.L.; Tong, L.; Au, C.T.; Luo, Z.Q.; Yin, S.F. Synthesis, characterization and anti-proliferative activity of heterocyclic hypervalent organoantimony compounds. Eur. J. Med. Chem. 2014, 79, 391–398. [Google Scholar] [CrossRef] [PubMed]
- El-Sawy, E.R.; Mandour, A.H.; El-Hallouty, S.M.; Shaker, K.H.; Abo-Salem, H.M. Synthesis, antimicrobial and anticancer activities of some new N-methylsulphonyl and N-benzenesulphonyl-3-indolyl heterocycles. 1st Cancer Update. Arab. J. Chem. 2013, 6, 67–78. [Google Scholar] [CrossRef]
- Mabkhot, Y.N.; Barakat, A.; Al-Majid, A.M.; Alshahrani, S.; Yousuf, S.; Choudhary, M.I. Synthesis, reactions and biological activity of some new bis-heterocyclic ring compounds containing sulphur atom. Chem. Cent. J. 2013, 7, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Alvárez-Builla, J.; Barluenga, J. Heterocyclic Compounds: An Introduction. Mod. Heterocycl. Chem. 2011, 1, 1–9. [Google Scholar]
- Top Prescription Drugs by U.S. Sales 2014|Statistic. Available online: http://www.statista.com/statistics/258010/top-branded-drugs-based-on-retail-sales-in-the-us/ (accessed on 28 May 2015).
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Hambley, T.W.; Hait, W.N. Is anticancer drug development heading in the right direction? Cancer Res. 2009, 69, 1259–1262. [Google Scholar] [CrossRef] [PubMed]
- Martins, P.; Rosa, D.; Fernandes, A.R.; Baptista, P.V. Nanoparticle Drug Delivery Systems : Recent Patents and Applications in Nanomedicine. Recent Pat. Nanomed. 2014, 3, 1–14. [Google Scholar] [CrossRef]
- Conde, J.; Ambrosone, A.; Sanz, V.; Hernandez, Y.; Marchesano, V.; Tian, F.; Child, H.; Berry, C.C.; Ibarra, M.R.; Baptista, P.V.; Tortiglione, C.; Fuente, J.M. De Design of Multifunctional Gold Nanoparticles for in Vitro and in Vivo Gene Silencing. ACS Nano 2012, 6, 8316–8324. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.; Doria, G.; Baptista, P. Noble metal nanoparticles applications in cancer. J. Drug Deliv. 2012, 2012, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Market Opportunities in Nanotechnology Drug Delivery. Available online: http://www.cientifica.com/research/white-papers/market-opportunities-in-nanotechnology-drug-delivery/ (accessed on 28 May 2015).
- Research, C. for D.E. and New Drugs at FDA: CDER’s New Molecular Entities and New Therapeutic Biological Products. Available online: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/default.htm (accessed on 1 June 2015).
- Click2Drug—Encyclopedia—Chemical Compounds—Most Frequent Rings in FDA Approved Drugs. Available online: http://www.click2drug.org/encyclopedia/chemistry/fda-based-rings.html (accessed on 5 June 2015).
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.A.S.; Dar, B.A.; Pradhan, V.; Farooqui, M. Chemistry and biology of indoles and indazoles: a mini-review. Mini Rev. Med. Chem. 2013, 13, 1792–1800. [Google Scholar] [PubMed]
- Kaushik, N.K.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C.H.; Verma, A.K.; Choi, E.H. Biomedical importance of indoles. Molecules 2013, 18, 6620–6662. [Google Scholar] [CrossRef] [PubMed]
- Sherer, C.; Snape, T.J. Heterocyclic scaffolds as promising anticancer agents against tumours of the central nervous system: Exploring the scope of indole and carbazole derivatives. Eur. J. Med. Chem. 2015, 97, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Brancale, A.; Silvestri, R. Indole, a core nucleus for potent inhibitors of tubulin polymerization. Med. Res. Rev. 2007, 27, 209–238. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Mehndiratta, S.; Nepali, K.; Gupta, M.K.; Koul, S.; Sharma, P.R.; Saxena, A.K.; Dhar, K.L. Novel indole-bearing combretastatin analogues as tubulin polymerization inhibitors. Org. Med. Chem. Lett. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Hsu, P.C.; Chen, M.Y.; Li, W.S.; More, S.V.; Lu, K.T.; Wang, Y.C. The novel indole compound SK228 induces apoptosis and FAK/Paxillin disruption in tumor cell lines and inhibits growth of tumor graft in the nude mouse. Int. J. Cancer 2012, 131, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Joshi, S.; Singh, D. Imidazole: Having versatile biological activities. J. Chem. 2013, 2013, 1–12. [Google Scholar] [CrossRef]
- Sharma, G.V.M.; Ramesh, A.; Singh, A.; Srikanth, G.; Jayaram, V.; Duscharla, D.; Jun, J.H.; Ummanni, R.; Malhotra, S.V. Imidazole derivatives show anticancer potential by inducing apoptosis and cellular senescence. Med. Chem. Commun. 2014, 5, 1751–1760. [Google Scholar] [CrossRef]
- Hou, J.; Zhao, W.; Huang, Z.-N.; Yang, S.-M.; Wang, L.-J.; Jiang, Y.; Zhou, Z.-S.; Zheng, M.-Y.; Jiang, J.-L.; Li, S.-H.; et al. Evaluation of Novel N -(piperidine-4-yl)benzamide Derivatives as Potential Cell Cycle Inhibitors in HepG2 Cells. Chem. Biol. Drug Des. 2014, 1–9. [Google Scholar]
- Khan, I.; Ibrar, A.; Abbas, N. Triazolothiadiazoles and triazolothiadiazines-Biologically attractive scaffolds. Eur. J. Med. Chem. 2013, 63, 854–868. [Google Scholar] [CrossRef] [PubMed]
- Husain, A.; Rashid, M.; Shaharyar, M.; Siddiqui, A.A.; Mishra, R. Benzimidazole clubbed with triazolo-thiadiazoles and triazolo-thiadiazines: New anticancer agents. Eur. J. Med. Chem. 2013, 62, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Husain, A.; Rashid, M.; Mishra, R.; Parveen, S.; Shin, D.S.; Kumar, D. Benzimidazole bearing oxadiazole and triazolo-thiadiazoles nucleus: Design and synthesis as anticancer agents. Bioorg. Med. Chem. Lett. 2012, 22, 5438–5444. [Google Scholar] [CrossRef] [PubMed]
- Kamel, M.M.; Megally Abdo, N.Y. Synthesis of novel 1,2,4-triazoles, triazolothiadiazines and triazolothiadiazoles as potential anticancer agents. Eur. J. Med. Chem. 2014, 86, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Mekhail, T.M.; Markman, M. Paclitaxel in cancer therapy. Expert Opin. Pharmacother. 2002, 3, 755–766. [Google Scholar] [PubMed]
- Vrignaud, P.; Sémiond, D.; Lejeune, P.; Bouchard, H.; Calvet, L.; Combeau, C.; Riou, J.F.; Commerçon, A.; Lavelle, F.; Bissery, M.C. Preclinical antitumor activity of cabazitaxel, a semisynthetic taxane active in taxane-resistant tumors. Clin. Cancer Res. 2013, 19, 2973–2983. [Google Scholar] [CrossRef] [PubMed]
- Devriese, L.A.; Mergui-Roelvink, M.; Wanders, J.; Jenner, A.; Edwards, G.; Reyderman, L.; Copalu, W.; Peng, F.; Marchetti, S.; Beijnen, J.H.; et al. Eribulin mesylate pharmacokinetics in patients with solid tumors receiving repeated oral ketoconazole. Investig. New Drugs 2013, 31, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Yadagiri, B.; Holagunda, U.D.; Bantu, R.; Nagarapu, L.; Kumar, C.G.; Pombala, S.; Sridhar, B. Synthesis of novel building blocks of benzosuberone bearing coumarin moieties and their evaluation as potential anticancer agents. Eur. J. Med. Chem. 2014, 79, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Kontogiorgis, C.; Detsi, A.; Hadjipavlou-litina, D. Coumarin-Based Drugs : A Patent Review (2008–present ). Expert Opin. Ther. Pat. 2012, 22, 437–454. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Kohli, S.; Sandhu, S.; Bansal, Y.; Bansal, G. Coumarin: A Promising Scaffold for Anticancer Agents. Anticancer Agents Med. Chem. 2015. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, J.H.; Huang, Q.; Lu, L.; Min, W. Novel Action and Mechanism of Auranofin in Inhibition of Vascular Endothelial Growth Factor Receptor-3-Dependent Lymphangiogenesis|BenthamScience. Anti-Cancer Agents 2014, 14, 946–954. [Google Scholar] [CrossRef]
- Liu, N.; Li, X.; Huang, H.; Zhao, C.; Liao, S.; Yang, C.; Liu, S.; Song, W.; Lu, X.; Lan, X.; et al. Clinically used antirheumatic agent auranofin is a proteasomal deubiquitinase inhibitor and inhibits tumor growth. Oncotarget 2014, 5, 5453–5471. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Lee, J.; Berek, J.; Hu, M. Auranofin displays anticancer activity against ovarian cancer cells through FOXO3 activation independent of p53. Int. J. Oncol. 2014, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Murti, Y.; Mishra, P. Synthesis and evaluation of flavanones as anticancer agents. Indian J. Pharm. Sci. 2014, 76, 163–166. [Google Scholar] [PubMed]
- Khanam, H. Shamsuzzaman Bioactive Benzofuran derivatives: A review. Eur. J. Med. Chem. 2014, 97, 483–504. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Jo, H.; Park, H.-J.; Sateesh Kumar, A.; Lee, J.; Yun, J.; Kim, Y.; Han, S.; Jung, J.-K.; Cho, J.; et al. Design, synthesis, and biological evaluation of benzofuran- and 2,3-dihydrobenzofuran-2-carboxylic acid N-(substituted)phenylamide derivatives as anticancer agents and inhibitors of NF-κB. Bioorg. Med. Chem. Lett. 2015, 25, 2545–2549. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.A.R.; Bomfim, I.D.S.; Cavalcanti, B.C.; Pessoa, C.; Goncalves, R.S.B.; Wardell, J.L.; Wardell, S.M.S.V.; de Souza, M.V.N. Mefloquine-Oxazolidine Derivatives: A New Class of Anticancer Agents. Chem. Biol. Drug Des. 2014, 83, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Andrade, S.F.; Teixeira, C.S.; Ramos, J.P.; Lopes, M.S.; Pádua, R.M.; Oliveira, M.C.; Souza-Fagundes, E.M.; Alves, R.J. Synthesis of a novel series of 2,3,4-trisubstituted oxazolidines designed by isosteric replacement or rigidification of the structure and cytotoxic evaluation. Med. Chem. Commun. 2014, 5, 1693–1699. [Google Scholar] [CrossRef]
- Khatik, G.L.; Kaur, J.; Kumar, V.; Tikoo, K.; Nair, V.A. 1,2,4-Oxadiazoles: A new class of anti-prostate cancer agents. Bioorg. Med. Chem. Lett. 2012, 22, 1912–1916. [Google Scholar] [CrossRef] [PubMed]
- Valente, S.; Trisciuoglio, D.; de Luca, T.; Nebbioso, A.; Labella, D.; Lenoci, A.; Bigogno, C.; Dondio, G.; Miceli, M.; Brosch, G.; et al. 1,3,4-Oxadiazole-containing histone deacetylase inhibitors: Anticancer activities in cancer cells. J. Med. Chem. 2014, 57, 6259–6265. [Google Scholar] [CrossRef] [PubMed]
- García-Valverde, M.; Torroba, T. Special Issue: Sulfur-Nitrogen Heterocycles. Molecules 2005, 10, 318–320. [Google Scholar] [CrossRef]
- Marcos, C.F.; Polo, C.; Rakitin, O.A.; Rees, C.W.; Torroba, T. From Hiinig’s Base to Bis([l,2]dithiolo)-[1,4]thiazines in One Pot: The Fast Route to Highly Sulfurated Heterocycles. Angew. Chem. Int. Ed. Engl. 1997, 36, 281–283. [Google Scholar]
- Makki, M.S.T.; Abdel-rahman, R.M.; El-Shahawi, M.S. Synthesis of New Bioactive Sulfur Compounds Bearing Heterocyclic Moiety and Their Analytical Applications. Int. J. Chem. 2011, 3, 181–192. [Google Scholar] [CrossRef]
- Toohey, J.; Cooper, A. Thiosulfoxide (Sulfane) Sulfur: New Chemistry and New Regulatory Roles in Biology. Molecules 2014, 19, 12789–12813. [Google Scholar] [CrossRef] [PubMed]
- Said, M.; Elshihawy, H. Synthesis, anticancer activity and structure-activity relationship of some anticancer agents based on Cyclopenta (b) thiophene scaffold. Pak. J. Pharm. Sci. 2014, 27, 885–892. [Google Scholar] [PubMed]
- Ghorab, M.M.; Bashandy, M.S.; Alsaid, M.S. Novel thiophene derivatives with sulfonamide, isoxazole, benzothiazole, quinoline and anthracene moieties as potential anticancer agents. Acta Pharm. 2014, 64, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.S.; Kramer, S.; Talley, J.J.; Penning, T.; Collins, P.; Graneto, M.J.; Seibert, K.; Koboldt, C.M.; Masferrer, J.; Zweifel, B. Synthesis and activity of sulfonamide-substituted 4,5-diaryl thiazoles as selective cyclooxygenase-2 inhibitors. Bioorg. Med. Chem. Lett. 1999, 9, 1171–1174. [Google Scholar] [CrossRef]
- Rudolph, J.; Theis, H.; Hanke, R.; Endermann, R.; Johannsen, L.; Geschke, F. seco-Cyclothialidines: new concise synthesis, inhibitory activity toward bacterial and human DNA topoisomerases, and antibacterial properties. J. Med. Chem. 2001, 44, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.H.; Shi, Y.X.; Ma, Y.; Zhang, C.Y.; Dong, W.L.; Pan, L.; Wang, B.L.; Li, B.J.; Li, Z.M. Synthesis, antifungal activities and 3D-QSAR study of N-(5-substituted-1,3,4-thiadiazol-2-yl)cyclopropanecarboxamides. Eur. J. Med. Chem. 2009, 44, 2782–2786. [Google Scholar] [CrossRef] [PubMed]
- Bell, F.W.; Cantrell, A.S.; Högberg, M.; Jaskunas, S.R.; Johansson, N.G.; Jordan, C.L.; Kinnick, M.D.; Lind, P.; Morin, J.M.; Noréen, R. Phenethylthiazolethiourea (PETT) compounds, a new class of HIV-1 reverse transcriptase inhibitors. 1. Synthesis and basic structure-activity relationship studies of PETT analogs. J. Med. Chem. 1995, 38, 4929–4936. [Google Scholar] [CrossRef] [PubMed]
- Laczkowski, K.Z.K.M.; Switalska, M.; Wietrzyk, J.; Baranowska, A.L.; Berta, F.; Paneth, A.; Plech, T. Synthesis and in Vitro Antiproliferative Activity of Thiazole-Based Nitrogen Mustards: The Hydrogen Bonding Interaction between Model Systems and Nucleobases. Anti-Cancer Agents 2014, 14, 1271–1281. [Google Scholar] [CrossRef]
- Penthala, N.R.; Sonar, V.N.; Horn, J.; Leggas, M.; Yadlapalli, J.S.K.B.; Crooks, P.A. Synthesis and evaluation of a series of benzothiophene acrylonitrile analogs as anticancer agents. MedChemComm 2012, 4, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Molica, S. The emerging role of ibrutinib in the treatment of chronic lymphocytic leukemia. Expert Rev. Hematol. 2013, 4, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Fiorcari, S.; Brown, W.S.; McIntyre, B.W.; Estrov, Z.; Maffei, R.; O’Brien, S.; Sivina, M.; Hoellenriegel, J.; Wierda, W.G.; Keating, M.J.; et al. The PI3-kinase delta inhibitor idelalisib (GS-1101) targets integrin-mediated adhesion of chronic lymphocytic leukemia (CLL) cell to endothelial and marrow stromal cells. PLoS ONE 2013, 8, e83830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fact Sheets by Population. Available online: http://globocan.iarc.fr/Pages/fact_sheets_population.aspx (accessed on 8 June 2015).
- Cadoo, K.A.; Gucalp, A.; Traina, T.A. Palbociclib: An evidence-based review of its potential in the treatment of breast cancer. Dove Press 2014, 4, 123–133. [Google Scholar]
- Home—PubChem Compound—NCBI. Available online: http://www.ncbi.nlm.nih.gov/pccompound (accessed on 10 September 2015).
- Noori, H.R.; Spanagel, R. In silico pharmacology: Drug design and discovery’s gate to the future. Silico Pharmacol. 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.F.; Swanson, S.M.; Williams, J.A. Molecular modelling and drug design. Pharmacol. Ther. 2000, 85, 113–121. [Google Scholar] [CrossRef]
- Chaniyara, R.; Tala, S.; Chen, C.W.; Lee, P.C.; Kakadiya, R.; Dong, H.; Marvania, B.; Chen, C.H.; Chou, T.C.; Lee, T.C.; et al. Synthesis and antitumor evaluation of novel Benzo[d]pyrrolo[2,1-b]thiazole derivatives. Eur. J. Med. Chem. 2012, 53, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Baptista, P.; Fernandes, A.; Figueiredo, S.; Vinhas, R.; Cordeiro, M.; Carlos, F.; Mendo, S. Gold nanoparticle-based theranostics: Disease diagnostics and treatment using a single nanomaterial. Nanobiosen. Dis. Diagn. 2015, 4, 11. [Google Scholar] [CrossRef]
- Xu, X.; Ho, W.; Zhang, X.; Bertrand, N.; Farokhzad, O. Cancer nanomedicine: From targeted delivery to combination therapy. Trends Mol. Med. 2015, 21, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Sagnella, S.M.; McCarroll, J.A.; Kavallaris, M. Drug delivery: Beyond active tumour targeting. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control. Release 2015, 200, 138–157. [Google Scholar] [CrossRef] [PubMed]
- Estanqueiro, M.; Amaral, M.H.; Conceição, J.; Sousa Lobo, J.M. Nanotechnological carriers for cancer chemotherapy: The state of the art. Colloids Surf. B Biointerfaces 2015, 126, 631–648. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gu, F.X.; Chan, J.M.; Wang, A.Z.; Langer, R.S.; Farokhzad, O.C. Nanoparticles in Medicine : Therapeutic Applications and Developments. Clin. Pharmacol. Ther. 2008, 83, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.Z.; Langer, R.; Farokhzad, O.C. Nanoparticle delivery of cancer drugs. Annu. Rev. Med. 2012, 63, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J. Complement activation-related pseudoallergy: A new class of drug-induced acute immune toxicity. Toxicology 2005, 216, 106–121. [Google Scholar] [CrossRef] [PubMed]
- Parhi, P.; Mohanty, C.; Sahoo, S.K. Nanotechnology-based combinational drug delivery: An emerging approach for cancer therapy. Drug Discov. Today 2012, 17, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Nazir, S.; Hussain, T.; Ayub, A.; Rashid, U.; MacRobert, A.J. Nanomaterials in combating cancer: Therapeutic applications and developments. Nanomedicine 2014, 10, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Gillies, E.R.; Fréchet, J.M.J. Dendrimers and dendritic polymers in drug delivery. Drug Discov. Today 2005, 10, 35–43. [Google Scholar] [CrossRef]
- Kesharwani, P.; Iyer, A.K. Recent advances in dendrimer-based nanovectors for tumor-targeted drug and gene delivery. Drug Discov. Today 2014, 20, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Sanna, V.; Pala, N.; Sechi, M. Targeted therapy using nanotechnology: Focus on cancer. Int. J. Nanomed. 2014, 9, 467–483. [Google Scholar]
- Chari, R.V.J.; Miller, M.L.; Widdison, W.C. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew. Chem. Int. Ed. 2014, 53, 3796–3827. [Google Scholar] [CrossRef] [PubMed]
- Elzoghby, A.O.; Samy, W.M.; Elgindy, N.A. Albumin-based nanoparticles as potential controlled release drug delivery systems. J. Control. Release 2012, 157, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Cabral, R.M.; Baptista, P.V. The Chemistry and Biology of Gold Nanoparticle-Mediated Photothermal Therapy: Promises and Challenges. Nano Life 2013, 3, 1330001. [Google Scholar] [CrossRef]
- Peralta, D.V.; Heidari, Z.; Dash, S.; Tarr, M.A. Hybrid Paclitaxel and Gold Nanorod-Loaded Human Serum Albumin Nanoparticles for Simultaneous Chemotherapeutic and Photothermal Therapy on 4T1 Breast Cancer Cells. ACS Appl. Mater. Interfaces 2015, 7, 7101–7111. [Google Scholar] [CrossRef] [PubMed]
- Zolot, R.S.; Basu, S.; Million, R.P. Antibody–drug conjugates. Nat. Rev. Drug Discov. 2013, 12, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A. Antibody drug conjugates. Curr. Opin. Oncol. 2014, 26, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. MAbs 2014, 6, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Dinutuximab: First Global Approval. Drugs 2015, 75, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G. Nivolumab: Another weapon in the growing immunotherapy arsenal. Lancet Oncol. 2015, 16, 350–351. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Flygare, J.A.; Pillow, T.H.; Aristoff, P. Antibody-Drug Conjugates for the Treatment of Cancer. Chem. Biol. Drug Des. 2013, 81, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Ornes, S. Antibody–drug conjugates. Proc. Natl. Acad. Sci. USA 2013, 110, 13695. [Google Scholar] [CrossRef] [PubMed]
- Casi, G.; Neri, D. Antibody-drug conjugates: Basic concepts, examples and future perspectives. J. Control. Release 2012, 161, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Muggia, F.M.; Hainsworth, J.D.; Jeffers, S.; Miller, P.; Groshen, S.; Tan, M.; Roman, L.; Uziely, B.; Muderspach, L.; Garcia, A.; et al. Phase II study of liposomal doxorubicin in refractory ovarian cancer: Antitumor activity and toxicity modification by liposomal encapsulation. J. Clin. Oncol. 1997, 15, 987–993. [Google Scholar] [PubMed]
- Khemapech, N.; Oranratanaphan, S.; Termrungruanglert, W.; Lertkhachonsuk, R.; Vasurattana, A. Salvage chemotherapy in recurrent platinum-resistant or refractory epithelial ovarian cancer with Carboplatin and distearoylphosphatidylcholine pegylated liposomal Doxorubicin (lipo-dox®). Asian Pac. J. Cancer Prev. 2013, 14, 2131–2135. [Google Scholar] [CrossRef] [PubMed]
- Batist, G.; Ramakrishnan, G.; Rao, C.S.; Chandrasekharan, A.; Gutheil, J.; Guthrie, T.; Shah, P.; Khojasteh, A.; Nair, M.K.; Hoelzer, K.; et al. Reduced cardiotoxicity and preserved antitumor efficacy of liposome-encapsulated doxorubicin and cyclophosphamide compared with conventional doxorubicin and cyclophosphamide in a randomized, multicenter trial of metastatic breast cancer. J. Clin. Oncol. 2001, 19, 1444–1454. [Google Scholar] [PubMed]
- Tejada-Berges, T.; Granai, C.O.; Gordinier, M.; Gajewski, W. Caelyx/Doxil for the treatment of metastatic ovarian and breast cancer. Expert Rev. Anticancer Ther. 2002, 2, 143–150. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency—Find Medicine—Myocet. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000297/human_med_000916.jsp&mid=WC0b01ac058001d124 (accessed on 27 August 2015).
- European Medicines Agency—Find medicine—Caelyx. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000089/human_med_000683.jsp&mid=WC0b01ac058001d124 (accessed on 27 August 2015).
- Awada, A.; Garcia, A.A.; Chan, S.; Jerusalem, G.H.M.; Coleman, R.E.; Huizing, M.T.; Mehdi, A.; O’Reilly, S.M.; Hamm, J.T.; Barrett-Lee, P.J.; et al. Two schedules of etirinotecan pegol (NKTR-102) in patients with previously treated metastatic breast cancer: A randomised phase 2 study. Lancet Oncol. 2013, 14, 1216–1225. [Google Scholar] [CrossRef]
- Feldman, E.J.; Lancet, J.E.; Kolitz, J.E.; Ritchie, E.K.; Roboz, G.J.; List, A.F.; Allen, S.L.; Asatiani, E.; Mayer, L.D.; Swenson, C.; et al. First-in-man study of CPX-351: A liposomal carrier containing cytarabine and daunorubicin in a fixed 5:1 molar ratio for the treatment of relapsed and refractory acute myeloid leukemia. J. Clin. Oncol. 2011, 29, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Sarris, A.H.; Hagemeister, F.; Romaguera, J.; Rodriguez, M.A.; McLaughlin, P.; Tsimberidou, A.M.; Medeiros, L.J.; Samuels, B.; Pate, O.; et al. Liposomal vincristine in relapsed non-Hodgkin’s lymphomas: Early results of an ongoing phase II trial. Ann. Oncol. 2000, 11, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.; Larguinho, M.; Cordeiro, A.; Raposo, L.R.; Costa, P.M.; Santos, S.; Diniz, M.S.; Fernandes, A.R.; Baptista, P. V Gold-nanobeacons for gene therapy: Evaluation of genotoxicity, cell toxicity and proteome profiling analysis. Nanotoxicology 2014, 8, 521–532. [Google Scholar] [CrossRef] [PubMed]
- R. Fernandes, A.; Viana Baptista, P. Nanotechnology for Cancer Diagnostics and Therapy – An Update on Novel Molecular Players. Curr. Cancer Ther. Rev. 2013, 164–172. [Google Scholar] [CrossRef]
- Zhao, F.; Zhao, Y.; Liu, Y.; Chang, X.; Chen, C.; Zhao, Y. Cellular uptake, intracellular trafficking, and cytotoxicity of nanomaterials. Small 2011, 7, 1322–1337. [Google Scholar] [CrossRef] [PubMed]
- Iversen, T.G.; Skotland, T.; Sandvig, K. Endocytosis and intracellular transport of nanoparticles: Present knowledge and need for future studies. Nano Today 2011, 6, 176–185. [Google Scholar] [CrossRef]
- Shukla, R.; Bansal, V.; Chaudhary, M.; Basu, A.; Bhonde, R.R.; Sastry, M. Biocompatibility of gold nanoparticles and their endocytotic fate inside the cellular compartment: A microscopic overview. Langmuir 2005, 21, 10644–10654. [Google Scholar] [CrossRef] [PubMed]
- Murugan, M.; Anthony, K.J.P.; Jeyaraj, M.; Rathinam, N.K.; Gurunathan, S. Biofabrication of gold nanoparticles and its biocompatibility in human breast adenocarcinoma cells (MCF-7). J. Ind. Eng. Chem. 2013, 20, 1713–1719. [Google Scholar] [CrossRef]
- Alkilany, A.M.; Murphy, C.J. Toxicity and cellular uptake of gold nanoparticles: What we have learned so far? J. Nanopart. Res. 2010, 12, 2313–2333. [Google Scholar] [CrossRef] [PubMed]
- Coradeghini, R.; Gioria, S.; García, C.P.; Nativo, P.; Franchini, F.; Gilliland, D.; Ponti, J.; Rossi, F. Size-dependent toxicity and cell interaction mechanisms of gold nanoparticles on mouse fibroblasts. Toxicol. Lett. 2013, 217, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Patlolla, A.; Knighten, B.; Tchounwou, P. Multi-walled carbon nanotubes induce cytotoxicity, genotoxicity and apoptosis in normal human dermal fibroblast cells. Ethn. Dis. 2010, 20, 1–17. [Google Scholar]
- Kumarathasan, P.; Breznan, D.; Das, D.; Salam, M.A.; Siddiqui, Y.; MacKinnon-Roy, C.; Guan, J.; de Silva, N.; Simard, B.; Vincent, R. Cytotoxicity of carbon nanotube variants: A comparative: In vitro exposure study with A549 epithelial and J774 macrophage cells. Nanotoxicology 2015, 9, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Chithrani, B.D.; Ghazani, A.A.; Chan, W.C.W. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 2006, 6, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Chithrani, D.B. Intracellular uptake, transport, and processing of gold nanostructures. Mol. Membr. Biol. 2010, 27, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Oh, N.; Park, J.H. Endocytosis and exocytosis of nanoparticles in mammalian cells. Int. J. Nanomed. 2014, 9, 51–63. [Google Scholar]
- Win, K.Y.; Feng, S.-S. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials 2005, 26, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Nativo, P.; Prior, I.A.; Brust, M. Uptake and intracellular fate of surface-modified gold nanoparticles. ACS Nano 2008, 2, 1639–1644. [Google Scholar] [CrossRef] [PubMed]
- Sahay, G.; Alakhova, D.Y.; Kabanov, A.V. Endocytosis of nanomedicines. J. Control. Release 2010, 145, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Farhangrazi, Z.S. Just so stories: The random acts of anti-cancer nanomedicine performance. Nanomedicine 2014, 8, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Taurin, S.; Nehoff, H.; Greish, K. Anticancer nanomedicine and tumor vascular permeability; Where is the missing link? J. Control. Release 2012, 164, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Toy, R.; Peiris, P.M.; Ghaghada, K.B.; Karathanasis, E. Shaping cancer nanomedicine: The effect of particle shape on the in vivo journey of nanoparticles. Nanomedicine (Lond). 2014, 9, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y. Nanotechnology platforms and physiological challenges for cancer therapeutics. Nanomedicine 2007, 3, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, C.; Meade, Th.J.; Petrosko, S.H.; Stegh, A.H. Nanotechnology-Based Precision Tools for the Detection and Treatment of Cancer; Springer: Phoenix, AZ, USA, 2015; Volume 166. [Google Scholar]
- Naguib, Y.W.; Cui, Z. Nanomedicine: The promise and challenges in cancer chemotherapy. Adv. Exp. Med. Biol. 2014, 811, 207–233. [Google Scholar] [PubMed]
- Doane, T.L.; Burda, C. The unique role of nanoparticles in nanomedicine: Imaging, drug delivery and therapy. Chem. Soc. Rev. 2012, 41, 2885. [Google Scholar] [CrossRef] [PubMed]
- Fubini, B.; Ghiazza, M.; Fenoglio, I. Physico-chemical features of engineered nanoparticles relevant to their toxicity. Nanotoxicology 2010, 4, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Kettiger, H.; Schipanski, A.; Wick, P.; Huwyler, J. Engineered nanomaterial uptake and tissue distribution: from cell to organism. Int. J. Nanomed. 2013, 8, 3255–3269. [Google Scholar]
- Stone, V.; Johnston, H.; Schins, R.P.F. Development of in vitro systems for nanotoxicology: methodological considerations. Crit. Rev. Toxicol. 2009, 39, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Hamburg, M.A. Science and regulation. FDA’s approach to regulation of products of nanotechnology. Science 2012, 336, 299–300. [Google Scholar] [CrossRef] [PubMed]
- Desai, N. Challenges in development of nanoparticle-based therapeutics. AAPS J. 2012, 14, 282–295. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, P.; Jesus, J.; Santos, S.; Raposo, L.R.; Roma-Rodrigues, C.; Baptista, P.V.; Fernandes, A.R. Heterocyclic Anticancer Compounds: Recent Advances and the Paradigm Shift towards the Use of Nanomedicine’s Tool Box. Molecules 2015, 20, 16852-16891. https://doi.org/10.3390/molecules200916852
Martins P, Jesus J, Santos S, Raposo LR, Roma-Rodrigues C, Baptista PV, Fernandes AR. Heterocyclic Anticancer Compounds: Recent Advances and the Paradigm Shift towards the Use of Nanomedicine’s Tool Box. Molecules. 2015; 20(9):16852-16891. https://doi.org/10.3390/molecules200916852
Chicago/Turabian StyleMartins, Pedro, João Jesus, Sofia Santos, Luis R. Raposo, Catarina Roma-Rodrigues, Pedro Viana Baptista, and Alexandra R. Fernandes. 2015. "Heterocyclic Anticancer Compounds: Recent Advances and the Paradigm Shift towards the Use of Nanomedicine’s Tool Box" Molecules 20, no. 9: 16852-16891. https://doi.org/10.3390/molecules200916852