
Influenza Neuraminidase Inhibitors: Synthetic Approaches, Derivatives and Biological Activity


Abstract
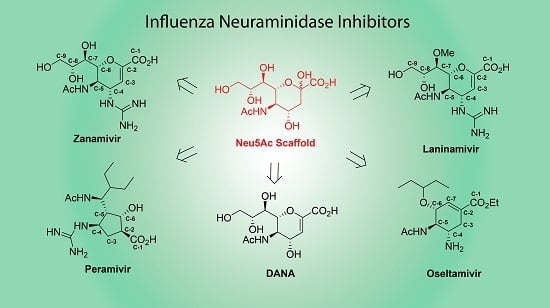
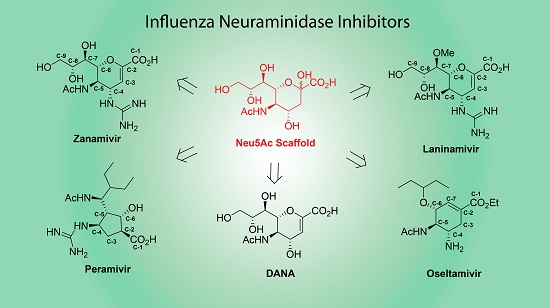
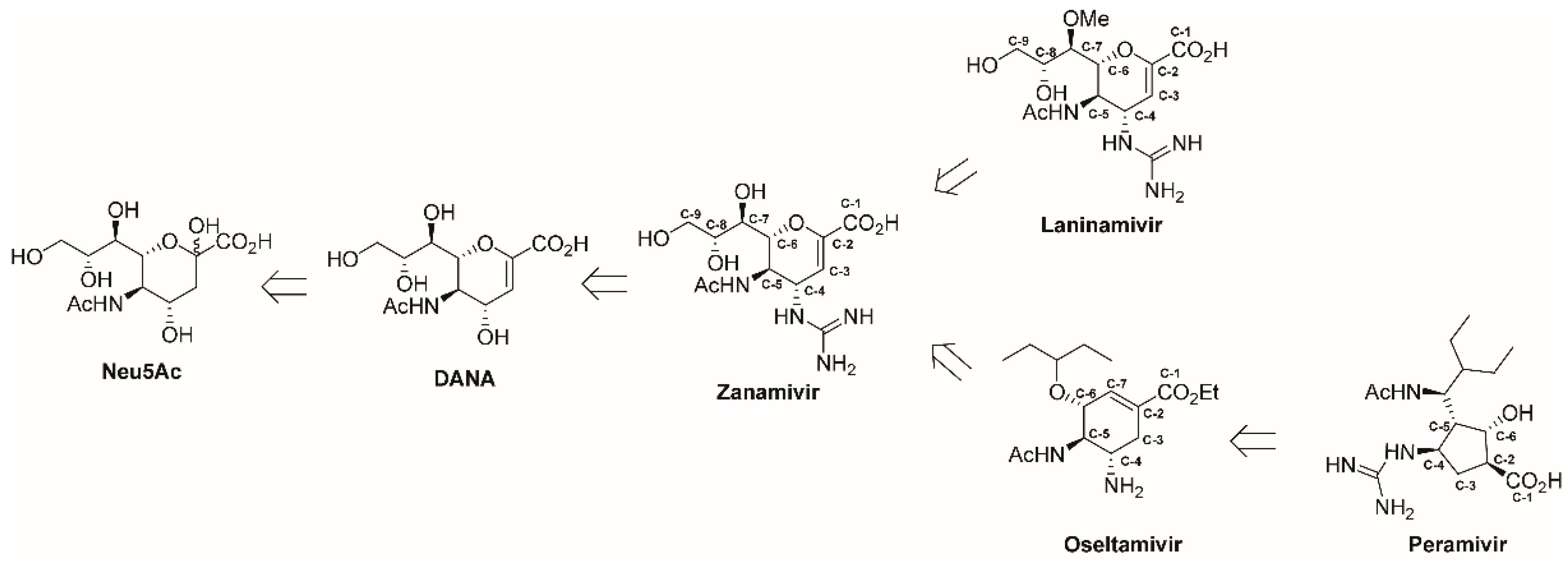
:
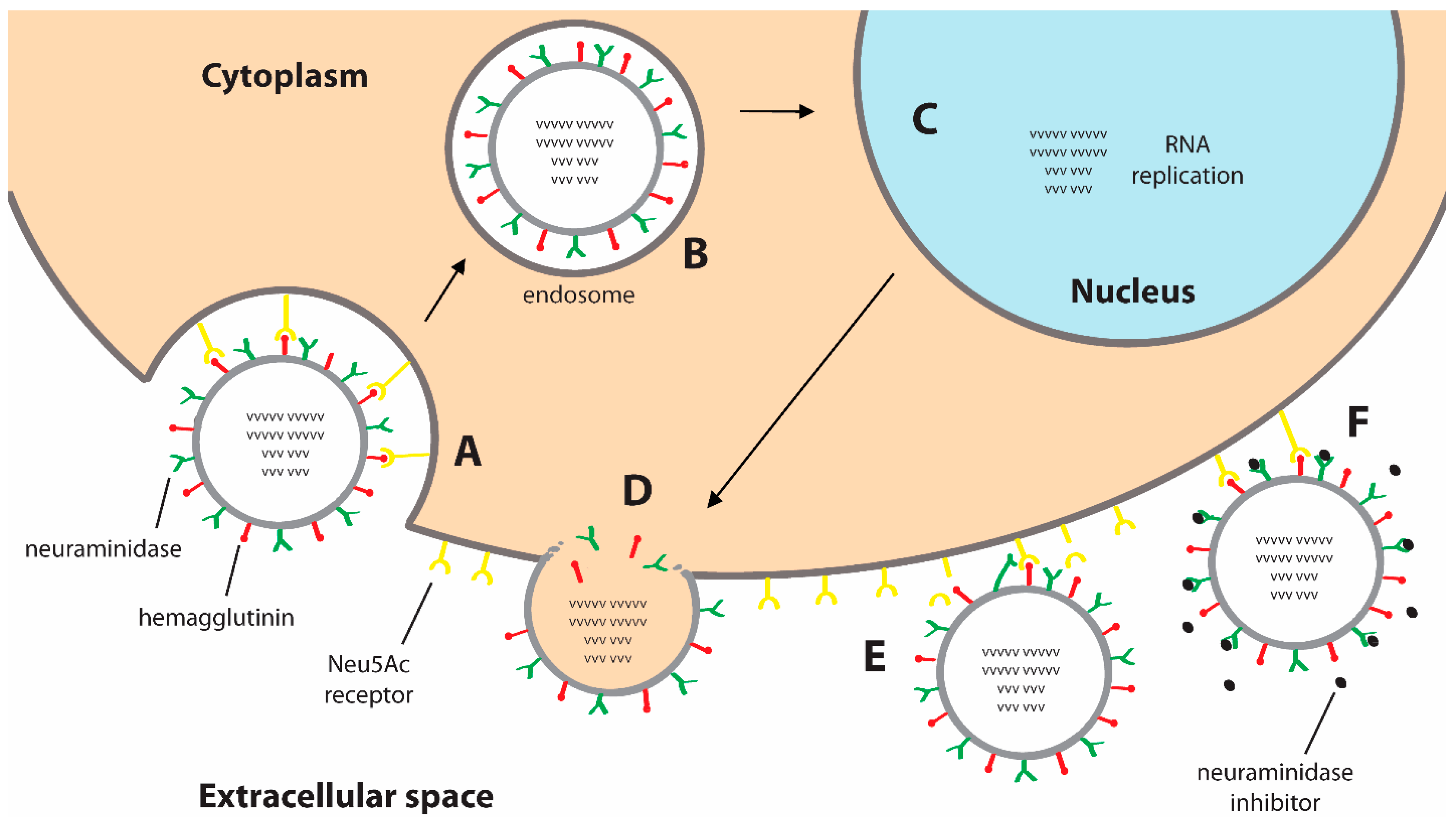
1. Introduction
2. Zanamivir, Laninamivir and Other Derivatives
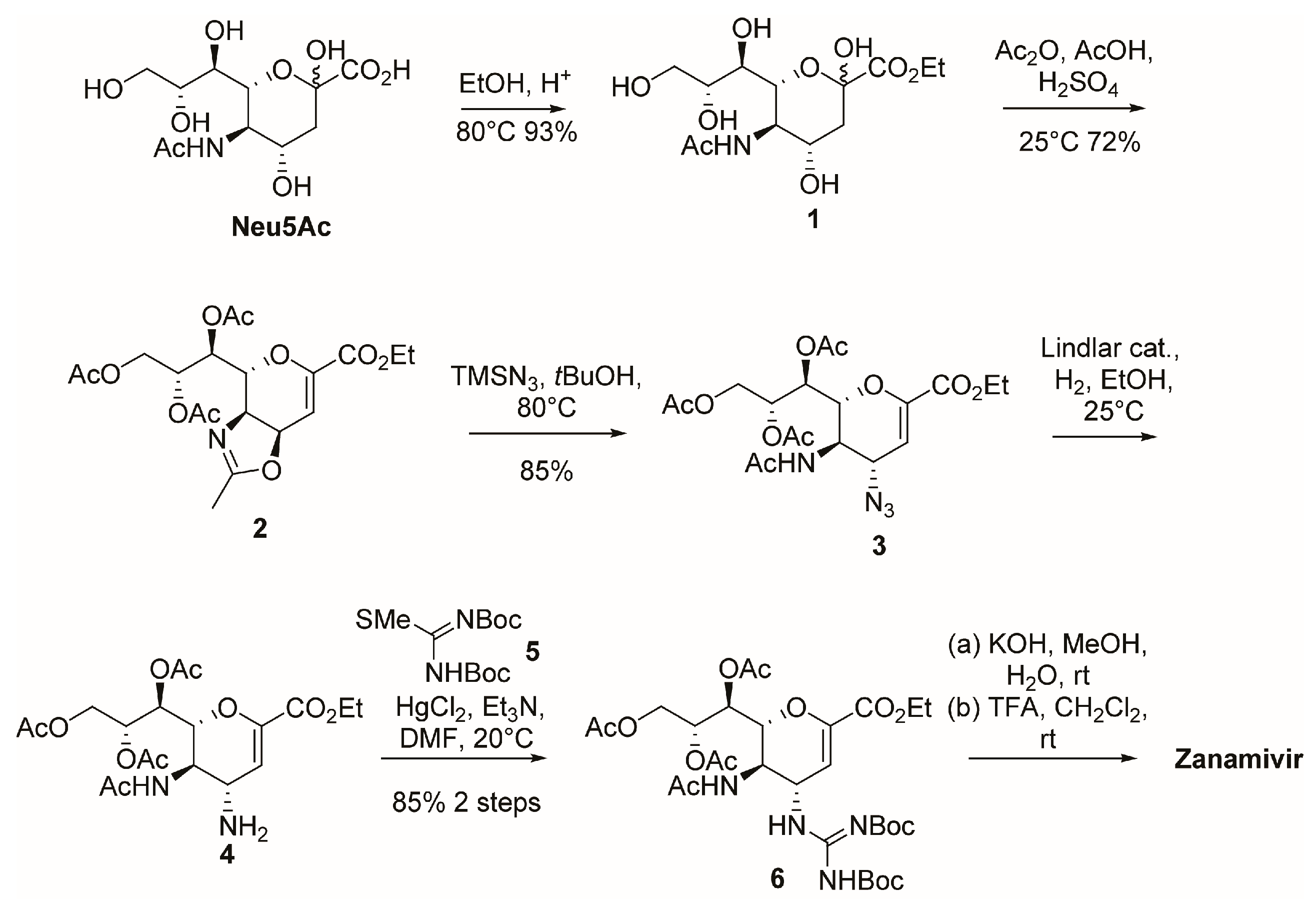
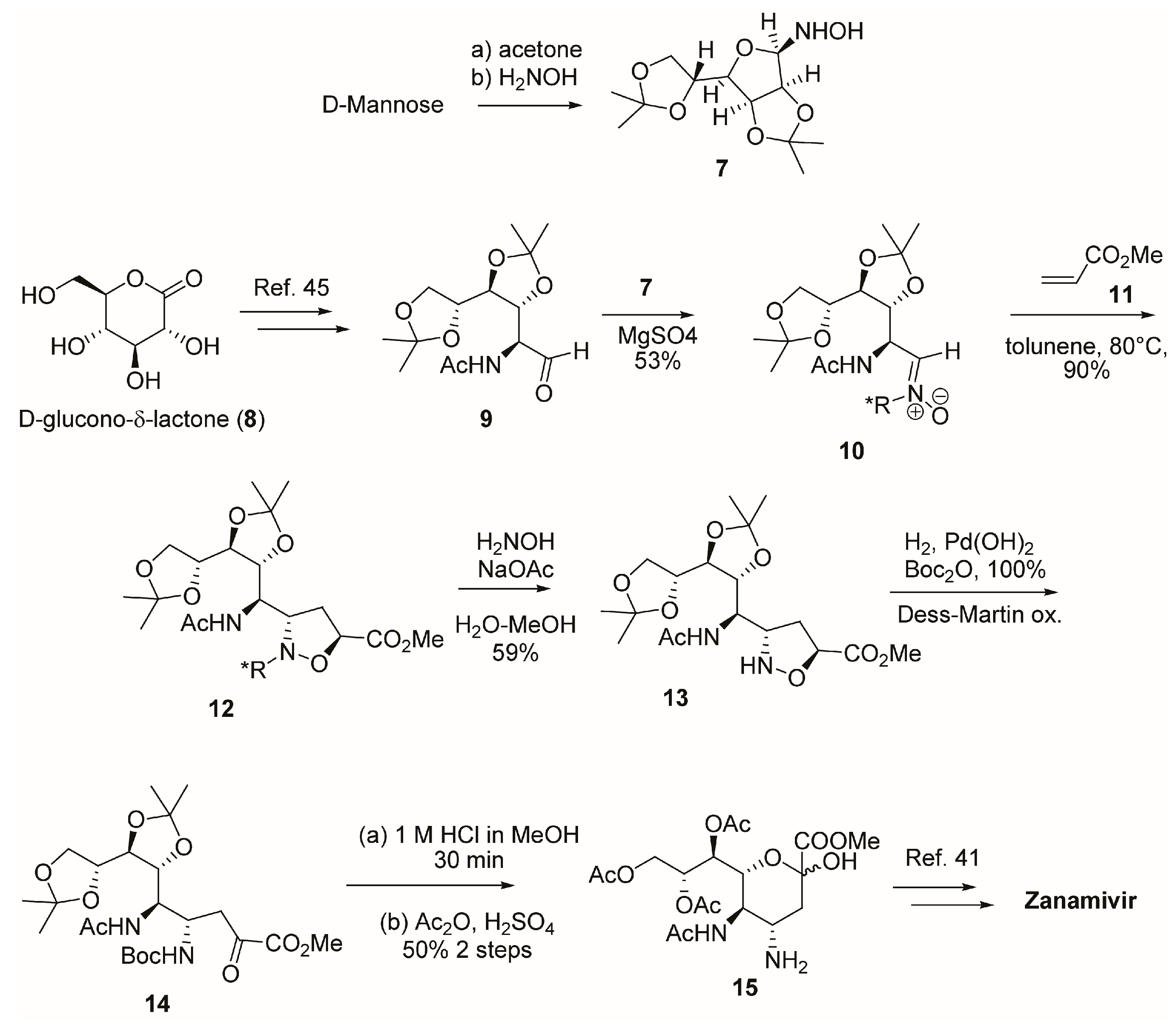
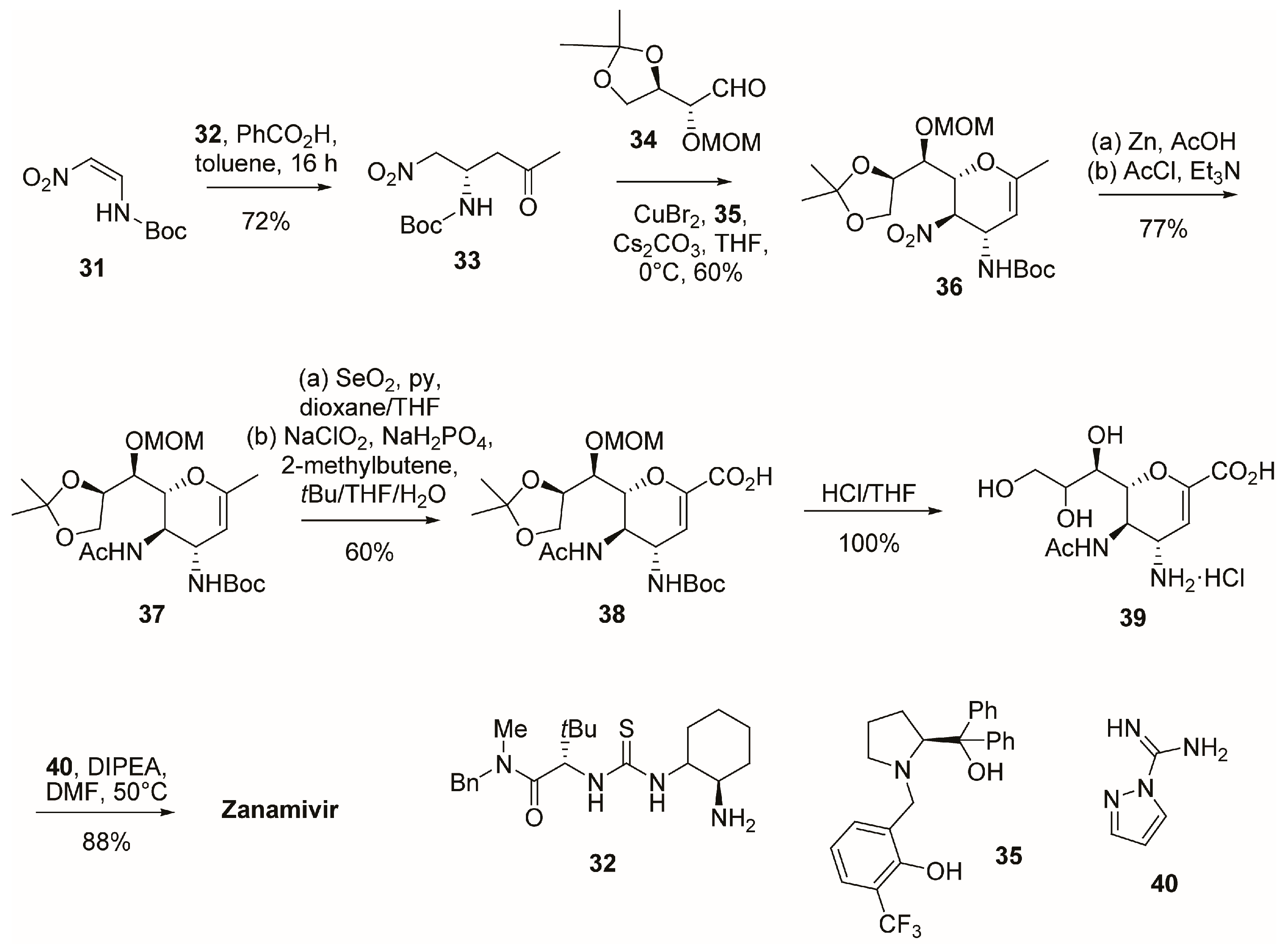
2.1. Synthesis of Zanamivir
2.2. C-1 Modifications
2.3. C-4 Modifications
2.4. C-5 Modifications
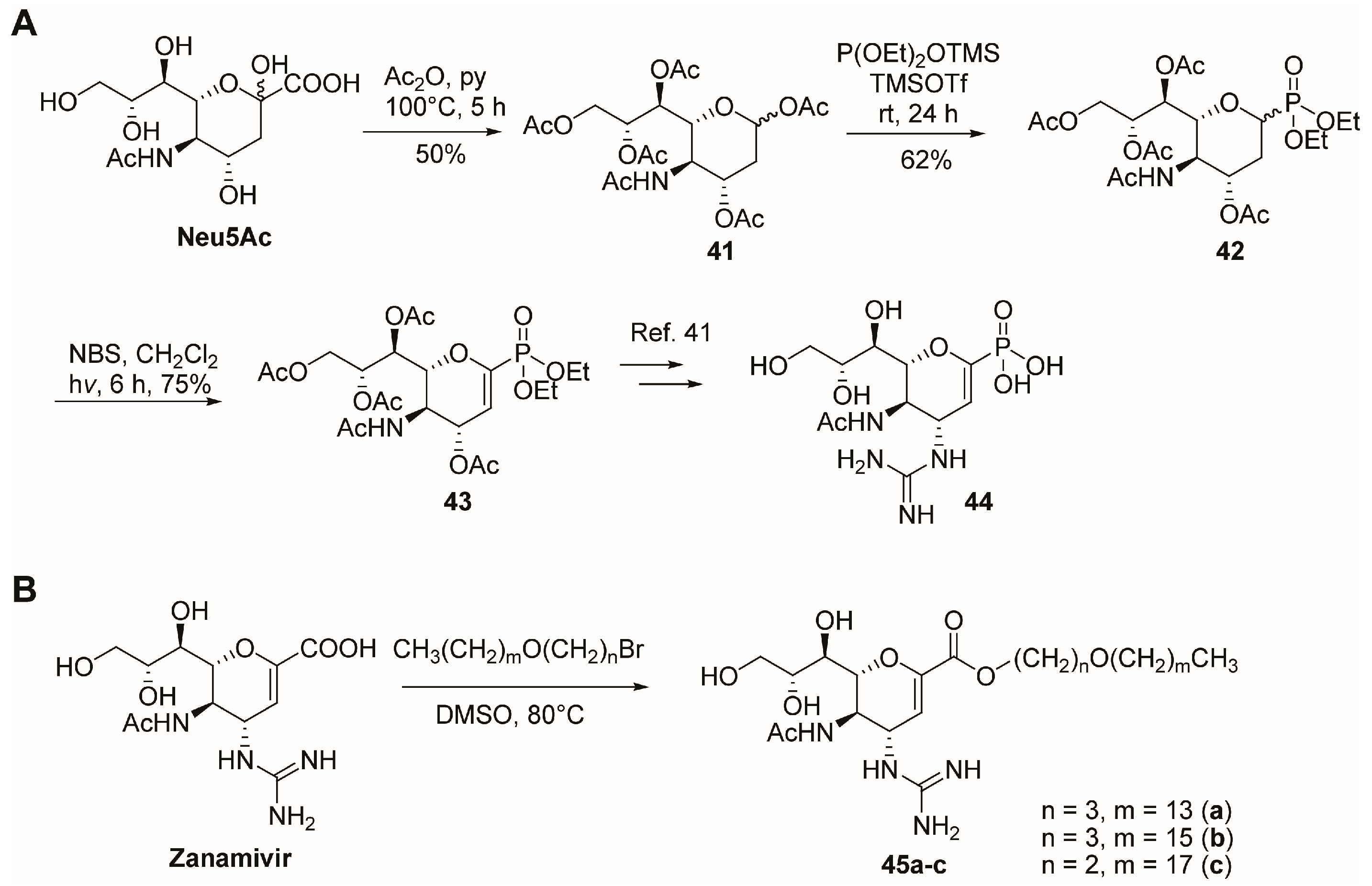
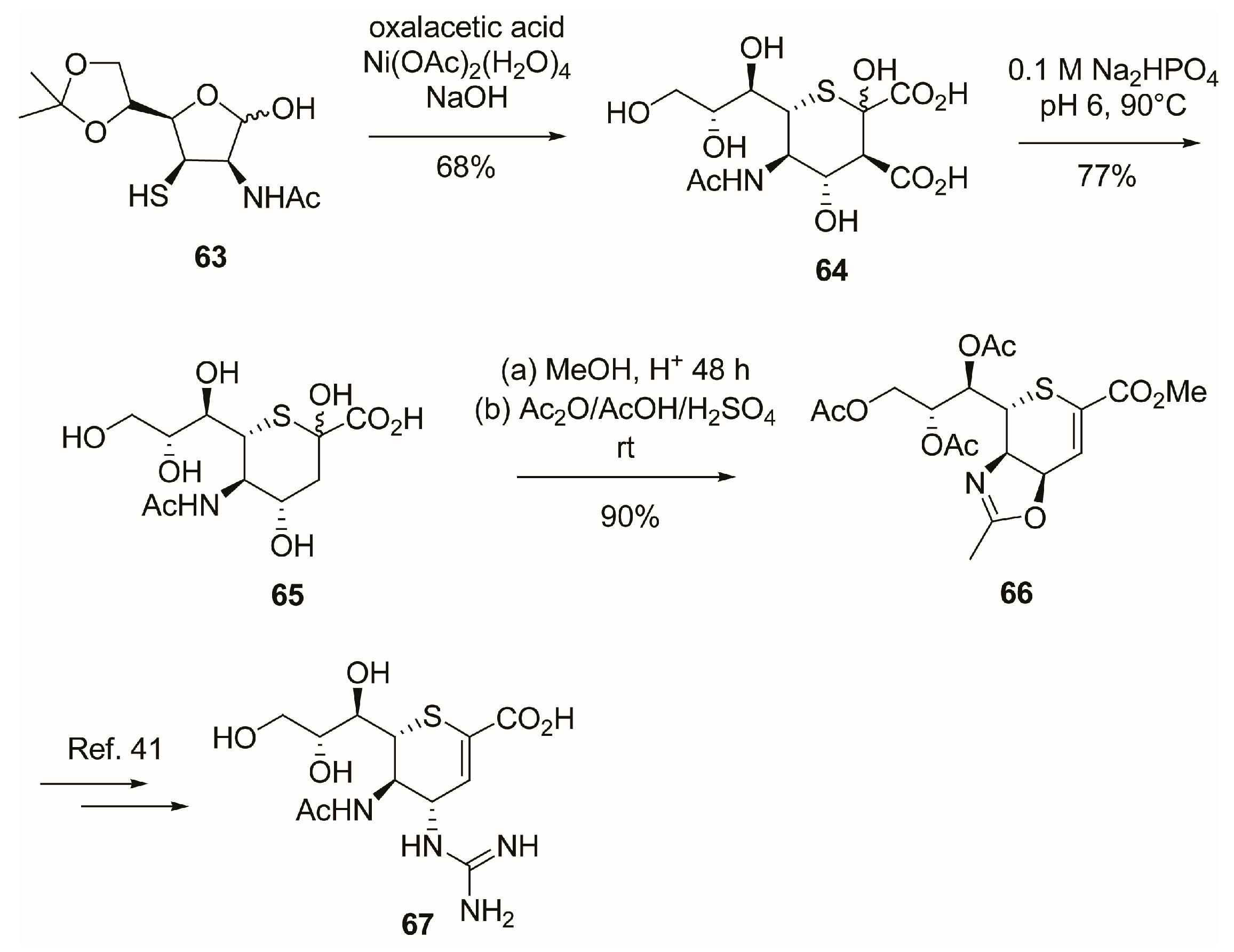
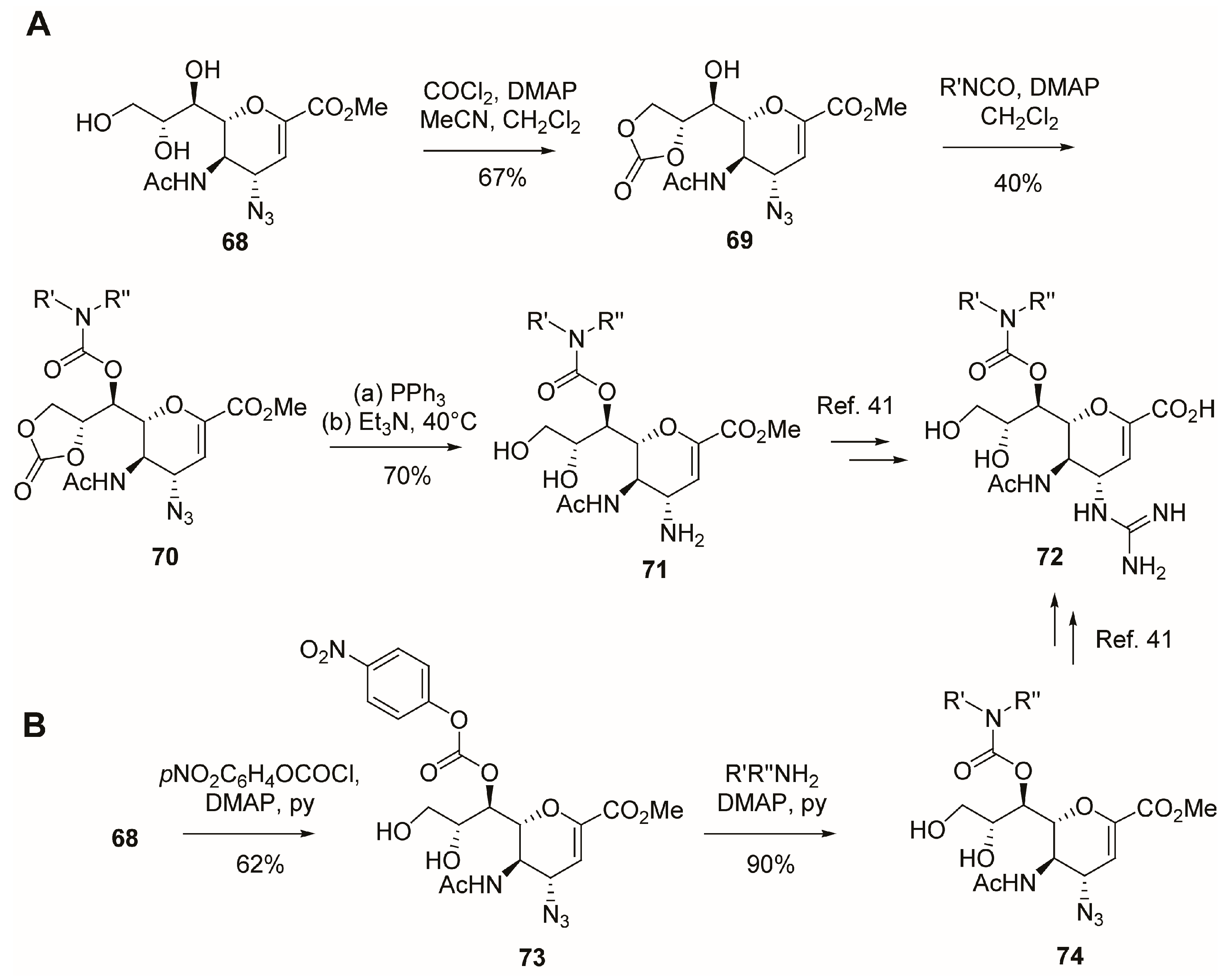
2.5. C-6 Modifications
2.6. C-7 Modifications (Laninamivir)
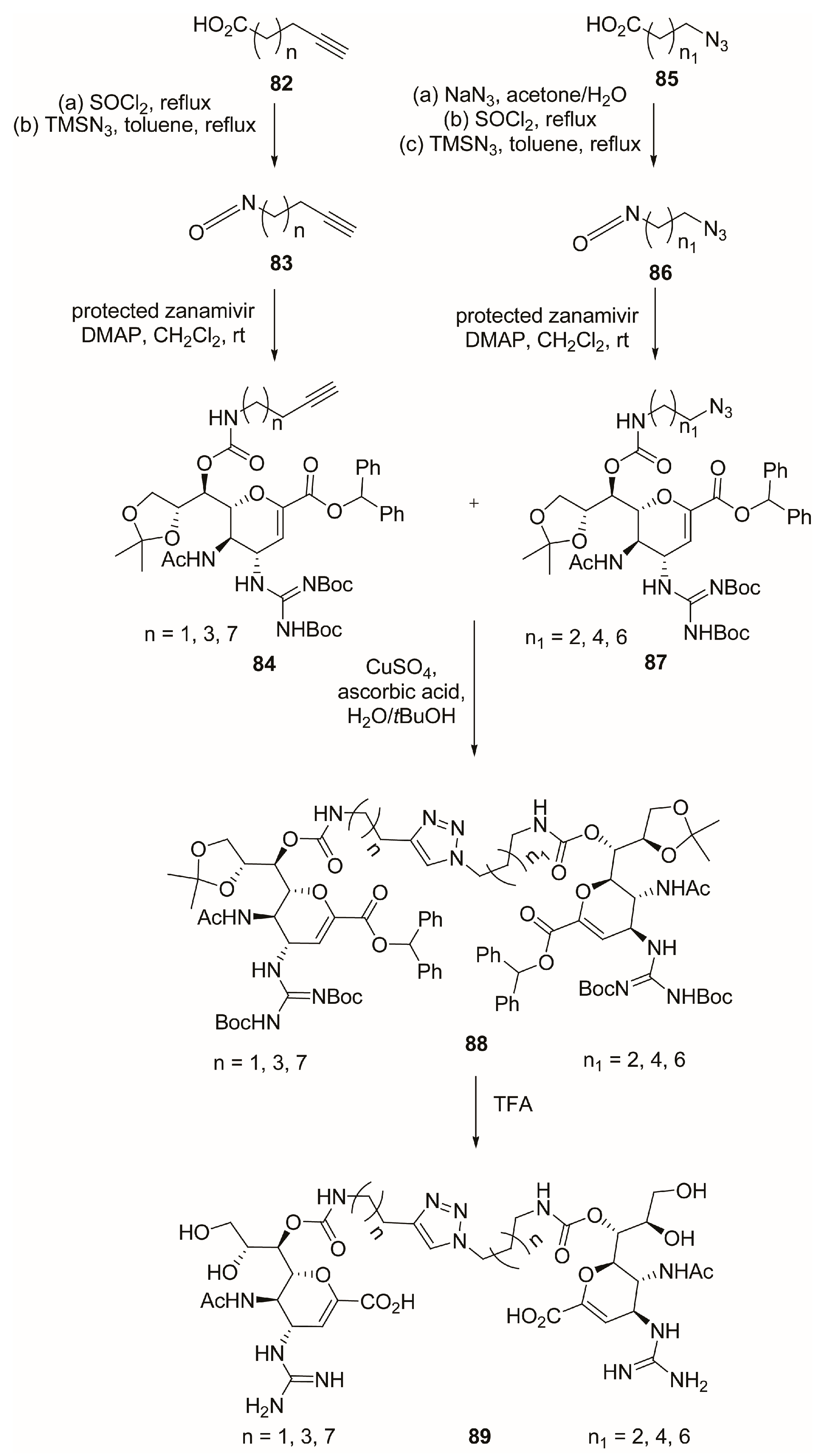
2.7. C-9 Modifications
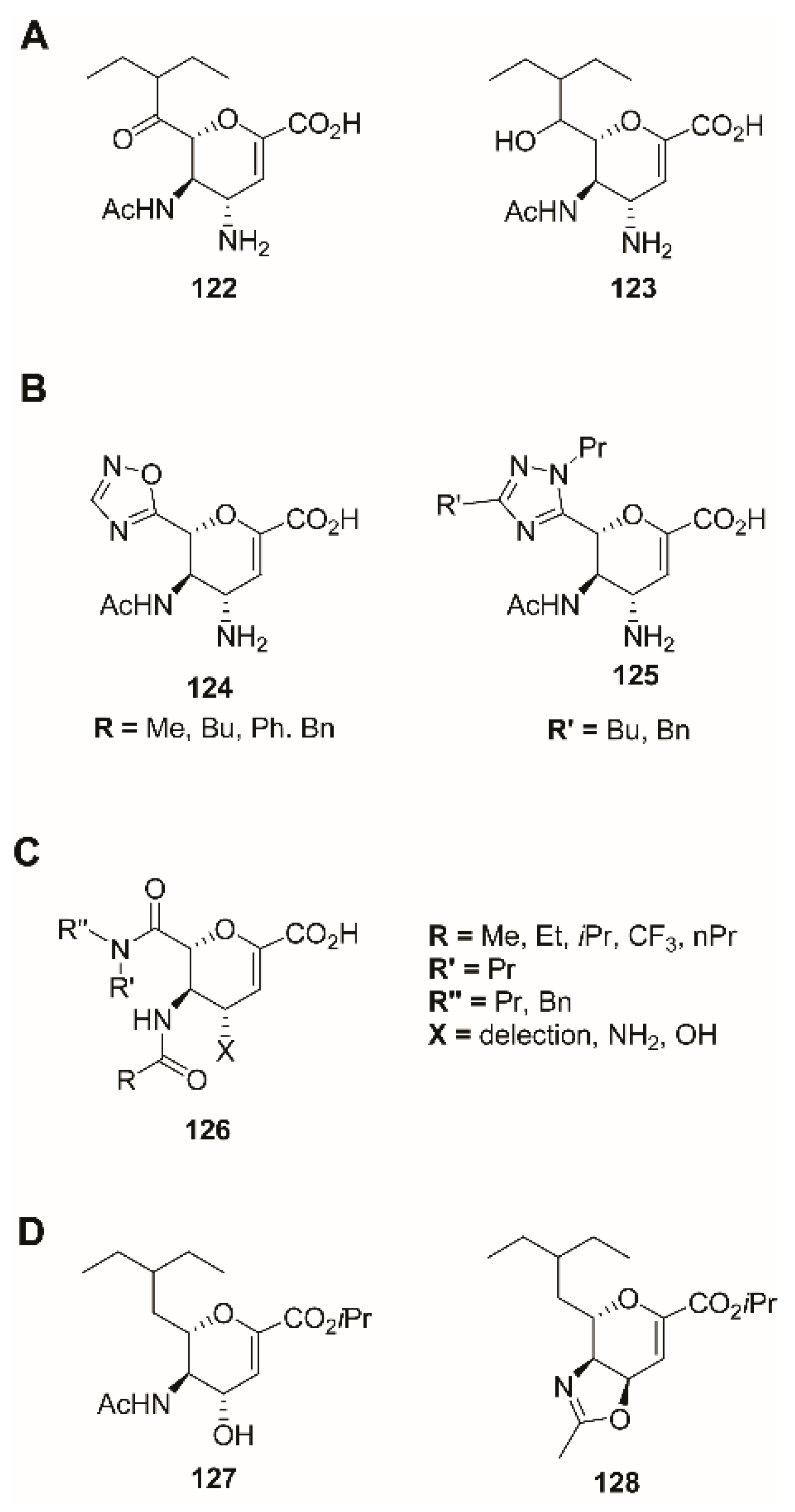
2.8. Other Modifications
3. Oseltamivir
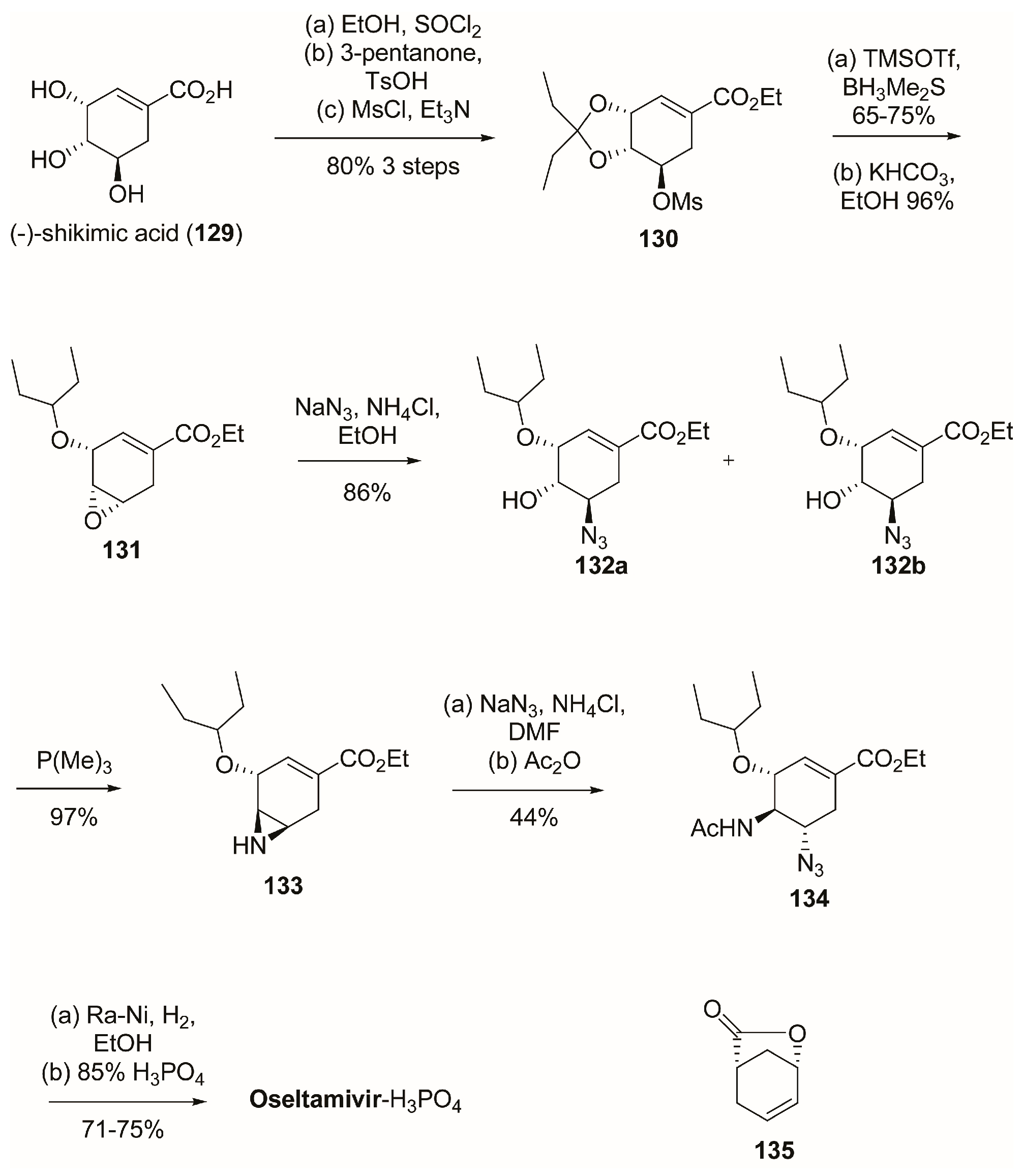
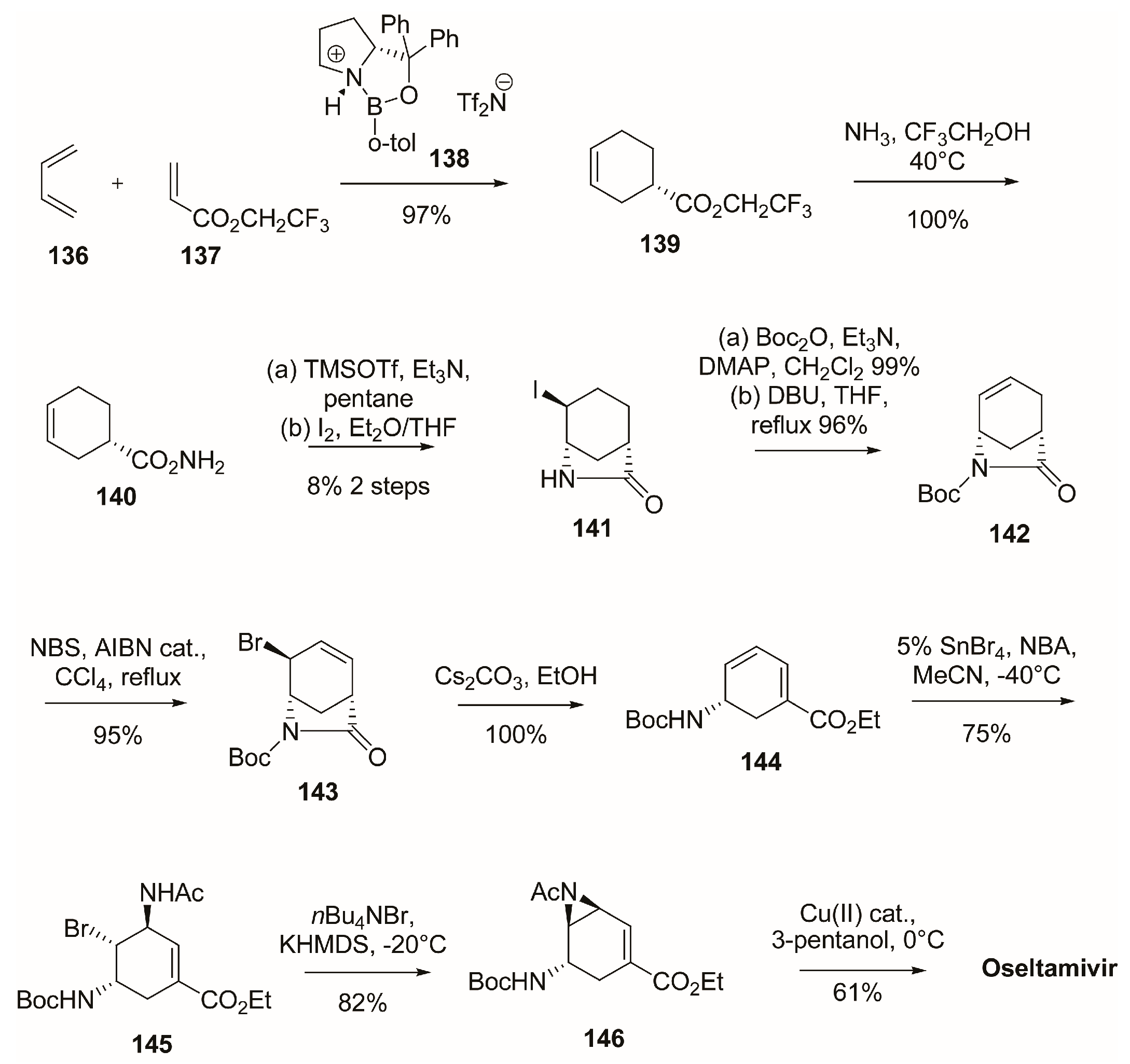
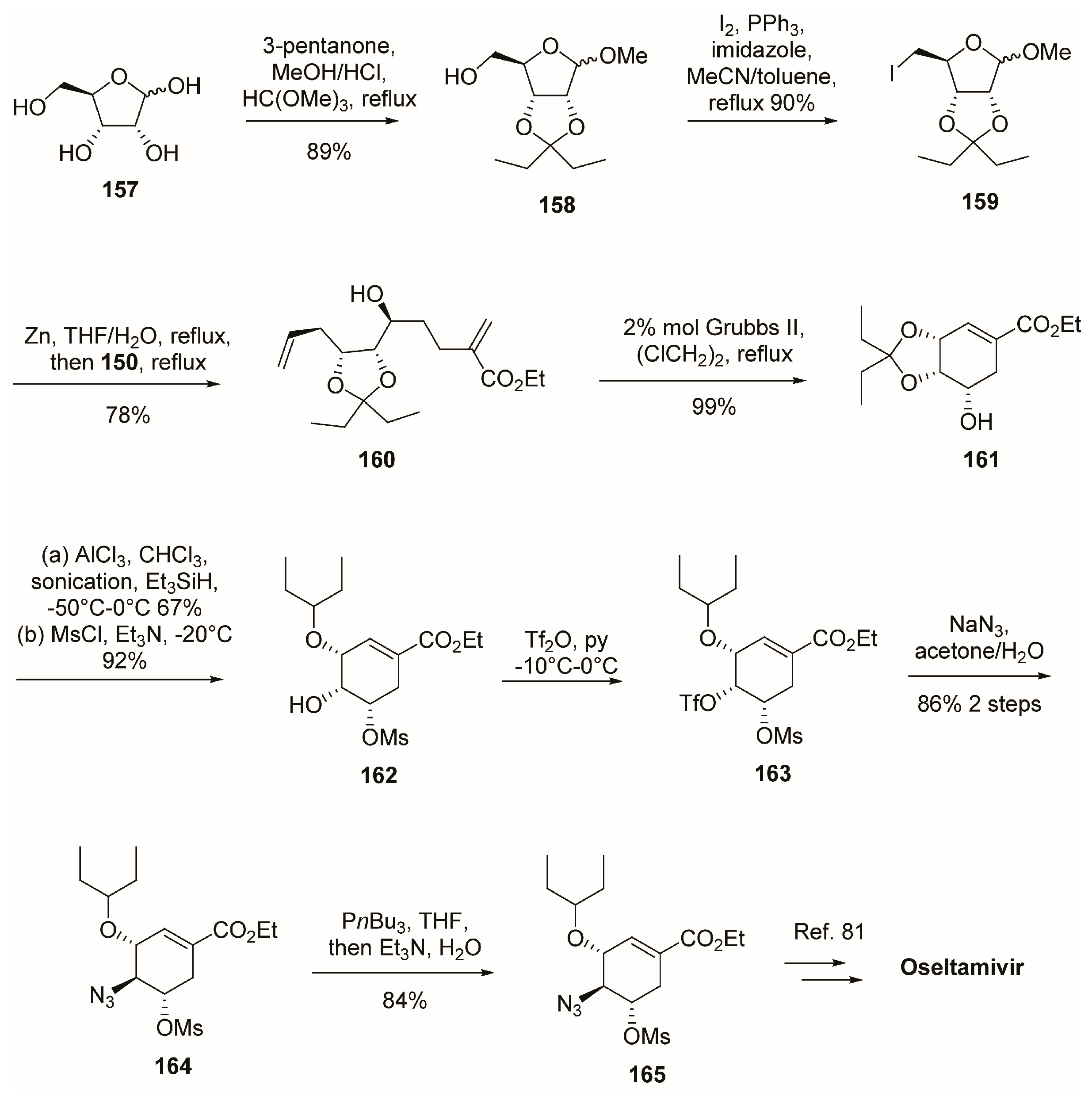
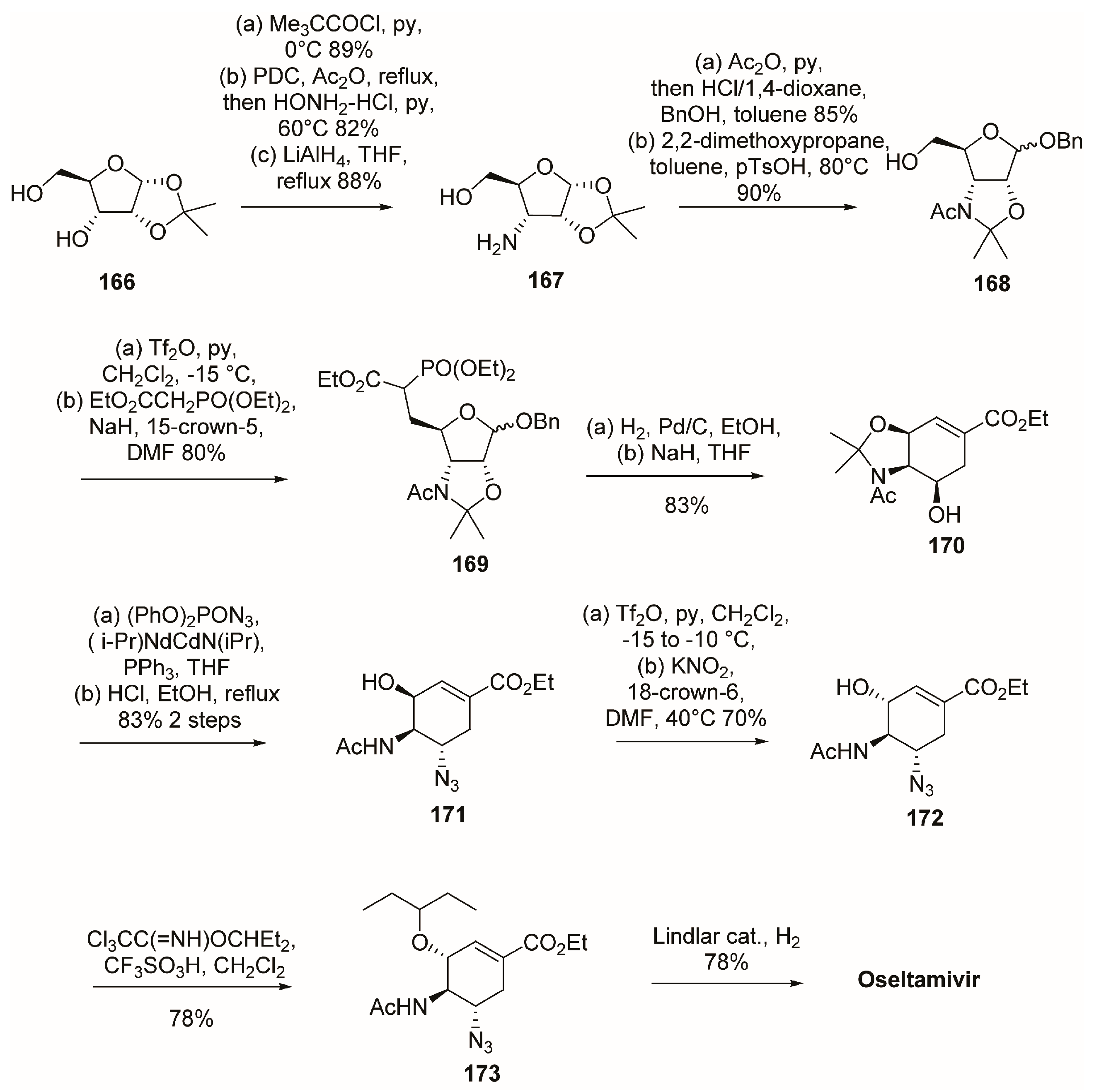
3.1. Synthesis of Oseltamivir
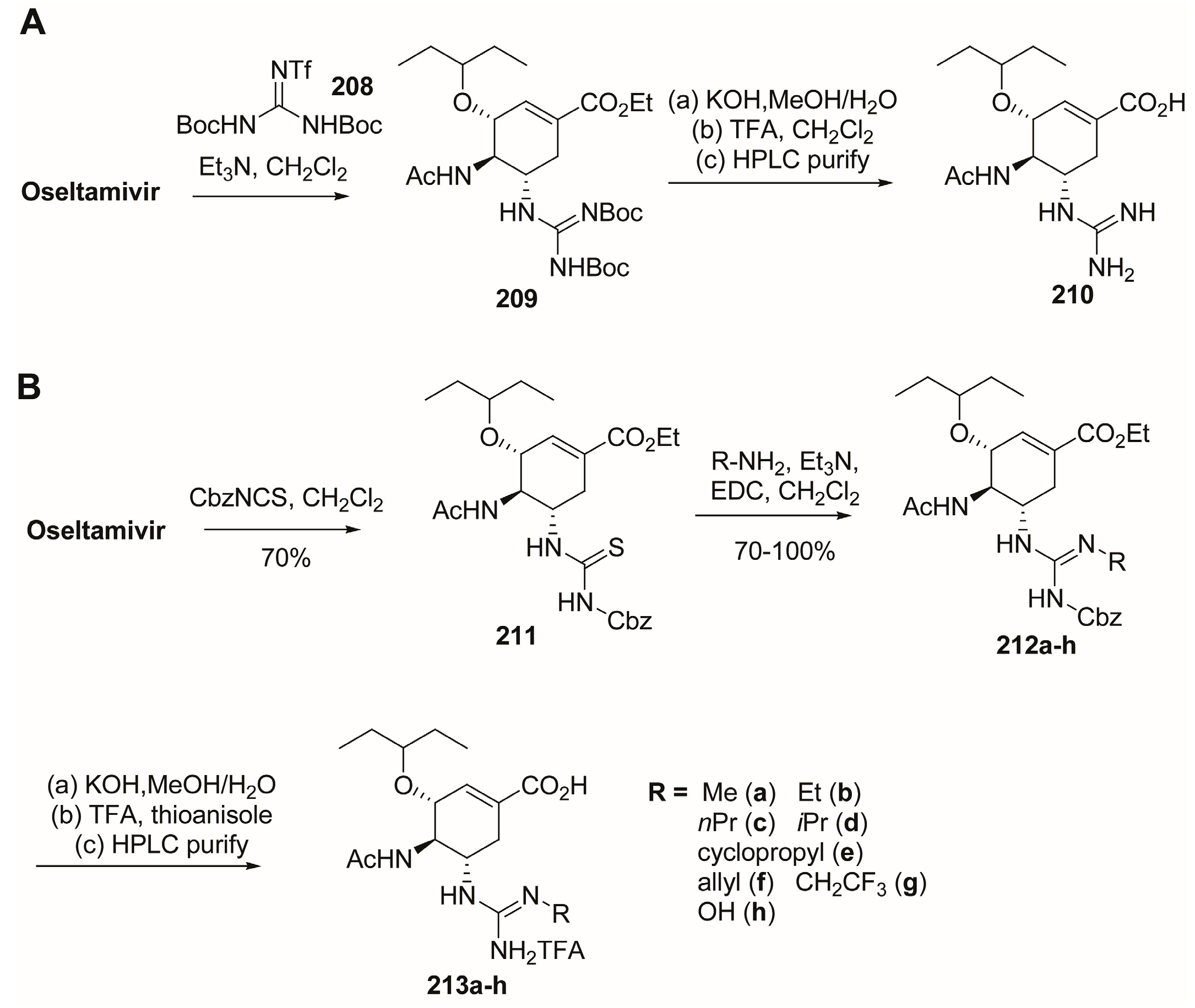
3.2. C-1 Modifications
3.3. C-4 Modifications
3.4. C-5 Modifications
3.5. C-6 Modifications
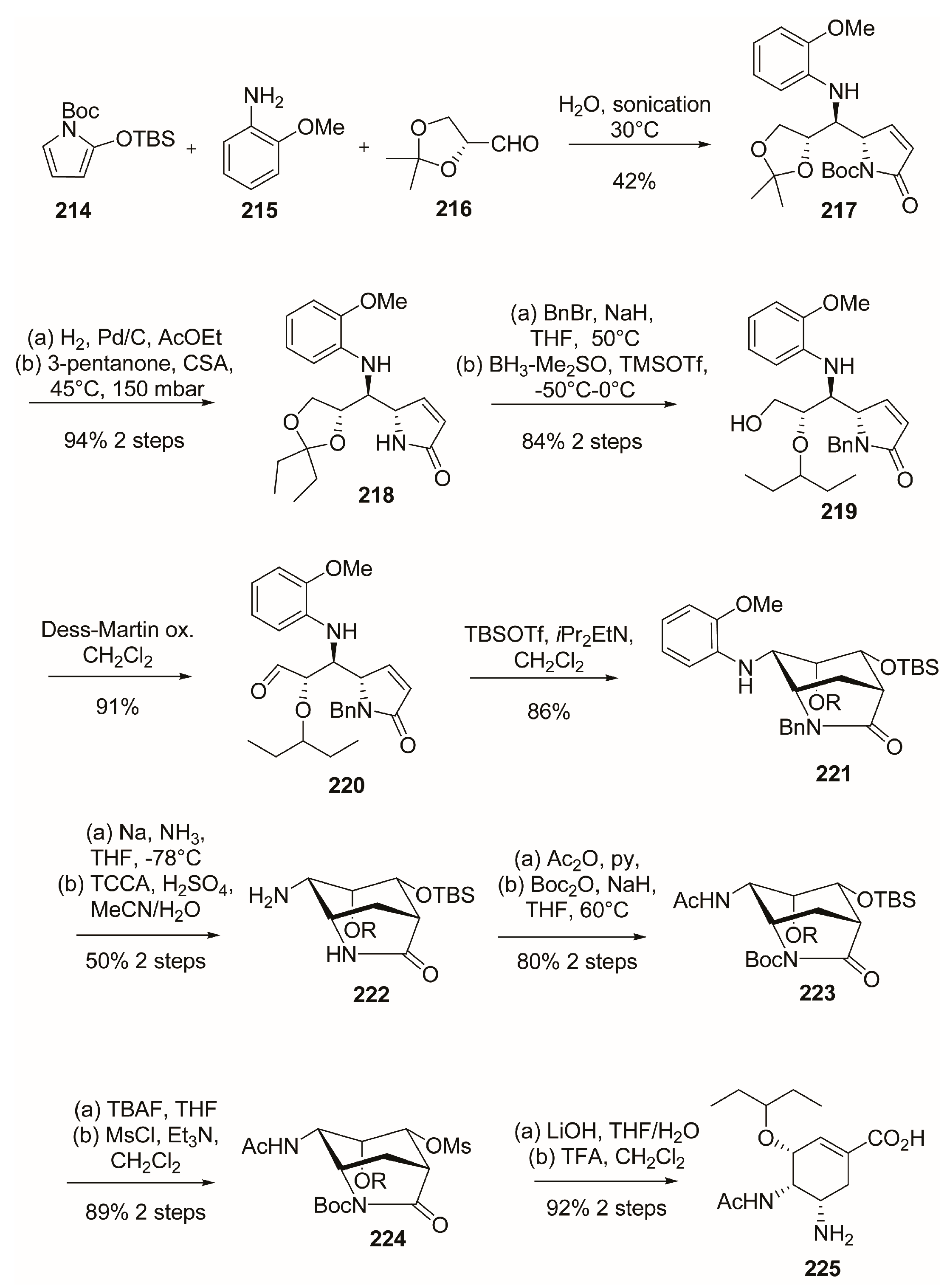
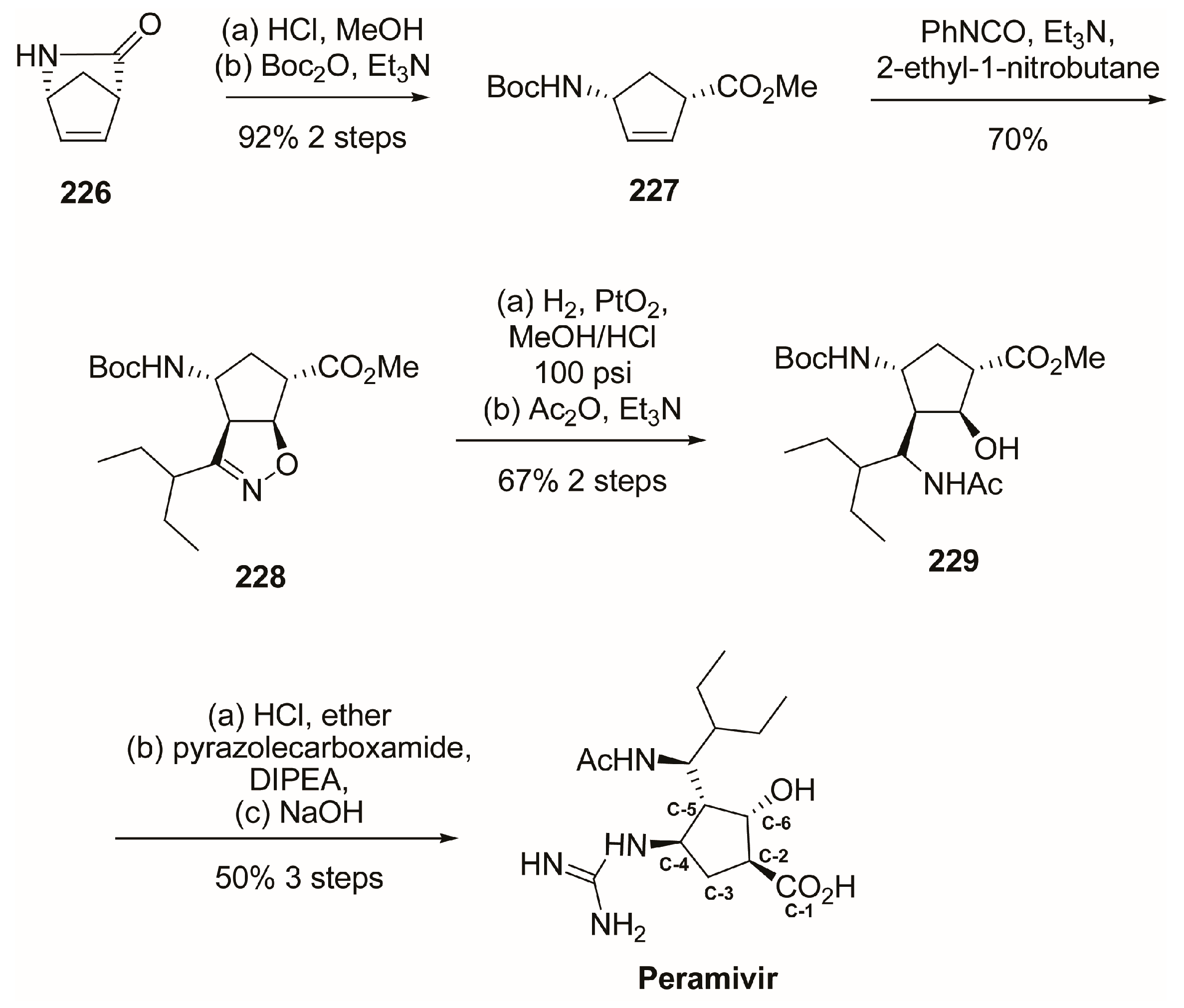
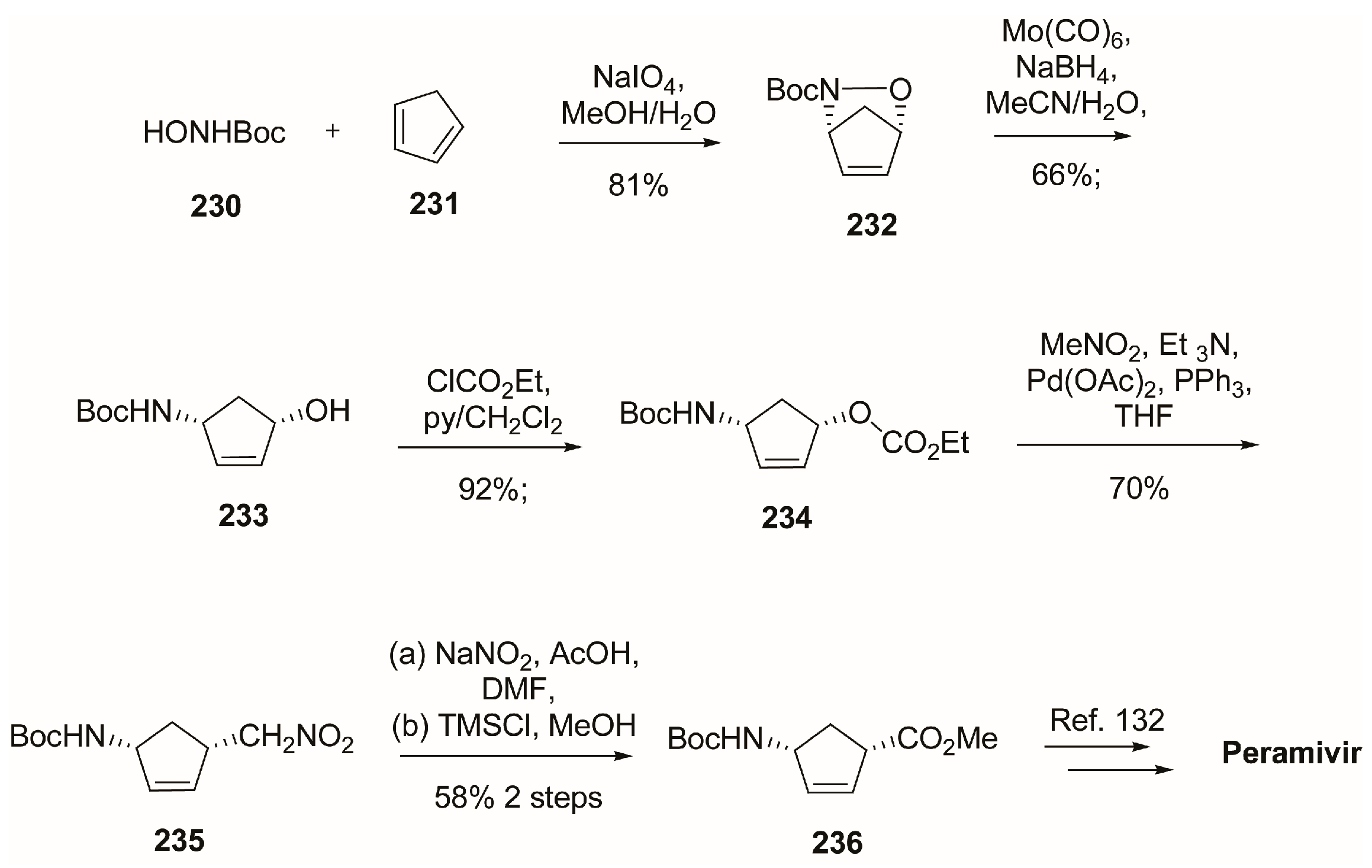
4. Peramivir
4.1. Synthesis of Peramivir
4.2. C-4 Modifications
4.3. C-5 Modifications
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Thomas, R.E. Are influenza-associated morbidity and mortality estimates for those ≥ 65 in statistical databases accurate, and an appropriate test of influenza vaccine effectiveness? Vaccine 2014, 32, 6884–6901. [Google Scholar] [CrossRef] [PubMed]
- Szucs, T. The socio-economic burden of influenza. J. Antimicrob. Chemother. 1999, 44, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Vonitzstein, M.; Wu, W.Y.; Kok, G.B.; Pegg, M.S.; Dyason, J.C.; Jin, B.; Phan, T.V.; Smythe, M.L.; White, H.F.; Oliver, S.W.; et al. Rational design of potent sialidase-based inhibitors of influenza-virus replication. Nature 1993, 363, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Zambon, M.C. Epidemiology and pathogenesis of influenza. J. Antimicrob. Chemother. 1999, 44, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Spanakis, N.; Pitiriga, V.; Gennimata, V.; Tsakris, A. A review of neuraminidase inhibitor susceptibility in influenza strains. Expert Rev. Anti-Infect. Ther. 2014, 12, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Bolton, C.E.; Bush, A.; Hurst, J.R.; Kotecha, S.; McGarvey, L. Lung consequences in adults born prematurely. Thorax 2015, 70, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Pizzorno, A.; Abed, Y.; Boivin, G. Influenza virus resistance to neuraminidase inhibitors. Antivir. Res. 2013, 98, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.J. The Viral Network: A Pathography of H1N1 Influenza Pandemic. Am. Anthropol. 2016, 118, 202–203. [Google Scholar] [CrossRef]
- Xiong, X.; McCauley, J.W.; Steinhauer, D.A. Receptor Binding Properties of the Influenza Virus Hemagglutinin as a Determinant of Host Range. In Influenza Pathogenesis and Control—Volume I; Compans, R.W., Oldstone, M.B.A., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 385, pp. 63–91. [Google Scholar]
- Varghese, J.N.; McKimmbreschkin, J.L.; Caldwell, J.B.; Kortt, A.A.; Colman, P.M. The structure of the complex between influenza-virus neuraminidase and sialic-acid, the viral receptor. Proteins 1992, 14, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Rossman, J.S.; Lamb, R.A. Influenza virus assembly and budding. Virology 2011, 411, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.H.; Fanning, T.G.; Janczewski, T.A.; Taubenberger, J.K. Characterization of the 1918 “Spanish” influenza virus neuraminidase gene. Proc. Natl. Acad. Sci. USA 2000, 97, 6785–6790. [Google Scholar] [CrossRef] [PubMed]
- Varghese, J.N.; Laver, W.G.; Colman, P.M. Structure of the influenza virus glycoprotein antigen neuraminidase at 2.9 A resolution. Nature 1983, 303, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhu, X.; Dwek, R.A.; Stevens, J.; Wilson, I.A. Structural Characterization of the 1918 Influenza Virus H1N1 Neuraminidase. J. Virol. 2008, 82, 10493–10501. [Google Scholar] [CrossRef] [PubMed]
- Colman, P.M.; Varghese, J.N.; Laver, W.G. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature 1983, 303, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Lam, M.K.-H.; Zhang, Q.; Elderfield, R.; Barclay, W.S.; Shaw, P.-C. The Functional Study of the N-Terminal Region of Influenza B Virus Nucleoprotein. PLoS ONE 2015, 10, e0137802. [Google Scholar] [CrossRef] [PubMed]
- Jongkon, N.; Sangma, C. Receptor recognition mechanism of human influenza A H1N1 (1918), avian influenza A H5N1 (2004), and pandemic H1N1 (2009) neuraminidase. J. Mol. Model. 2012, 18, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.-M.; Lai, J.C.C.; Haselhorst, T.; Choy, K.T.; Yen, H.-L.; Peiris, J.S.M.; von Itzstein, M.; Nicholls, J.M. Investigation of the binding and cleavage characteristics of N1 neuraminidases from avian, seasonal, and pandemic influenza viruses using saturation transfer difference nuclear magnetic resonance. Influenza Other Respir. Viruses 2014, 8, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Ushirogawa, H.; Ohuchi, M. Novel antiviral activity of neuraminidase inhibitors against an avian influenza A virus. Virol. J. 2011, 8, 411. [Google Scholar] [CrossRef] [PubMed]
- Wohlbold, T.J.; Krammer, F. In the Shadow of Hemagglutinin: A Growing Interest in Influenza Viral Neuraminidase and Its Role as a Vaccine Antigen. Viruses 2014, 6, 2465–2494. [Google Scholar] [CrossRef] [PubMed]
- Alame, M.M.; Massaad, E.; Zaraket, H. Peramivir: A Novel Intravenous Neuraminidase Inhibitor for Treatment of Acute Influenza Infections. Front. Microbiol. 2016, 7, 450. [Google Scholar] [CrossRef] [PubMed]
- Shie, J.-J.; Fang, J.-M. Phosphonate Congeners of Oseltamivir and Zanamivir as Effective Anti-influenza Drugs: Design, Synthesis and Biological Activity. J. Chin. Chem. Soc. 2014, 61, 127–141. [Google Scholar] [CrossRef]
- Wade, R.C. ‘Flu’ and structure-based drug design. Structure 1997, 5, 1139–1145. [Google Scholar] [CrossRef]
- Oxford, J.S.; Novelli, P.; Sefton, A.; Lambkin, R. New millennium antivirals against pandemic and epidemic influenza: The neuraminidase inhibitors. Antivir. Chem. Chemother. 2002, 13, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Chamni, S.; De-Eknamkul, W. Recent progress and challenges in the discovery of new neuraminidase inhibitors. Expert Opin. Ther. Pat. 2013, 23, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Chand, P. Recent advances in the discovery and synthesis of neuraminidase inhibitors. Expert Opin. Ther. Pat. 2005, 15, 1009–1025. [Google Scholar] [CrossRef]
- Burmeister, W.P.; Henrissat, B.; Bosso, C.; Cusack, S.; Ruigrok, R.W.H. Influenza-B virus neuraminidase can synthetize its own inhibitor. Structure 1993, 1, 19–26. [Google Scholar] [CrossRef]
- Eyer, L.; Hruska, K. Antiviral agents targeting the influenza virus: A review and publication analysis. Vet. Med. 2013, 58, 113–185. [Google Scholar]
- Burnham, A.J.; Baranovich, T.; Govorkova, E.A. Neuraminidase inhibitors for influenza B virus infection: Efficacy and resistance. Antivir. Res. 2013, 100, 520–534. [Google Scholar] [CrossRef] [PubMed]
- Chairat, K.; Tarning, J.; White, N.J.; Lindegardh, N. Pharmacokinetic Properties of Anti-Influenza Neuraminidase Inhibitors. J. Clin. Pharmacol. 2013, 53, 119–139. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-K.; Tsai, C.-H.; Shie, J.-J.; Fang, J.-M. From neuraminidase inhibitors to conjugates: A step towards better anti-influenza drugs? Future Med.Chem. 2014, 6, 757–774. [Google Scholar] [CrossRef] [PubMed]
- Feng, E.; Ye, D.; Li, J.; Zhang, D.; Wang, J.; Zhao, F.; Hilgenfeld, R.; Zheng, M.; Jiang, H.; Liu, H. Recent Advances in Neuraminidase Inhibitor Development as Anti-influenza Drugs. ChemMedChem 2012, 7, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Kamali, A.; Holodniy, M. Influenza treatment and prophylaxis with neuraminidase inhibitors: A review. Infect. Drug Resist. 2013, 6, 187–198. [Google Scholar] [PubMed]
- Muthuri, S.G.; Venkatesan, S.; Myles, P.R.; Leonardi-Bee, J.; Al Khuwaitir, T.S.A.; Al Mamun, A.; Anovadiya, A.P.; Azziz-Baumgartner, E.; Baez, C.; Bassetti, M.; et al. Effectiveness of neuraminidase inhibitors in reducing mortality in patients admitted to hospital with influenza A H1N1pdm09 virus infection: A meta-analysis of individual participant data. Lancet Respir. Med. 2014, 2, 395–404. [Google Scholar] [CrossRef]
- Moscona, A. Drug therapy—Neuraminidase inhibitors for influenza. N. Engl. J. Med. 2005, 353, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, S.; Zell, R.; Schwemmle, M.; Herold, S. Influenza, a One Health paradigm-Novel therapeutic strategies to fight a zoonotic pathogen with pandemic potential. Int. J. Med. Microbiol. 2014, 304, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Neumann, G.; Noda, T.; Kawaoka, Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 2009, 459, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Mitrasinovic, P.M. Advances in the Structure-Based Design of the Influenza A Neuraminidase Inhibitors. Curr. Drug Targets 2010, 11, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Sano, K.; Ito, M.; Saito, M.; Hidari, K.; Suzuki, T.; Suzuki, Y.; Tanaka, K. Synthesis of 2-deoxy-2,3-didehydro-N-acetylneuraminic acid analogues modified at the C-4 and C-9 positions and their behaviour towards sialidase from influenza virus and pig liver membrane. Carbohydr. Res. 2001, 330, 31–41. [Google Scholar] [CrossRef]
- Smith, B.J.; Colman, P.M.; von Itzstein, M.; Danylec, B.; Varghese, J.N. Analysis of inhibitor binding in influenza virus neuraminidase. Protein Sci. 2001, 10, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Vonitzstein, M.; Wu, W.Y.; Jin, B. The synthesis of 2,3-dehydro-2,4-dideoxy-4-guanidinyl-N-acetylneuraminic acid—A potent influenza virus sialidase inhibitor. Carbohydr. Res. 1994, 259, 301–305. [Google Scholar] [CrossRef]
- Scheigetz, J.; Zamboni, R.; Bernstein, M.A.; Roy, B. A synthesis of 4-alpha-guanidino-2-deoxy-2,3-didehydro N-acetylneuraminic acid. Org. Prep. Proced. Int. 1995, 27, 637–644. [Google Scholar] [CrossRef]
- Chandler, M.; Bamford, M.J.; Conroy, R.; Lamont, B.; Patel, B.; Patel, V.K.; Steeples, I.P.; Storer, R.; Weir, N.G.; Wright, M.; et al. Synthesis of the potent infleunza neuraminidase inhibitor 4-guanidino Neu5Ac2en—X-ray molecular strucuture of 5-Acetamido-4-amino-2,6-anhydro-3,4,5-trideoxy-d-trideoxy-d-erythro-l-gluco-nononic acid. J. Chem. Soc. Perkin Trans. 1 1995, 1173–1180. [Google Scholar] [CrossRef]
- Zhu, X.B.; Wang, M.; Wang, S.Z.; Yao, Z.J. Concise synthesis of zanamivir and its C4-thiocarbamido derivatives utilizing a 3 + 2-cycloadduct derived from d-glucono-delta-lactone. Tetrahedron 2012, 68, 2041–2044. [Google Scholar] [CrossRef]
- Nitabaru, T.; Kumagai, N.; Shibasaki, M. Catalytic Asymmetric anti-Selective Nitroaldol Reaction En Route to Zanamivir. Angew. Chem. Int. Ed. 2012, 51, 1644–1647. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.S.; Zhong, J.K.; Li, Y.S.; Ma, D.W. Organocatalytic and Scalable Synthesis of the Anti-Influenza Drugs Zanamivir, Laninamivir, and CS-8958. Angew. Chem. Int. Ed. 2014, 53, 13885–13888. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.G.; Zhou, H.B.; Wu, Y.L.; Yao, Z.J. Synthesis of a new stable conformationally constrained 2,7-anhydrosialic acid derivative. J. Org. Chem. 2003, 68, 9528–9531. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.G.; Yan, S.; Wu, Y.L.; Yao, Z.J. Synthesis of 4-azido-4-deoxy-Neu5,7,8,9Ac(4)2en1Me. A key intermediate for the synthesis of GG167 from d-glucono-delta-lactone. Org. Lett. 2004, 6, 2269–2272. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.B.; Jacobsen, E.N. Highly enantioselective direct conjugate addition of ketones to nitroalkenes promoted by a chiral primary amine-thiourea catalyst. J. Am. Chem. Soc. 2006, 128, 7170–7171. [Google Scholar] [CrossRef] [PubMed]
- Luzzio, F.A. The Henry reaction: Recent examples. Tetrahedron 2001, 57, 915–945. [Google Scholar] [CrossRef]
- Vasella, A.; Wyler, R. Synthesis of a phosphonic acid analog of N-acetyl-2,3-didehydro-2-deoxyneuraminic acid, an inhibitor of Vibrio chlolerae sialidase. Helv. Chim. Acta 1991, 74, 451–463. [Google Scholar] [CrossRef]
- Shie, J.J.; Fang, J.M.; Lai, P.T.; Wen, W.H.; Wang, S.Y.; Cheng, Y.S.E.; Tsai, K.C.; Yang, A.S.; Wong, C.H. A Practical Synthesis of Zanamivir Phosphonate Congeners with Potent Anti-influenza Activity. J. Am. Chem. Soc. 2011, 133, 17959–17965. [Google Scholar] [CrossRef] [PubMed]
- Udommaneethanakit, T.; Rungrotmongkol, T.; Frecer, V.; Seneci, P.; Miertus, S.; Bren, U. Drugs Against Avian Influenza A Virus: Design of Novel Sulfonate Inhibitors of Neuraminidase N1. Curr. Pharm. Des. 2014, 20, 3478–3487. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Y.; Wang, B.; Zhao, L.X.; Li, Y.H.; Shao, H.Y.; Yi, H.; You, X.F.; Li, Z.R. Synthesis and anti-influenza activities of carboxyl alkoxyalkyl esters of 4-guanidino-Neu5Ac2en (zanatnivir). Bioorg. Med. Chem. Lett. 2007, 17, 4851–4854. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Gervay-Hague, J. Synthesis of C-4 and C-7 triazole analogs of zanamivir as multivalent sialic acid containing scaffolds. Carbohydr. Res. 2007, 342, 1636–1650. [Google Scholar] [CrossRef] [PubMed]
- .Zhao, Q.J.; Lou, Y.Y.; Xiong, R.S.; Li, H.H.; Shen, J.S. Regioselective synthesis of 4azido-Neu2en5,7Ac(2)1Me and its intramolecular transformation to 4azido-Neu2en5,9Ac(2)1Me. Carbohydr. Res. 2008, 343, 2459–2462. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Sato, K.; Kitani, S.; Suzuki, T.; Maki, N.; Suzuki, Y.; Sato, M. 2-Deoxy-2,3-didehydro-N-acetylneuraminic acid analogues structurally modified at the C-4 position: Synthesis and biological evaluation as inhibitors of human parainfluenza virus type 1. Bioorg. Med. Chem. 2006, 14, 7893–7897. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Chang, T.C.; Das, A.; Fang, M.Y.; Hung, H.C.; Hsu, K.C.; Yang, J.M.; von Itzstein, M.; Mong, K.K.T.; Hsu, T.A.; et al. Synthesis of acylguanidine zanamivir derivatives as neuraminidase inhibitors and the evaluation of their bio-activities. Org. Biomol. Chem. 2013, 11, 3943–3948. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.J.; Shin, W.J.; Li, N.; Tang, W.; Feng, E.G.; Li, J.; He, P.L.; Zuo, J.P.; Kim, H.; Nam, K.Y.; et al. Synthesis of C-4-modified zanamivir analogs as neuraminidase inhibitors and their anti-AIV activities. Eur. J. Med. Chem. 2012, 54, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.J.; Li, J.; Zhang, J.; Liu, H.; Jiang, H.L. Simultaneous stereoselective 4-amination with cyclic secondary amines and 2-O-deacetylation of peracetylated sialic acid derivatives. Tetrahedron Lett. 2007, 48, 4023–4027. [Google Scholar] [CrossRef]
- Smith, P.W.; Starkey, I.D.; Howes, P.D.; Sollis, S.L.; Keeling, S.P.; Cherry, P.C.; vonItzstein, M.; Wu, W.Y.; Jin, B. Synthesis and influenza virus sialidase inhibitory activity of analogues of 4-guanidino-Neu5Ac2en (GG167) with modified 5-substituents. Eur. J. Med. Chem. 1996, 31, 143–150. [Google Scholar] [CrossRef]
- Kok, G.B.; Campbell, M.; Mackey, B.; vonItzstein, M. Synthesis and biological evaluation of sulfur isosteres of the potent influenza virus sialidase inhibitors 4-amino-4-deoxy- and 4-deoxy-4-guanidino-Neu5Ac2en. J. Chem. Soc. Perkin Trans. 1 1996, 2811–2815. [Google Scholar] [CrossRef]
- Andrews, D.M.; Cherry, P.C.; Humber, D.C.; Jones, P.S.; Keeling, S.P.; Martin, P.F.; Shaw, C.D.; Swanson, S. Synthesis and influenza virus sialidase inhibitory activity of analogues of 4-Guanidino-Neu5Ac2en (Zanamivir) modified in the glycerol side-chain. Eur. J. Med. Chem. 1999, 34, 563–574. [Google Scholar] [CrossRef]
- Schreiner, E.; Christian, R.; Zbiral, E. Structural variations of N-acetylneuraminic acid. 15. Synthesis of 9-deoxy-N-acetylneuraminic, 7,9-dideoxy-N-acetylneuraminic, and 4,7,9-trideoxy-N-acetylneuraminic acids and their behaviour towards CMP-sialate synthase. Liebigs Annalen Der Chem. 1990, 93–97. [Google Scholar] [CrossRef]
- Haldar, J.; de Cienfuegos, L.A.; Tumpey, T.M.; Gubareva, L.V.; Chen, J.Z.; Klibanov, A.M. Bifunctional Polymeric Inhibitors of Human Influenza A Viruses. Pharm. Res. 2010, 27, 259–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, T.; Masuda, T.; Yoshida, S.; Arai, M.; Kobayashi, Y.; Yamashita, M. Synthesis and anti-influenza virus activity of 4-guanidino-7-substituted Neu5Ac2en derivatives. Bioorg. Med. Chem. Lett. 2002, 12, 1921–1924. [Google Scholar] [CrossRef]
- Ikematsu, H.; Kawai, N. Laninamivir octanoate: A new long-acting neuraminidase inhibitor for the treatment of influenza. Expert Rev. Anti-Infect. Ther. 2011, 9, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, H.; Hanaya, K.; Shoji, M.; Hada, N.; Sugai, T. A new route toward 2-acetamido-4-O-methyl-2-deoxy-d-mannopyranose from a Ferrier derivative of tri-O-acetyl-d-glucal, which contributes to aldolase-catalyzed synthesis of laninamivir (CS-8958). Tetrahedron 2013, 69, 7931–7935. [Google Scholar] [CrossRef]
- Masuda, T.; Yoshida, S.; Arai, M.; Kaneko, S.; Yamashita, M.; Honda, T. Synthesis and anti-influenza evaluation of polyvalent sialidase inhibitors bearing 4-guanidino-Neu5Ac2en derivatives. Chem. Pharm. Bull. 2003, 51, 1386–1398. [Google Scholar] [CrossRef] [PubMed]
- Fraser, B.H.; Hamilton, S.; Krause-Heuer, A.M.; Wright, P.J.; Greguric, I.; Tucker, S.P.; Draffan, A.G.; Fokin, V.V.; Sharpless, K.B. Synthesis of 1,4-triazole linked zanamivir dimers as highly potent inhibitors of influenza A and B. MedChemComm 2013, 4, 383–386. [Google Scholar] [CrossRef]
- Sriwilaijaroen, N.; Magesh, S.; Imamura, A.; Ando, H.; Ishida, H.; Sakai, M.; Ishitsubo, E.; Hori, T.; Moriya, S.; Ishikawa, T.; et al. A Novel Potent and Highly Specific Inhibitor against Influenza Viral N1–N9 Neuraminidases: Insight into Neuraminidase-Inhibitor Interactions. J. Med. Chem. 2016, 59, 4563–4577. [Google Scholar] [CrossRef] [PubMed]
- Bamford, M.J.; Pichel, J.C.; Husman, W.; Patel, B.; Storer, R.; Weir, N.G. Synthesis of 6-carbon, 7-carbon and 8-carbon sugar analogs of potent antiinfluenza 2,3-dehydro-2,3-dideoxy-N-acetylneuraminic acid-derivatives. J. Chem. Soc. Perkin Trans. 1 1995, 1181–1187. [Google Scholar] [CrossRef]
- Masuda, T.; Shibuya, S.; Arai, M.; Yoshida, S.; Tomozawa, T.; Ohno, A.; Yamashita, A.; Honda, T. Synthesis and anti-influenza evaluation of orally active bicyclic ether derivatives related to zanamivir. Bioorg. Med. Chem. Lett. 2003, 13, 669–673. [Google Scholar] [CrossRef]
- Smith, P.W.; Robinson, J.E.; Evans, D.N.; Sollis, S.L.; Howes, P.D.; Trivedi, N.; Bethell, R.C. Sialidase inhibitors related to zanamivir: Synthesis and biological evaluation of 4H-pyran 6-ether and ketone. Bioorg. Med. Chem. Lett. 1999, 9, 601–604. [Google Scholar] [CrossRef]
- Smith, P.W.; Sollis, S.L.; Howes, P.D.; Cherry, P.C.; Evans, D.N.; Bethell, R.C.; Fenton, R.; Morley, P.; Pateman, A.; Taylor, N.R. Novel inhibitors of influenza sialidases related to zanamivir (GG167). Abstr. Pap. Am. Chem. Soc. 1997, 214. [Google Scholar] [CrossRef]
- Wyatt, P.G.; Coomber, B.A.; Evans, D.N.; Jack, T.I.; Fulton, H.E.; Wonacott, A.J.; Colman, P.; Varghese, J. Sialidase inhibitors related to zanamivir. Further SAR studies of 4-amino-4H-pyran-2-carboxylic acid-6-propylamides. Bioorg. Med. Chem. Lett. 2001, 11, 669–673. [Google Scholar] [CrossRef]
- Soule, J.F.; Mathieu, A.; Norsikian, S.; Beau, J.M. Coupling the Petasis Condensation to an Iron(III) Chloride-Promoted Cascade Provides a Short Synthesis of Relenza Congeners. Org. Lett. 2010, 12, 5322–5325. [Google Scholar] [CrossRef] [PubMed]
- Chandler, M.; Conroy, R.; Cooper, A.W.J.; Lamont, R.B.; Scicinski, J.J.; Smart, J.E.; Storer, R.; Weir, N.G.; Wilson, R.D.; Wyatt, P.G. Approaches to carbocyclic analogs of the potent neuraminidase inhibitor 4-guanidino-Neu5Ac3en—X-ray molecular-structure of N-(1S,2S,6R)-2-azido-6-benzyloxymethyl-4-formylcyclohex-3-enyl acetamide. J. Chem. Soc. Perkin Trans. 1 1995, 1189–1197. [Google Scholar] [CrossRef]
- Bischofberger, N.W.; Kim, C.U.; Lew, W.; Liu, H.; Williams, M.A. Novel Selctive Inhibitors of Viral or Bactyerial Neuraminidases. EP0759917, 14 April 2000. [Google Scholar]
- Abrecht, S.; Harrington, P.; Iding, H.; Karpf, M.; Trussardi, R.; Wirz, B.; Zutter, U. The synthetic development of the anti-influenza neuraminidase inhibitor oseltamivir phosphate (Tamiflu(R)): A challenge for synthesis & process research. Chimia 2004, 58, 621–629. [Google Scholar]
- Rohloff, J.C.; Kent, K.M.; Postich, M.J.; Becker, M.W.; Chapman, H.H.; Kelly, D.E.; Lew, W.; Louie, M.S.; McGee, L.R.; Prisbe, E.J.; et al. Practical total synthesis of the anti-influenza drug GS-4104. J. Org. Chem. 1998, 63, 4545–4550. [Google Scholar] [CrossRef]
- Federspiel, M.; Fischer, R.; Hennig, M.; Mair, H.J.; Oberhauser, T.; Rimmler, G.; Albiez, T.; Bruhin, J.; Estermann, H.; Gandert, C.; et al. Industrial synthesis of the key precursor in the synthesis of the anti-influenza drug oseltamivir phosphate (Ro 64-0796/002, GS-4104-02): Ethyl (3R,4S,5S)-4,5-epoxy-3-(1-ethyl-propoxy)-cyclohex-1-ene-1-carboxylate. Org. Process Res. Dev. 1999, 3, 266–274. [Google Scholar] [CrossRef]
- Nie, L.D.; Wang, F.F.; Ding, W.; Shi, X.X.; Lu, X. A novel azide-free asymmetric synthesis of oseltamivir phosphate (Tamiflu) starting from Roche’s epoxide. Tetrahedron Asymmetry 2013, 24, 638–642. [Google Scholar] [CrossRef]
- Nie, L.D.; Shi, X.X. A novel asymmetric synthesis of oseltamivir phosphate (Tamiflu) from (−)-shikimic acid. Tetrahedron Asymmetry 2009, 20, 124–129. [Google Scholar] [CrossRef]
- Nie, L.D.; Shi, X.X.; Quan, N.; Wang, F.F.; Lu, X. Novel asymmetric synthesis of oseltamivir phosphate (Tamiflu) from (−)-shikimic acid via cyclic sulfite intermediates. Tetrahedron Asymmetry 2011, 22, 1692–1699. [Google Scholar] [CrossRef]
- Nie, L.D.; Ding, W.; Shi, X.X.; Quan, N.; Lu, X. A novel and high-yielding asymmetric synthesis of oseltamivir phosphate (Tamiflu) starting from (–)-shikimic acid. Tetrahedron Asymmetry 2012, 23, 742–747. [Google Scholar] [CrossRef]
- Kim, H.K.; Park, K.J.J. A new efficient synthesis of oseltamivir phosphate (Tamiflu) from (−)-shikimic acid. Tetrahedron Lett. 2012, 53, 1561–1563. [Google Scholar] [CrossRef]
- Karpf, M.; Trussardi, R. New, azide-free transformation of epoxides into 1,2-diamino compounds: Synthesis of the anti-influenza neuraminidase inhibitor oseltamivir phosphate (Tamiflu). J. Org. Chem. 2001, 66, 2044–2051. [Google Scholar] [CrossRef] [PubMed]
- Kalashnikov, A.I.; Sysolyatin, S.V.; Sakovich, G.V.; Sonina, E.G.; Shchurova, I.A. Facile method for the synthesis of oseltamivir phosphate. Russ. Chem. Bull. 2013, 62, 163–170. [Google Scholar] [CrossRef]
- Sullivan, B.; Carrera, I.; Drouin, M.; Hudlicky, T. Symmetry-Based Design for the Chemoenzymatic Synthesis of Oseltamivir (Tamiflu) from Ethyl Benzoate. Angew. Chem. Int. Ed. 2009, 48, 4229–4231. [Google Scholar] [CrossRef] [PubMed]
- Bromfield, K.M.; Graden, H.; Hagberg, D.P.; Olsson, T.; Kann, N. An iron carbonyl approach to the influenza neuraminidase inhibitor oseltamivir. Chem. Commun. 2007, 3183–3185. [Google Scholar] [CrossRef] [PubMed]
- Zutter, U.; Iding, H.; Spurr, P.; Wirz, B. New, efficient synthesis of oseltamivir phosphate (Tamiflu) via enzymatic desymmetrization of a meso-1,3-cyclohexanedicarboxylic acid diester. J. Org. Chem. 2008, 73, 4895–4902. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.; Babu, V.S. Enantioselective synthesis of oseltamivir phosphate. Tetrahedron 2011, 67, 2044–2050. [Google Scholar] [CrossRef]
- Shie, J.J.; Fang, J.M.; Wong, C.H. A concise and flexible synthesis of the potent anti-influenza agents tamiflu and tamiphosphor. Angew. Chem. Int. Ed. 2008, 47, 5788–5791. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Zhang, T. Development of a Concise Synthesis of (−)-Oseltamivir (Tamiflu). Chem. Eur. J. 2011, 17, 3630–3643. [Google Scholar] [CrossRef] [PubMed]
- Yeung, Y.Y.; Hong, S.; Corey, E.J. A short enantioselective pathway for the synthesis of the anti-influenza neuramidase inhibitor Oseltamivir from 1,3-butadiene and acrylic acid. J. Am. Chem. Soc. 2006, 128, 6310–6311. [Google Scholar] [CrossRef] [PubMed]
- Satoh, N.; Akiba, T.; Yokoshima, S.; Fukuyama, T. A practical synthesis of (−)-oseltamivir. Tetrahedron 2009, 65, 3239–3245. [Google Scholar] [CrossRef]
- Sun, H.; Lin, Y.J.; Wu, Y.L.; Wu, Y.K. A Facile Access to Antiflu Agent Tamiflu/Oseltamivir. Synlett 2009, 2473–2476. [Google Scholar]
- Morita, M.; Sone, T.; Yamatsugu, K.; Sohtome, Y.; Matsunaga, S.; Kanaia, M.; Yasuyoshi, W.B.; Shibasaki, M. A method for the synthesis of an oseltamivir PET tracer. Bioorg. Med. Chem. Lett. 2008, 18, 600–602. [Google Scholar] [CrossRef] [PubMed]
- Yamatsugu, K.; Kamijo, S.; Suto, Y.; Kanai, M.; Shibasaki, M. A concise synthesis of Tamiflu: Third generation route via the Diels-Alder reaction and the Curtius rearrangement. Tetrahedron Lett. 2007, 48, 1403–1406. [Google Scholar] [CrossRef]
- Rawat, V.; Dey, S.; Sudalai, A. Synthesis of the anti-influenza agent (−)-oseltamivir free base and (−)-methyl 3-epi-shikimate. Org. Biomol. Chem. 2012, 10, 3988–3990. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.S.; Kang, H.Y. Synthesis of (−)-Oseltamivir Phosphate (Tamiflu) Starting from cis-2,3-Bis(hydroxymethyl)aziridine. J. Org. Chem. 2012, 77, 8792–8796. [Google Scholar] [CrossRef] [PubMed]
- Cong, X.; Yao, Z.J. Ring-closing metathesis-based synthesis of (3R,4R,5S)-4-acetylamino-5-amino-3-hydroxycyclohex-1-ene-carboxylic acid ethyl ester: A functionalized cycloalkene skeleton of GS4104. J. Org. Chem. 2006, 71, 5365–5368. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic, M.; Ferjancic, Z.; Saicic, R.N. An aldol approach to the enantioselective synthesis of (−)-oseltamivir phosphate. Org. Biomol. Chem. 2011, 9, 6927–6929. [Google Scholar] [CrossRef] [PubMed]
- Osato, H.; Jones, I.L.; Chen, A.Q.; Chai, C.L.L. Efficient Formal Synthesis of Oseltamivir Phosphate (Tamiflu) with Inexpensive d-Ribose as the Starting Material. Org. Lett. 2010, 12, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Wichienukul, P.; Akkarasamiyo, S.; Kongkathip, N.; Kongkathip, B. An efficient synthesis of oseltamivir phosphate (Tamiflu) via a metal-mediated domino reaction and ring-closing metathesis. Tetrahedron Lett. 2010, 51, 3208–3210. [Google Scholar] [CrossRef]
- Kongkathip, B.; Akkarasamiyo, S.; Kongkathip, N. A new and efficient asymmetric synthesis of oseltamivir phosphate (Tamiflu) from d-glucose. Tetrahedron 2015, 71, 2393–2399. [Google Scholar] [CrossRef]
- Shie, J.J.; Fang, J.M.; Wang, S.Y.; Tsai, K.C.; Cheng, Y.S.E.; Yang, A.S.; Hsiao, S.C.; Su, C.Y.; Wong, C.H. Synthesis of Tamiflu and its phosphonate congeners possessing potent anti-influenza activity. J. Am. Chem. Soc. 2007, 129, 11892–11893. [Google Scholar] [CrossRef] [PubMed]
- Chuanopparat, N.; Kongkathip, N.; Kongkathip, B. A concise and practical synthesis of oseltamivir phosphate(Tamiflu) from d-mannose. Tetrahedron 2012, 68, 6803–6809. [Google Scholar] [CrossRef]
- Chen, C.A.; Fang, J.M. Synthesis of oseltamivir and tamiphosphor from N-acetyl-d-glucosamine. Org. Biomol. Chem. 2013, 11, 7687–7699. [Google Scholar] [CrossRef] [PubMed]
- Mandai, T.; Oshitari, T. Efficient Asymmetric Synthesis of Oseltamivir from d-Mannitol. Synlett 2009, 783–786. [Google Scholar] [CrossRef]
- Schmid, C.R.; Bradley, D.A. 2,3-O-(3-pentylidene)-d-glyceraldehyde and 2,3-O-(3-pentylidene)-l-glyceraldehyde—Convinient glyceraldehyde surrogates obtained via a novel periodate-based oxidation system. Synthesis 1992, 1992, 587–590. [Google Scholar] [CrossRef]
- Oshitari, T.; Mandai, T. Azide-Free Synthesis of Oseltamivir from l-Methionine. Synlett 2009, 5, 787–789. [Google Scholar]
- Ko, J.S.; Keum, J.E.; Ko, S.Y. A Synthesis of Oseltamivir (Tamiflu) Starting from d-Mannitol. J. Org. Chem. 2010, 75, 7006–7009. [Google Scholar] [CrossRef] [PubMed]
- Alagiri, K.; Furutachi, M.; Yamatsugu, K.; Kumagai, N.; Watanabe, T.; Shibasaki, M. Two Approaches toward the Formal Total Synthesis of Oseltamivir Phosphate (Tamiflu): Catalytic Enantioselective Three-Component Reaction Strategy and l-Glutamic Acid Strategy. J. Org. Chem. 2013, 78, 4019–4026. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.L.; Yu, S.Y.; Wang, Y.; Ma, D.W. Organocatalytic Michael Addition of Aldehydes to Protected 2-Amino-1-Nitroethenes: The Practical Syntheses of Oseltamivir (Tamiflu) and Substituted 3-Aminopyrrolidines. Angew. Chem. Int. Ed. 2010, 49, 4656–4660. [Google Scholar] [CrossRef] [PubMed]
- Rehak, J.; Hut’ka, M.; Latika, A.; Brath, H.; Almassy, A.; Hajzer, V.; Durmis, J.; Toma, S.; Sebesta, R. Thiol-Free Synthesis of Oseltamivir and Its Analogues via Organocatalytic Michael Additions of Oxyacetaldehydes to 2-Acylaminonitroalkenes. Synthesis 2012, 44, 2424–2430. [Google Scholar]
- Ishikawa, H.; Bondzic, B.P.; Hayashi, Y. Synthesis of (−)-Oseltamivir by Using a Microreactor in the Curtius Rearrangement. Eur. J. Org. Chem. 2011, 2011, 6020–6031. [Google Scholar] [CrossRef]
- Mukaiyama, T.; Ishikawa, H.; Koshino, H.; Hayashi, Y. One-Pot Synthesis of (−)-Oseltamivir and Mechanistic Insights into the Organocatalyzed Michael Reaction. Chem. Eur. J. 2013, 19, 17789–17800. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Li, Y.B.; Wang, R.B.; Li, F.Q.; Liu, C.; Chan, A.S.C.; Lu, G. A Practical and Azide-Free Synthetic Approach to Oseltamivir from Diethyl d-Tartrate. J. Org. Chem. 2010, 75, 3125–3128. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.M.; Zhao, Y.Y.; Ng, S.; Zhang, J.; Zeng, J.; Than, A.; Chen, P.; Liu, X.W. Sugar-Based Synthesis of Tamiflu and Its Inhibitory Effects on Cell Secretion. Chem. Eur. J. 2010, 16, 4533–4540. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, D.S. Formal Synthesis of Tamiflu: Conversion of Tamiflu into Tamiphosphor. Synlett 2012, 573–576. [Google Scholar] [CrossRef]
- Carbain, B.; Collins, P.J.; Callum, L.; Martin, S.R.; Hay, A.J.; McCauley, J.; Streicher, H. Efficient Synthesis of Highly Active Phospha-Isosteres of the Influenza Neuraminidase Inhibitor Oseltamivir. ChemMedChem 2009, 4, 335–337. [Google Scholar] [CrossRef] [PubMed]
- Adamska, A.; Olejniczak, A.B.; Zwolinski, K.; Szczepek, W.J.; Krol, E.; Szewczyk, B.; Grynkiewicz, G.; Lesnikowski, Z.J. Oseltamivir analog with boron cluster modulator. Acta Pol. Pharm. 2012, 69, 1218–1223. [Google Scholar] [PubMed]
- Saito, K.; Kanai, M. synthesis of a new oseltamivir derivative through late-stage catalytic C-H functionalization. Heterocycles 2012, 86, 1565–1574. [Google Scholar]
- Stankova, I.; Chuchkov, K.; Schmidtke, M.; Galabov, A. Synthesis and In Vitro Anti-influenza Activity of New Amino Acids and Peptidomimetics Derivatives of Oseltamivir and Rimantadine. Antivir. Res. 2011, 90, A76–A76. [Google Scholar] [CrossRef]
- Giorgi, M.E.; Piuselli, D.; Agusti, R.; de Lederkremer, R.M. Synthesis of oseltamivir conjugates with lactose analogs for inhibition studies on Trypanosoma cruzi trans-sialidase. Arkivoc 2011, 7, 260–271. [Google Scholar]
- Chochkova, M.; Ivanova, G.; Galabov, A.; Milkova, T. Amides of antiviral drug oseltamivir with antioxidant active aminoacids: Synthesis and biological activities. J. Pept. Sci. 2012, 18, S177–S177. [Google Scholar]
- Mooney, C.A.; Johnson, S.A.; ‘t Hart, P.; Quarles van Ufford, L.; de Haan, C.A.; Moret, E.E.; Martin, N.I. Oseltamivir Analogues Bearing N-Substituted Guanidines as Potent Neuraminidase Inhibitorsdx. J. Med. Chem. 2014, 57, 3154–3160. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.; Dell’Amico, L.; Battistini, L.; Curti, C.; Rivara, S.; Pala, D.; Kerry, P.S.; Pelosi, G.; Casiraghi, G.; Rassu, G.; et al. Synthesis, structure and inhibitory activity of a stereoisomer of oseltamivir carboxylate. Org. Biomol. Chem. 2014, 12, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Kongkamnerd, J.; Cappelletti, L.; Prandi, A.; Seneci, P.; Rungrotmongkol, T.; Jongaroonngamsang, N.; Rojsitthisak, P.; Frecer, V.; Milani, A.; Cattoli, G.; et al. Synthesis and in vitro study of novel neuraminidase inhibitors against avian influenza virus. Bioorg. Med. Chem. 2012, 20, 2152–2157. [Google Scholar] [CrossRef] [PubMed]
- Babu, Y.S.; Chand, P.; Bantia, S.; Kotian, P.; Dehghani, A.; El-Kattan, Y.; Lin, T.H.; Hutchison, T.L.; Elliott, A.J.; Parker, C.D.; et al. BCX-1812 (RWJ-270201): Discovery of a novel, highly potent, orally active, and selective influenza neuraminidase inhibitor through structure-based drug design. J. Med. Chem. 2000, 43, 3482–3486. [Google Scholar] [CrossRef] [PubMed]
- Sidwell, R.W.; Smee, D.F. Peramivir (BCX-1812, RWJ-270201): Potential new therapy for influenza. Expert Opin. Investig. Drugs 2002, 11, 859–869. [Google Scholar] [PubMed]
- Chand, P.; Bantia, S.; Kotian, P.L.; El-Kattan, Y.; Lin, T.H.; Babu, Y.S. Comparison of the anti-influenza virus activity of cyclopentane derivatives with oseltamivir and zanamivir in vivo. Bioorg. Med. Chem. 2005, 13, 4071–4077. [Google Scholar] [CrossRef] [PubMed]
- Bantia, S.; Arnold, C.S.; Parker, C.D.; Upshaw, R.; Chand, P. Anti-influenza virus activity of peramivir in mice with single intramuscular injection. Antivir. Res. 2006, 69, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Smee, D.F.; Huffman, J.H.; Morrison, A.C.; Barnard, D.L.; Sidwell, R.W. Cyclopentane neuraminidase inhibitors with potent in vitro anti-influenza virus activities. Antimicrob. Agents Chemother. 2001, 45, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.; Hong, J.; Sun, P.H.; Chen, J.X.; Chen, W.M. Facile Synthesis of the Neuraminidase Inhibitor Peramivir. Synth. Commun. 2013, 43, 2641–2647. [Google Scholar] [CrossRef]
- Mineno, T.; Miller, M.J. Stereoselective total synthesis of racemic BCX-1812 (RWJ-270201) for the development of neuraminidase inhibitors as anti-influenza agents. J. Org. Chem. 2003, 68, 6591–6596. [Google Scholar] [CrossRef] [PubMed]
- Bromba, C.M.; Mason, J.W.; Brant, M.G.; Chan, T.; Lunke, M.D.; Petric, M.; Boulanger, M.J.; Wulff, J.E. The de-guanidinylated derivative of peramivir remains a potent inhibitor of influenza neuraminidase. Bioorg. Med. Chem. Lett. 2011, 21, 7137–7141. [Google Scholar] [CrossRef] [PubMed]
- Chand, P.; Kotian, P.L.; Dehghani, A.; El-Kattan, Y.; Lin, T.H.; Hutchison, T.L.; Babu, Y.S.; Bantia, S.; Elliott, A.J.; Montgomery, J.A. Systematic structure-based design and stereoselective synthesis of novel multisubstituted cyclopentane derivatives with potent antiinfluenza activity. J. Med. Chem. 2001, 44, 4379–4392. [Google Scholar] [CrossRef] [PubMed]
- Hronowski, L.J.J.; Szarek, W.A. Regiospecific synthesis of cyclopentane analogs of (2′- and 3′-deoxy-threo-pentofuranosyl)-uracil and -2-thiouracil nucleosides. Can. J. Chem. Rev. Can. Chim. 1985, 63, 2787–2797. [Google Scholar] [CrossRef]
- Chand, P.; Babu, Y.S.; Bantia, S.; Rowland, S.; Dehghani, A.; Kotian, P.L.; Hutchison, T.L.; Ali, S.; Brouillette, W.; El-Kattan, Y.; Lin, T.H. Syntheses and neuraminidase inhibitory activity of multisubstituted cyclopentane amide derivatives. J. Med. Chem. 2004, 47, 1919–1929. [Google Scholar] [CrossRef] [PubMed]





































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laborda, P.; Wang, S.-Y.; Voglmeir, J. Influenza Neuraminidase Inhibitors: Synthetic Approaches, Derivatives and Biological Activity. Molecules 2016, 21, 1513. https://doi.org/10.3390/molecules21111513
Laborda P, Wang S-Y, Voglmeir J. Influenza Neuraminidase Inhibitors: Synthetic Approaches, Derivatives and Biological Activity. Molecules. 2016; 21(11):1513. https://doi.org/10.3390/molecules21111513
Chicago/Turabian StyleLaborda, Pedro, Su-Yan Wang, and Josef Voglmeir. 2016. "Influenza Neuraminidase Inhibitors: Synthetic Approaches, Derivatives and Biological Activity" Molecules 21, no. 11: 1513. https://doi.org/10.3390/molecules21111513