
Expedient Organocatalytic Syntheses of 4-Substituted Pyrazolidines and Isoxazolidines

Abstract
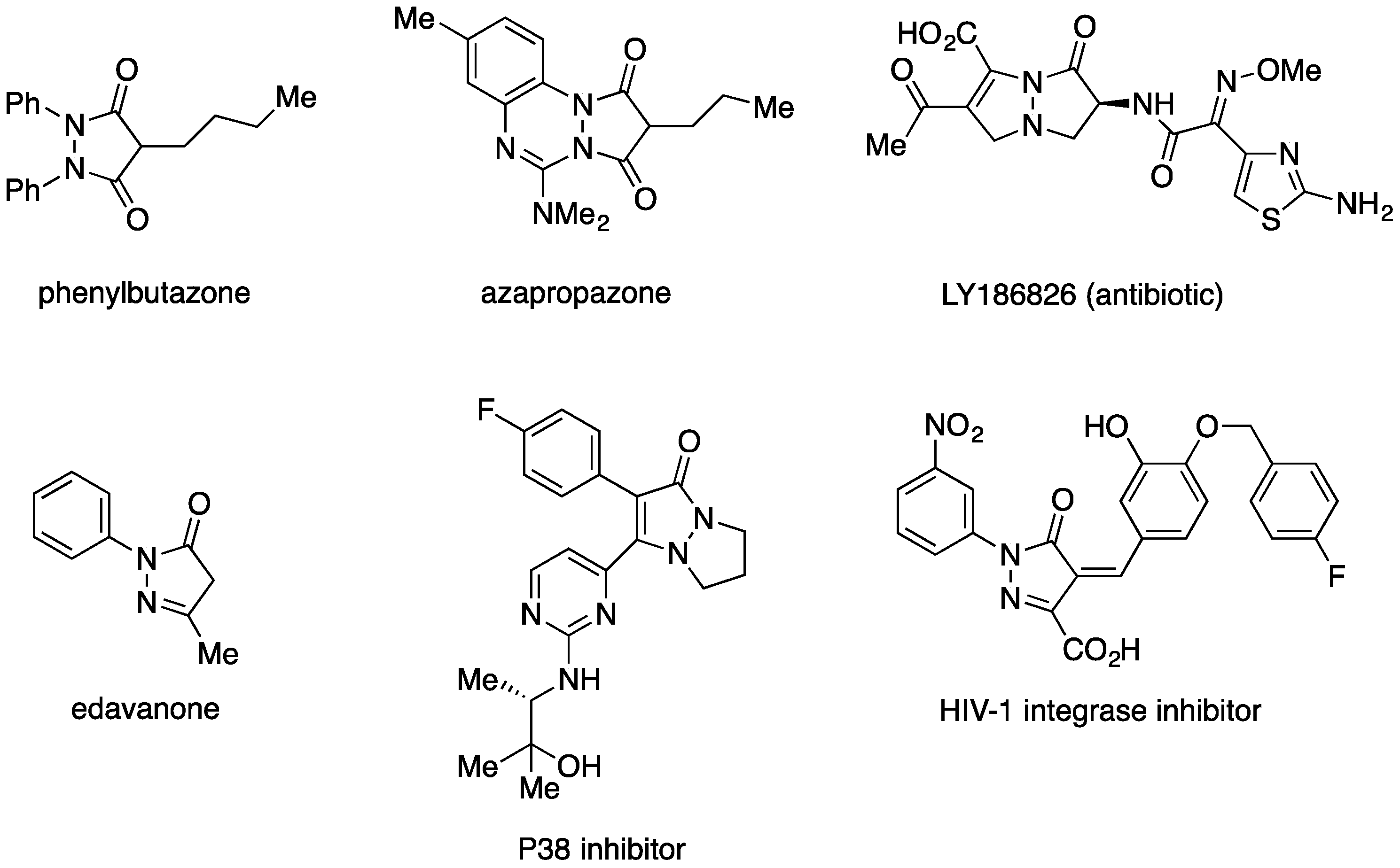
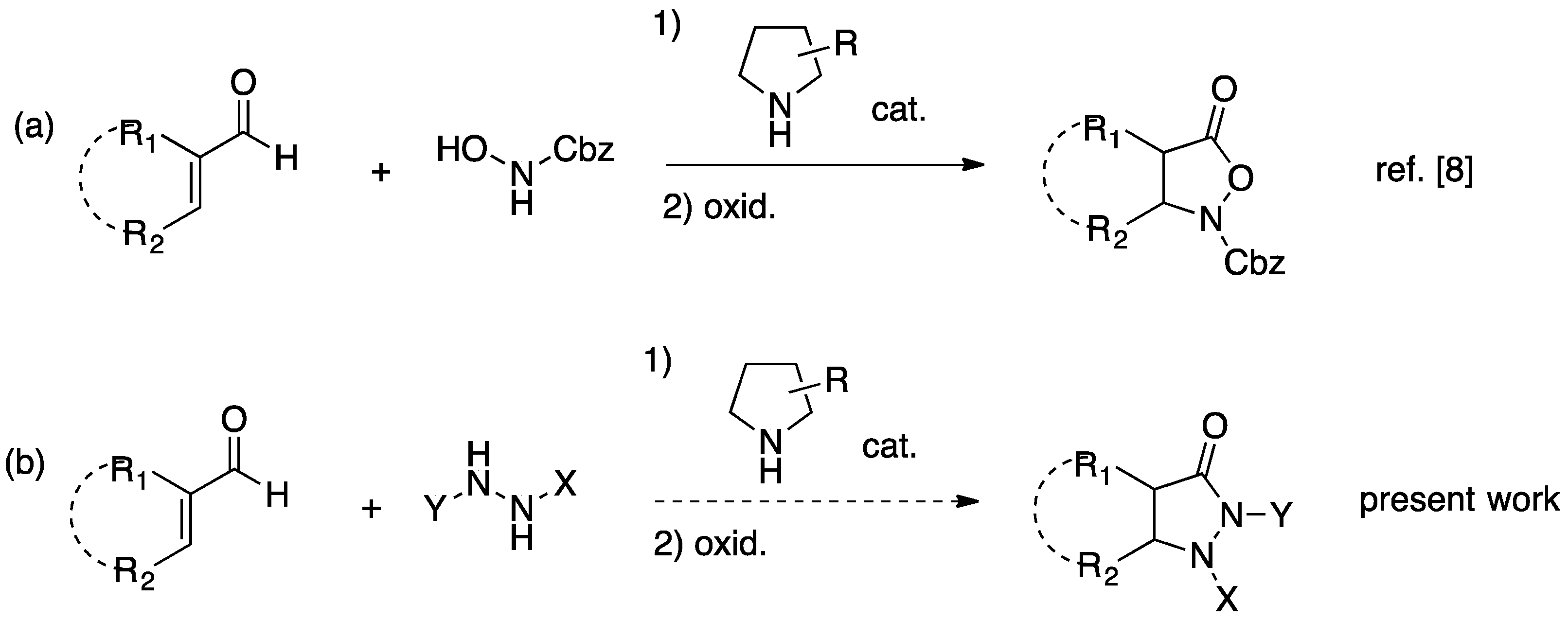
:1. Introduction
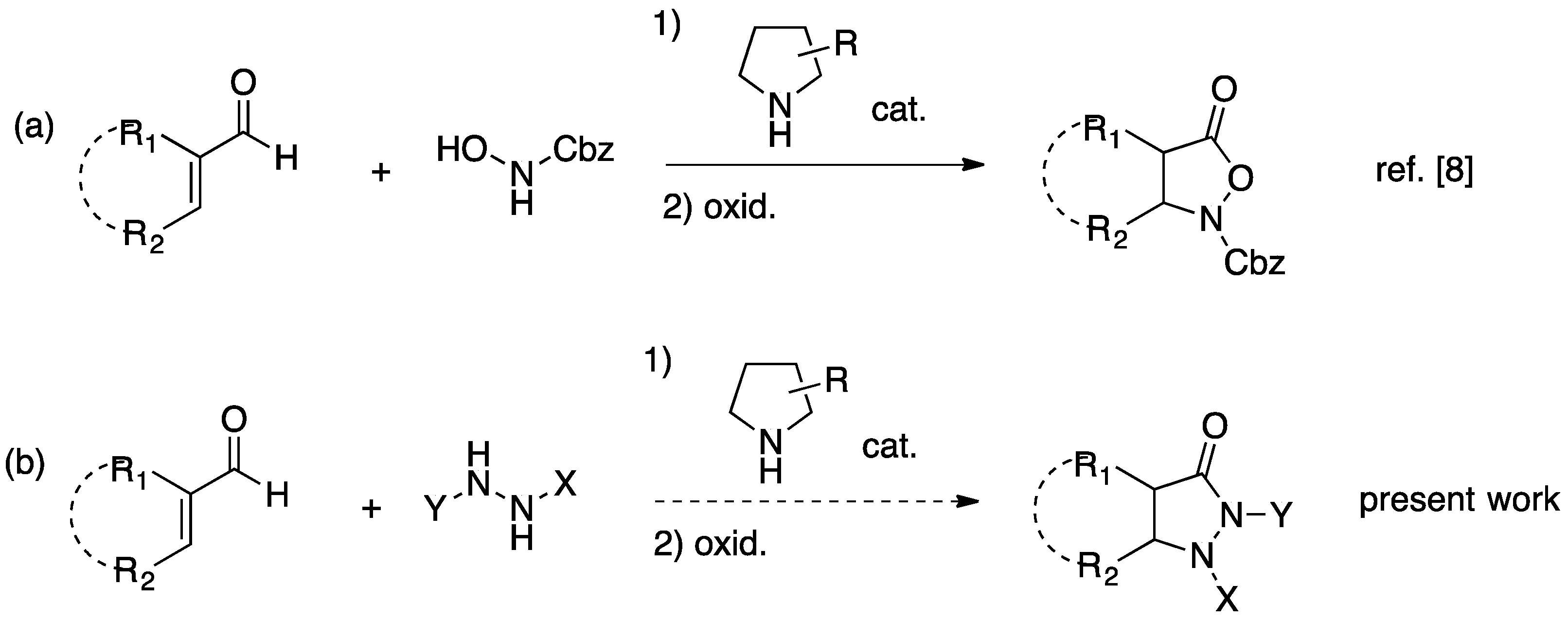
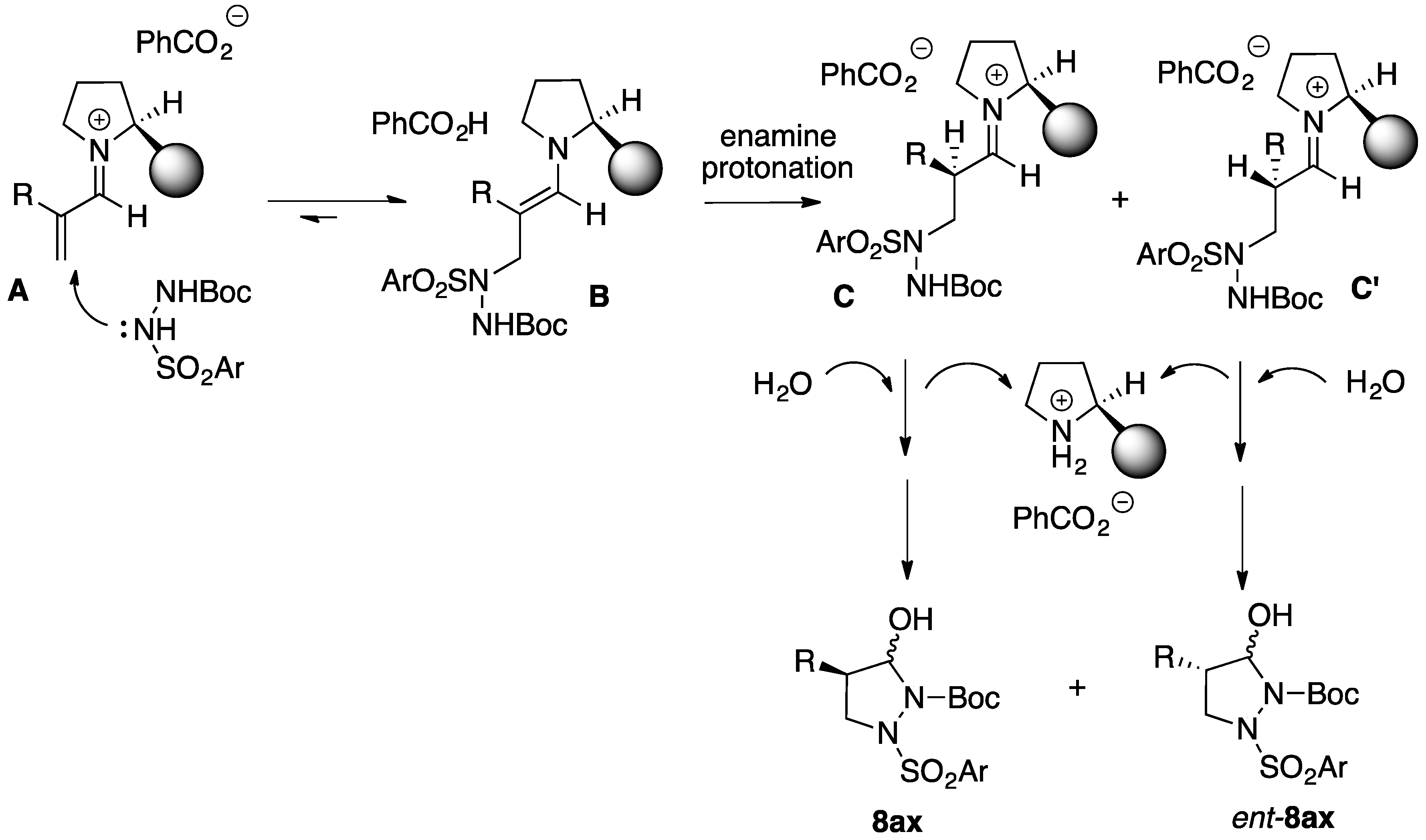
2. Results and Discussion
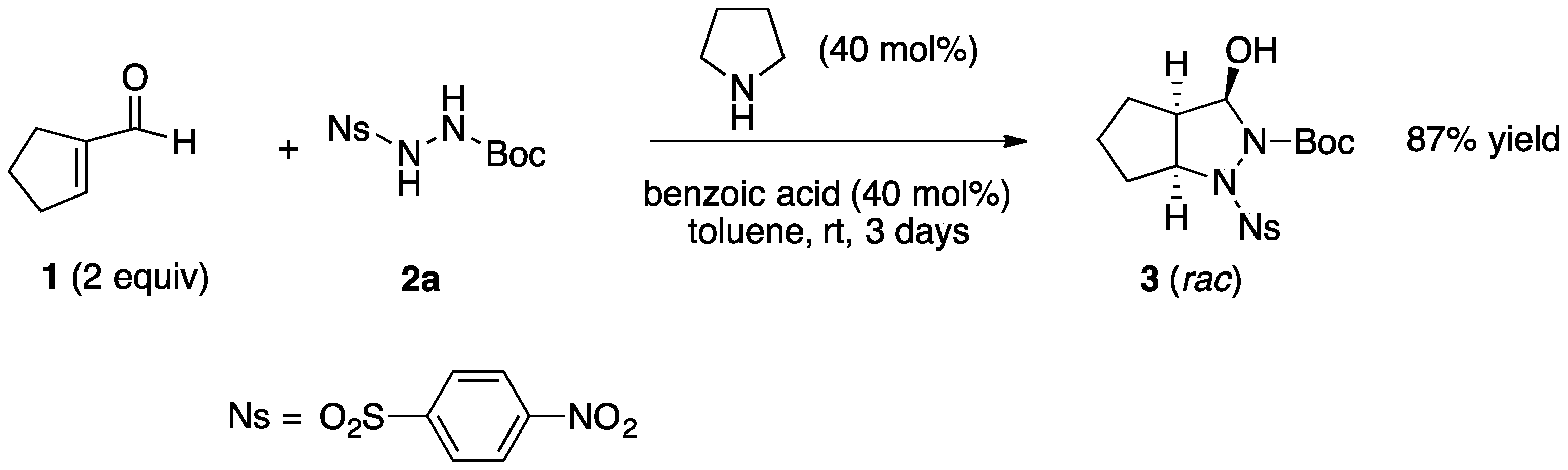
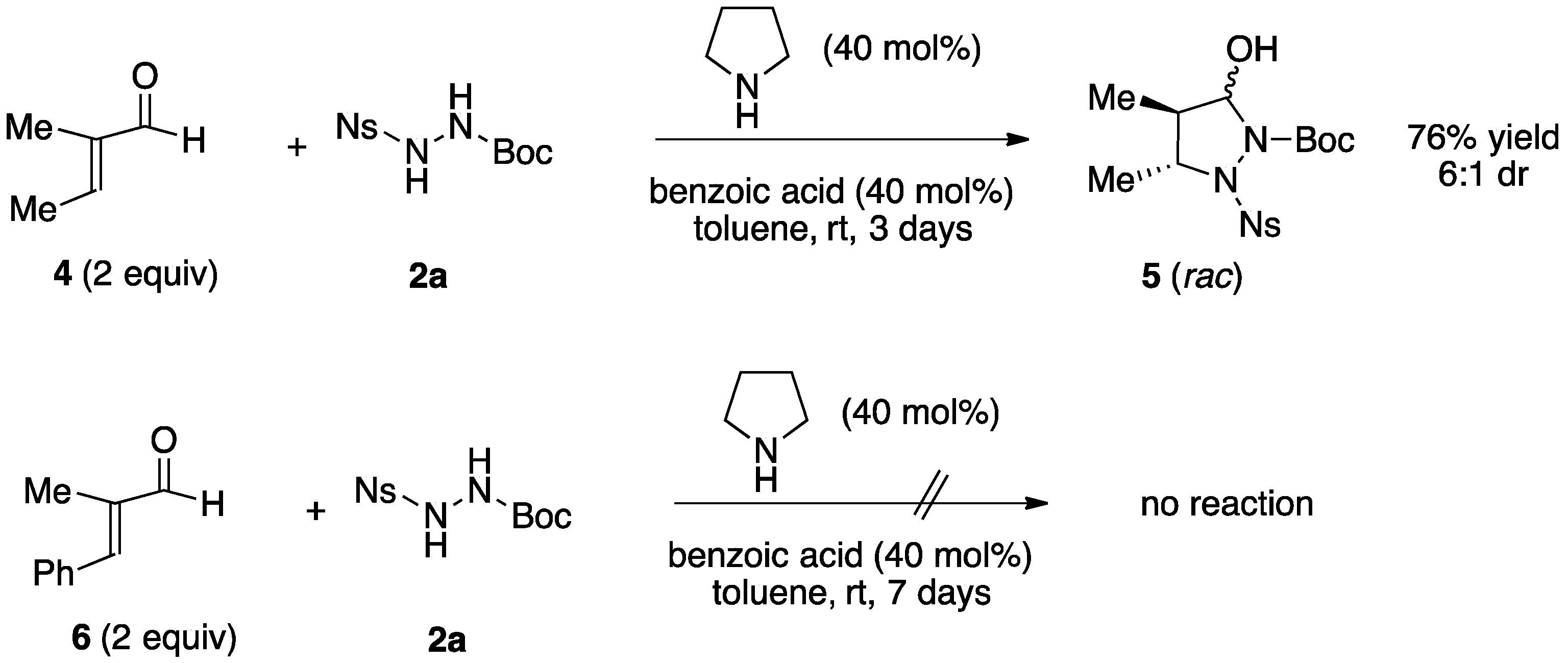
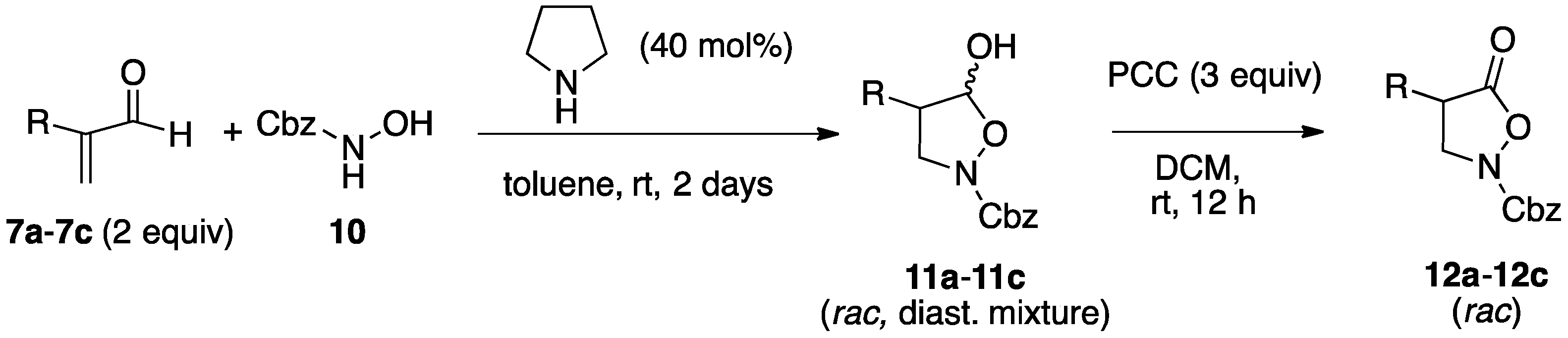
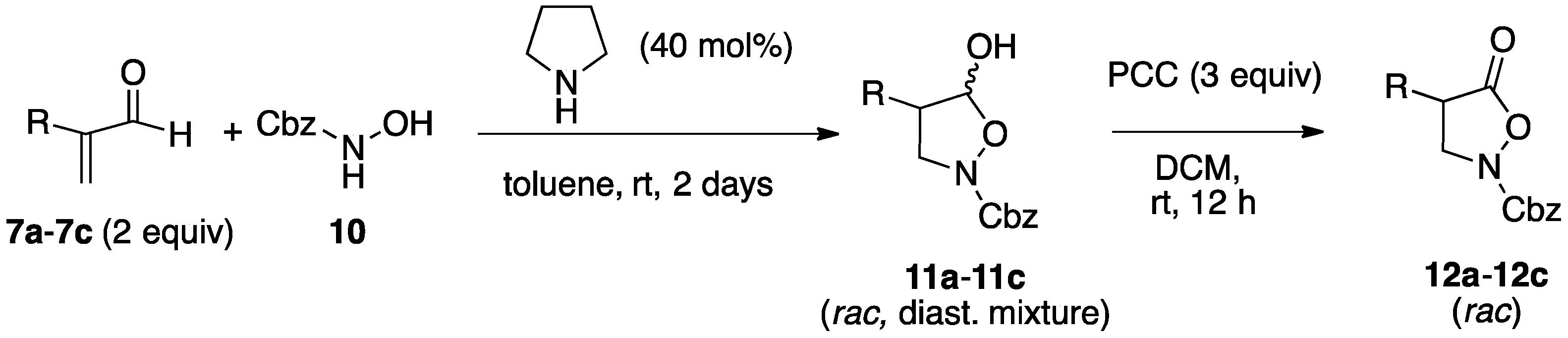
2.1. Organocatalytic Synthesis of 4-Substituted Pyrazolidinones and Isoxazolidinones
2.2. Primary Antimicrobial Screening
3. Experimental Section
3.1. General Information
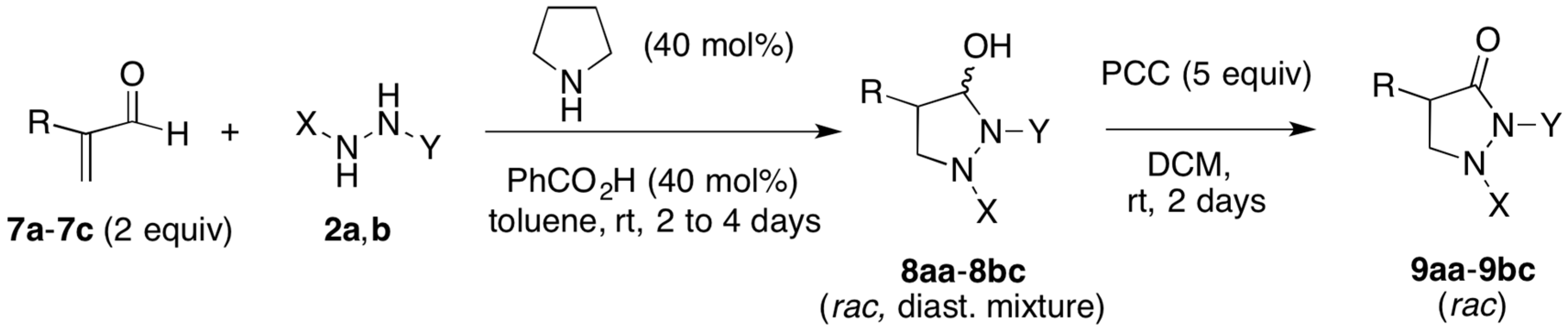
3.2. General Procedures for the Preparation of Pyrazolidines
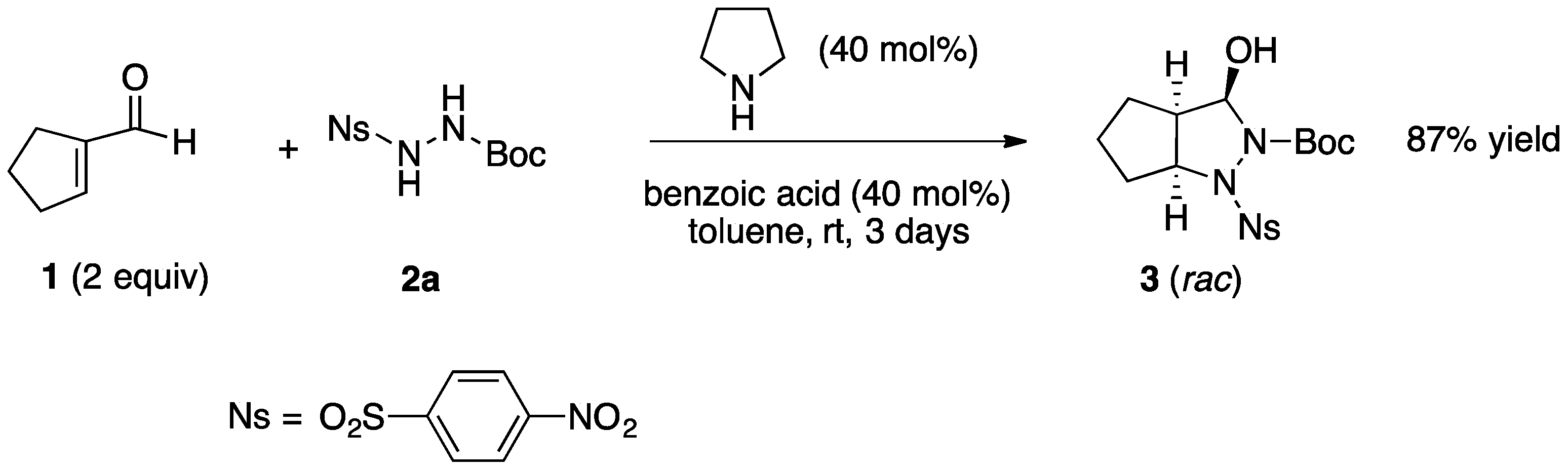
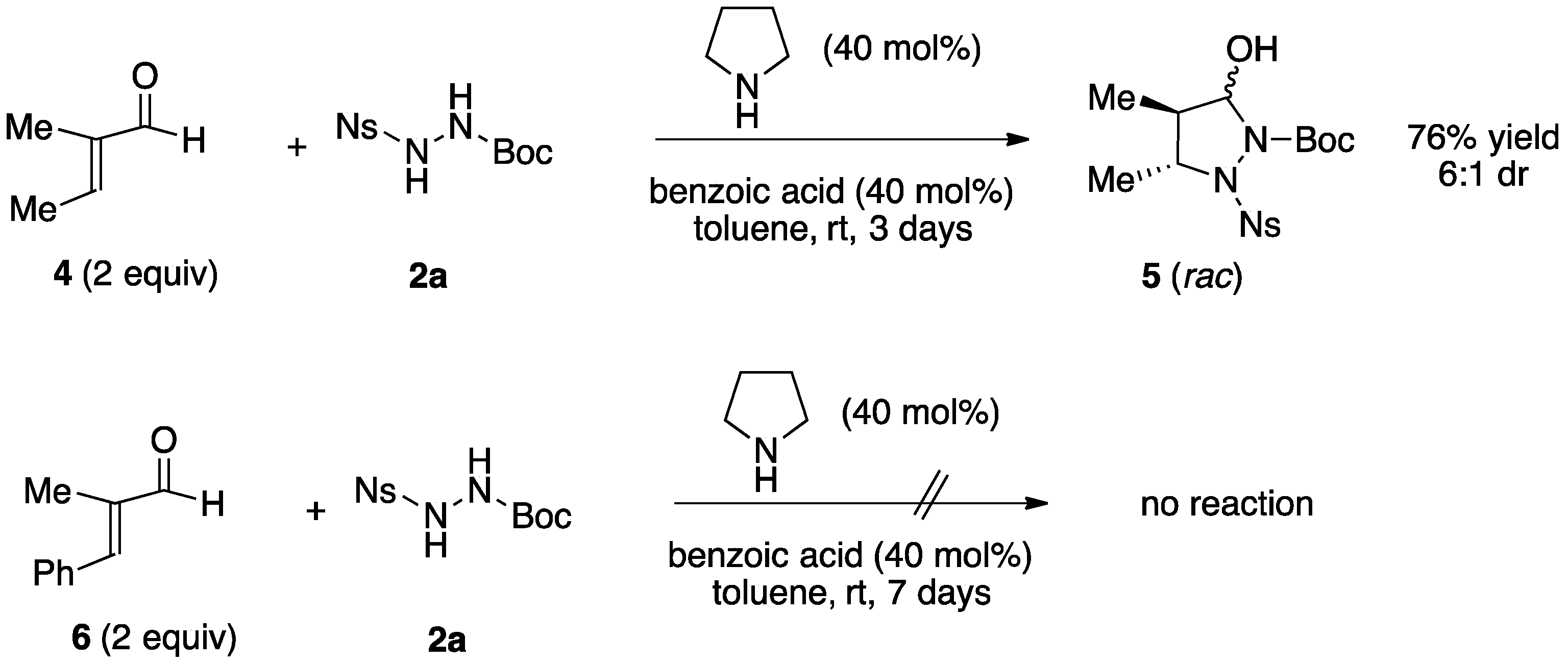
3.2.1. Racemic Pyrazolidinols from Enals and Hydrazines
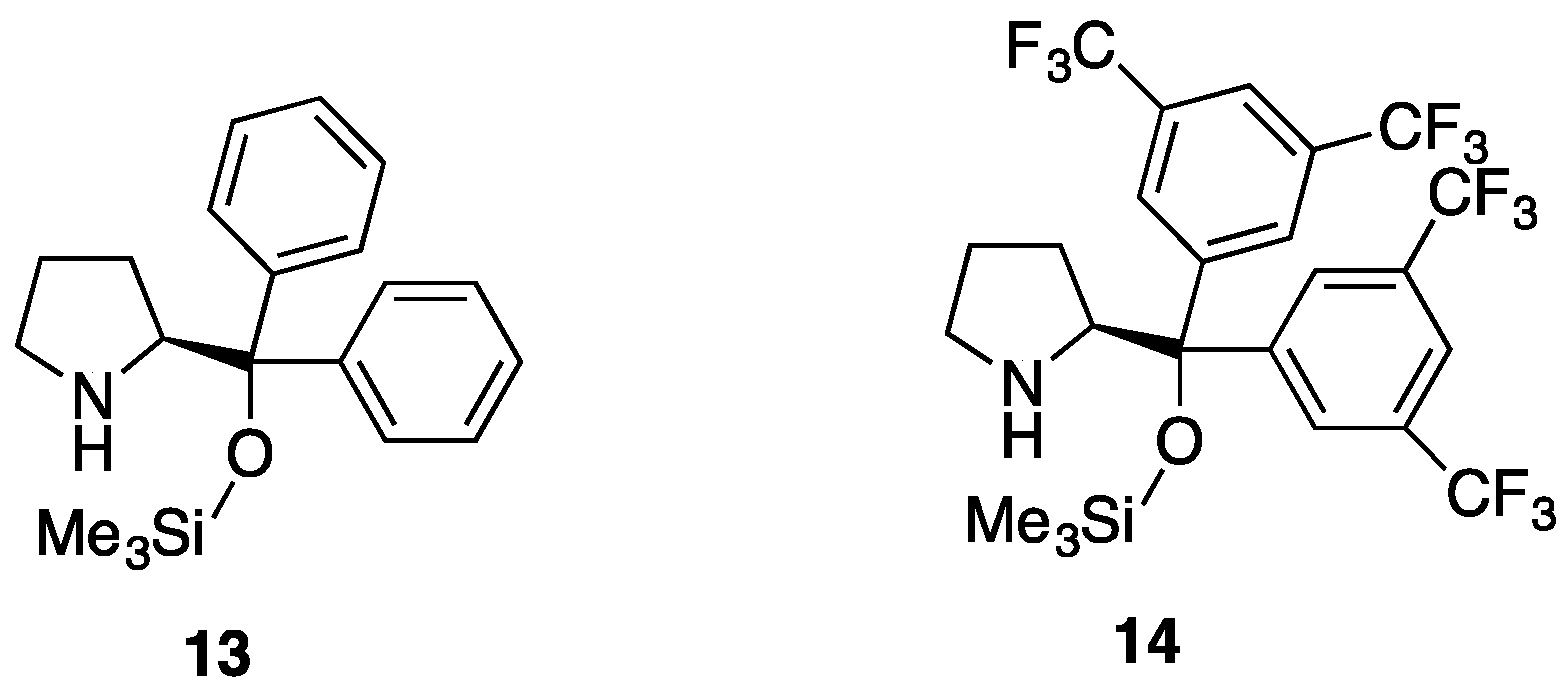
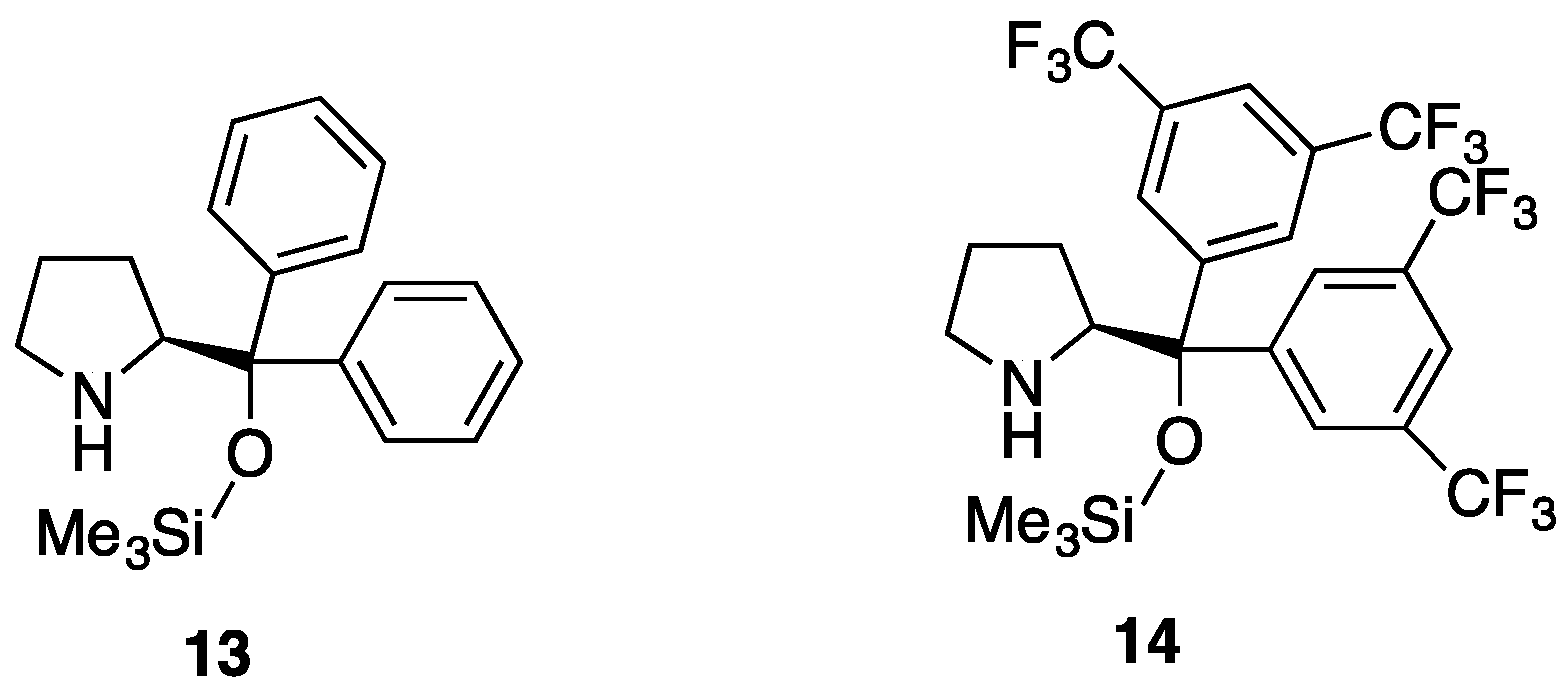
3.2.2. Asymmetric Synthesis of Pyrazolidinols from Enals and Hydrazines
3.2.3. Pyrazolidinones by Oxidation of Pyrazolidinols
3.3. General Procedures for the Preparation of Isoxazolidines
3.3.1. Racemic Isoxazolidinols from Enals and Hydroxylamine 10
3.3.2. Asymmetric Synthesis of Isoxazolidinols from Enals and Hydroxylamine 10
3.3.3. Isoxazolidinones by Oxidation of Isoxazolidinols
3.4. Characterization of the Products
3.5. Methods for the Biological Activity Studies
3.5.1. Sample Preparation
3.5.2. Antimicrobial Assay
Procedure
Analysis
3.5.3. Antifungal Assay
Procedure
Analysis
3.5.4. Antibiotic Standards Preparation and Quality Control
3.5.5. Materials
3.5.6. Microbial Strains
| Escherichia coli | ATCC 25922 (FDA control strain) |
| Klebsiella pneumoniae | ATCC 700603 (MDR) |
| Acinetobacter baumannii | ATCC 19606 (type strain) |
| Pseudomonas aeruginosa | ATCC 27853 (Quality control strain) |
| Staphylococcus aureus | ATCC 43300 (MRSA) |
| Candida albicans | ATCC 90028 (CLSI reference) |
| Cryptococcus neoformans | ATCC 208821 (H99—Type strain) |
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mixich, G. Isolierung, Struktur und Synthese des Metaboliten von Azapropazon-dihydrat. Helv. Chim. Acta 1972, 55, 1031–1038. [Google Scholar] [CrossRef]
- Bennett, G.B.; Houlihan, W.J.; Mason, R.B.; Roach, J.B., Jr. Synthesis and biological activity of a series of 1-aryl-3-pyrazolidinones. J. Med. Chem. 1976, 19, 715–717. [Google Scholar] [CrossRef] [PubMed]
- Jungheim, L.N.; Sigmund, S. 1,3-Dipolar cycloaddition reactions of pyrazolidinium ylides with acetylenes. Synthesis of a new class of antibacterial agents. J. Org. Chem. 1987, 52, 4007–4013. [Google Scholar] [CrossRef]
- Marchand-Baynaert, J.; Ghosez, L. Non-β-Lactam Analogs of Penicillins and Cephalosporins. In Recent Progress in the Chemical Synthesis of Antibiotics; Lukacs, G., Ohno, M., Eds.; Springer: Berlin/Heidelberg, Germany, 1990; pp. 726–794. [Google Scholar]
- Hadjipavlou-Litina, D. Review, reevaluation, and new results in quantitative structure-activity studies of anticonvulsants. Med. Res. Rev. 1998, 18, 91–119. [Google Scholar] [CrossRef]
- Coulognier, E.; Cartier, D.; Labia, R. Synthesis of pyrazolidinone antibacterial agents. Bioorg. Med. Chem. Lett. 1999, 9, 2205–2206. [Google Scholar] [CrossRef]
- Cusan, C.; Spalluto, G.; Prato, M.; Adams, M.; Bodensieck, A.; Bauer, R.; Tubaro, A.; Bernardi, P.; Da Ros, T. Synthesis and biological evaluation of new phenidone analogues as potential dual cyclooxygenase (COX-1 and COX-2) and human lipoxygenase (5-LOX) inhibitors. Farmaco 2005, 60, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, T.H.; Krueger, W.A.; Dieterich, H.-J.; Nohé, B. Activity of the lipoxygenase inhibitor 1-phenyl-3-pyrazolidinone (phenidone) and derivatives on the inhibition of adhesion molecule expression on human umbilical vascular endothelial cells. Biol. Targets. Ther. 2008, 2, 151–160. [Google Scholar] [CrossRef]
- Sachdeva, H.; Dwivedi, D.; Goyal, P. Green Chemical Synthesis and Analgesic Activity of Fluorinated Thiazolidinone, Pyrazolidinone, and Dioixanedione Derivatives. Org. Chem. Int. 2013, 2013, 976032. [Google Scholar] [CrossRef]
- Radhika, C.; Venkatesham, A.; Anantha Krishna Chaitanya, D. Synthesis and Biological activity of new 5-Methyl-3-Oxo-N2-[5′-Carbonyl-(4′-Aryl-6′-methyl)-1′,2′,3′,4′-tetrahydropyrimidine-2′-one]pyrazolidinones. J. Adv. Pharm. Educ. Res. 2014, 4, 66–70. [Google Scholar]
- Varvounis, G. Pyrazol-5-ones. Part IV: Synthesis and Applications. In Advances in Heterocyclic Chemistry; Katritzky, A.R., Ed.; Academic Press Inc.: London, UK, 2009; Volume 98, pp. 143–223. [Google Scholar]
- Brogden, R.N. Pyrazolone derivatives. Drugs 1986, 32, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.W.; Zeng, L.H.; Wu, J.; Fung, K.P. Myocardial protection of MCI-186 in rabbit ischemia-reperfusion. Life Sci. 2002, 71, 2249–2255. [Google Scholar] [CrossRef]
- Lange, J.H.M.; Kruse, G.G. Recent Advances in CB1 Receptor Antagonists. Curr. Opin. Drug Discov. Dev. 2004, 7, 498–506. [Google Scholar]
- Ali, M.A.; Shaharyar, M. Discovery of novel phenoxyacetic acid derivatives as antimycobacterial agents. Bioorg. Med. Chem. 2007, 15, 1896–1902. [Google Scholar] [CrossRef] [PubMed]
- Janin, Y.L. Antituberculosis drugs: Ten years of research. Bioorg. Med. Chem. 2007, 15, 2479–2513. [Google Scholar] [CrossRef] [PubMed]
- Pégurier, C.; Collart, P.; Danhaive, P.; Defays, S.; Gillard, M.; Gilson, F.; Kogej, T.; Pasau, P.; van Houtvin, N.; van Thuyne, M. Pyrazolone methylamino piperidine derivatives as novel CCR3 antagonists. Bioorg. Med. Chem. Lett. 2007, 17, 4228–4231. [Google Scholar] [CrossRef] [PubMed]
- Casas, J.S.; Castellano, E.E.; Ellena, J.; García-Tasende, M.S.; Peses-Paralle, M.L.; Sánchez, A.; Sánchez-González, A.; Sordo, J.; Touceda, A. New Pd(II) and Pt(II) complexes with N,S-chelated pyrazolonate ligands: Molecular and supramolecular structure and preliminary study of their in vitro antitumoral activity. J. Inorg. Biochem. 2008, 102, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Groselj, U.; Svete, J. Recent advances in the synthesis of polysubstituted 3-pyrazolidinones. Arkivoc 2015, 6, 175–205. [Google Scholar]
- Fernández, M.; Reyes, E.; Vicario, J.L.; Badía, L.; Carrillo, L. Organocatalytic Enantioselective Synthesis of Pyrazolidines, Pyrazolines and Pyrazolidinones. Adv. Synth. Catal. 2012, 354, 371–376. [Google Scholar] [CrossRef]
- Geng, Z.-G.; Chen, J.; Li, N.; Huang, X.-F.; Zhang, Y.; Zhang, Y.-W.; Wang, X.-W. Organocatalytic cascade aza-Michael/hemiacetal reaction between disubstituted hydrazines and α,β-unsaturated aldehydes: Highly diastereo- and enantioselective synthesis of pyrazolidine derivatives. Beilstein J. Org. Chem. 2012, 8, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, B.; Zhang, Y.; Jeret, M.; Wang, H.; Zheng, P.; Yang, S.; Song, A.-B.; Chi, Y.R. Enantioselective Nucleophilic β-Carbon-Atom Amination of Enals: Carbene-Catalyzed Formal [3 + 2] Reactions. Angew. Chem. Int. Ed. 2016, 55, 12280–12284. [Google Scholar] [CrossRef] [PubMed]
- Companyó, X.; Zea, A.; Alba, A.-N.R.; Mazzanti, A.; Moyano, A.; Rios, R. Organocatalytic synthesis of spiro compounds via a cascade Michael-Michael-aldol reaction. Chem. Commun. 2010, 46, 6953–6955. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, A.; Calbet, T.; Font-Bardía, M.; Moyano, A.; Rios, R. Organocatalytic enantioselective pyrazol-3-one addition to maleimides: Reactivity and stereochemical course. Org. Biomol. Chem. 2012, 10, 1645–1652. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Bao, X.; Qu, J.; Wang, B. Asymmetric tandem Michael addition/oxidation of pyrazolones with p-benzoquinone catalyzed by cinchona alkaloids. Tetrahedron Asymmetry 2015, 26, 1382–1387. [Google Scholar] [CrossRef]
- Pou, A.; Moyano, A. Stereoselective Organocatalytic Approach to α,β-Disubstituted-β-amino Acids: A Short Enantioselective Synthesis of Cispentacin. Eur. J. Org. Chem. 2013, 2013, 3103–3111. [Google Scholar] [CrossRef]
- Ibrahem, I.; Rios, R.; Vesely, J.; Zhao, G.-L.; Córdova, A. Organocatalytic asymmetric 5-hydroxyisoxazolidinone synthesis: A highly enantioselective route to β-amino acids. Chem. Commun. 2007, 8, 849–851. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.-L.; Lin, A.; Korotvicka, A.; Deiana, L.; Kullberg, M.; Córdova, A. Asymmetric Synthesis of Maraviroc (UK-427,857). Adv. Synth. Catal. 2010, 352, 2291–2298. [Google Scholar] [CrossRef]
- Fu, N.; Zhang, L.; Luo, S.; Cheng, J.-P. Chiral primary amine catalyzed asymmetric conjugate additions of azoles to α-substituted vinyl ketones. Org. Chem. Front. 2014, 1, 68–72. [Google Scholar] [CrossRef]
- Fu, N.; Zhang, L.; Luo, S.; Cheng, J.-P. Asymmetric Sulfa-Michael Addition to α-Substituted Vinyl Ketones Catalyzed by Chiral Primary Amine. Org. Lett. 2014, 16, 4626–4629. [Google Scholar] [CrossRef] [PubMed]
- Community for Open Antimicrobial Drug Discovery. Available online: http://www.co-add.org (accessed on 21 October 2016).
- Meyers, M.J.; Arhancet, G.B.; Hockerman, S.L.; Chen, X.; Long, S.A.; Mahoney, M.W.; Rico, J.R.; Garland, D.J.; Blinn, J.R.; Collins, J.T.; et al. Discovery of (3S,3aR)-2-(3-chloro-4-cyanophenyl)-3-cyclopentyl-3,3a,4,5-tetrahydro-2H-benzo[g]indazole-7-carboxylic acid (PF-3882845), an orally efficacious mineralocorticoid receptor (MR) antagonist for hypertension and nephropathy. J. Med. Chem. 2010, 53, 5979–6002. [Google Scholar] [CrossRef] [PubMed]
- Erkkilä, A.; Pihko, P.M. Mild Organocatalytic α-Methylenation of Aldehydes. J. Org. Chem. 2006, 71, 2538–2541. [Google Scholar] [CrossRef] [PubMed]
- Erkkilä, A.; Pihko, P.M. Rapid Organocatalytic Aldehyde-Aldehyde Condensation Reactions. Eur. J. Org. Chem. 2007, 4205–4216. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 3, 5, 8aa–8bc, 9aa–9bc, 11a–11c and 12a–12c are available from the authors.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Reactants | R | X | Y | Pyrazolidinol | Yield 1 | Reaction Time (Day) | dr 2 | Pyrazolidinone | Yield 1 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 7a, 2a | Me | Ns | Boc | 8aa | 93% | 4 | 4:1 | 9aa | 94% |
| 2 | 7b, 2a | PhCH2 | Ns | Boc | 8ab | 83% | 2 | >20:1 | 9ab | 98% |
| 3 | 7c, 2a | n-Hexyl | Ns | Boc | 8ac | 99.6% | 2 | 5:1 | 9ac | 99% |
| 4 | 7a, 2b | Me | Ts | Ts | 8ba | 88% | 2 | 8:1 | 9ba | 98% |
| 5 | 7b, 2b | PhCH2 | Ts | Ts | 8bb | 85% | 2 | 9:1 | 9bb | 97% |
| 6 | 7c, 2b | n-Hexyl | Ts | Ts | 8bc | 98% | 2 | 10:1 | 9bc | 98% |
| Entry | Reactants | R | Isoxazolidinol | Yield 1 | dr 2 | Isoxazolidinone | Yield 1 |
|---|---|---|---|---|---|---|---|
| 1 | 7a, 10 | Me | 11a | 83% | 2:1 | 12a | 98% |
| 2 | 7b, 10 | PhCH2 | 11b | 79% | 3:1 | 12b | 99% |
| 3 | 7c, 10 | n-Hexyl | 11c | 78% | 3:1 | 12c | 87% |
| Entry | Reactants | Catalyst | Pyrazolidinol | Yield 1 | dr 2 | Pyrazolidinone | Yield 1 | er 3 |
|---|---|---|---|---|---|---|---|---|
| 1 | 7a, 2a | 13 | 8aa | 82% | 4:1 | 9aa | 87% | 50:50 |
| 2 | 7a, 2a | 14 | 8aa | 99% | 3:1 | 9aa | 94% | 90:10 |
| 3 | 7b, 2a | 13 | 8ab | 77% | >20:1 | 9ab | 98% | 44:56 |
| 4 | 7b, 2a | 14 | 8ab | 98% | >20:1 | 9ab | 98% | 45:55 |
| 5 | 7b, 2b | 13 | 8bb | 85% | 9:1 | 9bb | 97% | - 4 |
| 6 | 7b, 2b | 14 | 8bb | 85% | 9:1 | 9bb | 97% | - 4 |
| 7 | 7a, 2b | 13 | 8ba | 84% | 8:1 | 9ba | 98% | 96:4 |
| 8 | 7a, 2b | 14 | 8ba | 0% 5 | 9ba | - | - | |
| 9 | 7c, 2b | 13 | 8bc | 93% | 10:1 | 9bc | 98% | 78:22 |
| 10 | 7c, 2b | 14 | 8bc | 77% | 10:1 | 9bc | 98% | 50:50 |
| Entry | S. aureus | E. coli | K. pneumoniae | A. baumannii | P. aeruginosa | C. albicans | C. neoformans |
|---|---|---|---|---|---|---|---|
| 1 | - | - | - | - | - | 12b | - |
| 2 | 8aa (rac) | - | - | - | - | - | - |
| 3 | 8aa (90:10 er) | - | - | - | - | - | - |
| 4 | - | - | - | - | - | - | 12a |
| 5 | - | - | - | - | 11c | - |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yousfi, T.; Elliott, A.; Hanane, M.; Merdes, R.; Moyano, A. Expedient Organocatalytic Syntheses of 4-Substituted Pyrazolidines and Isoxazolidines. Molecules 2016, 21, 1655. https://doi.org/10.3390/molecules21121655
Yousfi T, Elliott A, Hanane M, Merdes R, Moyano A. Expedient Organocatalytic Syntheses of 4-Substituted Pyrazolidines and Isoxazolidines. Molecules. 2016; 21(12):1655. https://doi.org/10.3390/molecules21121655
Chicago/Turabian StyleYousfi, Tarek, Alysha Elliott, Messiad Hanane, Rachid Merdes, and Albert Moyano. 2016. "Expedient Organocatalytic Syntheses of 4-Substituted Pyrazolidines and Isoxazolidines" Molecules 21, no. 12: 1655. https://doi.org/10.3390/molecules21121655