
An Expeditious and Greener Synthesis of 2-Aminoimidazoles in Deep Eutectic Solvents

 , , and
, , and
Abstract
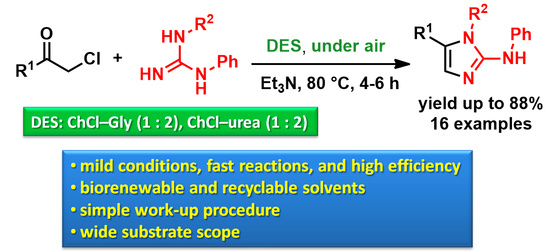
:
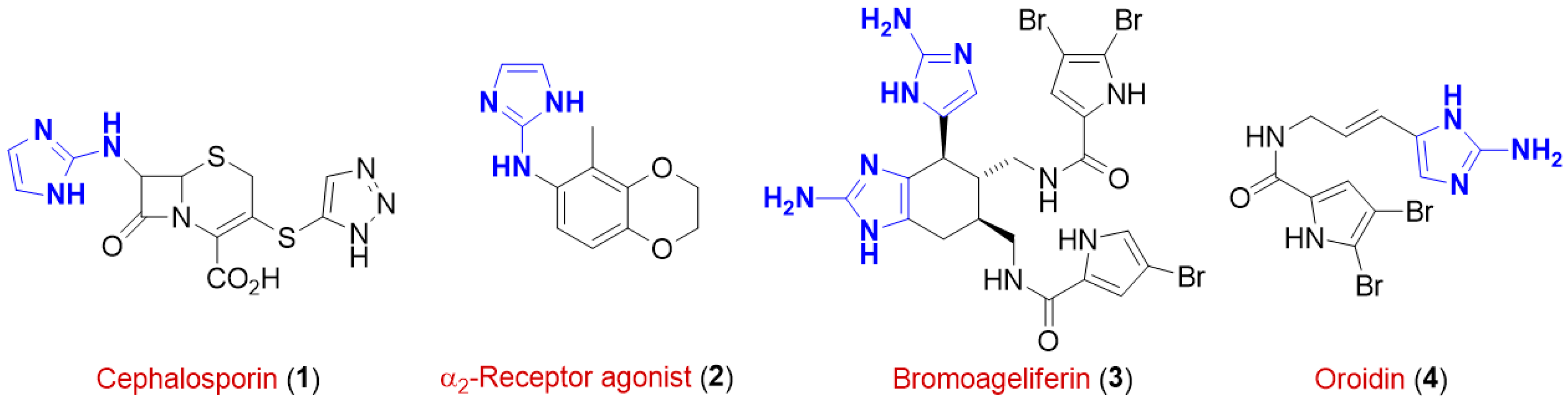
1. Introduction
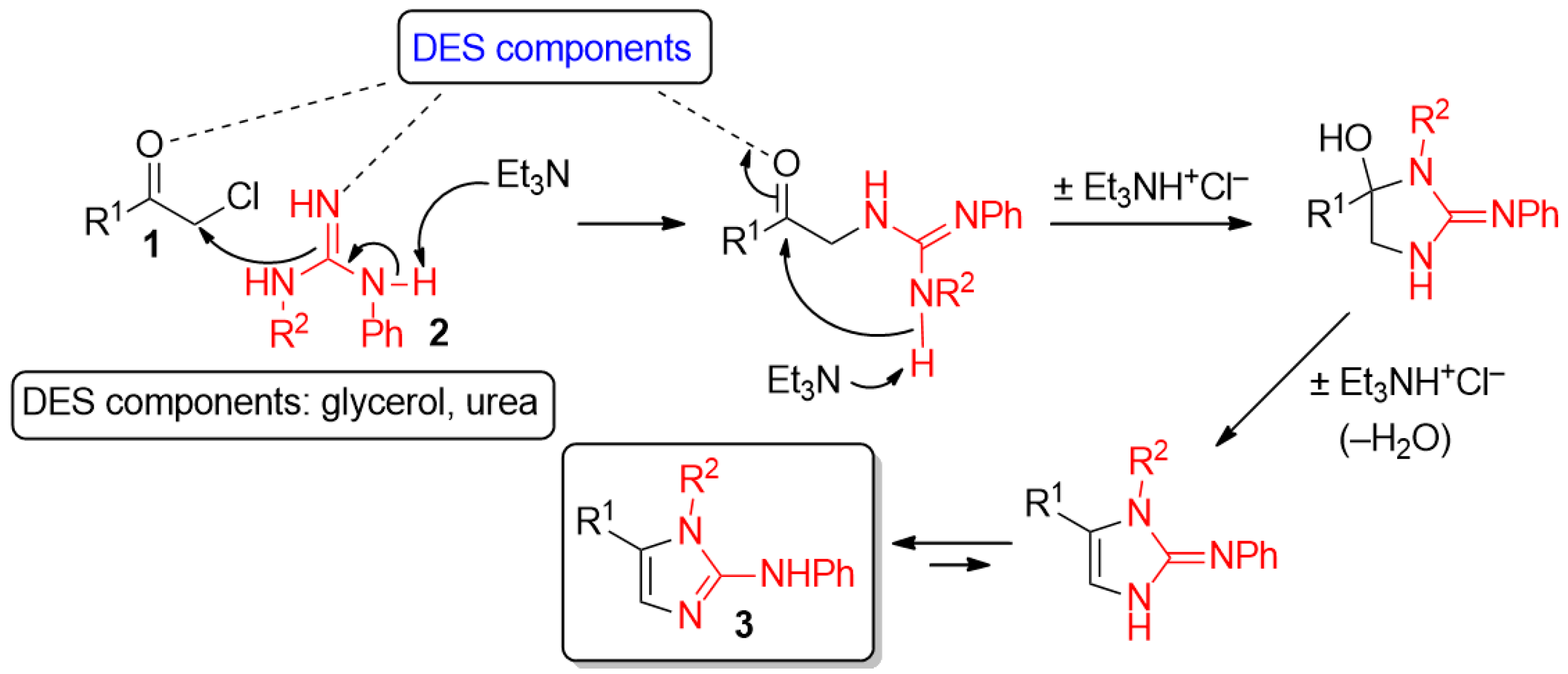
2. Results and Discussion
3. Materials and Methods
3.1. General Informations
3.2. Synthesis of 2-Aminoimidazoles 3a–g in the DES ChCl–Gly (1:2) (Table 1)
3.3. Synthesis of 2-Aminoimidazoles 3h,i in the DES ChCl–Gly (1:2) (Table 1)
3.4. Synthesis of 2-Aminoimidazoles 3a,b,d–g in the DES ChCl–Urea (1:2) (Table 2)
3.5. Synthesis of 2-Aminoimidazoles 3h in the DES ChCl–Urea (1:2) (Table 2)
3.6. Characterization Data of Compounds 3a–i (Table 1)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Ashcroft, C.P.; Dunn, P.J.; Hayler, J.D.; Wells, A.S. Survey of Solvent Usage in Papers Published in Organic Process Research & Development 1997–2012. Org. Process Res. Dev. 2015, 19, 740–747. [Google Scholar]
- Dunn, P.J.; Wells, A.S.; Williams, M.T. Future Trends for Green Chemistry in the Pharmaceutical Industry. In Green Chemistry in the Pharmaceutical Industry; Dunn, P.J., Wells, A.S., Williams, M.T., Eds.; Wiley-VCH: Weinheim, Germany, 2010; pp. 333–355. [Google Scholar]
- Jung, F.; Olivier, A.; Boucherot, D. A new approach to the synthesis of amino imidazoles application to the cephalosporin series. Tetrahedron Lett. 1989, 30, 2379–2382. [Google Scholar] [CrossRef]
- Munk, S.A.; Harcourt, D.A.; Arasasingham, P.N.; Burke, J.A.; Kharlamb, A.B.; Manlapaz, C.A.; Padillo, E.U.; Roberts, D.; Runde, E.; Williams, L.; et al. Synthesis and Evaluation of 2-(Arylamino)imidazoles as α2-Adrenergic Agonists. J. Med. Chem. 1997, 40, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Kitamura, H.; Yamaguchi, K.; Fukuzawa, S.; Kamijima, C.; Yazawa, K.; Kuramoto, M.; Wang, G.Y.S.; Fujitani, Y.; Uemura, D. Development of chemical substances regulating biofilm formation. Bull. Chem. Soc. Jpn. 1997, 70, 3061–3069. [Google Scholar] [CrossRef]
- Assmann, M.; Lichte, E.; Pawlik, J.R.; Kock, M. Chemical defenses of the Caribbean sponges Agelas wiedenmayeri and Agelas conifera. Mar. Ecol. Prog. Ser. 2000, 207, 255–262. [Google Scholar] [CrossRef]
- Tamm, M.; Randoll, S.; Bannenberg, T.; Herdtweck, E. Titanium complexes with imidazolin-2-iminato ligand. Chem. Commun. 2004, 876–877. [Google Scholar] [CrossRef] [PubMed]
- Tamm, M.; Randoll, S.; Herdtweck, E.; Kleigrewe, N.; Kehr, G.; Erker, G.; Rieger, B. Imidazolin-2-iminato titanium complexes: Synthesis, structure and use in ethylene polymerization catalysis. Dalton Trans. 2006, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Selig, P. Guanidine Organocatalysis. Synthesis 2013, 45, 703–718. [Google Scholar] [CrossRef]
- Sullivan, J.D.; Giles, R.L.; Looper, R.E. 2-Aminoimidazoles from Leucetta Sponges: Synthesis and Biology of an Important Pharmacophore. Curr. Bioact. Compd. 2009, 5, 39–78. [Google Scholar] [CrossRef]
- Žula, A.; Kikelj, D.; Ilaš, J. 2-Aminoimidazoles in medicinal chemistry. Mini Rev. Med. Chem. 2013, 13, 1921–1943. [Google Scholar] [CrossRef] [PubMed]
- Little, T.L.; Webber, S.E. A Simple and Practical Synthesis of 2-Aminoimidazoles. J. Org. Chem. 1994, 59, 7299–7305. [Google Scholar] [CrossRef]
- Lemrová, B.; Soural, M. Synthetic Strategies for Preparing Bicyclic Guanidines. Eur. J. Org. Chem. 2015, 1869–1886. [Google Scholar] [CrossRef]
- Gibbons, J.B.; Gligorich, K.M.; Welm, B.E.; Looper, R.E. Synthesis of the Reported Structures for Kealiinines B and C. Org. Lett. 2012, 14, 4734–4737. [Google Scholar] [CrossRef] [PubMed]
- Giles, R.L.; Sullivan, J.D.; Steiner, A.M.; Looper, R.E. Addition-Hydroamination Reactions of Propargyl Cyanamides: Rapid Access to Highly Substituted 2-Aminoimidazoles. Angew. Chem. Int. Ed. 2009, 48, 3116–3120. [Google Scholar] [CrossRef] [PubMed]
- Ermolat’ev, D.S.; Bariwal, J.B.; Steenackers, H.P.L.; De Keersmaecker, S.C.J.; Van der Eycken, E.V. Concise and Diversity-Oriented Route toward Polysubstituted 2-Aminoimidazole Alkaloids and Their Analogues. Angew. Chem. Int. Ed. 2010, 49, 9465–9468. [Google Scholar] [CrossRef] [PubMed]
- Zavesky, B.P.; Babij, N.R.; Wolfe, J.P. Synthesis of Substituted 2-Aminoimidazoles via Pd-Catalyzed Alkyne Carboamination Reactions. Application to the Synthesis of Preclathridine Natural Products. Org. Lett. 2014, 16, 4952–4955. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Eickhoff, J.A.; Zakarian, A. Synthesis of 2-Aminoazoles from Thioesters via α-Heterosubstituted Ketones by Copper-Mediated Cross-Coupling. J. Org. Chem. 2015, 80, 9989–9999. [Google Scholar] [CrossRef] [PubMed]
- Garlets, Z.J.; Silvi, M.; Wolfe, J.P. Synthesis of Cyclic Guanidines via Silver-Catalyzed Intramolecular Alkene Hydroamination Reactions of N-Allylguanidines. Org. Lett. 2016, 18, 2331–2334. [Google Scholar] [CrossRef] [PubMed]
- Abbott, A.P.; Capper, G.; Davies, D.L.; Rasheed, R.K.; Tambyrajah, V. Novel solvent properties of choline chloride/urea mixtures. Chem. Commun. 2003, 70–71. [Google Scholar] [CrossRef]
- Abbott, A.P.; Barron, J.C.; Ryder, K.S.; Wilson, D. Eutectic-based ionic liquids with metal-containing anions and cations. Chem. Eur. J. 2007, 13, 6495–6501. [Google Scholar] [CrossRef] [PubMed]
- Francisco, M.; van den Bruinhorst, A.; Kroon, M.C. Low-Transition-Temperature Mixtures (LTTMs): A New Generation of Designer Solvents. Angew. Chem. Int. Ed. 2013, 52, 3074–3085. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira Vigier, K.; Chatel, G.; Jérôme, F. Contribution of Deep Eutectic Solvents for Biomass Processing: Opportunities, Challenges, and Limitations. ChemCatChem 2015, 7, 1250–1260. [Google Scholar] [CrossRef]
- Smith, E.L.; Abbott, A.P.; Ryder, K.S. Deep Eutectic Solvents (DESs) and Their Applications. Chem. Rev. 2014, 114, 11060–11082. [Google Scholar] [CrossRef] [PubMed]
- Alonso, D.A.; Baeza, A.; Chinchilla, R.; Guillena, G.; Pastor, I.M.; Ramón, D.J. Deep Eutectic Solvents: The Organic Reaction Medium of the Century. Eur. J. Org. Chem. 2016, 4, 612–632. [Google Scholar] [CrossRef]
- Pena-Pereira, F.; Namieśnik, J. Ionic Liquids and Deep Eutectic Mixtures: Sustainable Solvents for Extraction Processes. ChemSusChem 2014, 7, 1784–1800. [Google Scholar] [CrossRef] [PubMed]
- Carriazo, D.; Serrano, M.C.; Gutiérrez, M.C.; Ferrer, M.L.; del Monte, F. Deep-eutectic solvents playing multiple roles in the synthesis of polymers and related materials. Chem. Soc. Rev. 2012, 41, 4996–5014. [Google Scholar] [CrossRef] [PubMed]
- Del Monte, F.; Carriazo, D.; Serrano, M.C.; Gutiérrez, M.C. Deep eutectic solvents in polymerizations: A greener alternative to conventional syntheses. ChemSusChem 2014, 7, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Mallardo, V.; Rizzi, R.; Sassone, F.C.; Mansueto, R.; Perna, F.M.; Salomone, A.; Capriati, V. Regioselective desymmetrization of diaryltetrahydrofurans via directed ortho-lithiation: An unexpected help from green chemistry. Chem. Commun. 2014, 50, 8655–8658. [Google Scholar] [CrossRef] [PubMed]
- Vidal, C.; García-Álvarez, J.; Hernán-Gómez, A.; Kennedy, A.R.; Hevia, E. Introducing Deep Eutectic Solvents to Polar Organometallic Chemistry: Chemoselective Addition of Organolithium and Grignard Reagents to Ketones in Air. Angew. Chem. Int. Ed. 2014, 53, 5969–5973. [Google Scholar] [CrossRef] [PubMed]
- Sassone, F.C.; Perna, F.M.; Salomone, A.; Florio, S.; Capriati, V. Unexpected lateral-lithiation-induced alkylative ring opening of tetrahydrofurans in deep eutectic solvents: Synthesis of functionalised primary alcohols. Chem. Commun. 2015, 51, 9459–9462. [Google Scholar] [CrossRef] [PubMed]
- García-Álvarez, J.; Hevia, E.; Capriati, V. Reactivity of Polar Organometallic Compounds in Unconventional Reaction Media: Challenges and Opportunities. Eur. J. Org. Chem. 2015, 6779–6799. [Google Scholar] [CrossRef] [Green Version]
- Cicco, L.; Sblendorio, S.; Mansueto, R.; Perna, F.M.; Salomone, A.; Florio, S.; Capriati, V. Water opens the door to organolithiums and Grignard reagents: Exploring and comparing the reactivity of highly polar organometallic compounds in unconventional reaction media towards the synthesis of tetrahydrofurans. Chem. Sci. 2016, 7, 1192–1199. [Google Scholar] [CrossRef]
- Rodríguez-Àlvarez, M.J.; Vidal, C.; Díez, J.; García-Àlvarez, J. Introducing deep eutectic solvents as biorenewable media for Au(I)-catalysed cycloisomerisation of γ-alkynoic acids: An unprecedented catalytic system. Chem. Commun. 2014, 50, 12927–12929. [Google Scholar] [CrossRef] [PubMed]
- Vidal, C.; Merz, L.; García-Àlvarez, J. Deep eutectic solvents: Biorenewable reaction media for Au(I)-catalysed cycloisomerisations and one-pot tandem cycloisomerisation/Diels-Alder reactions. Green Chem. 2015, 17, 3870–3878. [Google Scholar] [CrossRef]
- Mancuso, R.; Maner, A.; Cicco, L.; Perna, F.M.; Capriati, V.; Gabriele, B. Synthesis of thiophenes in a deep eutectic solvent: heterocyclodehydration and iodocyclization of 1-mercapto-3-yn-2-ols in a choline chloride/glycerol medium. Tetrahedron 2016, 72, 4239–4244. [Google Scholar] [CrossRef]
- Müller, C.R.; Meiners, I.; de María, P.D. Highly enantioselective tandem enzyme-organocatalyst crossed aldol reactions with acetaldehyde in deep-eutectic-solvents. RSC Adv. 2014, 4, 46097–46101. [Google Scholar] [CrossRef]
- Massolo, E.; Palmieri, S.; Benaglia, M.; Capriati, V.; Perna, F.M. Stereoselective organocatalysed reactions in deep eutectic solvents: Highly tunable and biorenewable reaction media for sustainable organic synthesis. Green Chem. 2016, 18, 792–797. [Google Scholar] [CrossRef]
- Martínez, R.; Berbegal, L.; Guillena, G.; Ramón, D.J. Bio-renewable enantioselective aldol reaction in natural deep eutectic solvents. Green Chem. 2016, 18, 1724–1730. [Google Scholar] [CrossRef]
- By running the reaction at a lower temperature (66 °C), compound 3a could be isolated only with a slight decrease of the reaction yield (80%) after 4 h.
- Singh, B.S.; Lobo, H.R.; Pinjari, D.V.; Jarag, K.J.; Pandit, A.B.; Shankarling, G.S. Comparative material study and synthesis of 4-(4-nitrophenyl)oxazol-2-amine via sonochemical and thermal method. Ultrason. Sonochem. 2013, 20, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Perkins, S.L.; Painter, P.; Colina, C.M. Experimental and Computational Studies of Choline Chloride-Based Deep Eutectic Solvents. J. Chem. Eng. Data 2014, 59, 3652–3662. [Google Scholar] [CrossRef]
- Hawkins, I.; Handy, S.T. Synthesis of aurones under neutral conditions using a deep eutectic solvent. Tetrahedron 2013, 69, 9200–9204. [Google Scholar] [CrossRef]
- Handy, S.; Lavender, K. Organic Synthesis in deep eutectic solvents: Paal-Knorr reactions. Tetrahedron Lett. 2013, 54, 4377–4379. [Google Scholar] [CrossRef]
- Nemati, F.; Hosseini, M.M.; Kiani, H. Glycerol as a green solvent for efficient, one-pot and catalyst free synthesis of 2,4,5-triaryl and 1,2,4,5-tetraaryl imidazole derivatives. J. Saudi Chem. Soc. 2013, in press. [Google Scholar] [CrossRef]
- Lu, J.; Li, X.-T.; Ma, E.-Q.; Mo, L.-P.; Zhang, Z.-H. Superparamagnetic CuFeO2 Nanoparticles in Deep Eutectic Solvent: An Efficient and Recyclable Catalytic System for the Synthesis of Imidazo(1,2-a)pyridines. ChemCatChem 2014, 6, 2854–2859. [Google Scholar] [CrossRef]
- Hu, H.-C.; Liu, Y.-H.; Li, B.-L.; Cui, Z.-S.; Zhang, Z.-H. Deep eutectic solvent based on choline chloride and malonic acid as an efficient and reusable catalytic system for one-pot synthesis of functionalized pyrroles. RSC Adv. 2015, 5, 7720–7728. [Google Scholar] [CrossRef]
- Liu, P.; Hao, J.-W.; Mo, L.-P.; Zhang, Z.-H. Recent advances in the application of deep eutectic solvents as sustainable media as well as catalysts in organic reactions. RSC Adv. 2015, 5, 48675–48704. [Google Scholar] [CrossRef]
- Liu, P.; Hao, J.; Zhang, Z.-H. A General, Efficient and Green Procedure for Synthesis of Dihydropyrimidine-5-carboxamides in Low Melting Betaine Hydrochloride/Urea Mixture. Chin. J. Chem. 2016, 34, 637–645. [Google Scholar] [CrossRef]
- It is worth noting that the use of weaker bases than Et3N has proved to be not effective. For example, once the cyclocondensation of α-chloroketone 1a with guanidine derivative 2a was carried out in a ChCl–Gly eutectic mixture in the presence of 1,4-diazabicyclo(2.2.2)octane (DABCO), the expected 2-AI 3a could be isolated in 67% yield only, the remaining material being the dehalogenated starting ketone, that is acetophenone. For a basicity scale, see: Frenna, V.; Vivona, N.; Consiglio, G.; Spinelli, D. Amine basicities in benzene and in water. J. Chem. Soc. Perkin Trans. 2 1985, 1865–1868. [Google Scholar] [CrossRef]
- Shao, J.; Chen, W.; Giulianotti, M.A.; Houghten, R.A.; Yu, Y. Palladium-Catalyzed C-H Functionalization Using Guanidine as a Directing Group: Ortho Arylation and Olefination of Arylguanidines. Org. Lett. 2012, 14, 5452–5455. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 3a–i are available from the authors.




| Entry | 1 | R1 | 2 | R2 | Solvent | Temperature | 3, Yield (%) b |
|---|---|---|---|---|---|---|---|
| 1 | 1a | Ph | 2a | Ph | DES | 80 | 3a, 85 |
| 2 | 1a | Ph | 2a | Ph | THF | reflux | 3a, 81 |
| 3 | 1b | t-Bu | 2a | Ph | DES | 80 | 3b, 74 |
| 4 | 1b | t-Bu | 2a | Ph | THF | reflux | 3b, 78 |
| 5 | 1c | Me | 2a | Ph | DES | 80 | 3c, 70 |
| 6 | 1c | Me | 2a | Ph | THF | reflux | 3c, 71 |
| 7 | 1d | 4-MeC6H4 | 2a | Ph | DES | 80 | 3d, 80 |
| 8 | 1d | 4-MeC6H4 | 2a | Ph | THF | reflux | 3d, 78 |
| 9 | 1e | 4-MeOC6H4 | 2a | Ph | DES | 80 | 3e, 79 |
| 10 | 1e | 4-MeOC6H4 | 2a | Ph | THF | reflux | 3e, 77 |
| 11 | 1f | 4-ClC6H4 | 2a | Ph | DES | 80 | 3f, 80 |
| 12 | 1f | 4-ClC6H4 | 2a | Ph | THF | reflux | 3f, 83 |
| 13 | 1g | 4-FC6H4 | 2a | Ph | DES | 80 | 3g, 83 |
| 14 | 1g | 4-FC6H4 | 2a | Ph | THF | reflux | 3g, 85 |
| 15 | 1a | Ph | 2b c | H | DES | 80 | 3h, 73 |
| 16 | 1a | Ph | 2b c | H | EtOH | reflux | 3h, 76 |
| 17 | 1b | t-Bu | 2b c | H | DES | 80 | 3i, 72 |
| 18 | 1b | t-Bu | 2b c | H | EtOH | reflux | 3i, 70 |

{kind=link}
{kind=link}
{kind=link}
| Entry | 1 | R1 | 2 | R2 | 3, Yield (%) |
|---|---|---|---|---|---|
| 1 | 1a | Ph | 2a | Ph | 3a, 75 b |
| 2 | 1b | t-Bu | 2a | Ph | 3b, 78 c |
| 3 | 1d | 4-MeC6H4 | 2a | Ph | 3d, 88 b |
| 4 | 1e | 4-MeOC6H4 | 2a | Ph | 3e, 84 b |
| 5 | 1f | 4-ClC6H4 | 2a | Ph | 3f, 76 b |
| 6 | 1g | 4-FC6H4 | 2a | Ph | 3g, 80 b |
| 7 | 1a | Ph | 2b | H | 3h, 65 c |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capua, M.; Perrone, S.; Perna, F.M.; Vitale, P.; Troisi, L.; Salomone, A.; Capriati, V. An Expeditious and Greener Synthesis of 2-Aminoimidazoles in Deep Eutectic Solvents. Molecules 2016, 21, 924. https://doi.org/10.3390/molecules21070924
Capua M, Perrone S, Perna FM, Vitale P, Troisi L, Salomone A, Capriati V. An Expeditious and Greener Synthesis of 2-Aminoimidazoles in Deep Eutectic Solvents. Molecules. 2016; 21(7):924. https://doi.org/10.3390/molecules21070924
Chicago/Turabian StyleCapua, Martina, Serena Perrone, Filippo Maria Perna, Paola Vitale, Luigino Troisi, Antonio Salomone, and Vito Capriati. 2016. "An Expeditious and Greener Synthesis of 2-Aminoimidazoles in Deep Eutectic Solvents" Molecules 21, no. 7: 924. https://doi.org/10.3390/molecules21070924