
Charge Transfer Enhancement in the D-π-A Type Porphyrin Dyes: A Density Functional Theory (DFT) and Time-Dependent Density Functional Theory (TD-DFT) Study

Abstract
:1. Introduction
2. Results and Discussion
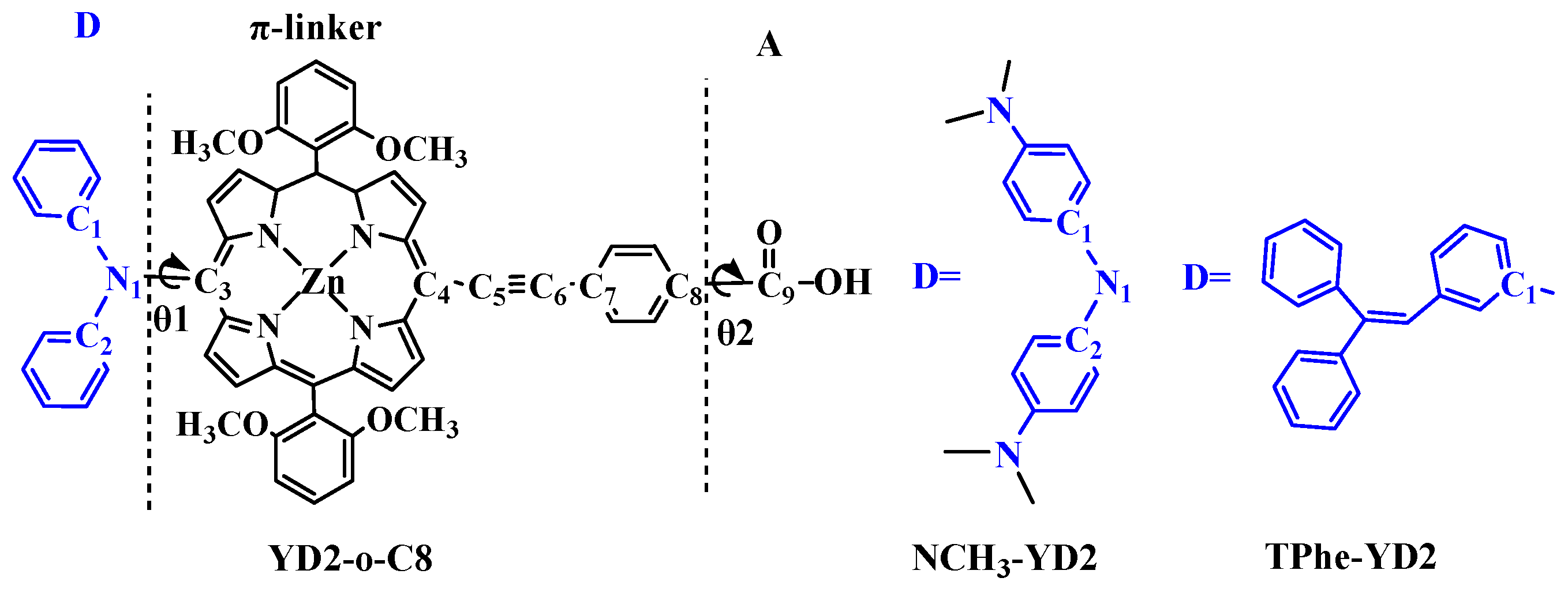
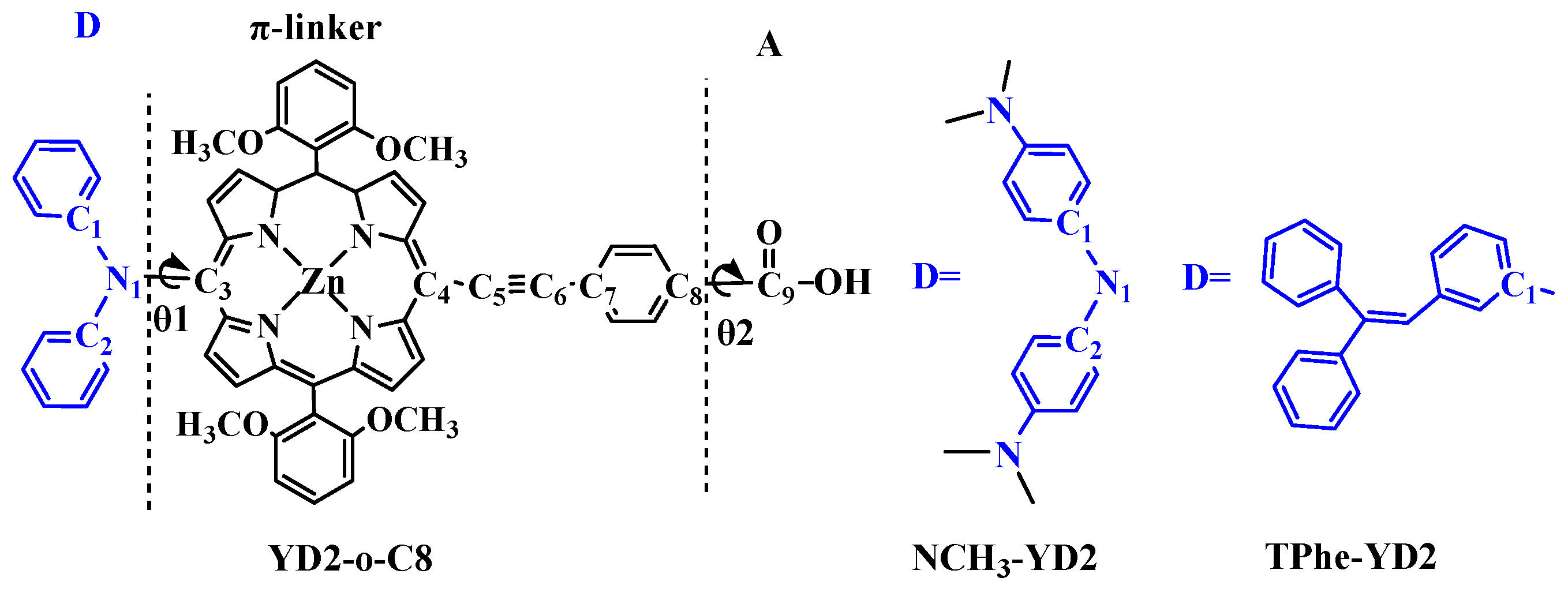
2.1. Geometric Structures of the Dyes
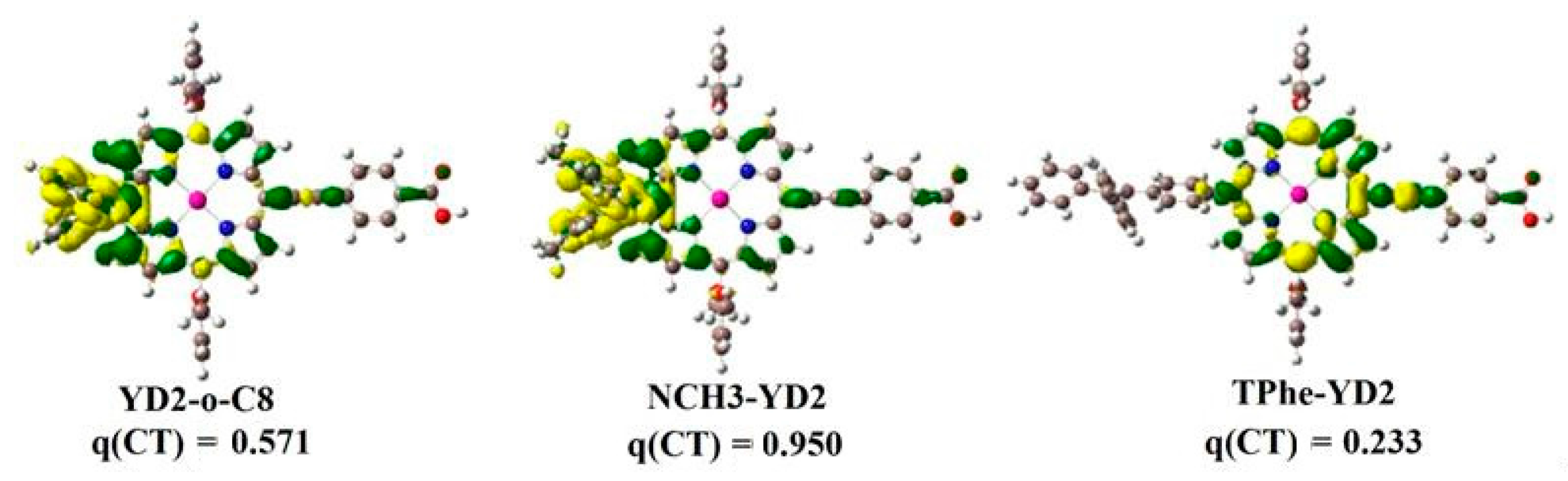
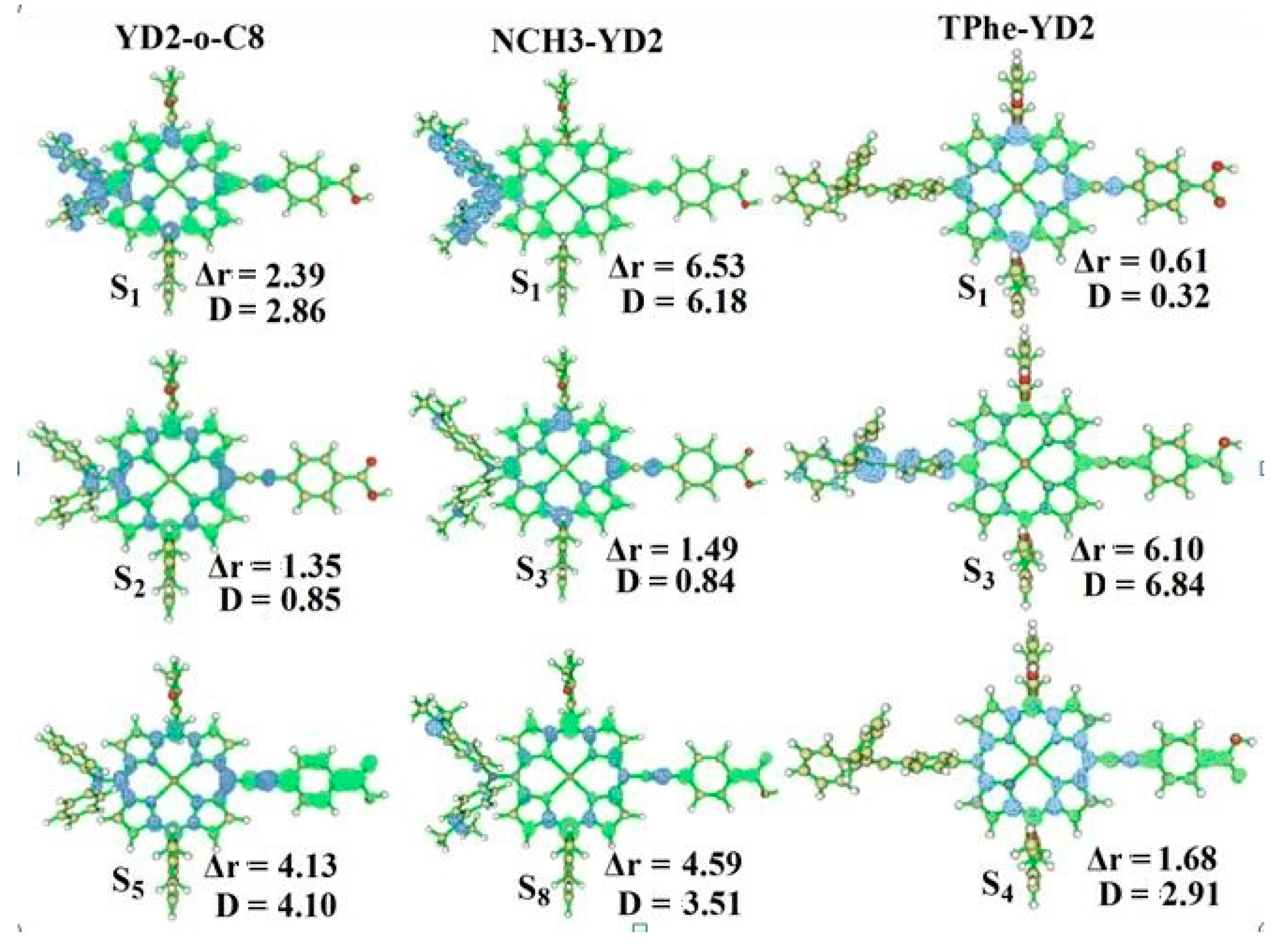
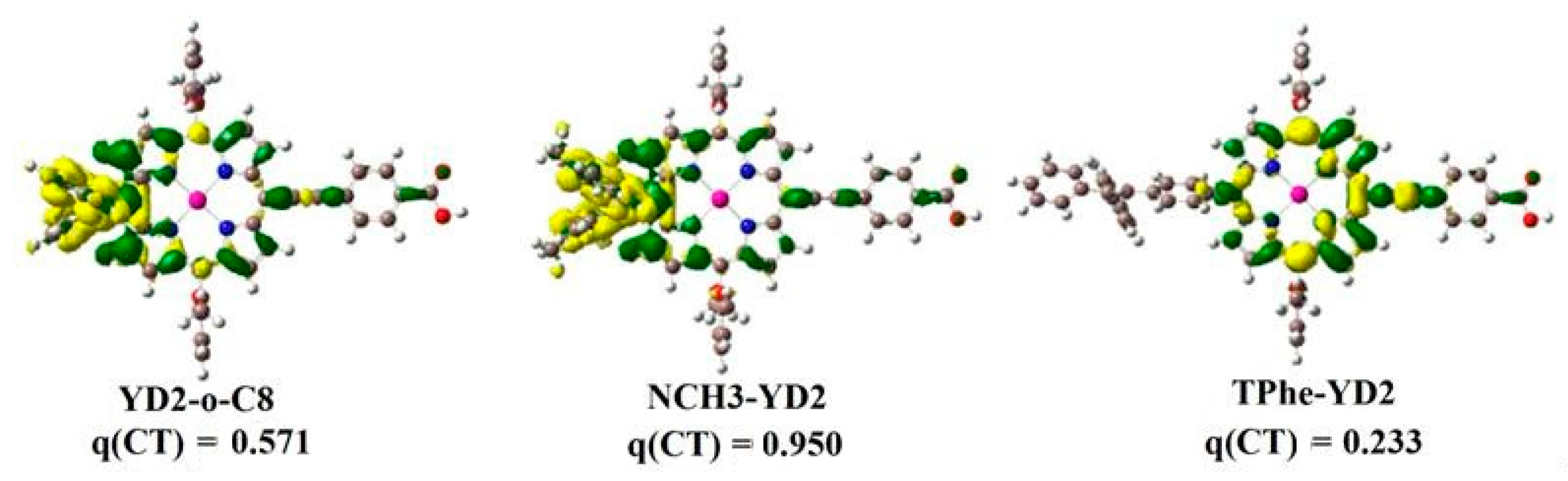
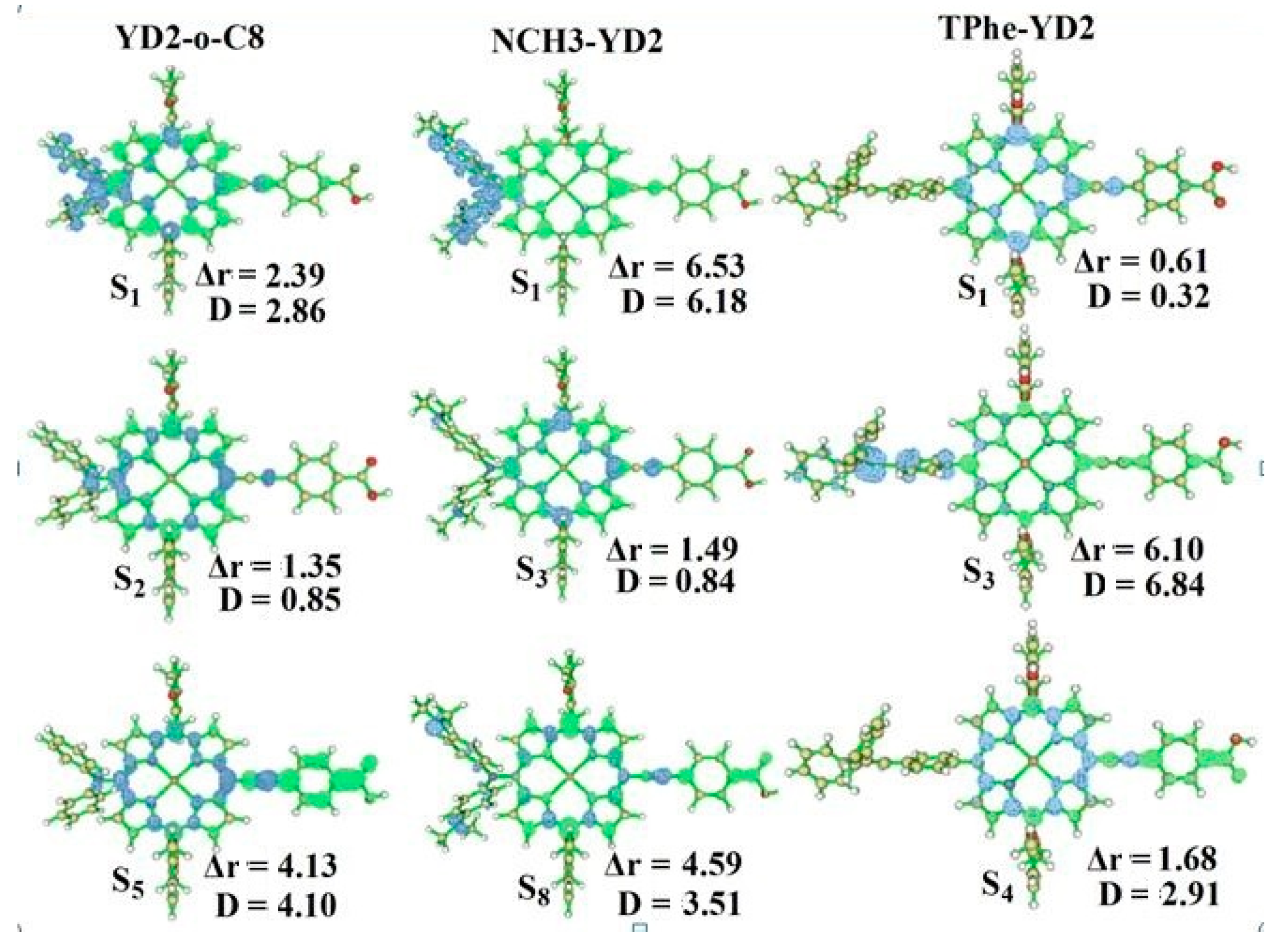
2.2. Intermolecular Charge Transfer
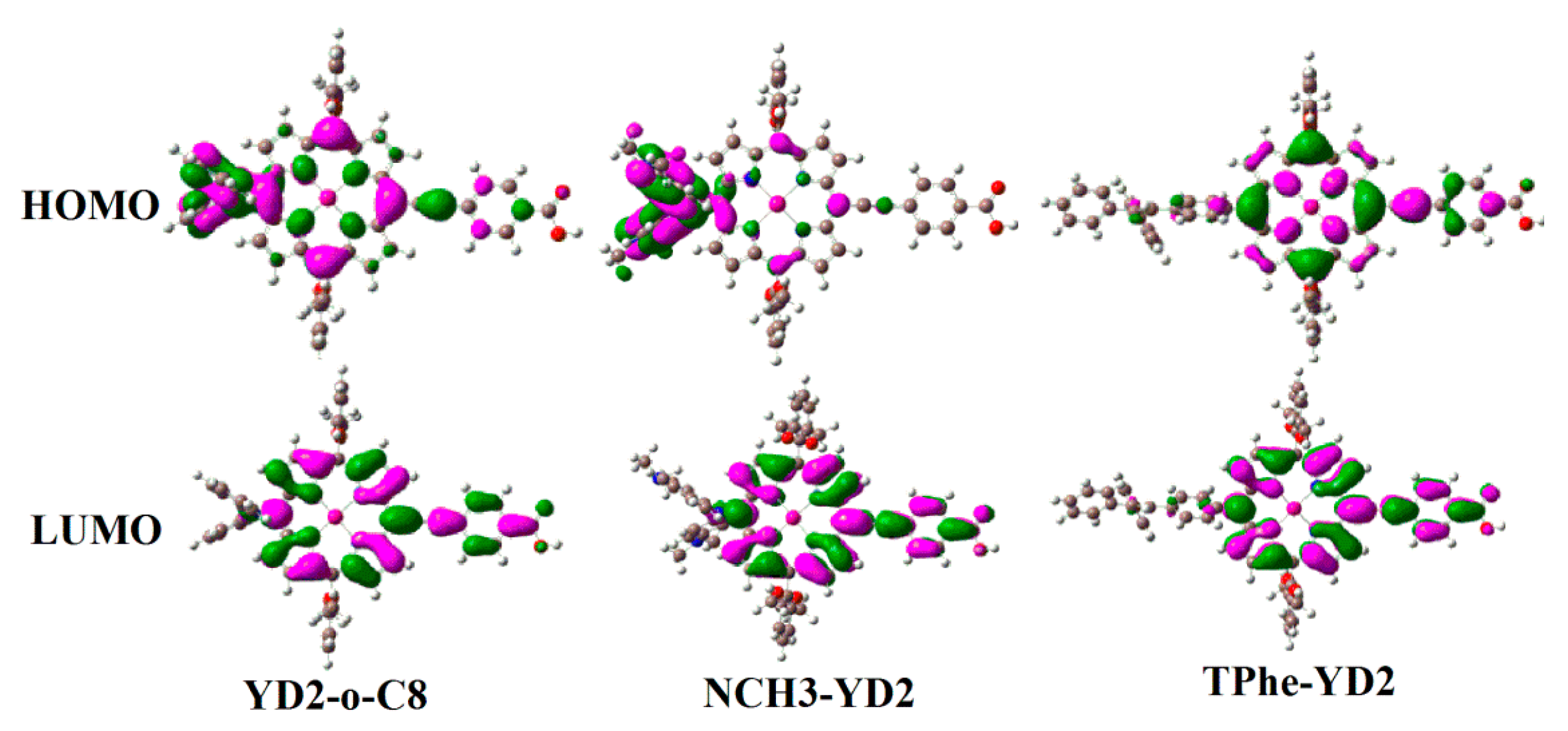
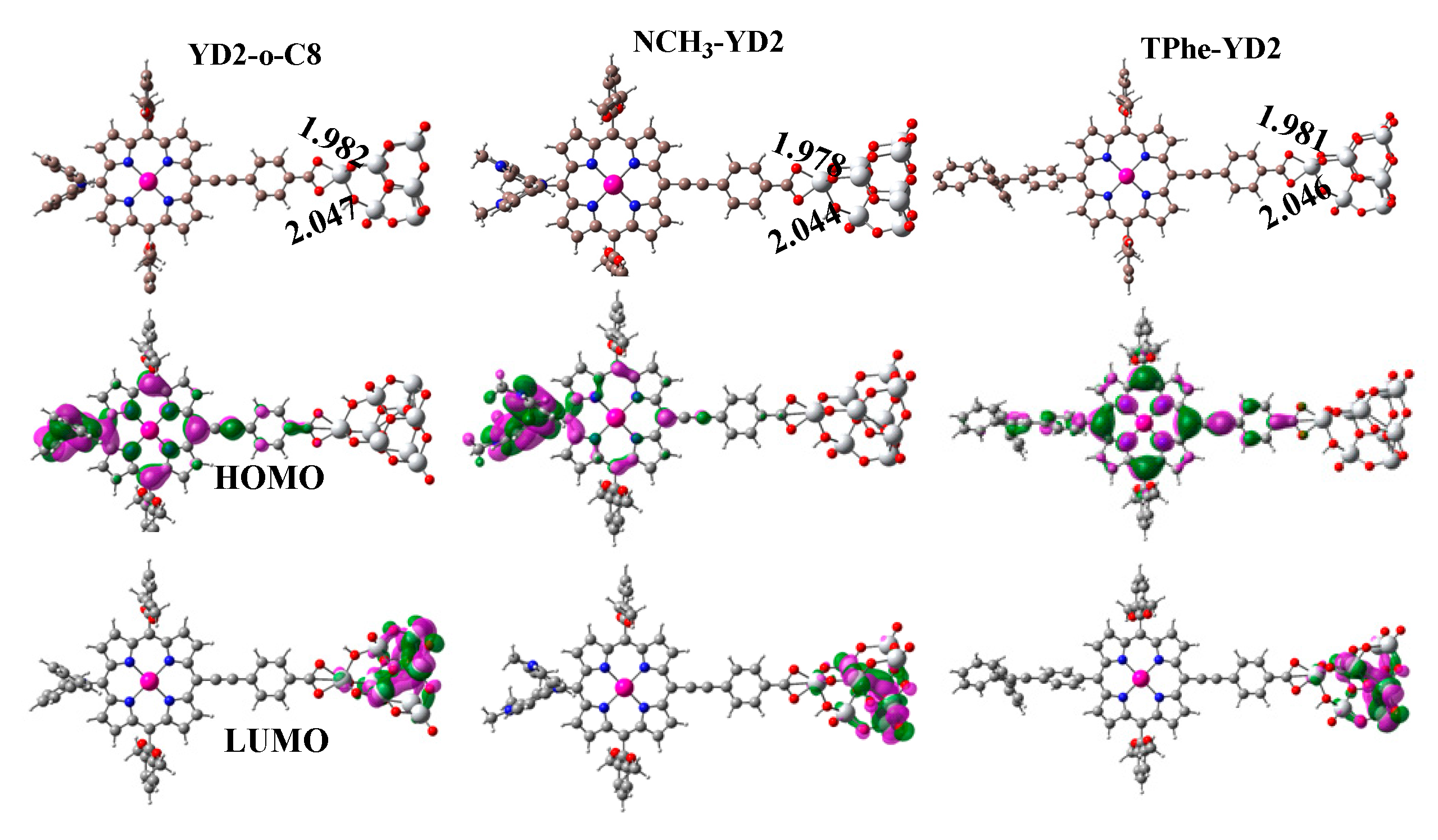
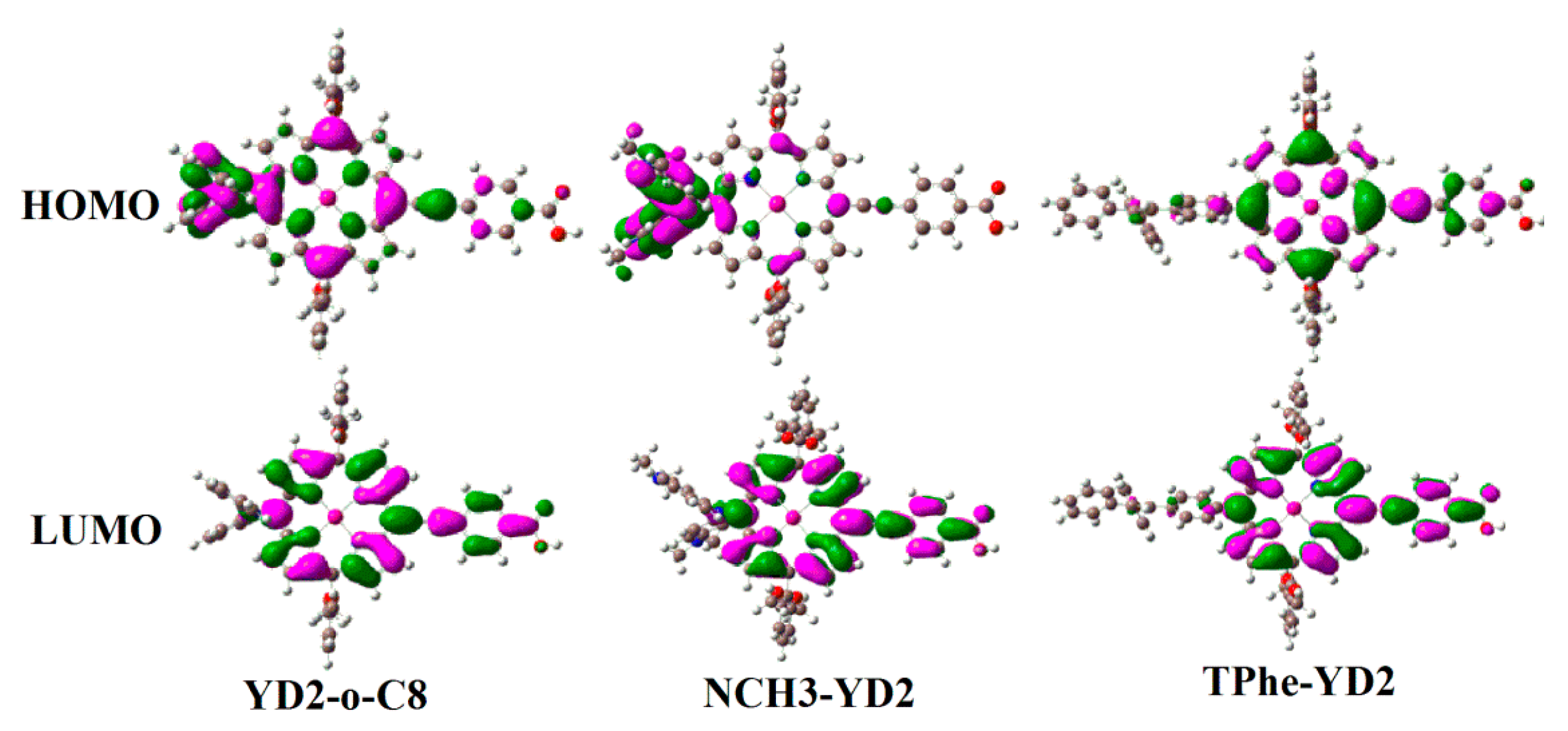
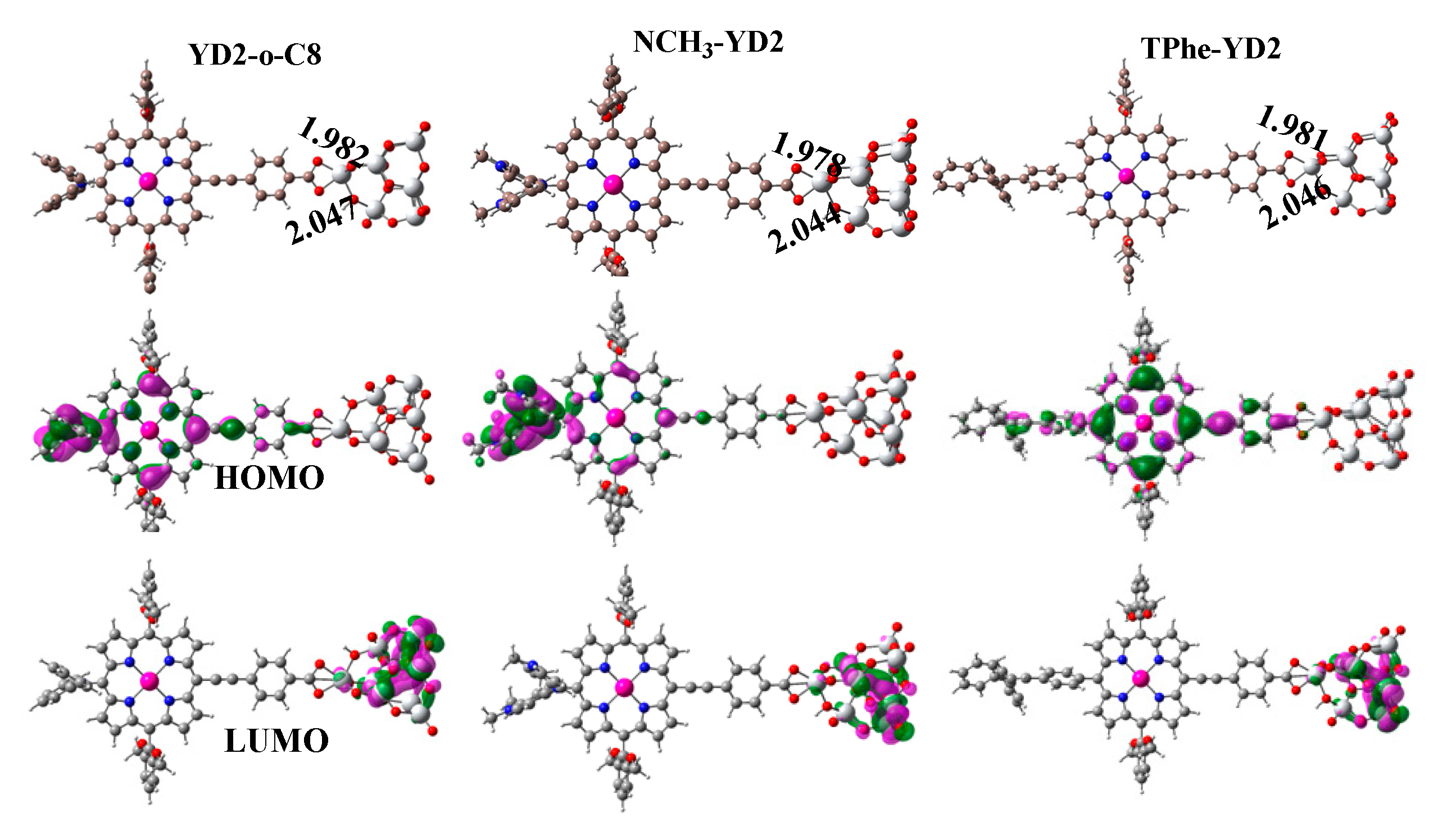
2.3. Frontier Molecular Orbitals
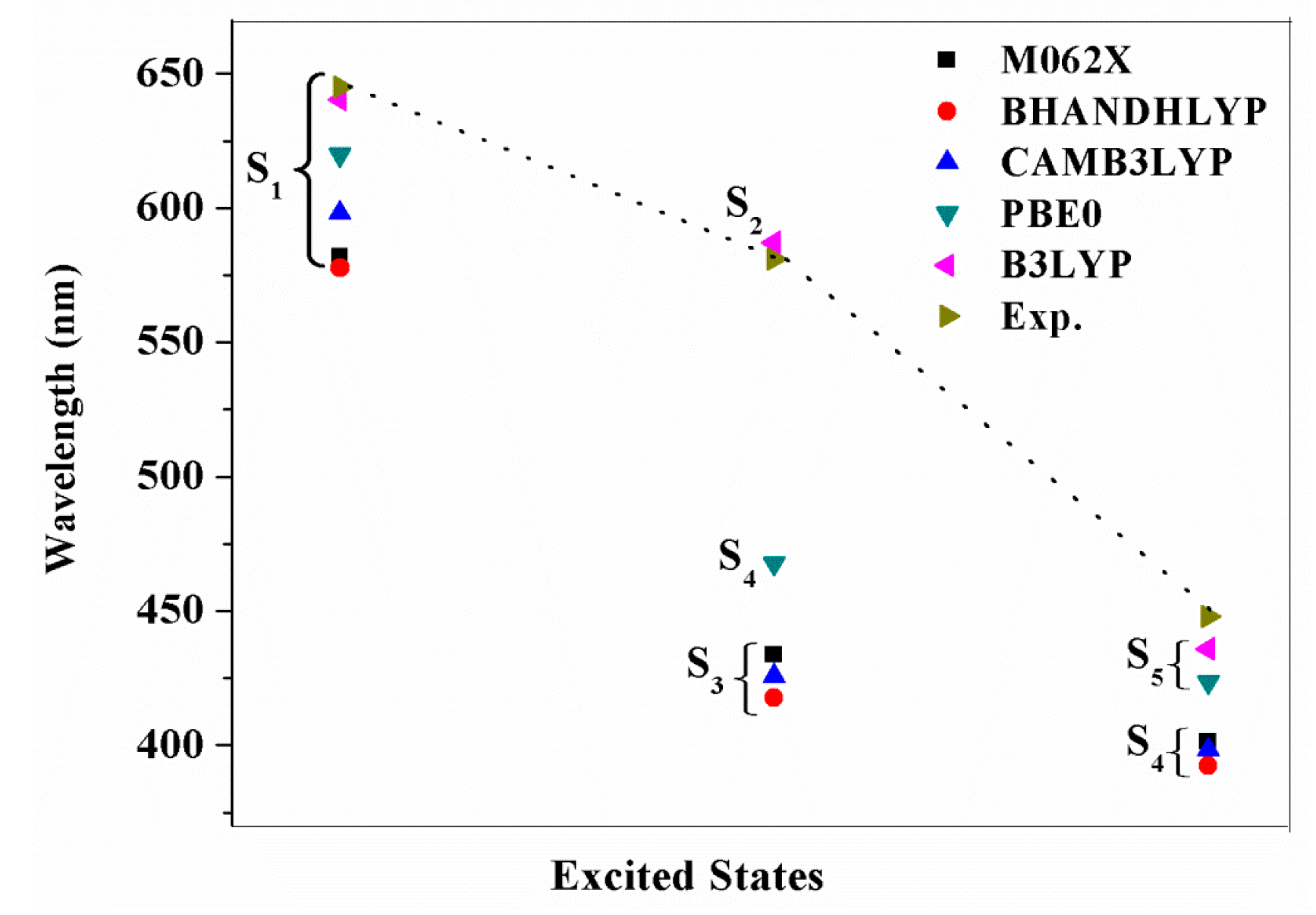
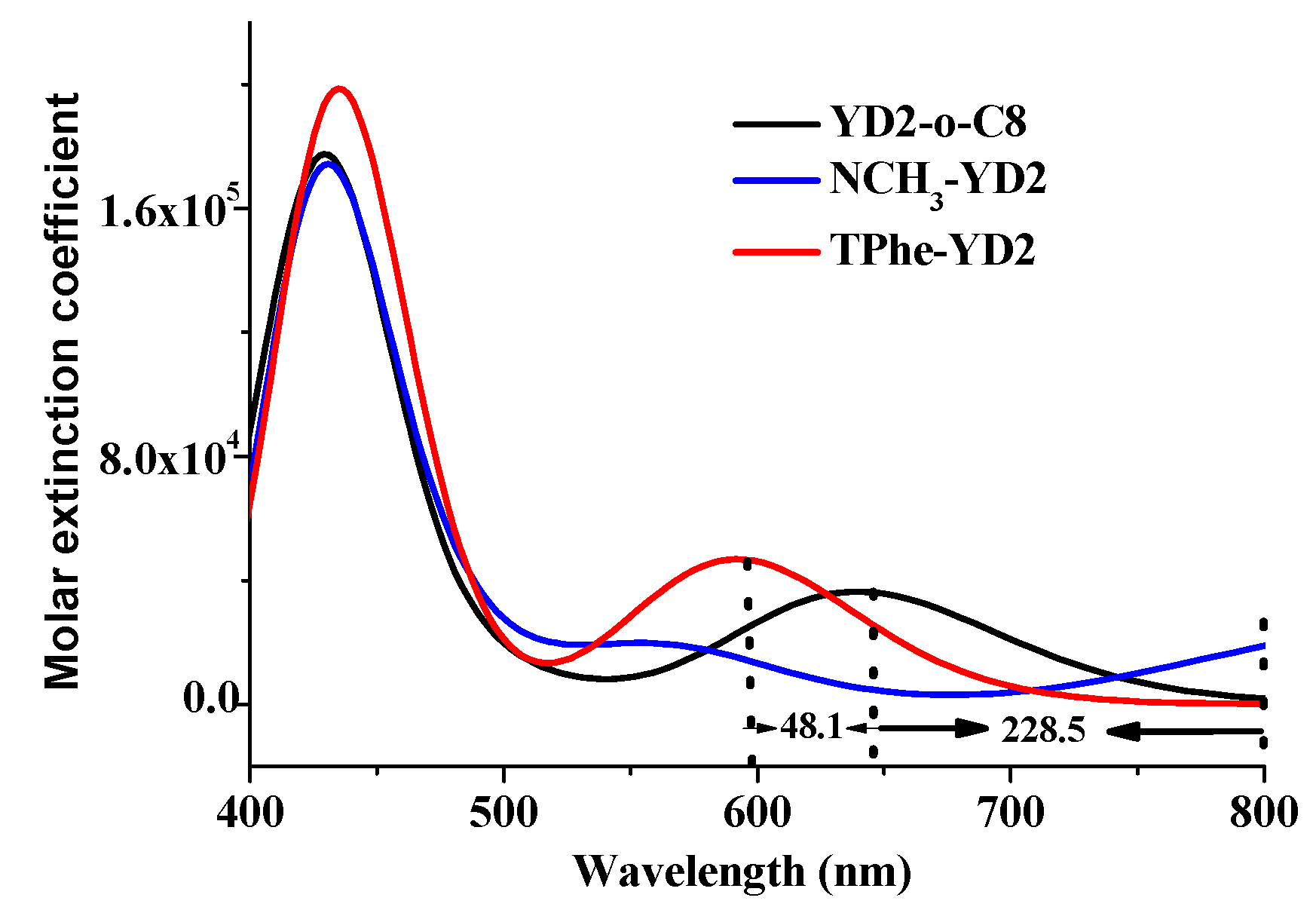
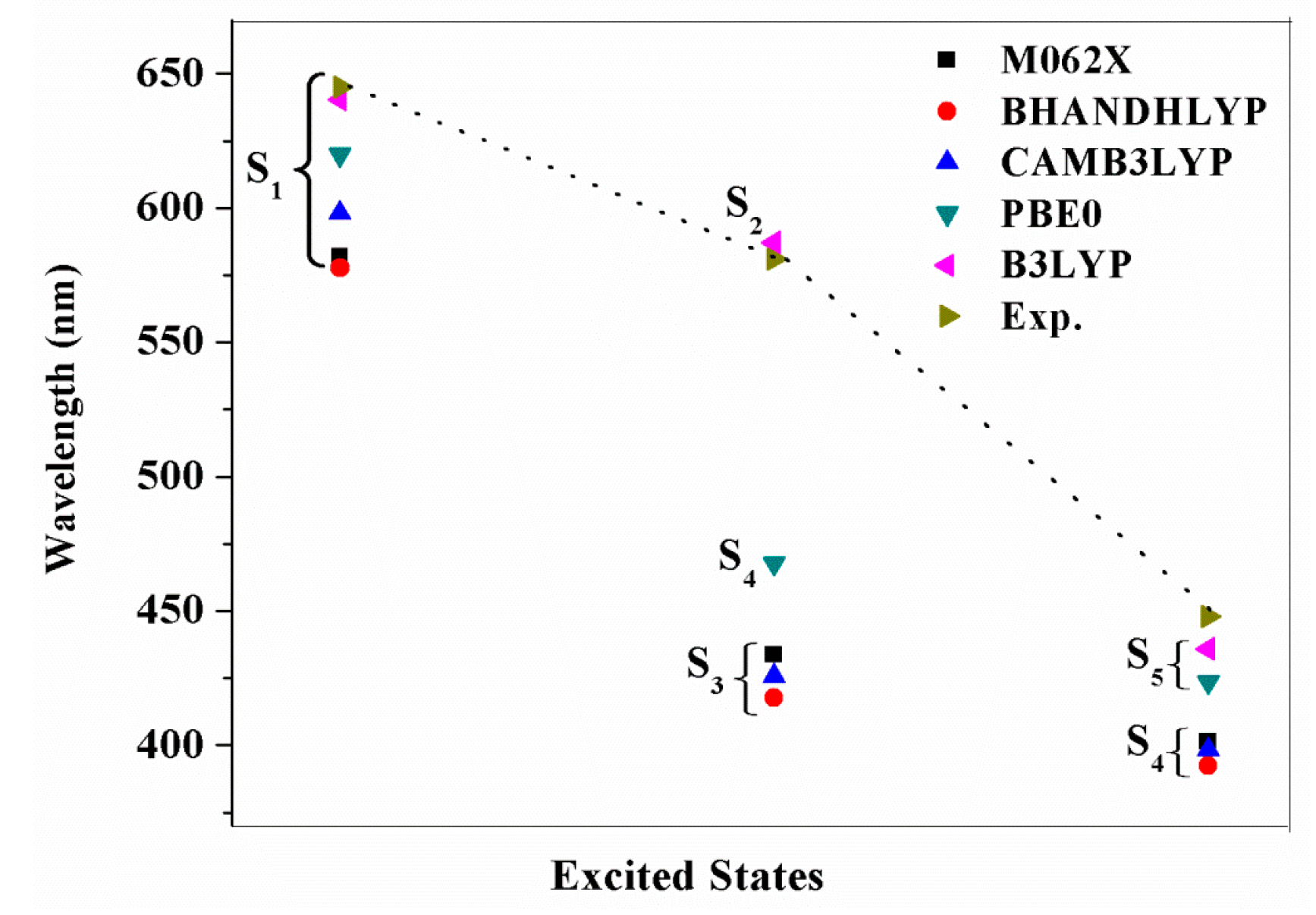
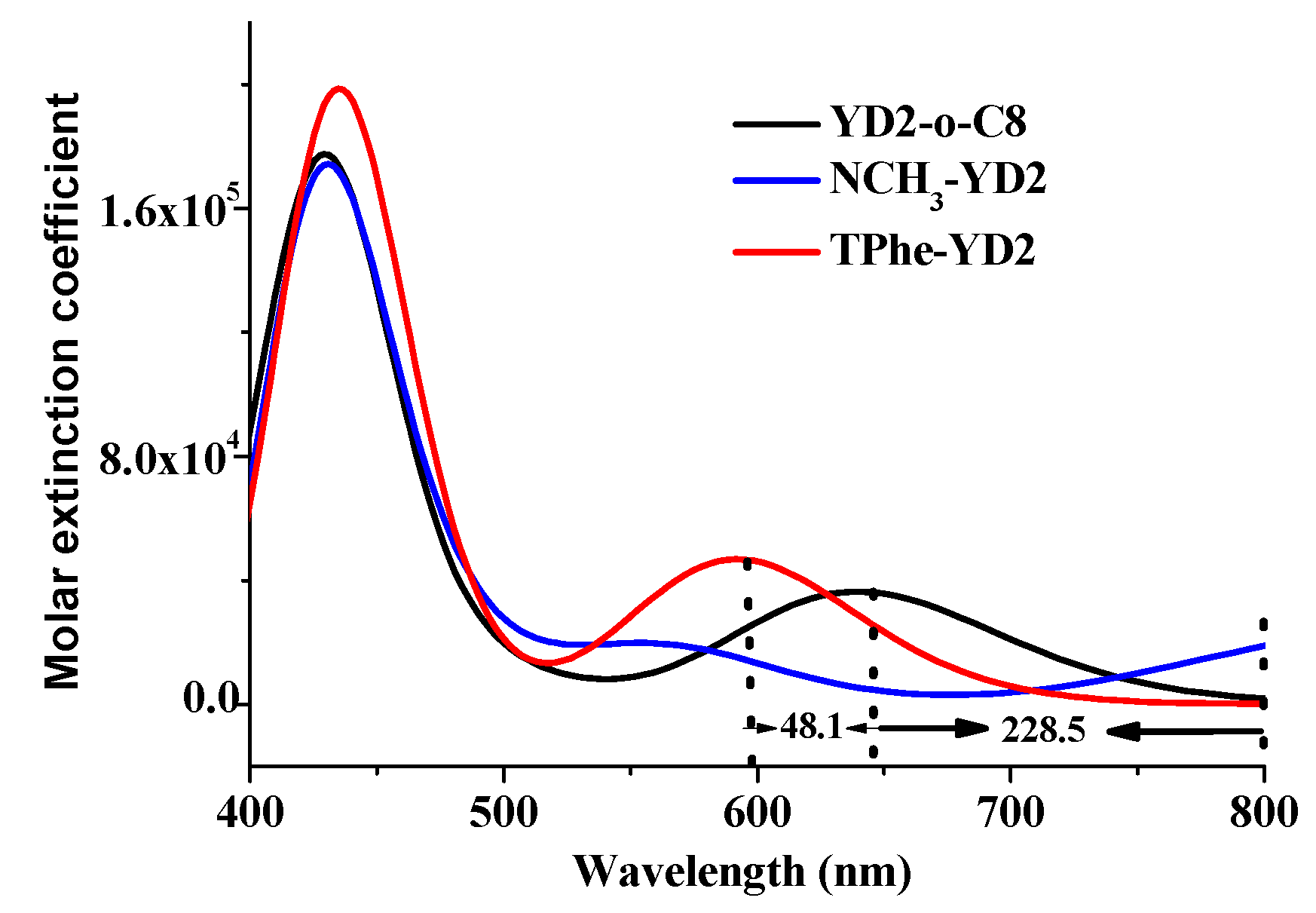
2.4. The Absorption Spectra
2.5. Structures and Properties of Dyes/TiO2 Complexes
2.6. Photoelectric Conversion Efficiency of Dyes
3. Computational Details
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- O’Reagen, B.; Grätzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. [Google Scholar]
- Ito, S.; Miura, H.; Uchida, S.; Takata, M.; Sumioka, K.; Liska, P.; Comte, P.; Péchy, P.; Grätzel, M. High-conversion-efficiency organic dye-sensitized solar cells with a novel indoline dye. Chem. Commun. 2008, 41, 5194–5196. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.H.; Kong, F.T.; Li, J.Z.; Fang, X.Q.; Li, Y.; Dai, S.Y.; Chen, Q.Q.; Zhang, X.X. Triphenylamine-based organic dyes with julolidine as the secondary electron donor for dye-sensitized solar cells. J. Power Sources 2013, 243, 131–137. [Google Scholar] [CrossRef]
- Nazeeruddin, M.K.; Angelis, F.D.; Fantacci, S.; Selloni, A.; Viscardi, G.; Liska, P.; Ito, S.; Takeru, B.; Grätzel, M. Combined Experimental and DFT-TDDFT Computational Study of Photoelectrochemical Cell Ruthenium Sensitizers. J. Am. Chem. Soc. 2005, 127, 16835–16847. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhu, W. Organic sensitizers from D–π–A to D–A–π–A: Effect of the internal electron-withdrawing units on molecular absorption, energy levels and photovoltaic performances. Chem. Soc. Rev. 2013, 42, 2039–2058. [Google Scholar] [CrossRef] [PubMed]
- Narayan, M.R. Review: Dye sensitized solar cells based on natural photosensitizers. Renew. Sustain. Energy Rev. 2012, 16, 208–215. [Google Scholar] [CrossRef]
- Zhang, J.; Kan, Y.-H.; Li, H.-B.; Geng, Y.; Wu, Y.; Su, Z.-M. How to design proper π-spacer order of the D-π-A dyes for DSSCs? A density functional response. Dyes Pigment. 2012, 95, 313–321. [Google Scholar] [CrossRef]
- Ding, W.-L.; Wang, D.-M.; Geng, Z.-Y.; Zhao, X.-L.; Xu, W.-B. Density functional theory characterization and verification of high-performance indoline dyes with D–A–π–A architecture for dye-sensitized solar cells. Dyes Pigment. 2013, 98, 125–135. [Google Scholar] [CrossRef]
- Kang, G.J.; Song, C.; Ren, X.F. Theoretical study of zinc porphyrin-based dyes for dye-sensitized solar cells. J. Photochem. Photobiol. A Chem. 2017, 333, 200–207. [Google Scholar] [CrossRef]
- Ren, X.-F.; Kang, G.-J.; He, Q.-Q. Triphenylamine-based indoline derivatives for dye-sensitized solar cells: A density functional theory investigation. J. Mol. Model. 2016, 22, 8. [Google Scholar] [CrossRef] [PubMed]
- Chiba, Y.; Islam, A.; Watanabe, Y.; Komiya, R.; Koide, N.; Han, L. Dye-sensitized solar cells with conversion efficiency of 11.1%. Jpn. J. Appl. Phys. 2006, 45, 24–28. [Google Scholar] [CrossRef]
- Grätzel, M. Solar Energy Conversion by Dye-Sensitized Photovoltaic Cells. Inorg. Chem. 2005, 44, 6841–6851. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.S.; Yanagida, M.; Sayama, K.; Sugihara, H. Electronic-Insulating Coating of CaCO3 on TiO2 Electrode in Dye-Sensitized Solar Cells: Improvement of Electron Lifetime and Efficiency. Chem. Mater. 2006, 18, 2912–2916. [Google Scholar] [CrossRef]
- Yella, A.; Lee, H.-W.; Tsao, H.N.; Yi, C.; Chandiran, A.K.; Khaja Nazeeruddin, M.; Diau, E.W.-G.; Yeh, C.-Y.; Zakeeruddin, S.M.; Grätzel, M. Porphyrin-Sensitized Solar Cells with Cobalt (II/III)—Based Redox Electrolyte Exceed 12 Percent Efficiency. Science 2011, 334, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.; Yella, A.; Gao, P.; Humphry-Baker, R.; Curchod, B.F.E.; Ashari-Astani, N.; Tavernelli, I.; Rothlisberger, U.; Nazeeruddin, M.K.; Grätzel, M. Dye-sensitized solar cells with 13% efficiency achieved through the molecular engineering of porphyrin sensitizer. Nat. Chem. 2014, 6, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Santhanamoorthi, N.; Lo, C.-M.; Jiang, J.-C. Molecular Design of Porphyrins for Dye-Sensitized Solar Cells: A DFT/TDDFT Study. J. Phys. Chem. Lett. 2013, 4, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, T.; Wu, C.X.; Guan, W.; Yan, L.K.; Su, Z.M. A theoretical design and investigation on Zn-porphyrin-polyoxometalate hybrids with different π-linkers for searching high performance sensitizers of p-type dye-sensitized solar cells. Dyes Pigment. 2016, 130, 168–175. [Google Scholar] [CrossRef]
- Kang, S.H.; Choi, I.T.; Kang, M.S.; Eom, Y.K.; Ju, M.J.; Hong, J.Y.; Kang, H.S.; Kim, H.Y. Novel D–π–A structured porphyrin dyes with diphenylamine derived electron-donating substituents for highly efficient dye-sensitized solar cells. J. Mater. Chem. A 2013, 1, 3977–3982. [Google Scholar] [CrossRef]
- Joly, D.; Pellejà, L.; Narbey, S.; Oswald, F.; Meyer, T.; Kervella, Y.; Maldivi, P.; Clifford, J.N.; Palomares, E.; Demadrille, R. Metal-free organic sensitizers with narrow absorption in the visible for solar cells exceeding 10% efficiency. Energy Environ. Sci. 2015, 8, 2010–2018. [Google Scholar] [CrossRef]
- Yan, C.C.; Ma, W.T.; Ren, Y.M.; Zhang, M.; Wang, P. Efficient Triarylamine–Perylene Dye-Sensitized Solar Cells: Influence of Triple-Bond Insertion on Charge Recombination. ACS Appl. Mater. Interfaces 2015, 7, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.-F.; Zhang, J.; Kang, G.-J. Theoretical Studies of Electronic Structure and Photophysical Properties of a Series of Indoline Dyes with Triphenylamine Ligand. J. Nanomater. 2015, 605728, 1–9. [Google Scholar] [CrossRef]
- Horiuchi, T.; Miura, H.; Sumioka, K.; Uchida, S. High Efficiency of Dye-Sensitized Solar Cells Based on Metal-Free Indoline Dyes. J. Am. Chem. Soc. 2004, 126, 12218–12219. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comp. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Lu, T. Generalized Charge Decomposition Analysis (GCDA) Method. J. Adv. Phys. Chem. 2015, 4, 111–124. [Google Scholar] [CrossRef]
- Sang-aroon, W.; Saekow, S.; Amornkitbamrung, V. Density functional theory study on the electronic structure of Monascus dyes as photosensitizer for dye-sensitized solar cells. J. Photochem. Photobiol. A 2012, 236, 35–40. [Google Scholar] [CrossRef]
- Asbury, J.B.; Hao, E.; Wang, Y.; Ghosh, H.N.; Lian, T. Ultrafast Electron Transfer Dynamics from Molecular Adsorbates to Semiconductor Nanocrystalline Thin Films. J. Phys. Chem. B 2001, 105, 4545–4557. [Google Scholar] [CrossRef]
- Srinivas, K.; Yesudas, K.; Bhanuprakash, K.; Rao, V.J.; Giribabu, L.A. Combined Experimental and Computational Investigation of Anthracene Based Sensitizers for DSSC: Comparison of Cyanoacrylic and Malonic Acid Electron Withdrawing Groups Binding onto the TiO2 Anatase (101) Surface. J. Phys. Chem. C 2009, 113, 20117–20126. [Google Scholar] [CrossRef]
- Xia, H.-Q.; Wang, J.; Bai, F.-Q.; Zhang, H.-X. Theoretical studies of electronic and optical properties of the triphenylamine-based organic dyes with diketopyrrolopyrrole chromophore. Dyes Pigment. 2015, 113, 87–95. [Google Scholar] [CrossRef]
- Chen, P.; Yum, J.H.; Angelis, F.D.; Mosconi, E.; Fantacci, S.; Moon, S.-J.; Baker, R.H.; Ko, J.; Nazeeruddin, M.K.; Grätzel, M. High Open-Circuit Voltage Solid-State Dye-Sensitized Solar Cells with Organic Dye. Nano Lett. 2009, 9, 2487–2492. [Google Scholar] [CrossRef] [PubMed]
- Pastore, M.; de Angelis, F. Aggregation of Organic Dyes on TiO2 in Dye-Sensitized Solar Cells Models: An ab Initio Investigation. ACS Nano 2010, 4, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Katoh, R.; Furube, A.; Yoshihara, T.; Hara, K.; Fujihashi, G.; Takano, S.; Murata, S.; Arakawa, H.; Tachiya, M. Doping Dependence of the Electronic Structure of La1-xNaxMnO3 by Resonant X-ray Emission and X-ray Absorption Spectroscopy. J. Phys. Chem. B 2004, 108, 4818–4822. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Xie, M.; Wang, J.; Ren, J.; Hao, L.; Bai, F.-Q.; Pan, Q.-J.; Zhang, H.-X. Theoretical study on a high-efficient porphyrin-sensitizer in a local electric field: How does the local electric field affects the performance of dye-sensitized solar cells? Org. Electron. 2015, 26, 164–175. [Google Scholar] [CrossRef]
- Gua, X.; Sun, Q. Computational study of porphyrin-based dyes with better performance. Phys. Chem. Chem. Phys. 2013, 15, 15434–15440. [Google Scholar] [CrossRef] [PubMed]
- Mendizabal, F.; Lopéz, A.; Arratia-Pérez, R.; Inostroza, N.; Linares-Flores, C. Interaction of YD2 and TiO₂ in dye-sensitized solar cells (DSSCs): A density functional theory study. J. Mol. Model. 2015, 21, 226. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Durant, J.P. Evaluation of transition state properties by density functional theory. Chem. Phys. Lett. 1996, 256, 595–602. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functional. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Sánchez-de-Armas, R.; López, J.O.; San-Miguel, M.A.; Sanz, J.F. Real-Time TD-DFT Simulations in Dye Sensitized Solar Cells: The Electronic Absorption Spectrum of Alizarin Supported on TiO2 Nanoclusters. J. Chem. Theory Comput. 2010, 6, 2856–2865. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-de-Armas, R.; San-Miguel, M.A.; Oviedo, J.; Marquez, A.; Sanz, J.F. Real-Time TD-DFT Simulations in Dye Sensitized Solar Cells: The Electronic Absorption Spectrum of Alizarin Supported on TiO2 Nanoclusters. Phys. Chem. Chem. Phys. 2011, 13, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-de-Armas, R.; San-Miguel, M.A.; Oviedo, J.; Sanz, J.F. Coumarin derivatives for dye sensitized solar cells: A TD-DFT study. Phys. Chem. Chem. Phys. 2012, 14, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Hagfeldt, A.; Grätzel, M. Light-Induced Redox Reactions in Nanocrystalline Systems. Chem. Rev. 1995, 95, 49–68. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.04; Gaussian: Wallingford, CT, USA, 2009. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | π-Linker | A | Torsion Angle (°) | ||||
|---|---|---|---|---|---|---|---|
| N1/C1–C3 | C4–C5 | C5–C6 | C6–C7 | C8–C9 | θ1 | θ2 | |
| YD2-o-C8 | 1.437 | 1.421 | 1.220 | 1.419 | 1.482 | −71.3 | 0.0 |
| NCH3-YD2 | 1.431 | 1.420 | 1.220 | 1.419 | 1.481 | −69.3 | 0.1 |
| TPhe-YD2 | 1.496 | 1.421 | 1.220 | 1.419 | 1.482 | 64.9 | 0.0 |
| Molecule | S0/NBO | S1/NBO | CDA | ECDA | ||||
|---|---|---|---|---|---|---|---|---|
| D | π-Linker | A | D | π-Linker | A | d–b | ||
| YD2-o-C8 | −0.181 | 0.185 | −0.004 | −0.185 | 0.193 | −0.008 | 0.056084 | 0.2005 |
| NCH3-YD2 | −0.139 | 0.146 | −0.007 | −0.183 | 0.194 | −0.011 | 0.057193 | 0.2047 |
| TPhe-YD2 | 0.007 | −0.002 | −0.005 | 0.010 | −0.002 | −0.008 | 0.057470 | 0.2019 |
| Molecule | HOMO − 1 | HOMO | LUMO | LUMO + 1 | Eg |
|---|---|---|---|---|---|
| YD2-o-C8 | −5.04 | −4.68 | −2.34 | −2.01 | 2.34 |
| Ref. [14] | −5.004 | −4.632 | −2.298 | −1.967 | 2.334 |
| NCH3-YD2 | −4.78 | −4.08 | −2.18 | −1.86 | 1.90 |
| TPhe-YD2 | −4.98 | −4.73 | −2.26 | −1.95 | 2.47 |
| Molecule | Excited States | Wavelength λ/nm (f) | Assignment Composition | Expermental [14] λ/nm (ε/103 M−1·cm−1) |
|---|---|---|---|---|
| YD2-o-C8 | S1 | 640.3 (0.4996) | 0.67 H → L | 645 (31) |
| S2 | 587.3 (0.0028) | 0.56 H → L + 1 −0.42 H − 1 → L | 581 (12) | |
| S5 | 436.0 (1.6381) | 0.49 H – 1 → L + 1 −0.40 H → L + 2 | 448 (212) | |
| NCH3-YD2 | S1 | 868.8 (0.3346) | 0.70 H → L | |
| S3 | 566.4 (0.2293) | 0.63 H − 1 → L | ||
| S8 | 429.3 (1.4459) | 0.53 H − 3 → L + 1 | ||
| TPhe-YD2 | S1 | 592.2 (0.6461) | 0.64 H → L | |
| S3 | 452.7 (0.5685) | 0.61 H − 2 → L | ||
| S4 | 432.7 (1.4308) | 0.45 H − 1 → L −0.32 H − 2 → L |
| Molecule | Eads (kcal/mol) | HOMO (eV) | LUMO (eV) | ECDA | q(CT) (e−) |
|---|---|---|---|---|---|
| YD2-o-C8 | 64.40 | −5.07 | −3.91 | 0.2541 | 2.34 |
| NCH3-YD2 | 65.13 | −4.40 | −3.83 | 0.2670 | 1.90 |
| TPhe-YD2 | 64.65 | −5.19 | −3.89 | 0.2559 | 2.72 |
| Molecule | λmax | ∆Ginject | LHE | f | ||
|---|---|---|---|---|---|---|
| YD2-o-C8 | 2.8438 | 2.26 | 5.10 | −1.74 | 0.98 | 1.6396 |
| NCH3-YD2 | 2.8878 | 1.65 | 4.54 | −2.35 | 0.96 | 1.4459 |
| TPhe-YD2 | 2.8657 | 2.30 | 5.17 | −1.70 | 0.96 | 1.4308 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, G.-J.; Song, C.; Ren, X.-F. Charge Transfer Enhancement in the D-π-A Type Porphyrin Dyes: A Density Functional Theory (DFT) and Time-Dependent Density Functional Theory (TD-DFT) Study. Molecules 2016, 21, 1618. https://doi.org/10.3390/molecules21121618
Kang G-J, Song C, Ren X-F. Charge Transfer Enhancement in the D-π-A Type Porphyrin Dyes: A Density Functional Theory (DFT) and Time-Dependent Density Functional Theory (TD-DFT) Study. Molecules. 2016; 21(12):1618. https://doi.org/10.3390/molecules21121618
Chicago/Turabian StyleKang, Guo-Jun, Chao Song, and Xue-Feng Ren. 2016. "Charge Transfer Enhancement in the D-π-A Type Porphyrin Dyes: A Density Functional Theory (DFT) and Time-Dependent Density Functional Theory (TD-DFT) Study" Molecules 21, no. 12: 1618. https://doi.org/10.3390/molecules21121618