
Combating Ebola with Repurposed Therapeutics Using the CANDO Platform


Abstract
:1. Introduction
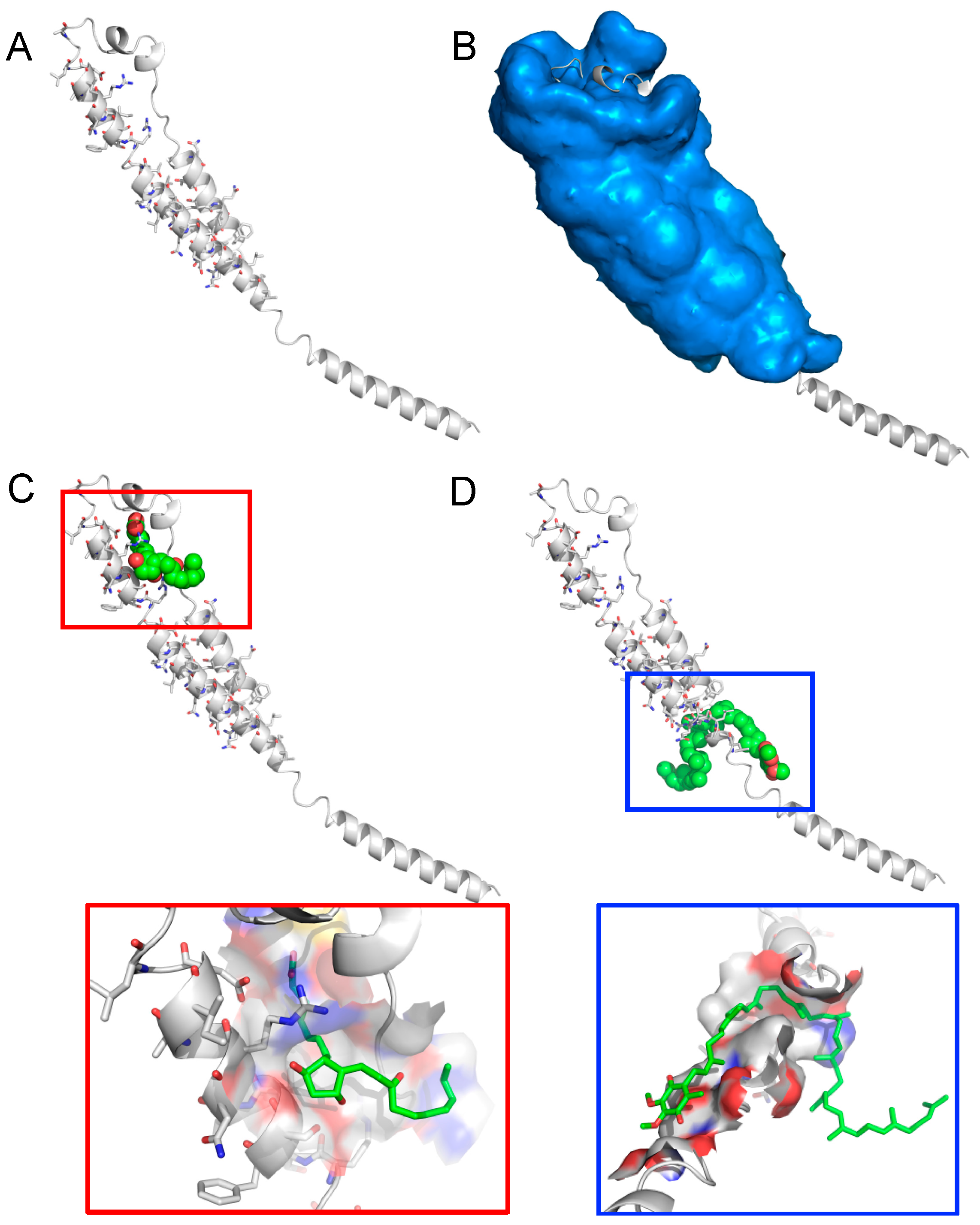
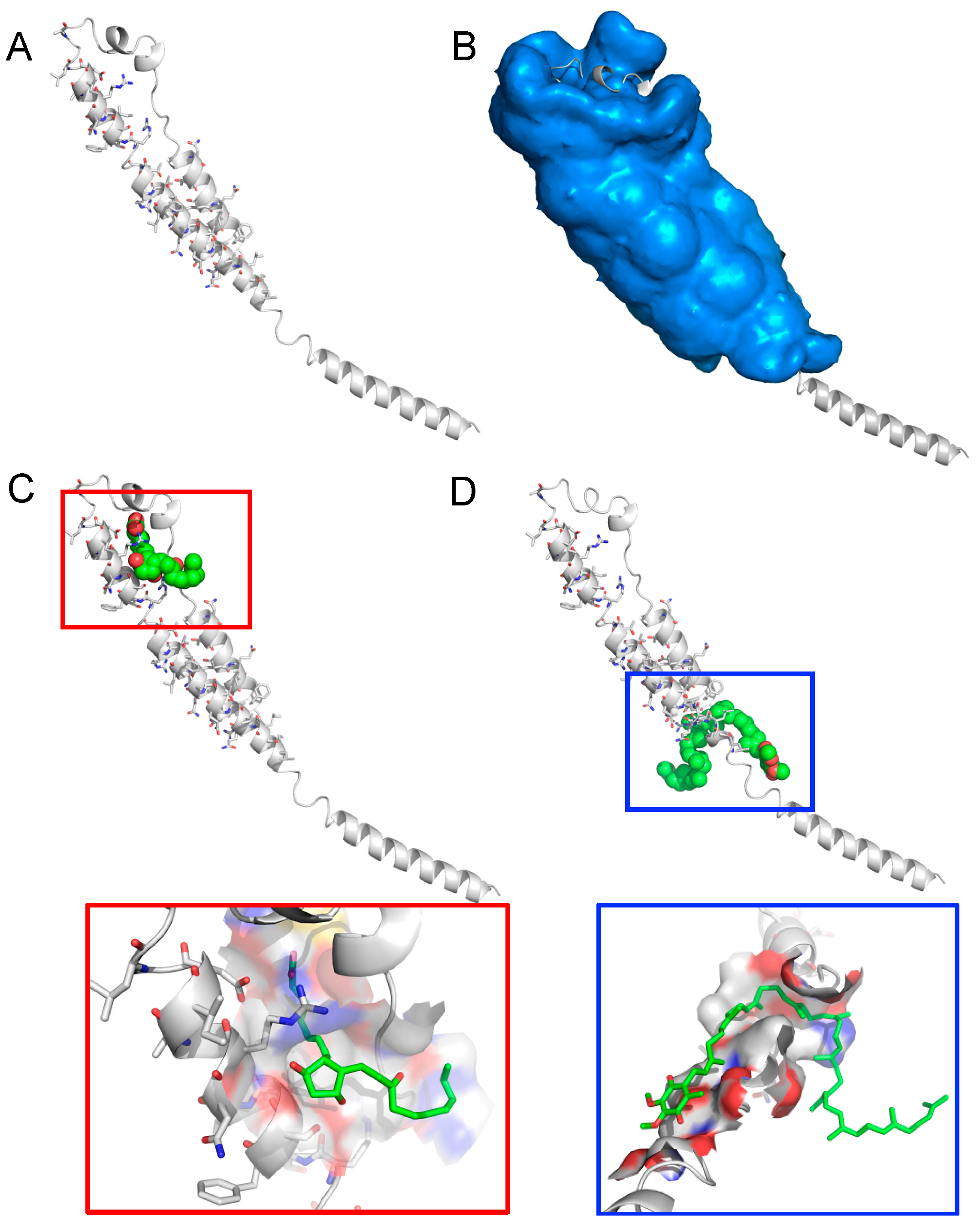
2. Results
3. Discussion
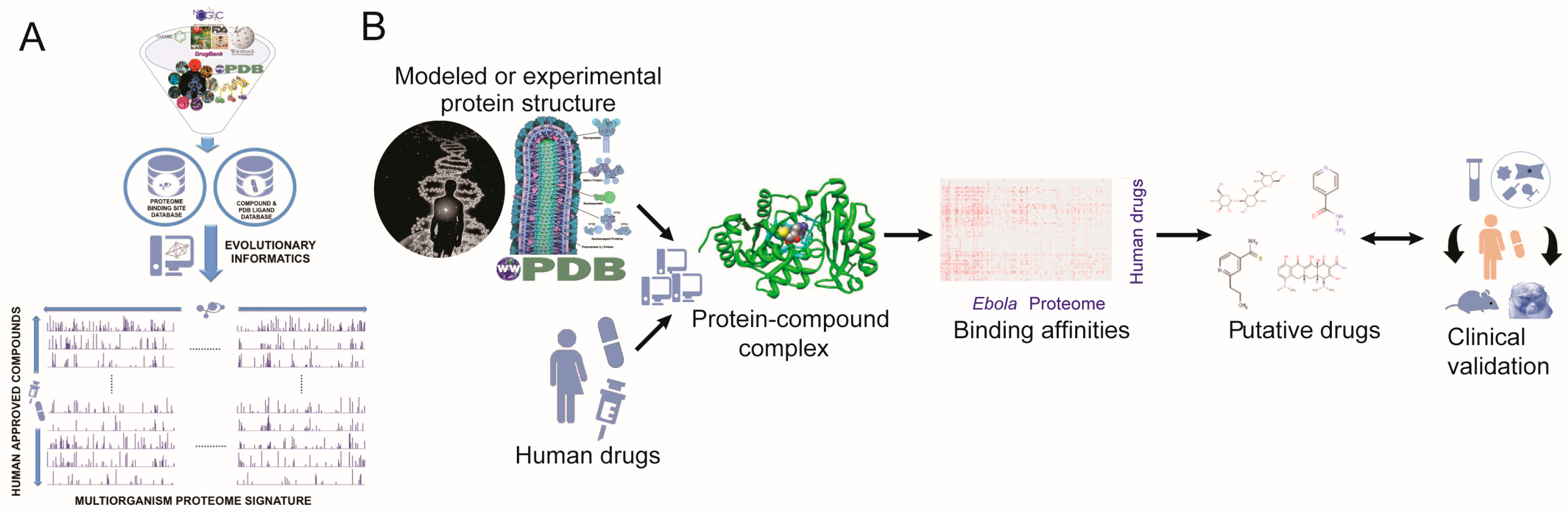
4. Materials and Methods
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Baize, S.; Pannetier, D.; Oestereich, L.; Rieger, T.; Koivogui, L.; Magassouba, N.; Soropogui, B.; Sow, M.S.; Keita, S.; de Clerck, H.; et al. Emergence of Zaire Ebola virus disease in Guinea. N. Engl. J. Med. 2014, 371, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- CDC 2014 Ebola Outbreak in West Africa—Case Counts—As of on Feb 7 2016. Available online: http://www.cdc.gov/vhf/ebola/outbreaks/2014-west-africa/case-counts.html (accessed on 10 February 2016).
- Gupta, R. Rethinking the development of Ebola treatments. Lancet Glob. Heal. 2014, 2, e563–e564. [Google Scholar] [CrossRef]
- Feldmann, H.; Geisbert, T.W. Ebola haemorrhagic fever. Lancet 2011, 377, 849–862. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Novel Drugs 2015 Summary; U.S. Food and Drug Administration: Silver Spring, MD, USA, 2016.
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [PubMed]
- Bonander, N.; Bill, R.M. Relieving the first bottleneck in the drug discovery pipeline: using array technologies to rationalize membrane protein production. Expert Rev. Proteomics 2009, 6, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, S.H.; Singh, K. XDR-TB, what is it; how is it treated; and why is therapeutic failure so high? Recent Pat. Antiinfect. Drug Discov. 2011, 6, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Horst, J.A.; Pieper, U.; Sali, A.; Zhan, L.; Chopra, G.; Samudrala, R.; Featherstone, J.D.B. Strategic Protein Target Analysis for Developing Drugs to Stop Dental Caries. Adv. Dent. Res. 2012, 24, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Sacks, L.V.; Behrman, R.E. Challenges, successes and hopes in the development of novel TB therapeutics. Future Med. Chem. 2009, 1, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Cote, T.R. Database identifies FDA-approved drugs with potential to be repurposed for treatment of orphan diseases. Br. Bioinform. 2011, 12, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Xie, L.; Li, W.W.; Bourne, P.E. SMAP-WS: A parallel web service for structural proteome-wide ligand-binding site comparison. Nucleic Acids Res. 2010, 38, W441–W444. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Williams, A.J.; Krasowski, M.D.; Freundlich, J.S. In silico repositioning of approved drugs for rare and neglected diseases. Drug Discov. Today 2011, 16, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Swamidass, S.J. Mining small-molecule screens to repurpose drugs. Br. Bioinform. 2011, 12, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Jenwitheesuk, E.; Samudrala, R. Identification of potential multitarget antimalarial drugs. JAMA 2005, 294, 1490–1491. [Google Scholar] [PubMed]
- Jenwitheesuk, E.; Horst, J.A.; Rivas, K.L.; Van Voorhis, W.C.; Samudrala, R. Novel paradigms for drug discovery: computational multitarget screening. Trends Pharmacol. Sci. 2008, 29, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Horst, J.A.; Laurenzi, A.; Bernard, B.; Samudrala, R. Computational Multitarget Drug Discovery. In Polypharmacology in Drug Discovery; Peters, J.-U., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Minie, M.; Chopra, G.; Sethi, G.; Horst, J.; White, G.; Roy, A.; Hatti, K.; Samudrala, R. CANDO and the infinite drug discovery frontier. Drug Discov. Today 2014, 19, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Dudley, J.T.; Deshpande, T.; Butte, A.J. Exploiting drug-disease relationships for computational drug repositioning. Br. Bioinform. 2011, 12, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Costin, J.M.; Jenwitheesuk, E.; Lok, S.M.; Hunsperger, E.; Conrads, K.A.; Fontaine, K.A.; Rees, C.R.; Rossmann, M.G.; Isern, S.; Samudrala, R.; et al. Structural optimization and de novo design of dengue virus entry inhibitory peptides. PLoS Negl. Trop. Dis. 2010, 4, e721. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, C.O.; Costin, J.M.; Rowe, D.K.; Lin, L.; Jenwitheesuk, E.; Samudrala, R.; Isern, S.; Michael, S.F. Viral entry inhibitors block dengue antibody-dependent enhancement in vitro. Antivir. Res. 2011, 89, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Chopra, G.; Samudrala, R. Multiscale modelling of relationships between protein classes and drug behavior across all diseases using the CANDO platform. Mini Rev. Med. Chem. 2015, 15, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Chopra, G.; Samudrala, R. Exploring Polypharmacology in Drug Discovery and Repurposing Using the CANDO Platform. Curr. Pharm. Des. 2016, 22, 3109–3123. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, D.E.; Siliciano, R.F.; Jacobs, W.R., Jr. Outwitting evolution: Fighting drug-resistant TB, malaria, and HIV. Cell 2012, 148, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, A.L.; Okumura, A.; Ferris, M.T.; Green, R.; Feldmann, F.; Kelly, S.M.; Scott, D.P.; Safronetz, D.; Haddock, E.; LaCasse, R.; et al. Host genetic diversity enables Ebola hemorrhagic fever pathogenesis and resistance. Science 2014, 346, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Kouznetsova, J.; Sun, W.; Martinez-Romero, C.; Tawa, G.; Shinn, P.; Chen, C.Z.; Schimmer, A.; Sanderson, P.; McKew, J.C.; Zheng, W.; et al. Identification of 53 compounds that block Ebola virus-like particle entry via a repurposing screen of approved drugs. Emerg. Microbes Infect. 2014, 3, e84. [Google Scholar] [CrossRef] [PubMed]
- Johansen, L.M.; DeWald, L.E.; Shoemaker, C.J.; Hoffstrom, B.G.; Lear-Rooney, C.M.; Stossel, A.; Nelson, E.; Delos, S.E.; Simmons, J.A.; Grenier, J.M.; et al. A screen of approved drugs and molecular probes identifies therapeutics with anti-Ebola virus activity. Sci. Transl. Med. 2015, 7, 290ra89. [Google Scholar] [CrossRef] [PubMed]
- Landry, J.J.; Pyl, P.T.; Rausch, T.; Zichner, T.; Tekkedil, M.M.; Stutz, A.M.; Jauch, A.; Aiyar, R.S.; Pau, G.; Delhomme, N.; et al. The genomic and transcriptomic landscape of a HeLa cell line. G3 2013, 3, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Ellis, D.S.; Simpson, I.H.; Francis, D.P.; Knobloch, J.; Bowen, E.T.; Lolik, P.; Deng, I.M. Ultrastructure of Ebola virus particles in human liver. J. Clin. Pathol. 1978, 31, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Ryabchikova, E.I.; Kolesnikova, L.V.; Luchko, S.V. An analysis of features of pathogenesis in two animal models of Ebola virus infection. J. Infect. Dis. 1999, 179, S199–S202. [Google Scholar] [CrossRef] [PubMed]
- Kash, J.C.; Muhlberger, E.; Carter, V.; Grosch, M.; Perwitasari, O.; Proll, S.C.; Thomas, M.J.; Weber, F.; Klenk, H.D.; Katze, M.G. Global suppression of the host antiviral response by Ebola- and Marburgviruses: Increased antagonism of the type I interferon response is associated with enhanced virulence. J. Virol. 2006, 80, 3009–3020. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Jenwitheesuk, E.; Samudrala, R. Improved prediction of HIV-1 protease-inhibitor binding energies by molecular dynamics simulations. BMC Struct. Biol. 2003, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Bernard, B.; Samudrala, R. A generalized knowledge-based discriminatory function for biomolecular interactions. Proteins Struct. Funct. Bioinform. 2009, 76, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2011, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhang, J.; Roy, A.; Zhang, Y. Automated protein structure modeling in CASP9 by I-TASSER pipeline combined with QUARK-based ab initio folding and FG-MD-based structure refinement. Proteins Struct. Funct. Bioinf. 2011, 79, 147–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chopra, G.; Summa, C.M.; Levitt, M. Solvent dramatically affects protein structure refinement. Proc. Natl. Acad. Sci. USA 2008, 105, 20239–20244. [Google Scholar] [CrossRef] [PubMed]
- Chopra, G.; Kalisman, N.; Levitt, M. Consistent refinement of submitted models at CASP using a knowledge-based potential. Proteins 2010, 78, 2668–2678. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.P.G.L.M.; Levitt, M.; Chopra, G. KoBaMIN: A knowledge-based minimization web server for protein structure refinement. Nucleic Acids Res. 2012, 40, W323–W328. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Zhang, Y. Recognizing protein-ligand binding sites by global structural alignment and local geometry refinement. Structure 2012, 20, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not Available.
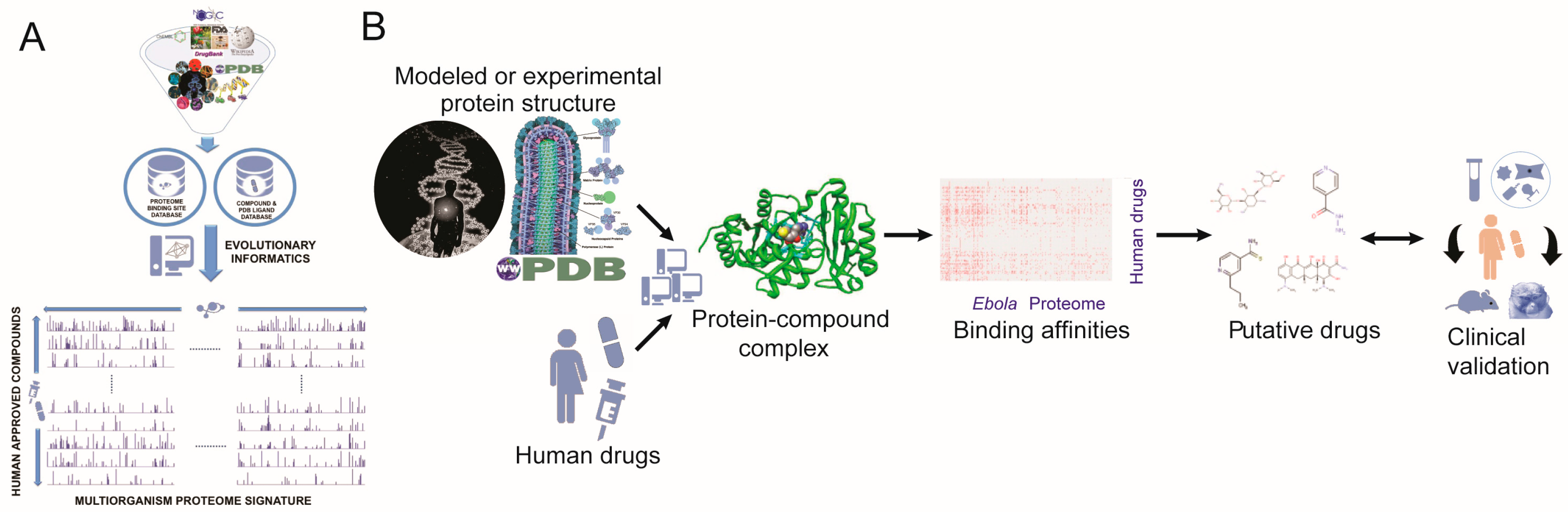

{kind=link}
{kind=link}
| Compound(s) | Interaction Score | Consensus Score (min) | Protein Target Identifiers |
|---|---|---|---|
| enfuvirtide | 2.0 | 7 | GP2, VP35, 1ebo-F |
| vancomycin, bleomycin | 2.0 | 10 | GP1,2, pre-sGP, SGP, SsGP |
| octreotide, lanreotide, somatostatin | 2.0 | 10 | GP1,2, pre-sGP, SGP, SsGP |
| ubidecarenone (CoQ10) | 1.6 | 7 | GP1,2, GP2, VP24, VP35, VP40, 1ebo-F |
| unoprostone | 1.3 | 10 | GP1,2, VP35, VP24, 1ebo-F |
| Compound(s) | Interaction Score | Consensus Score | Approved Indication | Mode of Action |
|---|---|---|---|---|
| Niclosamide * | 1.897 | 3 | helminthic infestation | inhibits parasite metabolism |
| Sertraline * | 1.897 | 1 | depression, anxiety | selective serotonin receptor inhibitor |
| Clomifene * | 1.897 | 1 | anovulation, oligoovulation | selective estrogen receptor modulator |
| Alverine | 1.897 | 1 | gastrointestinal muscle spasms | parasympathetic nervous system modulator |
| Aprindine | 1.897 | 1 | cardiac arrhythmia | sodium channel inhibitor |
| Mebendazole * | 1.897 | 1 | helminthic infestation | tubulin destabilizer |
| Salmeterol | 1.890 | 2 | asthma | beta 2 adrenergic receptor agonist |
| Topotecan | 1.823 | 1 | ovarian and lung cancers | DNA topoisomerase I inhibitor |
| Deslanoside * | 1.377 | 10 | cardiac arrhythmia | sodium-potassium channel blocker |
| Propafenone | 1.298 | 10 | cardiac arrhythmia | sodium channel blocker |
| Digoxin * | 1.067 | 1 | cardiac arrhythmia | sodium-potassium channel blocker |
| Proglumetacin | 1.058 | 1 | non-steroidal anti-inflammatory drug | cyclooxygenase-1 inhibitor |
| Posaconazole | 1.023 | 3 | fungal infection (aspergillus and candida) | membrane bound enzyme inhibitor |
| Raloxifene * | 0.843 | 2 | osteoporosis and breast cancer prevention | selective estrogen receptor modulator |
| Clarithromycin | 0.741 | 2 | bacterial infections | protein synthesis inhibitor |
| Clemastine * | 0.741 | 1 | allergies | H1 histamine receptor inhibitor |
| Colchicine | 0.741 | 1 | gout, pericarditis | microtubule inhibitor |
| Tamoxifen * | 0.741 | 1 | estrogen receptor positive breast cancer | Selective estrogen receptor modulator |
| Thiothixene | 0.741 | 1 | psychotic disorders, e.g., schizophrenia | dopamine antagonist |
| Daunorubicin | 0.714 | 1 | hematologic dyscrasia (acute lymphocytic leukemia, acute myeloid leukemia) | DNA topoisomerase II inhibitor |
| Dronedarone | 0.722 | 1 | cardiac arrhythmia | potassium channel blocker |
| Vincristine | 0.707 | 1 | hematologic dyscrasia (acute lymphocytic leukemia, acute myeloid leukemia) | microtubule inhibitor |
| Compound(s) | Interaction Score | Consensus Score | Approved Indication | Mode of Action |
|---|---|---|---|---|
| Niclosamide * | 1.897 | 3 | helminthic infestation | inhibits parasite metabolism |
| Quinestrol | 1.897 | 3 | hormone replacement therapy | synthetic steroidal estrogen receptor agonist |
| Sertraline * | 1.897 | 1 | depression, anxiety | selective serotonin receptor inhibitor |
| Clomifene * | 1.897 | 1 | anovulation, oligoovulation | selective estrogen receptor modulator |
| Propoxyphene | 1.897 | 1 | mild to moderate pain | opiate receptor binder |
| Atovaquone | 1.897 | 1 | pneumocystis Pneumonia | dihydroorotate dehydrogenase inhibitor |
| Azelastine | 1.897 | 1 | allergic rhinitis | H1 histamine receptor inhibitor |
| Danazol | 1.897 | 1 | endometriosis | androgen receptor competitive inhibitor |
| Mebendazole * | 1.897 | 1 | helminthic infestation | tubulin destabilizer |
| Hydroxyprogesterone | 1.823 | 2 | preterm labor | steroidal progesterone receptor agonist |
| Deslanoside * | 1.377 | 10 | cardiac arrhythmia | sodium-potassium channel blocker |
| Digoxin * | 1.067 | 1 | cardiac arrhythmia | sodium-potassium channel blocker |
| Ritonavir | 1.057 | 2 | HIV infection | protease inhibitor |
| Raloxifene * | 0.843 | 2 | osteoporosis and breast cancer prevention | selective estrogen receptor modulator |
| Ciclesonide | 0.843 | 1 | asthma, allergic rhinitis | glucocorticoid receptor agonist |
| Clemastine * | 0.741 | 1 | allergies | H1 histamine receptor inhibitor |
| Podofilox | 0.741 | 1 | skin warts caused by Human papilloma virus | tubulin polymerization inhibitor |
| Tamoxifen * | 0.741 | 1 | estrogen receptor + breast cancer | selective estrogen receptor modulator |
| Desloratadine | 0.741 | 1 | allergies | H1 histamine receptor inhibitor |
| Methdilazine | 0.741 | 1 | allergy symptoms, antiemetic | H1 histamine receptor antagonist |
| Chlorcyclizine | 0.741 | 1 | allergy symptoms, antiemetic | H1 histamine receptor antagonist |
| Azacitidine | 0.712 | 1 | myelodysplastic syndrome | DNA methyl transferase inhibitor |
| Terconazole | 0.711 | 1 | fungal infection | ERG11/CYP51 inhibitor |
| Bromocriptine | 0.707 | 1 | pituitary tumors, Parkinson’s disease | dopamine receptor agonist |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chopra, G.; Kaushik, S.; Elkin, P.L.; Samudrala, R. Combating Ebola with Repurposed Therapeutics Using the CANDO Platform. Molecules 2016, 21, 1537. https://doi.org/10.3390/molecules21121537
Chopra G, Kaushik S, Elkin PL, Samudrala R. Combating Ebola with Repurposed Therapeutics Using the CANDO Platform. Molecules. 2016; 21(12):1537. https://doi.org/10.3390/molecules21121537
Chicago/Turabian StyleChopra, Gaurav, Sashank Kaushik, Peter L. Elkin, and Ram Samudrala. 2016. "Combating Ebola with Repurposed Therapeutics Using the CANDO Platform" Molecules 21, no. 12: 1537. https://doi.org/10.3390/molecules21121537