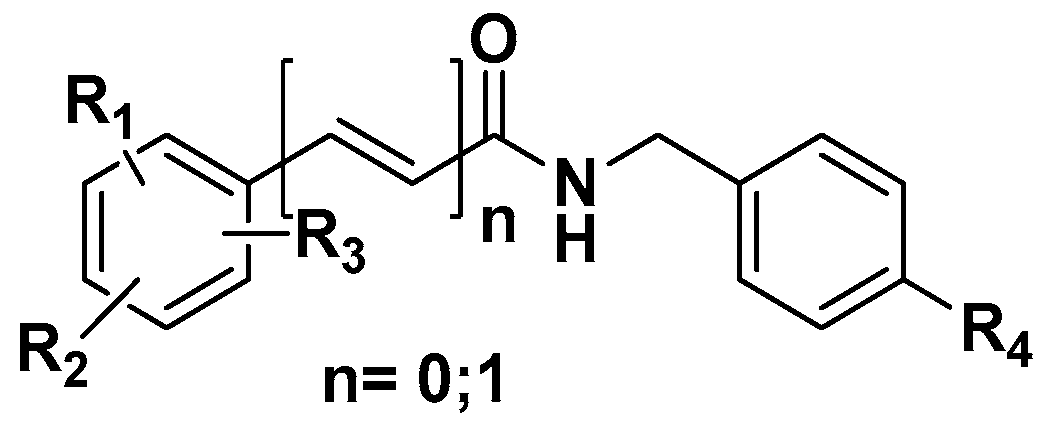
3.2.1. General Preparation of N-(4-Halobenzyl)amides
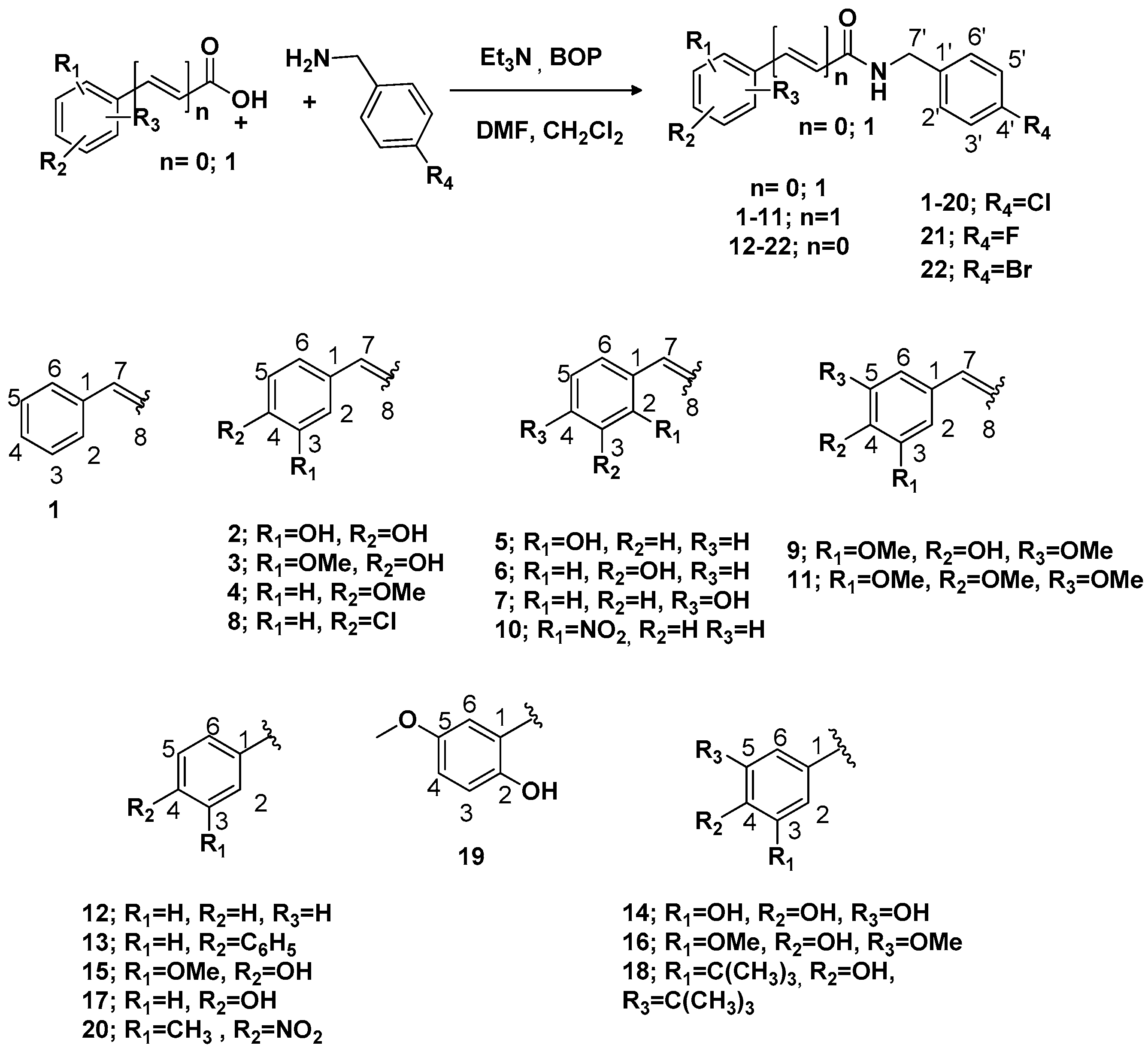
Procedure 1: In a 100 mL flask equipped with magnetic stirring, the organic acid (1.35 mmol, 200 mg) was dissolved in dimethylformamide (DMF, 2.7 mL) and trimethylamine (0.14 mL, 1.35 mmol). The solution was cooled in an ice bath (0 °C). Then, 4-chlorobenzylamine (1.35 mmol) was added. Soon after a 1.35 mmol solution of BOP in CH
2Cl
2 (10 mL) was added to the flask. The reaction was stirred at 0 °C for 30 min, and then for an additional period, at room temperature for 2 h. After the reaction, the CH
2Cl
2 was removed under reduced pressure and the solution was poured into a separatory funnel containing water (10 mL) and EtOAc (10 mL). The product was extracted with EtOAc (3 × 10 mL). The organic phase was washed sequentially with 1 N HCl, water, 1 M NaHCO
3 and water (10 mL of each); dried with Na
2SO
4, filtered and concentrated in a rotavapor. The amide was purified by gel chromatography on a silica gel column using as the mobile phase an EtOAc:Hex mixture gradient of increasing polarity [
11]. The following compounds were prepared by this procedure:
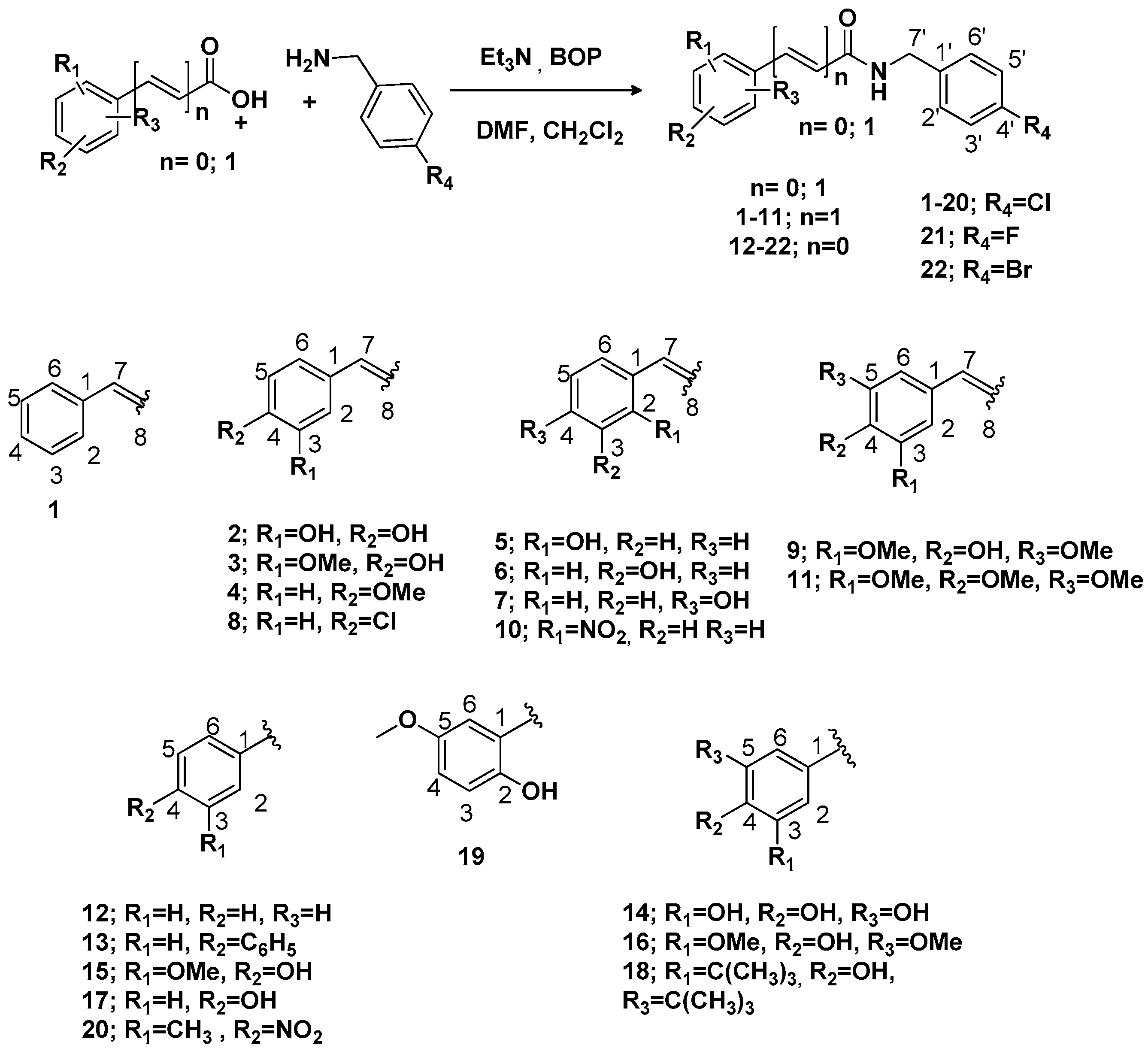
N-(4-Chlorobenzyl)cinnamamide (
1). Crystalline solid; 75% yield (274 mg), m.p.: 152–156 °C, IR ν
max (cm
−1): 3253 (N-H), 3080 (CH sp
2), 1654 (C=O), 1616 and 1489 (aromatic C=C), 1040 (stretching C-Cl),
1H-NMR (DMSO-
d6): 7.71 (d,
J = 16 Hz, 1H, H-7); 7.55–7.50 (m, 2H, H-2, H-6); 7.44–7.35 (m, 3H, H-3); 7.32–7.29 (m, 4H, H-2′, H-3′, H-5′, H-6′); 6.45 (d,
J = 16 Hz, 1H, H-8); 6.03 (bs, 1H, O=C-NH); 4.57 (d,
J = 4.0 Hz, H-7′).
13C-NMR (DMSO-
d6) 166.0 (C=O); 141.9 (C-7); 136.9 (C-1′); 134.8 (C-1); 133.5 (C-4′); 130.0 (C-2′, C-6′); 129.4 (C-3′, C-5′); 129.0 (C-3, C-5, C-6); 128.0 (C-2, C-4); 120.2 (C-8); 43.3 (C-7′) [
16].
(E)-N-(4-Chlorobenzyl)-3-(3,4-dihydroxyphenyl)acrylamide (
2). Dark amorphous solid; yield: 70% (228 mg), m.p.: 103–105 °C, IR ν
max (cm
−1): 3460 and 3406 (OH) 3230 (NH), 3018 (CH sp
2), 1651 (C=O) 1602 and 1490 (aromatic C=C), 1089 (C-Cl stretch).
1H-NMR (CD
3OD): 7.55 (d,
J = 15.7 Hz, 1H; H-7), 7.44–7.38 (m, 4H; H-2′, H-3′, H-5′, H-6′); 7.13 (d,
J = 1.7 Hz, 1H; H-2); 7.02 (dd,
J = 8.2, 1.8 Hz, 1H; H-5), 6.88 (d,
J = 8.1 Hz, 1H; H-6), 6.52 (d,
J = 15.7 Hz, 1H, H-8), 4.55 (bs, 2H; H-7′).
13C-NMR (CD
3OD): 169.2 (C=O); 148.8 (C-4); 146.7 (C-3); 142.8 (C-7); 138.9 (C-1′); 131,4 (C-4′); 130.1 (C-2′, C-6′); 129.6 (C-3′; C-5′); 128.2 (C-1); 122.2 (C-6); 118.0 (C-8); 115.0 (C-2); 116.4 (C-5); 43.5 (C-7′) [
16]. HRMS (MALDI) calculated for C
16H
14ClNNaO
3 [M + Na]
+: 326.0560; found 326.0561.
(
E)-N-(4-Chlorobenzyl)-3-(4-hydroxy-3-methoxyphenyl)acrylamide (
3). White amorphous solid; yield: 81% (273 mg), m.p. 129–131 °C, IR ν
max (cm
−1): 3414 (OH) 3275 (NH), 3010 (CH sp
2), 1645 (C=O), 1612 and 1460 (aromatic C=C), 1039 (C-Cl stretch).
1H-NMR (DMSO-
d6): 7.59 (d,
J = 16 Hz, 1H; H-7) 7.28 (dd,
J = 2.0 Hz and 6.0 Hz, 2H; H-2′, H-6′); 7.22 (dd,
J = 2.0 Hz, 6.0 Hz, 2H, H-3′, H-5′) 7.13 (s, 1H; H-2); 6.95 (d,
J = 8.1 Hz; 1H; H-5); 6.87 (d,
J = 8.1 Hz; 1H; H-6); 6.27(d,
J = 16 Hz, 1H; H-8); 4.36 (d,
J = 6.0 Hz, 2H, H-7′); 3.79 (
s, 3H, OCH
3).
13C-NMR (DMSO-
d6): 165.5 (C=O), 148.4 (C-3), 147.8 (C-4), 139.6 (C-7), 138.7 (C-1′), 131.3 (C-4′), 129.2 (C-2′, C-6′), 128.3 (C-3′, C-5′), 126.3 (C-1), 121.7 (C-6), 118.6 (C-8), 115.7 (C-5), 110.8 (C-2), 55.6 (OCH
3), 41.6 (C-7′) [
16]. HRMS (MALDI) calculated for C
17H
16ClNO
3 [M + H]
+: 318.0887; found 318.0870.
(E)-N-(4-Chlorobenzyl)-3-(4-methoxyphenyl)acrylamide (
4). Crystalline solid; yield: 91% (311 mg), m.p. 148–150 °C, IR ν
max (cm
−1): 3282 (N-H), 3041 (C-H sp
2), 1647 (C=O), 1602 and 1462 (aromatic C=C), 1029 (stretching C-Cl).
1H-NMR (DMSO-
d6): 8.59 (t,
J = 6.0 Hz, 1H, O=C-NH), 7.50 (t,
J = 7.3 Hz, 2H; H-2; H-6), 7.44–7.18 (m, 5H; H-7, H-2′, H-3′, H-5′, H-6′), 6.97 (d,
J = 8.7 Hz, 2H, H-3, H-5), 6.54 (d,
J = 15.7 Hz, 1H; H-8), 4.38 (d,
J = 6.0 Hz, 2H; H-7′), 3.77 (s, 3H, OCH
3).
13C-NMR (DMSO-
d6): 165.5 (C=O); 160.4 (C-4); 139.0 (C-7); 138.7 (C-1′); 131.4 (C-4′); 129.3 (C-2; C-6, C-2′; C-6′); 128.3 (C-3′, C-5′); 127.4 (C-1); 119.4 (C-8); 114.5 (C-3, C-5); 55.3 (OCH
3); 41.7 (C-7′) [
16]. HRMS (MALDI) calculated for C
16H
14ClNO
2 [M + H]
+: 324.0767; found 324.0771.
(E)-N-(4-Chlorobenzyl)-3-(2-hydroxyphenyl)acrylamide (
5). Yellow amorphous solid; yield: 79% (277 mg), m.p.: 165–171 °C. ν
max IR (cm
−1): 3369 (OH), 3072 (C-H sp
2), 1649 (C=O), 1589 and 1458 (aromatic C=C), 1093 (C-Cl stretch).
1H-NMR (DMSO-
d6) 10.00 (bs, 1H, OH), 8.64 (t,
J = 5.83 Hz, 1H; O=C-NH), 7.78 (d,
J = 15.92 Hz; 1H; H-7), 7.49–7.15 (m, 6H; H-6, H-2′, H-3′, H-5′, H-6′), 6.91–6.76 (m, 2H; H-4, H-5), 6.73 (d,
J = 15.92 Hz, 1H; H-8), 4.47 (d,
J = 5.86 Hz, 2H; H-7′).
13C-NMR (DMSO-
d6) 165.8 (C=O); 156.4 (C-2); 138.8 (C-1′); 135.2 (C-4); 135.0; 130.6 (C-4′); 129.3(C-2′, C-6′); 128.4 (C-6); 128.3 (C-3′, C-5′); 121.6 (C-1); 121.3 (C-5); 119.4 (C-8); 116.2; 41.7 (C-7′) [
16]. HRMS (MALDI) calculated for C
16H
14ClNO
2 [M + H]
+: 288.0781; found 288.0785.
(
E)-N-(4-Chlorobenzyl)-3-(3-hydroxyphenyl)acrylamide (
6). Yellow crystalline solid; yield: 76% (268 mg), m.p.: 140–143 °C ν
max IR (cm
−1): 3460 (OH), 3075 (CH sp
2), 1649 (C=O), 1591 and 1448 (aromatic C=C), 1089 (C-Cl stretch).
1H-NMR (DMSO-
d6): 9.61 (s, 1H, OH), 8.67 (t,
J = 5.9 Hz, 1H, O=C-NH), 7.47–7.14 (m, 6H; H-5, H-7, H-2′, H-3′, H-5′ e H-6′), 7.03–6.92 (m, 2H; H-4, H-6), 6.84–6.70 (m, 1H; H-2), 6.60 (d,
J = 15.8 Hz, 1H; H-8), 4.38 (d,
J = 5.9 Hz, 2H; H-7′).
13C-NMR (DMSO-
d6): 165.1 (C=O); 157.7 (C-3); 139.4 (C-7); 138.5 (C-1′); 136.1 (C-1); 131.4 (C-4′); 130.0 (C-5); 129.3 (C-2′, C-6′); 128.3 (C-3′, C-5′); 121.7 (C-8); 118.8 (C-6); 116.8 (C-2); 113.8 (C-4); 41.7 (C-7′) [
16]. HRMS (MALDI) calculated for C
16H
14ClNNaO
2 [M + Na]
+: 310.0611; found 310.0619.
(E)-N-(4-Chlorobenzyl)-3-(4-hydroxyphenyl)acrylamide (
7). Crystalline solid; yield: 63% (221 mg), m.p. 157–160 °C. IV ν
max (cm
−1): 3479 and 3369 (OH), 3072 (C-H sp
2), 1647 (C=O), 1589 and 1458 (aromatic C=C), 1093 (stretching C-Cl).
1H-NMR (DMSO-
d6): 9.87 (bs, 1H, OH); 8.53 (t,
J = 5.87 Hz, 1H, O=C-NH); 7.45–7.36 (m, 5H, H-2, H-6, H-7, H-2′, H-6′); 7.35–7.25 (m, 3H; H-2; H-3′e H-5′); 6.77 (d,
J = 8.5 Hz, 2H; H-3 e H-5); 6.44 (d,
J = 15.73 Hz, 1H; H-8); 4.36 (d,
J = 6.0 Hz, 2H; H-7′).
13C-NMR (DMSO-
d6) 165.6 (C=O); 159.0 (C-4); 139.4 (C-7); 138.8 (C-1′); 131.4 (C-4′); 129.4 (C-2, C-6); 129.3 (C-2′, C-6′); 128.3 (C-3′, C-5′); 125.9 (C-1); 118.3 (C-8); 115.8 (C-3, C-5); 41.6 (C-7′) [
16]. HRMS (MALDI) calculated for C
16H
14ClNNaO
2 [M + Na]
+: 310.0611; found 310.0596.
(E)-N-(4-Chlorobenzyl)-3-(4-chlorophenyl) acrylamide (
8). Crystalline solid; yield: 71% (240 mg), m.p. 157–161 °C. IV ν
max (cm
−1): 3414 (N-H), 3041 (C-H sp
2), 1651 (C=O), 1614 and 1487 (aromatic C=C), 1089 (stretching C-Cl).
1H-NMR (DMSO-
d6): 8.70 (t,
J = 6.0 Hz, 1H, O=C-NH), 7.64–7.46 (m, 4H, H-2′, H-3′, H-5′, H-6′), 7.45–7.25 (m, 5H; H-2, H-6, H-3, H-5, H-7), 6.69 (d,
J = 15.8 Hz, 1H; H-8), 4.39 (d,
J = 6.0 Hz, 2H).
13C-NMR (DMSO-
d6) 164.9 (C=O); 138.5 (C-1′); 137.9 (C-7); 134.0 (C-4); 133.8 (C-1); 131.5 (C-4′); 129.3 (C-2, C-6, C-2′ e C-6′); 129.0 (C-3, C-5); 128.3 (C-3′, C-5′);122.7 (C-8); 41.7 (C-7′) [
16]. HRMS (MALDI) calculated for C
16H
13Cl
2NNaO [M + Na]
+: 328.0272; found 328.0273.
(E)-N-(4-Chlorobenzyl)-3-(4-hydroxy-3,5-dimethoxyphenyl)-acrylamide (
9). Yellow amorphous solid; yield: 60% (193 mg), m.p.: 182–185 °C, IR ν
max (cm
−1): 3414 (OH) and 3358 or(NH), 3000 (CH sp
2), 1658 (C=O), 1624 and 1458 (aromatic C=C), 1091 (C-Cl stretch).
1H-NMR (DMSO-
d6) 8.52 (t,
J = 6.0 Hz; 1H, O=C-N
H), 7.28–7.48 (m, 5H; H-7, H-2′, H-3′, H-5′, H-6′), 6.86 (s, 2H; H-2, H-6), 6.55 (d,
J = 15.8, 1H; H-8), 4.37 (d,
J = 6 Hz, 2H; H-7′) 3.79 (s, 6H; OCH
3).
13C-NMR (DMSO-
d6) 165.6 (C=O); 148.1 (C-3, C-5); 140.0 (C-7) 138.7 (C-4); 137.4 (C-1′); 131.4; C-4′); 129.2 (C-2′, C-6′); 128.4 (C-3′, C-5′); 125.3 (C-1); 119.1 (C-8); 105.3 (C-2, C-6); 56.0 (OCH
3); 41.7 (C-7′) [
16]. HRMS (MALDI) calculated for C
18H
18ClNNaO
4 [M + Na]
+: 370.0822; found 370.0813.
(E)-N-(4-Chlorobenzyl)-3-(2-nitrophenyl)-acrylamide (
10). White crystalline solid; yield: 79% (260 mg), m.p.: 164–167 °C, IR ν
max (cm
−1): 3290 (NH), 3030 (CH sp
2), 1651 (C=O), 1624 and 1458 (aromatic C=C), 1525 and 1342 (C=O), 1091 (C-Cl stretch).
1H-NMR (DMSO-
d6): 8.82 (t,
J = 4.7 Hz, 1H, O=C-NH), 8.05 (d,
J = 8.0 Hz, 1H; H-3), 7.78–7.75 (m, 2H; H-6, H-7), 7.72–7.57 (
m, 2H; H-4, H-5), 7.43–7.27 (m, 4H, H-2′, H-3′, H-5′ e H-6′), 6.67 (d,
J = 5.6 Hz, 1H; H-8), 4.39 (d,
J = 5.9 Hz, 1H; H-7′).
13C-NMR (DMSO-
d6): 164.3 (C=O); 148.4 (C-2); 138.3 (C-1′); 134.3 (C-7); 133.9 (C-5); 131.6 (C-4′); 130.4 (C-4); 130.0 (C-1); 129.4 (C-2′, C-6′); 128.8 (C-6); 128.4 (C-3′, C-5′); 126.6 (C-3); 124.7 (C-8); 41.8 (C-7′) [
16]. HRMS (MALDI) calculated for C
16H
13ClN
2O
3 [M + H]
+: 317.0683; found 317.0683.
(E)-N-(4-Chlorobenzyl)-3-(3,4,5-trimethoxyphenyl) acrylamide (
11). Crystalline solid; yield: 86% (260 mg), m.p. 146–150 °C, IR ν
max (KBr, cm
−1): 3290 (N-H), 3070 (C-H sp
2), 1651 (C=O), 1614 and 1415 (aromatic C=C), 1029 (stretching C-Cl).
1H-NMR (DMSO-
d6): 8.60 (t,
J = 5.9 Hz, 1H; O=C-NH), 7.46–7.26 (m, 5H; H-7, H-2′, H-3′, H-5′, H-6′), 6.90 (s, 2H; H-2, H-6), 6.64 (d,
J = 15.7 Hz, 1H; H-8), 4.38 (d,
J = 5.9 Hz, 1H; H-7′), 3.80 (s, 6H;
m-OCH
3). 3.68 (s, 3H;
p-OCH
3).
13C-NMR (DMSO-
d6) 165.2 (C=O); 153.1 (C-3, C-5); 139.4 (C-7); 138.7 (C-4); 138.6 (C-1′); 131.4 (C-4′); 130.5 (C-1); 129.2 (C-2′, C-6′); 128.4 (C-3′, C-5′); 121.3 (C-8); 105.1 (C-2, C-6); 60.2 (C-4-OCH
3); 55.9 (C-3,5-OCH
3); 41.7 (C-7′) [
16]. HRMS (MALDI) calculated for C
19H
20ClNO
4 ([M + H]
+: 384.0979, found 384.0913.
N-(4-Chlorobenzyl) benzamide (
12). Crystalline solid; yield: 65% (260 mg), m.p. 136–139 °C, IR ν
max (cm
−1): 3300 (N-H), 3082 (C-H sp
2), 1637 (C=O), 1618 and 1490 (aromatic C=C), 1091 (stretching C-Cl).
1H-NMR (DMSO-
d6): 9.10 (t,
J = 5.9 Hz, 1H, O=C-NH), 7.89 (dd,
J = 8.0 e 1.6 Hz, 2H; H-2, H-6), 7.64–7.09 (m, 7H; H-3, H-4, H-5, H-2′, H-3′, H-5′, H-6′), 4.46 (d,
J = 6.0 Hz, 2H; H-7′).
13C-NMR (DMSO-
d6): 166.4 (C=O); 138.8 (C-1′); 134.2 (C-1); 131.4 (C-4); 131.3 (C-2′, C-6′); 129.1 (C-3′, C-5′); 128.3 (C-3, C-5); 127.3 (C-2, C-6); 42.1 (C-7′) [
16].
N-(4-Chlorobenzyl)-[1,1′-biphenyl]
-4-carboxamide (
13). The product was prepared according to procedure 1. White amorphous solid; yield: 56% (198 mg), m.p. 222–228 °C, IR ν
max (cm
−1): 3271 (N-H), 3078 (C-H sp
2), 1633 (C=O), 1606 and 1487 (aromatic C=C), 1089 (stretching C-Cl).
1H-NMR (DMSO-
d6): 9.15 (t,
J = 6.1 Hz, 1H, O=C-NH), 7.99 (d,
J = 8.3 Hz, 2H; H-2, H-6), 7.84–7.65 (m, 4H; H-3, H-5, H-2″, H-6″), 7.56–7.29 (m, 7H; H-2′, H-3′, H-5′, H-6′, H-3″ H-4″, H-5″), 4.48 (d,
J = 5.9 Hz, 2H).
13C-NMR (DMSO-
d6) 166.1 (C=O); 143.0 (C-4); 139.2 (C-1″); 138.8 (C-1′); 133.0 (C-1); 131.4 (C-4′); 129.2 (C-2′, C-6′); 128.4 (C-2, C-6); 128.1 (C-9, C-11); 127.0 (C-3′, C-5′); 126.7 (C-10) (C-3, C-5, C-8, C-12); 42.1 (C-7′) [
16]. HRMS (MALDI) calculated for C
20H
16ClNO
3 [M + H]
+: 322.0988; found 322.0969.
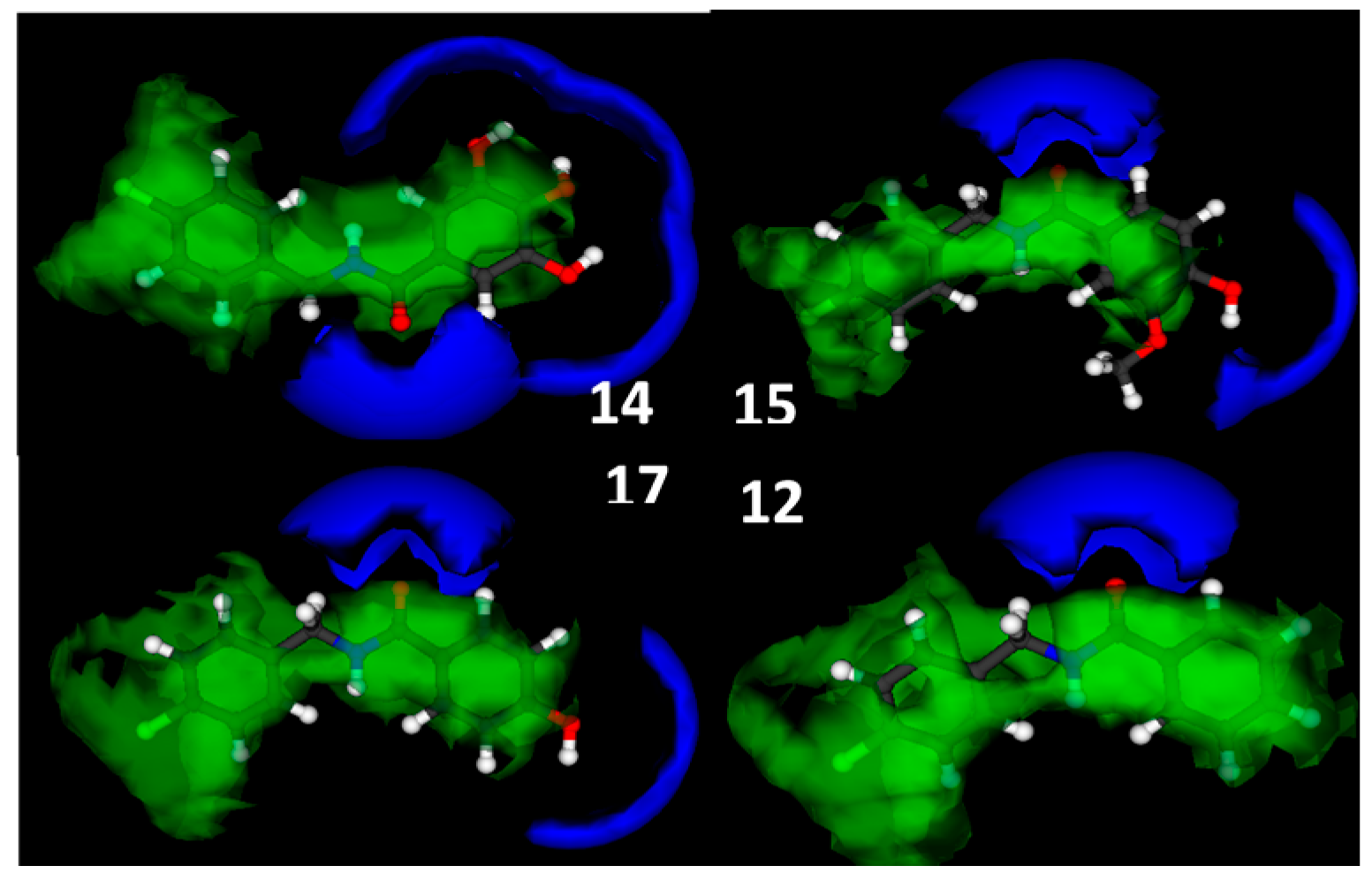
N-(4-Chlorobenzyl)-3,4,5-trihydroxybenzamide (
14). Yellow amorphous solid; yield: 21% (73 mg), m.p. 96–100 °C, IR ν
max (cm
−1): 3400 (OH), 3400 (NH), 3000 (CH sp
2), 1614 (C=O), 1589 and 1494 (aromatic C=C), 1043 (stretching C-Cl).
1H-NMR (MeOD): 8.12 (s, 1H; O=C-NH), 7.53–7.15 (m, 4H, H-2′, H-3′, H-5′, H-6), 6.85 (s, 2H; H-2, H-6), 4.58 (s, 2H, H-7′), 4.46 (s, 2H;
m-OH), 4.35 (s, 1H,
p-OH).
13C-NMR (MeOD): 170.5 (C=O), 146.7 (C-3, C-5), 139.4 (C-4), 136.1 (C-1), 134.2 (C-4), 130.5 (C-5′, C-6′), 130.1 (C-3′, C-5′), 125.9 (C-1), 107.8 (C-2, C-6), 43.6 (C-7′) [
16]. HRMS (MALDI) calculated for C
16H
12ClNNaO
3 [M + Na]
+: 316.0353; found 316.0373.
N-(4-Chlorobenzyl)-4-hydroxy-3-methoxybenzamide (
15). White amorphous solid; yield: 44% (151 mg), m.p.: 75–77 °C, IR ν
max (cm
−1): 3319 (O-H) 3251 (N-H), 3000 (C-H sp
2), 1639 (C=O), 1589 and 1487 (aromatic C=C), 1091 (stretching C-Cl).
1H-NMR (DMSO-
d6): 8.12 (d,
J = 9.2 Hz, 1H; O=C-NH), 7.56–7.22 (m, 7H; H-2, H-4, H-6, H-3′, H-5′, H-2′, H-6′), 4.52 (s, 2H), 3.88 (s, 1H; OCH
3).
13C-NMR (DMSO-
d6): 169.8 (C=O); 151.3 (C-3); 148.8 (C-4); 139.3 (C-1′); 133.8 (C-4′); 130.1 (C-2′, C-6′); 129.5 (C-5′, C-3′); 126.5 (C-1); 122.1 (C-5); 115.8 (C-6); 111.9 (C-2); 56.4 (3-OMe); 43.8 (C-7′) [
16]. HRMS (MALDI) calculated for C
16H
16ClNO
4 [M + Na]
+: 316.0530; found 316.0543.
N-(4-Chlorobenzyl)-4-hydroxy-3,5-dimethoxybenzamide (
16). White amorphous solid; yield: 50% (164 mg), m.p. 110–115 °C, IR ν
max (KBr, cm
−1): 3493 (OH), 3277 (NH), 3084 (CH sp
2), 1666 (C=O), 1597 and 1492 (aromatic C=C), 1016 (stretching C-Cl).
1H-NMR (MeOD): 8.15 (s, 1H, O=C-NH), 7.43 (s, 1H, OH), 7.35–7.27 (m, 4H; H-2′, H-3′, H-5′, H-6′), 7.25–7.15 (m, 2H; H-2, H-6), 4.54 (s, 2H, H-7′), 3.88 (
s, 6H; OCH
3).
13C-NMR (MeOD): 149.0 (C-3, C-5), 169.8 (C=O), 139.3 (C-4), 133.8 (C-1′), 130.1 (C-2′, C-6′), 129.5 (C-3′, C-5′), 125.3 (C-4′), 106.4 (C-1), 106.0 (C-2, C-6), 43.9 (C-7′), 56.8 (OCH
3) [
16]. HRMS (MALDI) calculated for C
16H
16ClNNaO
4 [M + Na]
+: 346.0636; found 346.0666.
N-(4-Chlorobenzyl)-4-hydroxybenzamide (
17). White amorphous solid; yield: 73% (246 mg), m.p. 189–192 °C, IR ν
max (cm
−1): 3450 (OH), 3122 (NH), 3055 (CH sp
2), 1631 (C=O), 1593 and 1440 (aromatic C=C), 1085 (stretching C-Cl).
1H-NMR (DMSO-
d6): 8.83 (t,
J = 5.9 Hz, 1H; O=C-NH), 7.75 (d,
J = 8.6 Hz, 2H, H-2, H-6), 7.59–7.43 (m, 4H, H-2′; H-3′, H-5′, H-6′), 6.80 (d,
J = 8.8 Hz, 2H, H-3, H-5), 4.42 (d,
J = 6.0 Hz, 2H; H-7′).
13C-NMR (DMSO-
d6): 166.0 (C=O); 160.3 (C-4); 139.2 (C-1′); 133.4 (C-4′); 130.9 (C-2, C-6); 129.3 (C-2′, C-6′); 128.3 (C-3′, C-5′); 124.9 (C-1); 114.9 (C-3, C-5); 41.7 (C-7′) [
16]. HRMS (MALDI) calculated for C
14H
12ClNNaO
2 [M + Na]
+: 286.0425; found 286.0432.
3,5-Di-tert-butyl-N-(4-chlorobenzyl)-4-hydroxybenzamide (
18). White amorphous solid; yield: 54% (203 mg), m.p. 184–186 °C, IR ν
max (cm
−1): 3450 (OH) and 3236 (NH), 3066 (CH sp
2), 1680 (C=O), 1544 and 1431 (aromatic C=C), 1012 (stretching C-Cl).
1H-NMR (DMSO-
d6): 8.89 (t,
J = 6.0 Hz, 1H; O=C-NH), 6.09–5.94 (m, 1H), 7.70–7.54 (m, 2H, H-2, H-6), 7.54–7.20 (m, 4H, H-2′, H-3′, H-5′, H-6′), 4.42 (d,
J = 5.9 Hz, 3H; H-7′), 1.39 (s, 18H; C(CH
3)
3).
13C-NMR (DMSO-
d6): 167.0 (C=O); 156.9 (C-4); 140.0 (C-1′); 138.3 (C-3, C-5); 131.2 (C-4′); 129.2 (C-2′, C-6′); 128.3 (C-5′, C-3′); 125.3 (C-1); 124.2 (C-2, C-6); 42.0 (C-7′); 34.7 (3,5-(
C(CH
3)
3; 30.3 (3,5-(C(
CH
3)
3) [
16] HRMS (MALDI) calculated for C
22H
28ClNO
3 [M + H]
+: 374.1877; found 374.1877.
N-(4-Chlorobenzil)-2-hydroxy-5-methoxybenzamide (
19). Crystalline solid; yield: 60% (180 mg), m.p. 137–140 °C, IR ν
max (cm
−1): 3360 (O-H and N-H), 3076 (C-H sp
2), 1651 (C=O), 1598 and 1435 (aromatic C=C), 1045 (stretching C-Cl).
1H-NMR (DMSO-
d6): 9.35 (t,
J = 5.8 Hz, 1H, O=C-NH), 7.48–7.26 (m, 5H, H-6, H-2′, H-3′, H-5′, H-6′), 7.04 (dd,
J = 9.0, 3.0 Hz, 1H; H-3), 6.85 (d,
J = 9.0 Hz, 1H; H-4), 4.49 (d,
J = 5.9 Hz, 2H, H-7′), 3.72 (s, 3H; OCH
3).
13C-NMR (50 MHz): 168.7 (C=O); 154.0 (C-5); 151.7 (C-2); 115.2 (C-1); 138.2 (C-1′); 131.6 (C-4′); 129.3 (C-2′, C-6′); 128.5 (C-5′, C-3′); 121.2 (C-3); 118.4 (C-4); 111.3 (C-6); 55.8 (OCH
3); 41.9 (C-7′) [
16]. HRMS (MALDI) calculated for C
15H
14ClNO
3 [M + H]
+: 327.0512; found 327.0516.
N-(4-Chlorobenzyl)-3-methyl-4-nitrobenzamide (
20). Crystalline solid; yield: 41% (143 mg), m.p. 148–151 °C, IR ν
max (cm
−1): 3278 (NH), 3080 (CH sp
2), 1637 (C=O), 1587 and 1423 (aromatic C=C), 1521 and 1355 (NO
2 arom), 1089 (stretch C-Cl).
1H-NMR (DMSO-
d6): 9.31 (t,
J = 5.8 Hz, 1H, O=C-NH), 8.06 (dd,
J = 8.4, 1H; H-5), 7.96 (s, 1H, H-2), 7.89 (dd,
J = 8.4, 1.7 Hz, 1H; H-6), 7.42–7.31 (m, 4H, H-2′, H-3′, H-5′, H-6′), 4.47 (dd,
J = 5.9, 1.7 Hz, 2H, H-7′), 2.56–2.51 (m, 3H; CH
3).
13C-NMR (DMSO-
d6): 164.8 (C=O); 150.5 (C-4); 138.3 (C-1); 138.1 (C-1′); 132.9 (C-4′); 131.8 (C-2′, C-6′); 131.5 (C-3); 129.3 (C-3′, C-5′); 128.4 (C-2); 126.2 (C-5); 124.6 (C-6); 42.3 (C-7′); 19.5 (
CH
3) [
16]. HRMS (MALDI) calculated for C
15H
13ClN
2NaO
3 [M + Na]
+: 327.0512; found 327.0516.
N-(4-Fluorobenzyl)-4-hydroxy-3-methoxybenzamide (
21). 4-Fluorbenzylamine was used as the reagent. White amorphous solid; yield: 21% (90 mg), m.p. 161–165 °C, IR ν
max (cm
−1): 3304 (O-H), 3078 (N-H), 1631 (C=O), 1593 and 1423 (aromatic C=C), 1116 (C-F stretch).
1H-NMR (CDCl
3): 9.60 (s, 1H; OH), 8.83 (t,
J = 5.8 Hz, 1H; NH), 7.52–7.28 (m, 4H; H-2, H-6, H-2′, H-6′), 7.22–7.05 (m, 2H; H-3′, H-5′), 6.81 (d,
J = 8.2 Hz, 1H; H-5), 4.43 (d,
J = 5.8 Hz, 2H; H-7′), 3.80 (s, 3H; OMe).
13C-NMR (CDCl
3): 166.0 (C=O), 161.1 (d,
J = 242.0 Hz; C-4′), 149.6 (C-4), 147.2 (C-3), 136.2 (C-1′), 129.2 (C-2′, C-6′), 125.3 (C-1), 120.9 (C-6), 114.9 (C-3′, C-5′), 115.2 (C-5), 111.3 (C-2), 55.7 (OMe), 42.0 (C-7′) [
16]. HRMS (MALDI) calculated for C
15H
14FNNaO
3 [M + Na]
+: 298.0855; found 298.0882.
N-(4-Bromobenzyl)-4-hydroxy-3-methoxybenzamide (
22). 4-Bromobenzylamine was used as reactant. Red amorphous solid; yield: 63% (252 mg), m.p. 119–122 °C. IV ν
max (cm
−1): 3304 (OH), 3078 (NH), 3003 (CH sp
2), 1631 (C=O), 1593 and 1423 (aromatic C=C), 1072 (stretching C-Br).
1H-NMR (DMSO-
d6): 7.43 (d,
J = 8.4 Hz, 3H; H-2, H-3′, H-5′), 7.29–7.12 (m, 3H; H-6, H-2′, H-6), 6.88 (d,
J = 8.2 Hz, 1H; H-5), 6.65 (t,
J = 5.2 Hz, 1H; NH), 6.21 (s, 1H; OH), 4.54 (d,
J = 5.8 Hz, 2H, H-7′), 3.88 (s, 3H; OMe).
13C-NMR (DMSO-
d6): 167.1 (C=O), 148.9 (C-4), 146.7 (C-3), 137.4 (C-1′), 131.7 (C-3′, C-5′), 129.4 (C-2′, C-6′), 126.1 (C-1), 119.8 (C-6, C-4′), 113.9 (C-5), 110.4 (C-2), 56.0 (OMe), 43.4 (C-7′) [
16]. HRMS (MALDI) calculated for C
15H
14BrNO
3 [M]
+: 335.0157; found 335.0156.
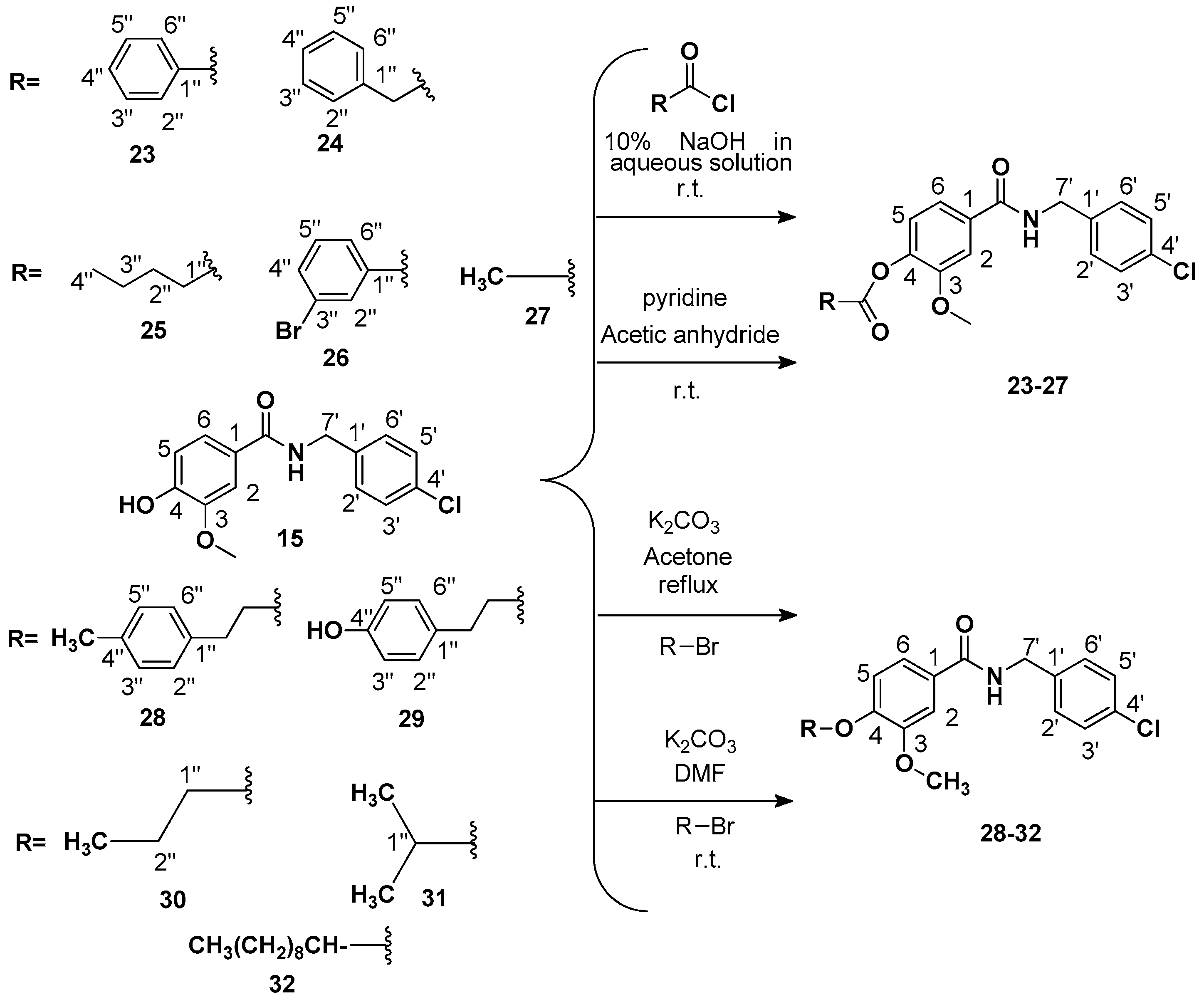
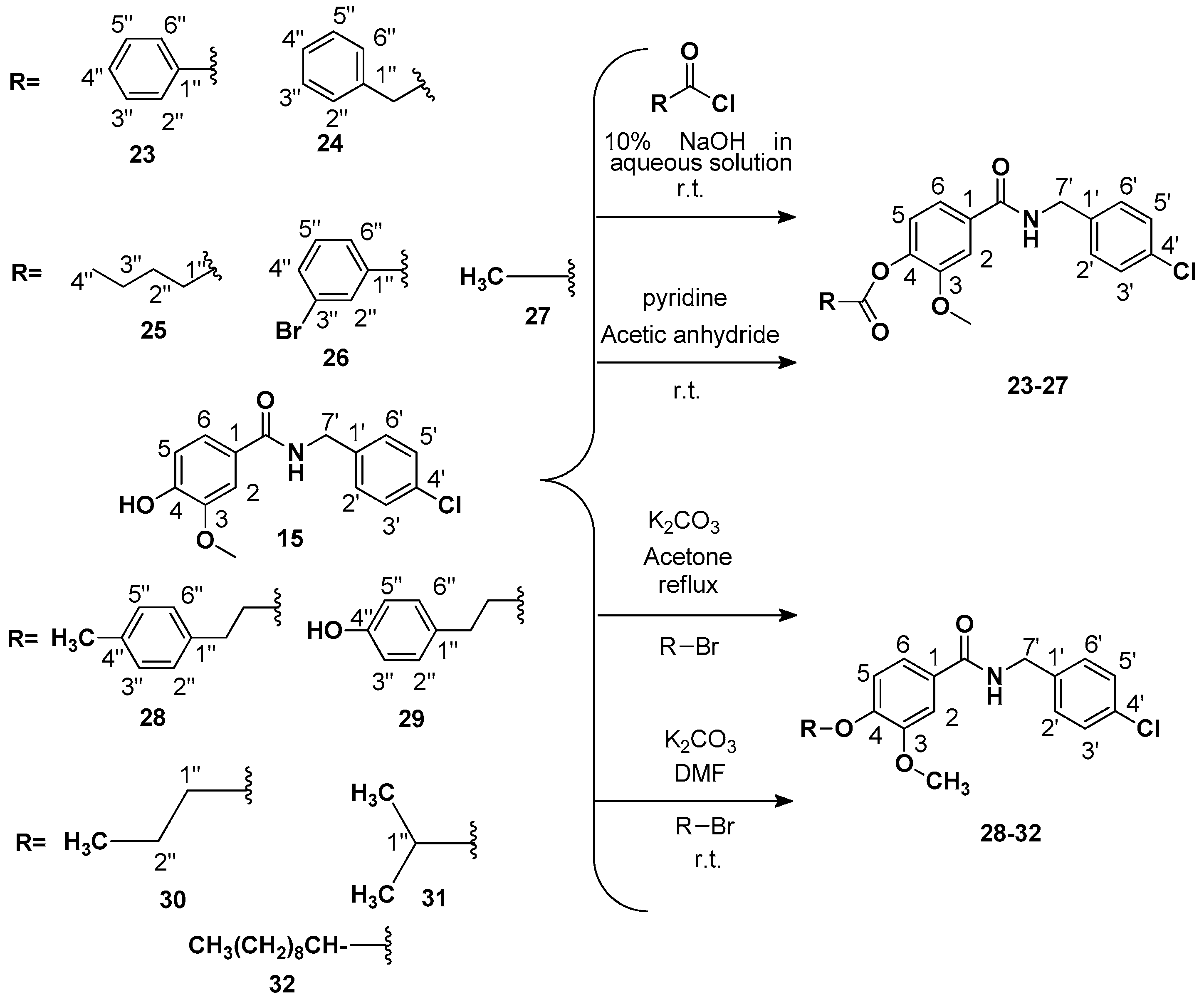
Procedure 2: Preparation of 4-((4-chlorobenzyl) carbamoyl)-2-methoxyphenyl benzoate (
23): To a 100 mL flask equipped with magnetic stirring, was added a sodium hydroxide solution 10% (0.51 mL, 0.1275 mmol NaOH) 0.100 g of the amide
15 (0.3400 mmol). Then, benzoyl chloride (0.04 mL, 0.343 mmol) was added in drops. The reaction was subjected to constant agitation for a period of one hour at room temperature. The reaction mixture was then poured into a separation funnel and extraction was completed with dichloromethane (3 × 15 mL), organic phase treated with sodium carbonate (Na
2CO
3) 5% (2 × 5 mL), and dried with anhydrous sodium sulfate. After filtration, the solution was concentrated under reduced pressure [
18]. The product was purified by silica gel column chromatography using a mixture EtOAc:Hex (65:35) as mobile phase to give a crystalline solid compound, yield: 76% (103 mg), m.p. 122–125 °C, IR ν
max (cm
−1): 3334 (N-H), 3000 (C-H sp
2), 1734 and 1637 (C=O), 1607 and 1425 (aromatic C=C), 1089 (stretching C-Cl).
1H-NMR (CDCl
3): 9.17 (t,
J = 5.9 Hz, 1H, NH), 8.13 (d,
J = 7.1 Hz, 2H, H-2″, H-6″), 7.84–7.20 (m, 10H; H-2, H-5, H-6, H-2, H-3′, H-5′, H-6′, H-3″, H-4″, H-5″), 4.50 (d,
J = 5.8 Hz, 2H; H-7′). 3.81 (s,
J = 6.0 Hz, 3H; OMe).
13C-NMR (CDCl
3): 165.9 (O=C-O), 164.2 (O=C-N), 151.2 (C-3), 142.1 (C-4), 139.1 (C-1′), 134.6 (C-2′), 133.6 (C-1), 134.6 (C-6′), 131.1 (C-4′), 130.3 (C-2″, C-4″, C-6″), 129.6 (C-3″, C-5″), 128.8 (C-1″), 128.7 (C-3′, C-5′), 123.4 (C-5), 120.4 (C-6), 112.2 (C-2), 42.5 (C-7′), 56.4 (OMe) [
19]. HRMS (MALDI) calculated for C
22H
18ClNNaO
4 [M + Na]
+: 418.0822; found 418.0809. The following compounds were similarly prepared:
4-((4-Chlorobenzyl)carbamoyl)-2-methoxyphenyl 2-phenylacetate (
24). Phenylacetyl chloride (0.05 mL, 0.343 mmol) was used. The reaction was subjected to constant agitation for a period of two hours at room temperature and finally extracted. Crystalline solid; yield: 78% (110 mg), m.p.: 141–145 °C, IR ν
max (cm
−1): 3331 (N-H), 3050 (C-H sp
2), 1761 and 1633 (C=O), 1602 and 1456 (aromatic C=C), 1033 (stretching C-Cl).
1H-NMR (CDCl
3): 9.12 (t,
J = 5.9 Hz, 1H; NH), 7.61 (d,
J= 1.7 Hz, 1H; H-2), 7.52 (dd,
J = 8.2, 1.8 Hz, 1H; H-6), 7.43–7.26 (m, 10H; H-2′, H-3′, H-5′, H-6′, H-2″, H-3″, H-4″, H-5″, H-6″), 7.20 (d,
J = 8.2 Hz, 1H; H-5), 4.47 (d,
J = 5.9 Hz, 2H; H-7′), 3.98 (s, 2H, CH
2-C
6H
5), 3.80 (s, 3H; OMe).
13C-NMR (CDCl
3): 169.6 (O=C-O), 165.7 (O=C-N), 150.9 (C-3), 142.0 (C-4), 138.9 (C-1′), 134.1 (C-1″), 133.3 (C-4′), 131.6 (C-1), 129.8 (C-2′, C-6′), 128.7 (C-2″, C-6″), 128.6 (C-3″, C-5″), 127.3 (C-4″), 127.3 (C-3′, C-5′), 122.9 (C-5), 120.2 (C-6), 112.0 (C-2), 56.3 (OCH
3), 42.4 (C-7′), 40.09 (
CH
2-C
6H
5) [
20]. HRMS (MALDI) calculated for C
23H
20ClNNaO
4 [M + Na]
+: 432.0979; found 432.0914.
4-((4-Chlorobenzyl)carbamoyl)-2-methoxyphenyl valerate (
25). Valeryl chloride (0.05 mL, 0.343 mmol) was used. The reaction was subjected to constant agitation for a period of five hours at room temperature and finally extracted. White amorphous solid; yield: 43% (55 mg), m.p. 97–100 °C, IR ν
max (cm
−1): 3253 (N-H), 3088 (C-H sp
2), 1761 and 1631 (C=O), 1602 and 1450 (aromatic C=C), 1031 (stretching C-Cl).
1H-NMR (CDCl
3): 7.44 (s, 1H; H-2); 7.31–7.15 (m, 5H; H-6, H-2′, H-3′, H-5′, H-6′), 6.96 (d,
J = 8.2 Hz, 1H; H-5), 6.79 (t,
J = 5.5 Hz, 1H; NH), 4.49 (d,
J = 5.8 Hz, 2H; H-7′), 3.78 (s, 3H; OCH
3), 2.56 (t,
J = 7.4 Hz, 2H; H-1″), 1.71 (quint,
J = 7.7 Hz, 2H; H-2″), 1.42 (sex,
J = 7.3 Hz, 2H; H-3″), 0.94 (t,
J = 7.2 Hz, 3H; Me).
13C-NMR (CDCl
3): 171.7 (O=C-O), 166.6 (O=C-N), 151.3 (C-3), 142.4 (C-4), 136.7 (C-1′), 133.2 (C-1), 132.8 (C-4′), 129.1 (C-2′, C-6′), 128.7 (C-3′, C-5′), 122.6 (C-5), 118.7 (C-6), 111.8 (C-2), 55.9 (OMe), 43.3 (C-7′), 33.7 (C-1″), 26.9 (C-2″), 22.1 (C-3″), 13.7 (Me) [
20]. HRMS (MALDI) calculated for C
20H
22ClNNaO
4 [M + Na]
+: 398.1135; found 398.1118.
4-((4-Chlorobenzyl) carbamoyl)-2-methoxyphenyl 3-bromobenzoate (
26). 3-Bromobenzoyl chloride (0.05 mL, 0.343 mmol) was employed. The reaction was subjected to constant agitation for a period of one hour at room temperature and finally extracted. White amorphous solid; yield: 86% (140 mg), m.p. 143–145 °C. IV νmax (cm
−1): 3334 (N-H), 3050 (C-H sp
2), 1734 and 1637 (C=O), 1602 and 1490 (aromatic C=C), 1089 (stretching C-Cl).
1H-NMR (CDCl
3): 8.30 (t,
J = 1.7 Hz, 1H; H-2″), 8.14–8.03 (m, 1H; H-4″), 7.81–7.71 (m, 1H; H-6″), 7.51 (d,
J = 1.8 Hz, 1H; H-2), 7.44–7.15 (m, 6H; H-6, H-2′, H-3′, H-5′, H-60′, H-5″), 7.09 (d,
J = 8.2 Hz, 1H; H-5), 6.81 (t,
J = 5.7 Hz, 1H; NH), 4.53 (d,
J = 5.8 Hz, 2H; H-7′), 3.80 (s, 3H; OMe).
13C-NMR (CDCl
3): 166.6 (O=C-O), 163.2 (O=C-N), 151.4 (C-3), 142.2 (C-4), 136.7 (C-4″), 136.6 (C-1′), 133.3 (C-1), 133.2 (C-1″), 133.2 (C-2″), 130.7 (C-4′), 130.1 (C-5″), 129.1 (C-2′, C-6′), 128.8 (C-3′, C-5′, C-6′′), 122.7 (C-3″), 122.6 (C-5), 118.7 (C-6), 112.0 (C-2), 43.4 (C-7′), 56.0 (OCH
3) [
21]. HRMS (MALDI) calculated for C
20H
22ClNO
4 [M]
+: 495.9927; found 495.9912.
Procedure 3: Preparation of 4-((4-chlorobenzyl) carbamoyl)-2-methoxyphenyl acetate (
27). For the acetylation of
15, to a 50 mL flask, equipped with magnetic stirrer was added the chlorinated vanillic amide
15 (0.1000 g, 0.034 mmol), pyridine (0.13 mL, 0.15924 mmol) and acetic anhydride (0.08 mL, 0.8111 mmol). The reaction mixture was subjected to constant magnetic stirring for 24 h. The first step of the reaction product extraction was then carried out by pouring into ice water (30 mL) in a separation funnel using ethyl acetate as extractor solvent (3 × 10 mL). The organic phase was treated with saturated copper sulfate solution (3 × 20 mL). The ethyl acetate phase was washed with water (3 × 30 mL) and dried with anhydrous sodium sulfate (Na
2SO
4). Subsequently, the organic phase was filtered and concentrated by rotary evaporation [
22]. The product was purified by silica gel column chromatography using EtOAc:Hex (65:35) as the mobile phase system to give a crystalline solid; yield: 98% (159 mg), m.p. 117–119 °C, IR ν
max (cm
−1): 3446 (N-H), 3088 (C-H sp
2), 1770 and 1635 (C=O), 1602 and 1421 (aromatic C=C), 1089 (stretching C-Cl).
1H-NMR (CDCl
3): 7.43 (d,
J = 1.9 Hz, 1H; H-2), 7.33–7.12 (m, 4H; H-2, H-3′, H-5′, H-6′), 7.03–6.86 (m, 1H; H-6), 6.88–6.69 (m, 1H; H-5) ,4.47 (d,
J = 5.8 Hz, 2H, H-7′), 3.77 (s, 3H; Me), 2.27 (s, 3H; Me).
13C-NMR (CDCl
3): 168.8 (O=C-O), 166.6 (O=C-N), 151.3 (C-3), 142.3 (C-4), 136.6 (C-1′), 133.2 (C-1), 132.9 (C-4′), 129.0 (C-2′, C-6′), 128.7 (C-3′, C-5′), 122.6 (C-5), 118.7 (C-6), 111.8 (C-2), 55.9 (OMe), 43.3 (C-7′), 20.6 (Me) [
19]. HRMS (MALDI) calculated for C
17H
16ClNNaO
4 [M + Na]
+: 356.0666; found 356.0664.
Procedure 4: Preparation of N-(4-chlorobenzyl)-3-methoxy-4-(4-methylphenetoxy)benzamide (
28). In a 100 mL flask equipped with magnetic stirring vanillic amide
15 (0.1000 g, 0.3400 mmol) was added to a solution of acetone (4 mL) together with K
2CO
3 (0.1388 g, 1.0046 mmol) and 4-methylbenzyl bromide (0.08 mL, 0.6011 mmol) under reflux (60 °C) for 16 h. After the reaction, the solvent was removed under reduced pressure. A solution of CH
2Cl
2:H
2O was poured onto the product, placed in a separation funnel and extracted with three portions of CH
2Cl
2 (10 mL each). The organic phase was washed 1 N NaOH (3 × 10 mL) and dried with Na
2SO
4, filtered and concentrated by rotary evaporation. The product was purified by silica gel column chromatography using EtOAc:Hex (65:35) as the mobile phase system to give a white amorphous solid; yield: 30% (42 mg), m.p. 126–129 °C, IR ν
max (cm
−1): 3290 (N-H), 3001 (C-H sp
2), 1629 (C=O), 1600 and 1490 (aromatic C=C), 1029 (stretching C-Cl).
1H-NMR (CDCl
3): 7.44 (s, 1H; H-2), 7.32–7.07 (m, 9H; H-6, H-2′, H-3′, H-5′, H-6′, H-2″, H-3″, H-5″, H-6″), 6.80 (d,
J = 8.4 Hz, 1H; H-5), 6.49 (t,
J = 5.2 Hz, 1H, NH), 4.56 (d,
J = 5.8 Hz, 2H; H-7′), 4.18 (t,
J = 7.6 Hz, 2H; CH
2-O), 3.88 (s, 3H; OCH
3), 3.11 (t,
J = 7.6 Hz, 2H; C
H2-C
6H
5).
13C-NMR (CDCl
3): 167.0 (O=C-N), 151.2 (C-3), 149.3 (C-4), 136.9 (C-1′), 136.2 (C-4″), 134.3 (C-1″), 133.3 (C-4′), 129.2 (C-2′, C-6′), 129.1 (C-3″, C-5″), 128.9 (C-3′, C-5′), 128.8 (C-2″, C-6″), 126.7 (C-1), 119.3 (C-6), 111.6 (C-2), 111.1 (C-5), 69.9 (CH
2-O), 56.1 (OMe), 43.3 (C-7′), 35.1 (CH
2-C
6H
5), 21.0 (Me) [
23]. HRMS (MALDI) calculated for C
24H
24ClNNaO
3 [M + Na]
+: 432.1342; found 432.1328.
Procedure 5: Preparation of N-(4-chlorobenzyl)-4-(4-hydroxyphenoxy)-3-methoxybenzamide (
29). In a 100 mL flask equipped with magnetic stirring, vanillic amide
15 (0.1000 g, 0.3400 mmol) was added to a solution of DMF (3.43 mL), K
2CO
3 (0.0711 g, 0.5142 mmol), and 4-hydroxyphenyl bromide (0.0827 g, 0.4113 mmol), and left stirring for 24 h at room temperature. After the reaction, the product was extracted in a separation funnel with CH
2Cl
2 (3 × 10 mL). The extraction solution was washed with distilled water (3 × 10 mL), and then with 10% NaOH (10 mL). The solution was treated with Na
2SO
4, filtered, and concentrated by rotary evaporation [
24]. The product was purified by silica gel column chromatography using EtOAc:Hex (65:35) as the mobile phase system to afford a colorless oil; yield: 75% (106 mg), IR ν
max (cm
−1): 3350 (OH) 3016 (CH sp
2), 1645 (C=O), 1600 and 1489 (aromatic C=C), 1014 (stretching C-Cl).
1H-NMR (CDCl
3): 7.95 (s, 1H; NH), 7.44 (s, 1H; H-2), 7.30–6.94 (m, 8H; H-5, H-6, H-2′, H-3′, H-5′, H-6′, H-2″, H-6″), 6.79 (dd,
J = 8.5, 2.6 Hz, 2H; H-3″, H-5″), 4.54 (d,
J = 5.8 Hz, 2H; H-7′), 4.14 (t,
J = 7.6 Hz, 2H; CH
2-O), 3.82 (s, 3H; OMe), 3.04 (t,
J = 7.5 Hz, 2H; C
H2-C
6H
5).
13C-NMR (CDCl
3): 167.5 (O=C-N), 155.0 (C-4″), 151.3 (C-4), 149.2 (C-3), 136.7 (C-1′), 133.3 (C-4′), 130.0 (C-2′, C-6′), 129.1 (C-2″, C-6″), 128.8 (C-3′, C-5′), 128.9 (C-1″), 126.4 (C-1), 119.5 (C-6), 115.6 (C-3″, C-5″), 111.6 (C-2), 111.2 (C-5), 70.1 (CH
2-O), 56.1 (OCH
3), 43.4 (C-7′), 34.8 (
CH
2-C
6H
5) [
23]. HRMS (MALDI) calculated for C
23H
22ClNNaO
4 [M + Na]
+: 434.1135; found 434.1123.
Preparation of N-(4-chlorobenzyl)-3-methoxy-4-propoxybenzamide (
30). This product was prepared according to procedure 4, using 1-propyl bromide (0.05 mL, 0.4102 mmol) under reflux (60 °C), for 16 h and finally extracted to give after the silica gel column chromatography a white amorphous solid; yield: 54% (62 mg), m.p. 124–126 °C. ν
max IR (cm
−1): 3296 (N-H), 1631 (C=O), 1581 to 1450 (aromatic C=C), 1014 (C-Cl stretch).
1H-NMR (CDCl
3): 7.40 (d,
J = 1.8 Hz, 1H; H-2), 7.31–7.22 (m, 5H; H-6, H-2′, H-3′, H-5′, H-6′), 6.81 (d,
J = 8.4 Hz, 1H; H-5), 6.74 (bs, 1H; NH), 4.55 (d,
J = 5.8 Hz, 2H; H-7′), 3.98 (t,
J = 6.8 Hz, 2H; H-1″), 3.86 (s, 3H; OMe), 2.04–1.76 (m, 2H; H-2″), 1.03 (t,
J = 7.4 Hz, 3H; H-3″).
13C-NMR (CDCl
3): 167.0 (O=
C-N), 151.4 (C-4), 149.2 (C-3), 137.0 (C-1′), 133.1 (C-4′), 129.1 (C-2′, C-6′), 128.7 (C-3′, C-5′), 126.3 (C-1), 119.4 (C-6), 110.9 (C-2), 111.4 (C-5), 70.4 (C-1″), 56.0 (OCH
3), 43.3 (C-7′), 22.3 (C-2″), 10.3 (C-3″) [
25]. HRMS (MALDI) calculated for C
18H
20ClNNaO
3 [M + Na]
+: 356.1029; found 356.1038.
Preparation of N-(4-chlorobenzyl)-4-isopropoxy-3-methoxybenzamide (
31). This product was prepared according to procedure 5, using 2-propyl bromide (0.05 mL, 0.4102 mmol), at room temperature and left stirring for 24 h and finally extracted to give, after silica gel column chromatography purification using EtOAc:Hex as the gradient mobile phase system, increasing in polarity and an isocratic EtOAc:Hex (65:35) system after elution of the purified product a white amorphous solid; yield: 54% (62 mg), m.p. 142–147 °C. IV ν
max (cm
−1): 3284 (N-H), 3078 (C-H sp
2), 1631 (C=O), 1598 and 1455 (aromatic C=C), 1031 (stretching C-Cl).
1H-NMR (CDCl
3): 7.40 (d,
J = 1.8 Hz, 1H; H-6), 7.26–7.18 (m, 5H; H-2, H-2′, H-3′, H-5′, H-6′), 6.79 (d,
J = 8.4 Hz, 2H; H-5), 6.79 (bs,
J = 8.4 Hz, 2H; NH), 4.51 (sept,
J = 6.2 Hz, 1H; H-2″), 4.51 (d,
J = 5.7 Hz, 2H; H-7′), 3.81 (s, 3H; OMe), 1.33 (d,
J = 6.0 Hz, 6H; H-1″; H-3″).
13C-NMR (CDCl
3): 167.1 (O=C-N), 150.3 (C-4), 150.0 (C-3), 137.0 (C-1′), 133.2 (C-4′), 129.1 (C-2′, C-6′), 128.7 (C-3′, C-5′), 126.5 (C-1), 119.4 (C-6), 113.6 (C-5), 111.2 (C-2), 71.2 (C-1″), 56.0 (OCH
3), 43.3 (C-7′), 21.9 (Me) [
25]. HRMS (MALDI) calculated for C
18H
20ClNNaO
3 [M + Na]
+: 356.1059; found 356.1027.
Preparation of N-(4-chlorobenzyl)-4-(decyloxy)-3-methoxybenzamide (
32). The product was prepared according to procedure 4, using 1-decyl bromide (0.05 mL, 0.4102 mmol) under reflux (60 °C) for 16 h and finally extracted to give after silica gel column chromatography a white amorphous solid; yield: 81% (120 mg), m.p. 110–105 °C, IR ν
max (cm
−1): 3305 (N-H), 3000 (C-H sp
2), 1627 (C=O), 1602 and 1508 (aromatic C=C), 1035 (stretching C-Cl).
1H-NMR (CDCl
3): 7.42 (s, 1H; H-6), 7.26 (s, 5H; H-2, H-2′, H-3′, H-5′, H-6′), 6.54 (s, 1H; H-5), 4.56 (d,
J = 5.8 Hz, 2H; H-7′), 4.01 (t,
J = 6.8 Hz, 2H; CH
2-C
6H
5), 3.86 (s, 3H; OMe), 1.83 (dt,
J = 14.1, 6.9 Hz, 2H; CH
2-O), 1.74–1.15 (m, 14H; (CH
2)
7), 0.86 (t,
J = 6.1 Hz, 3H; CH
3).
13C-NMR (CDCl
3): 167.2 (O=C-N), 151.7 (C-4), 149.4 (C-3), 137.1 (C-1′), 133.4 (C-4′), 129.3 (C-2′, C-6′), 128.9 (C-3′, C-5′), 126.5 (C-1), 119.5 (C-6), 111.6 (C-2), 111.1 (C-5), 69.2 (CH
2O), 56.2 (OMe), 43.5 (C-7′), 32.0 (
CH
2CH
2O), 29.7–22.8 ((
CH
2)
7CH
2O), 14.3 (
CH
3). [
26]. HRMS (MALDI) calculated for C
18H
20ClNaNO
3 [M + Na]
+: 454.2125; found 454.2119.


 and
and 


{kind=link}
{kind=link}
{kind=link}
{kind=link}



