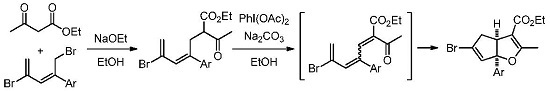
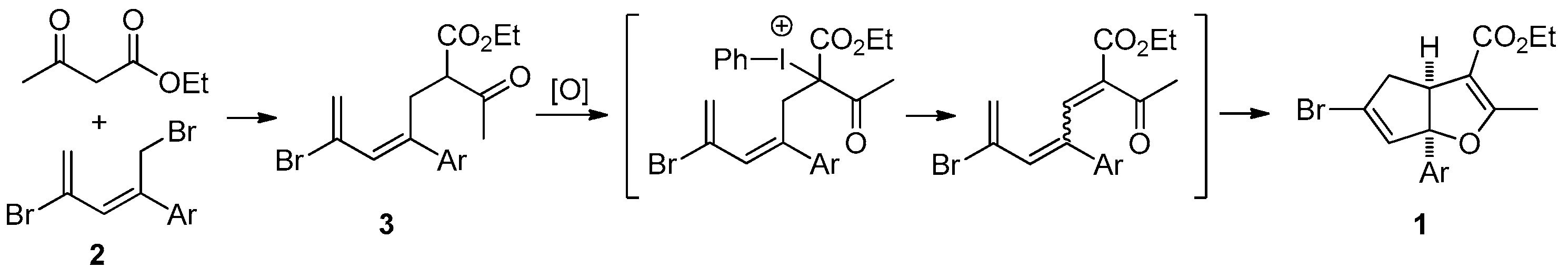
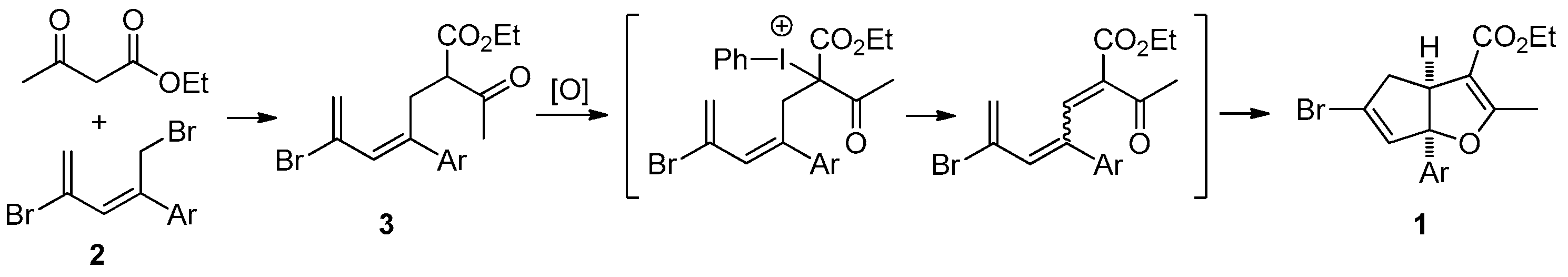
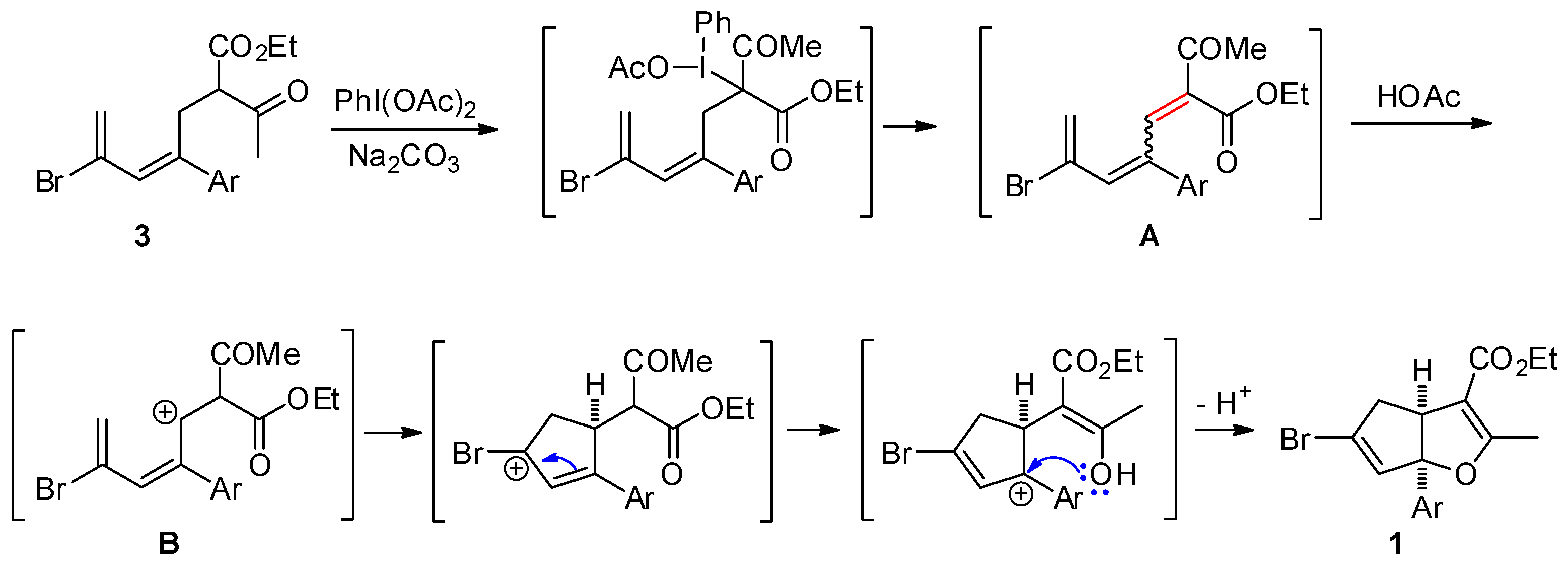
3.2.1. General Procedure for the Synthesis of Cyclopenta[b]furans 1
A mixture of bromodiene 2 (0.3 mmol), ethyl acetoacetate (0.3 mmol, 39 mg), and NaOEt (0.4 mmol, 27 mg) in EtOH (2 mL) was stirred at reflux overnight. The mixture was cooled to ambient temperature (25–28 °C) and transferred to a separatory funnel, followed by addition of Et2O (5 mL) and water (10 mL). The aqueous layer was back extracted with Et2O (2 mL × 2). The combined organic layers were washed with brine (5 mL), dried over MgSO4, and concentrated in a rotary evaporator. The residue was purified by a silica gel plug using Et2O/hexanes (10/1, 10 mL) as the eluent, followed by concentration to give the bromoester 3. To a solution of bromoester 3 (0.3 mmol, based on bromo-diene 2) in EtOH (1.2 mL) was added Na2CO3 (0.6 mmol, 64 mg) and PhI(OAc)2 (0.6 mmol, 193 mg) at ambient temperature. The resulting mixture was heated to reflux under N2 for 3 h. The reaction was then cooled room temperature and H2O (2 mL) was added. Et2O (5 mL) was added and the mixture was transferred to a separatory funnel. The aqueous layer was back extracted with Et2O (5 mL × 2). The combined organic layers were washed with 2 N HCl (2 mL), dried over Na2SO4, filtered, and concentrated in a rotary evaporator. The residue was purified by silica gel chromatography using Et2O/hexanes (1/10) as eluent to give the title products as yellow oils. Using this general procedure, the following title compounds were obtained:
Ethyl 5-bromo-2-methyl-6a-phenyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (1a). Yield 55 mg (72%). TLC (Et2O/hexanes (1:10)) Rf = 0.44; 1H-NMR (CDCl3) δ 1.25 (t, J = 7.2 Hz, 3H), 2.28 (d, J = 1.5 Hz, 3H), 2.83 (dt, J = 17.4, 2.1 Hz, 1H), 3.15 (ddd, J = 17.4, 7.8, 2.1 Hz, 1H), 3.74 (dt, J = 7.8, 1.5 Hz, 1H), 4.09–4.20 (m, 2H), 5.98 (t, J = 2.1 Hz, 1H), 7.25–7.35 (m, 5H); 13C-NMR (CDCl3) δ 14.3 (CH3), 14.4 (CH3), 46.9 (CH2), 53.9 (CH), 59.6 (CH2), 100.8 (C) 105.8 (C), 124.5 (CH × 2), 128.0 (CH), 128.6 (CH × 2), 129.1 (C), 131.9 (CH), 142.0 (C), 165.5 (C), 167.0 (C); IR (neat) 2977, 1698, 1644 cm−1; EI-MS m/z (rel intensity) 350 ([M + 2]+, 10), 348 ([M]+, 10), 302 (29), 223 (100); HRMS [M]+ calcd. for C17H17BrO3: 348.0361, found 348.0366.
Ethyl 5-bromo-6a-(4-bromophenyl)-2-methyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (1b). Yield 100 mg (78%). TLC (Et2O/hexanes (1:10)) Rf = 0.43; 1H-NMR (CDCl3) δ 1.26 (t, J = 7.2 Hz, 3H), 2.28 (d, J = 1.5 Hz, 3H), 2.80 (dt, J = 17.4, 1.8 Hz, 1H), 3.14 (ddd, J = 17.4, 7.8, 1.8 Hz, 1H), 3.69 (dt, J = 7.8, 1.5 Hz, 1H), 4.06–4.28 (m, 2H), 5.93 (t, J = 1.5 Hz, 1H), 7.15 (d, J = 7.8 Hz, 2H), 7.46 (d, J = 7.8 Hz, 2H); 13C-NMR (CDCl3) δ 14.2 (CH3), 14.4 (CH3), 46.8 (CH2), 53.9 (CH), 59.7 (CH2), 100.3 (C) 105.9 (C), 122.0 (C), 126.3 (CH × 2), 129.6 (C), 131.5 (CH), 131.7 (CH × 2), 141.1 (C), 165.4 (C), 166.8 (C); IR (neat) 2976, 1703, 1488 cm−1; EI-MS m/z (rel intensity) 428 ([M + 2]+, 19), 426 ([M]+, 10), 382 (59), 301 (100); HRMS [M]+ calcd. for C17H16Br2O3: 425.9466, found 425.9462.
Ethyl 5-bromo-6a-(4-chlorophenyl)-2-methyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (1c). Yield 104 mg (90%). TLC (Et2O/hexanes (1:10)) Rf = 0.43; 1H-NMR (CDCl3) δ 1.25 (t, J = 7.2 Hz, 3H), 2.28 (d, J = 1.5 Hz, 3H), 2.83 (dt, J = 17.4, 2.1 Hz, 1H), 3.13 (ddd, J = 17.4, 7.8, 2.1 Hz, 1H), 3.68 (dt, J = 7.8, 1.5 Hz, 1H), 4.09–4.20 (m, 2H), 5.93 (t, J = 2.1 Hz, 1H), 7.20 (d, J = 8.7 Hz, 2H), 7.31 (d, J = 8.7 Hz, 2H); 13C-NMR (CDCl3) δ 14.2 (CH3), 14.3 (CH3), 46.8 (CH2), 53.9 (CH), 59.6 (CH2), 100.2 (C) 105.9 (C), 125.9 (CH × 2), 128.7 (CH × 2), 129.5 (C), 131.5 (CH), 133.8 (C), 140.5 (C), 165.3 (C), 166.7 (C); IR (neat) 2977, 1700, 1646 cm−1; EI-MS m/z (rel intensity) 384 ([M + 2]+, 15), 382 ([M]+, 12), 338 (44), 257 (100); HRMS [M]+ calcd. for C17H16BrClO3: 381.9971, found 381.9978.
Ethyl 5-bromo-2-methyl-6a-(p-tolyl)-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (1d). Yield 76 mg (70%). TLC (Et2O/hexanes (1:10)) Rf = 0.51; 1H-NMR (CDCl3) δ 1.25 (t, J = 7.2 Hz, 3H), 2.28 (d, J = 1.5 Hz, 3H), 2.32 (s, 3H), 2.82 (dt, J = 17.4, 1.8 Hz, 1H), 3.13 (ddd, J = 17.4, 7.8, 1.8 Hz, 1H), 3.72 (dt, J = 7.8, 1.5 Hz, 1H), 4.09–4.20 (m, 2H), 5.97 (t, J = 1.8 Hz, 1H), 7.15 (s, 4H); 13C-NMR (CDCl3) δ 14.3 (CH3), 14.4 (CH3), 21.0 (CH3), 46.8 (CH2), 53.8 (CH), 59.5 (CH2), 100.7 (C) 105.8 (C), 124.4 (CH × 2), 128.9 (C), 129.2 (CH × 2), 131.9 (CH), 137.8 (C), 139.0 (C), 165.6 (C), 167.0 (C); IR (neat) 2979, 1702, 1644 cm−1; EI-MS m/z (rel intensity) 364 ([M + 2]+, 5), 362 ([M]+, 6), 316 (19), 237 (100); HRMS [M]+ calcd. for C18H19BrO3: 362.0518, found 362.0515.
Ethyl 5-bromo-6a-(4-methoxyphenyl)-2-methyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (1e). Yield 74 mg (65%). TLC (Et2O/hexanes (1:10)) Rf = 0.48; 1H-NMR (CDCl3) δ 1.25 (t, J = 7.2 Hz, 3H), 2.27 (d, J = 1.5 Hz, 3H), 2.81 (dt, J = 17.4, 1.8 Hz, 1H), 3.12 (ddd, J = 17.4, 7.8, 1.8 Hz, 1H), 3.71 (dt, J = 7.8, 1.5 Hz, 1H), 3.78 (s, 3H), 4.06–4.22 (m, 2H), 5.97 (t, J = 1.8 Hz, 1H), 6.85 (d, J = 8.7 Hz, 2H), 7.18 (d, J = 8.7 Hz, 2H); 13C-NMR (CDCl3) δ 14.3 (CH3), 14.4 (CH3), 46.8 (CH2), 53.7 (CH), 55.3 (CH3), 59.5 (CH2), 100.6 (C), 105.8 (C), 113.9 (CH × 2), 125.8 (CH × 2), 128.8 (C), 131.9 (CH), 134.1 (C), 159.3 (C), 165.6 (C), 166.9 (C); IR (neat) 2981, 1698, 1643 cm−1; EI-MS m/z (rel intensity) 380 ([M + 2]+, 6), 378 ([M]+, 6), 332 (20), 253 (100); HRMS [M]+ calcd. for C18H19BrO4: 378.0467, found 378.0458.
Ethyl 5-bromo-6a-(3-methoxyphenyl)-2-methyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (1f). Yield 72 mg (63%). TLC (Et2O/hexanes (1:10)) Rf = 0.45; 1H-NMR (CDCl3) δ 1.24 (t, J = 7.2 Hz, 3H), 2.28 (d, J = 1.5 Hz, 3H), 2.82 (dt, J = 17.4, 1.8 Hz, 1H), 3.14 (ddd, J = 17.4, 7.8, 1.8 Hz, 1H), 3.73 (dt, J = 7.8, 1.8 Hz, 1H), 3.79 (s, 3H), 4.03–4.24 (m, 2H), 5.95 (t, J = 1.8 Hz, 1H), 6.79–6.86 (m, 3H), 7.24–7.29 (m, 1H); 13C-NMR (CDCl3) δ 14.2 (CH3), 14.3 (CH3), 46.8 (CH2), 53.8 (CH), 55.2 (CH3), 59.5 (CH2), 100.6 (C) 105.8 (C), 110.7 (CH), 112.7 (CH), 116.7 (CH), 129.0 (C), 129.7 (CH), 131.8 (CH), 143.6 (C), 159.7 (C), 165.5 (C), 166.9 (C); IR (neat) 2981, 1702, 1644 cm−1; EI-MS m/z (rel intensity) 380 ([M + 2]+, 10), 378 ([M]+, 10), 332 (21), 253 (100); HRMS [M]+ calcd. for C18H19BrO4: 378.0467, found 378.0461.
Ethyl 5-bromo-6a-(3-bromophenyl)-2-methyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (1g). Yield 112 mg (87%). TLC (Et2O/hexanes (1:10)) Rf = 0.53; 1H-NMR (CDCl3) δ 1.24 (t, J = 7.2 Hz, 3H), 2.28 (d, J = 1.5 Hz, 3H), 2.83 (dt, J = 17.7, 2.1 Hz, 1H), 3.15 (ddd, J = 17.7, 7.8, 2.1 Hz, 1H), 3.70 (dt, J = 7.8, 2.1 Hz, 1H), 4.06–4.21 (m, 2H), 5.95 (t, J = 2.1 Hz, 1H), 7.15–7.20 (m, 2H), 7.37–7.41 (m, 2H); 13C-NMR (CDCl3) δ 14.2 (CH3), 14.3 (CH3), 46.7 (CH2), 53.9 (CH), 59.6 (CH2), 99.9 (C) 105.8 (C), 122.7 (C), 123.7 (CH), 127.6 (CH), 129.6 (CH), 130.1 (CH), 131.0 (CH), 131.3 (CH), 144.2 (C), 165.2 (C), 166.6 (C); IR (neat) 2979, 1704, 1643 cm−1; EI-MS m/z (rel intensity) 428 ([M + 2]+, 19), 426 ([M]+, 10), 382 (59), 301 (100); HRMS [M]+ calcd. for C17H16Br2O3: 425.9466, found 425.9460.
Ethyl 5-bromo-2-methyl-6a-(naphthalen-2-yl)-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (1h). Yield 77 mg (64%). TLC (Et2O/hexanes (1:10)) Rf = 0.48; 1H-NMR (CDCl3) δ 1.26 (t, J = 7.2 Hz, 3H), 2.35 (d, J = 1.5 Hz, 3H), 2.90 (dt, J = 17.4, 1.8 Hz, 1H), 3.22 (ddd, J = 17.4, 7.8, 1.8 Hz, 1H), 3.84 (dt, J = 7.8, 1.8 Hz, 1H), 4.11–4.23 (m, 2H), 6.09 (t, J = 1.8 Hz, 1H), 7.35 (dd, J = 8.4, 1.8 Hz, 1H), 7.44–7.51 (m, 2H), 7.74 (s, 1H), 7.80–7.85 (m, 3H); 13C-NMR (CDCl3) δ 14.3 (CH3), 14.4 (CH3), 46.9 (CH2), 53.7 (CH), 59.6 (CH2), 100.9 (C) 105.9 (C), 112.6 (CH), 123.1 (CH), 126.2 (CH), 126.4 (CH), 127.6 (CH), 128.1 (CH), 128.7 (CH), 129.3 (C), 131.8 (CH), 132.8 (C), 132.9 (C), 139.0 (C), 165.5 (C), 167.0 (C); IR (neat) 3058, 1700, 1637 cm−1; EI-MS m/z (rel intensity) 400 ([M + 2]+, 11), 398 ([M]+, 11), 273 (99), 202 (100); HRMS [M]+ calcd. for C21H19BrO3: 398.0518, found 398.0516.
3.2.2. General Procedure for the Suzuki Reactions of Cyclopenta[b]furans 1
To a solution of cyclopenta[b]furan 1 (0.5 mmol) in dioxane/H2O (5 mL/1 mL) was added Cs2CO3 (1.5 mmol, 489 mg), PhB(OH)2 (0.75 mmol, 91 mg), and Pd(dppf)Cl2 (0.05 mmol, 37 mg) at ambient temperature. The resulting mixture was vacuumed and purged with N2 for 3 times. The reaction was heated to 100 °C under N2 for 2 h. The reaction was then cooled room temperature and concentrated in a rotary evaporator. EtOAc (5 mL) and H2O (1 mL) were added and the mixture was transferred to a separatory funnel. The aqueous layer was back extracted with EtOAc (5 mL × 2). The combined organic layers were dried over Na2SO4, filtered, and concentrated in a rotary evaporator. The residue was purified by silica gel chromatography to give the title product. The following compounds were prepared using this method:
Ethyl 2-methyl-5,6a-diphenyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (4a). Yield 149 mg (86%). A yellow oil; TLC (EtOAc/hexanes (1:10)) Rf = 0.50; 1H-NMR (CDCl3) δ 1.29 (t, J = 7.2 Hz, 3H), 2.31 (s, 3H), 3.02 (dt, J = 17.1, 2.1 Hz, 1H), 3.29 (ddd, J = 17.1, 7.8, 2.1 Hz, 1H), 3.88 (dt, J = 7.8, 1.5 Hz, 1H), 4.11–4.26 (m, 2H), 6.23 (t, J = 2.1 Hz, 1H), 7.26–7.39 (m, 8H), 7.54 (d, J = 7.8 Hz, 2H); 13C-NMR (CDCl3) δ 14.4, 40.2, 52.8, 59.3, 102.3, 106.4, 124.6, 125.5, 126.4, 127.5, 128.4, 128.5, 134.7, 143.1, 146.5, 165.9, 166.6; IR (neat) 2977, 1698, 1644 cm−1; EI-MS m/z (rel intensity) 346 ([M]+, 12), 300 (100), 257 (30), 229 (47); HRMS [M]+ calcd. for C23H22O3: 346.1569, found 346.1561.
Ethyl 6a-([1,1′-biphenyl]-4-yl)-2-methyl-5-phenyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (4b). Yield 180 mg (85%). A colorless solid, m.p. 118–120 °C; TLC (EtOAc/hexanes (1:10)) Rf = 0.35; 1H-NMR (CDCl3) δ 1.28 (t, J = 7.2 Hz, 3H), 2.31 (s, 3H), 3.02 (dt, J = 17.1, 1.8 Hz, 1H), 3.31 (ddd, J = 17.1, 6.0, 2.1 Hz, 1H), 3.91 (dt, J = 6.0, 1.8 Hz, 1H), 4.10–4.25 (m, 2H), 6.25 (t, J = 1.8 Hz, 1H), 7.30–7.45 (m, 8H), 7.53–7.55 (m, 6H); 13C-NMR (CDCl3) δ 14.3, 40.1, 52.8, 59.3, 102.2, 106.4, 125.0, 125.4, 126.3, 126.9, 127.1, 127.2, 128.3, 128.4, 128.6, 134.6, 140.4, 140.5, 142.1, 146.5, 165.8, 166.5; IR (neat) 3031, 1700, 1637 cm−1; EI-MS m/z (rel intensity) 422 ([M]+, 10), 376 (100), 333 (13), 305 (23); HRMS [M]+ calcd. for C29H26O3: 422.1882, found 422.1878.
Ethyl 6a-(4-chlorophenyl)-2-methyl-5-phenyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (4c). Yield 164 mg (86%). A yellow oil; TLC (EtOAc/hexanes (1:10)) Rf = 0.43; 1H-NMR (CDCl3) δ 1.31 (t, J = 7.2 Hz, 3H), 2.32 (s, 3H), 3.04 (dt, J = 17.1, 2.1 Hz, 1H), 3.30 (ddd, J = 17.1, 7.8, 2.1 Hz, 1H), 3.84 (dt, J = 7.8, 1.8 Hz, 1H), 4.13–4.29 (m, 2H), 6.19 (t, J = 1.8 Hz, 1H), 7.27–7.40 (m, 7H), 7.54 (d, J = 7.8 Hz, 2H); 13C-NMR (CDCl3) δ 14.4, 40.1, 52.9, 59.4, 101.8, 106.4, 124.6, 125.0, 126.1, 128.4, 128.5, 128.6, 133.3, 134.5, 141.7, 146.9, 165.7, 166.4; IR (neat) 2979, 1698, 1643 cm−1; EI-MS m/z (rel intensity) 382 ([M + 2]+, 4), 380 ([M]+, 10), 334 (100), 228 (36); HRMS [M]+ calcd. for C23H21ClO3: 380.1179, found 380.1184.
Ethyl 2-methyl-5-phenyl-6a-(p-tolyl)-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (4d). Yield 151 mg (84%). A yellow oil; TLC (EtOAc/hexanes (1:10)) Rf = 0.60; 1H-NMR (CDCl3) δ 1.31 (t, J = 7.2 Hz, 3H), 2.32 (s, 3H), 2.36 (s, 3H), 3.03 (dt, J = 17.1, 2.1 Hz, 1H), 3.30 (ddd, J = 17.1, 7.8, 2.1 Hz, 1H), 3.88 (dt, J = 7.8, 1.8 Hz, 1H), 4.17–4.25 (m, 2H), 6.24 (t, J = 1.8 Hz, 1H), 7.18 (d, J = 8.1 Hz, 2H), 7.25–7.40 (m, 5H), 7.56 (d, J = 8.1 Hz, 2H); 13C-NMR (CDCl3) δ 14.4, 14.5, 21.0, 40.2, 52.8, 59.3, 102.4, 106.4, 124.6, 125.6, 126.4, 128.4, 128.5, 129.1, 134.8, 137.3, 140.2, 146.3, 166.0, 166.6; IR (neat) 2977, 2923, 1693 cm−1; EI-MS m/z (rel intensity) 360 ([M]+, 11), 314 (100), 243 (27), 228 (20); HRMS [M]+ calcd. for C24H24O3: 360.1725, found 360.1721.
Ethyl 6a-(4-methoxyphenyl)-2-methyl-5-phenyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (4e). Yield 154 mg (82%). A red oil; TLC (EtOAc/hexanes (1:10)) Rf = 0.50; 1H-NMR (CDCl3) δ 1.29 (t, J = 7.2 Hz, 3H), 2.29 (s, 3H), 2.99 (dt, J = 17.1, 1.8 Hz, 1H), 3.26 (ddd, J = 17.1, 6.6, 2.1 Hz, 1H), 3.79 (s, 3H), 3.83 (dt, J = 6.6, 1.8 Hz, 1H), 4.13–4.24 (m, 2H), 6.22 (t, J = 1.8 Hz, 1H), 6.88 (d, J = 6.6 Hz, 2H), 7.25–7.38 (m, 5H), 7.53 (d, J = 7.8 Hz, 2H); 13C-NMR (CDCl3) δ 14.4, 14.5, 40.1, 52.7, 55.2, 59.4, 102.2, 106.4, 113.7, 125.5, 125.9, 126.4, 128.4, 128.5, 134.8, 135.3, 146.3, 159.0, 166.0, 166.6; IR (neat) 2931, 1696, 1644 cm−1; EI-MS m/z (rel intensity) 376 ([M]+, 18), 330 (100), 287 (19), 259 (33); HRMS [M]+ calcd. for C24H24O4: 376.1675, found 376.1672.
Ethyl 6a-(3-methoxyphenyl)-2-methyl-5-phenyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (4f). Yield 152 mg (81%). A yellow oil; TLC (EtOAc/hexanes (1:10)) Rf = 0.44; 1H-NMR (CDCl3) δ 1.30 (t, J = 7.2 Hz, 3H), 2.32 (s, 3H), 3.02 (dt, J = 17.1, 2.1 Hz, 1H), 3.30 (ddd, J = 17.1, 7.8, 2.1 Hz, 1H), 3.80 (s, 3H), 3.88 (dt, J = 7.8, 1.8 Hz, 1H), 4.16–4.24 (m, 2H), 6.22 (t, J = 1.8 Hz, 1H), 6.80–6.84 (m, 1H), 6.92–6.95 (m, 2H), 7.25–7.39 (m, 4H), 7.55 (d, J = 8.1 Hz, 2H); 13C-NMR (CDCl3) δ 14.4, 40.2, 52.8, 55.2, 59.4, 102.2, 106.5, 110.7, 112.4, 116.9, 125.4, 126.4, 128.4, 128.5, 129.5, 134.7, 144.8, 146.5, 159.7, 165.9, 166.5; IR (neat) 3056, 2979, 1693 cm−1; EI-MS m/z (rel intensity) 376 ([M]+, 18), 330 (100), 287 (48), 259 (41); HRMS [M]+ calcd. for C24H24O4: 376.1675, found 376.1680.
Ethyl 6a-([1,1′-biphenyl]-3-yl)-2-methyl-5-phenyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (4g). Yield 180 mg (85%). A red oil; TLC (EtOAc/hexanes (1:10)) Rf = 0.53; 1H-NMR (CDCl3) δ 1.33 (t, J = 7.2 Hz, 3H), 2.38 (s, 3H), 3.10 (dt, J = 17.1, 1.8 Hz, 1H), 3.38 (ddd, J = 17.1, 6.0, 2.1 Hz, 1H), 3.99 (dt, J = 6.0, 1.8 Hz, 1H), 4.20–4.28 (m, 2H), 6.32 (t, J = 1.8 Hz, 1H), 7.34–7.49 (m, 8H), 7.53–7.63 (m, 6H); 13C-NMR (CDCl3) δ 14.4, 14.5, 40.3, 52.9, 59.4, 102.4, 106.5, 123.4, 123.6, 125.5, 126.4, 126.5, 127.2, 127.3, 128.4, 128.5, 128.7, 128.9, 134.7, 141.0, 141.5, 143.7, 146.6, 165.9, 166.6; IR (neat) 3060, 2981, 1706 cm−1; EI-MS m/z (rel intensity) 422 ([M]+, 13), 376 (100), 333 (23), 305 (27); HRMS [M]+ calcd. for C29H26O3: 422.1882, found 422.1884.
Ethyl 2-methyl-6a-(naphthalen-2-yl)-5-phenyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (4h). Yield 167 mg (84%). A yellow oil; TLC (EtOAc/hexanes (1:10)) Rf = 0.38; 1H-NMR (MHz, CDCl3) δ 1.31 (t, J = 7.2 Hz, 3H), 2.38 (s, 3H), 3.09 (dt, J = 17.1, 1.8 Hz, 1H), 3.37 (ddd, J = 17.1, 7.8, 1.8 Hz, 1H), 3.98 (d, J = 7.8 Hz, 1H), 4.14–4.29 (m, 2H), 6.34 (t, J = 1.8 Hz, 1H), 7.33–7.49 (m, 6H), 7.58–7.61 (m, 2H), 7.75–7.85 (m, 4H); 13C-NMR (CDCl3) δ 14.5, 14.6, 40.4, 52.9, 59.5, 102.6, 106.7, 123.2, 125.5, 126.1, 126.3, 126.5, 127.6, 128.2, 128.5, 128.6, 132.8, 133.1, 134.8, 140.4, 146.9, 166.1, 166.8; IR (neat) 3056, 2977, 1698 cm−1; EI-MS m/z (rel intensity) 396 ([M]+, 22), 350 (100), 307 (23), 279 (53); HRMS [M]+ calcd. for C27H24O3: 396.1725, found 396.1733.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}