
Antidiarrheal Thymol Derivatives from Ageratina glabrata. Structure and Absolute Configuration of 10-Benzoyloxy-8,9-epoxy-6-hydroxythymol Isobutyrate

 ,
,  and
and
Abstract
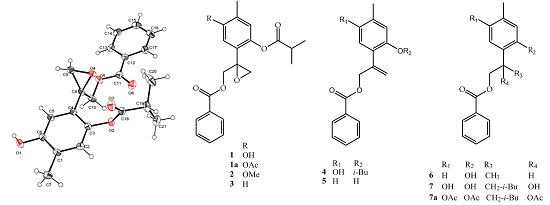
:
1. Introduction
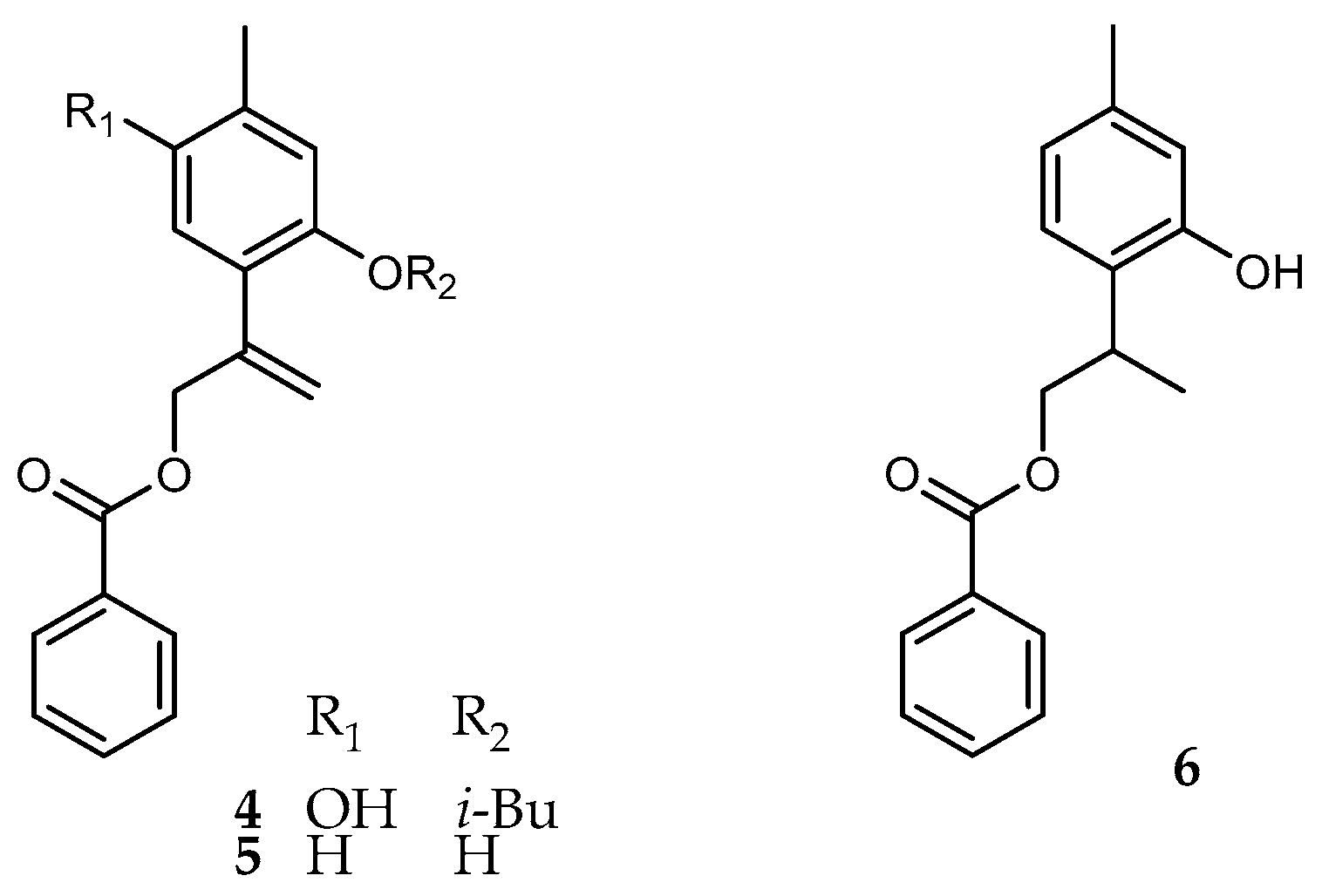
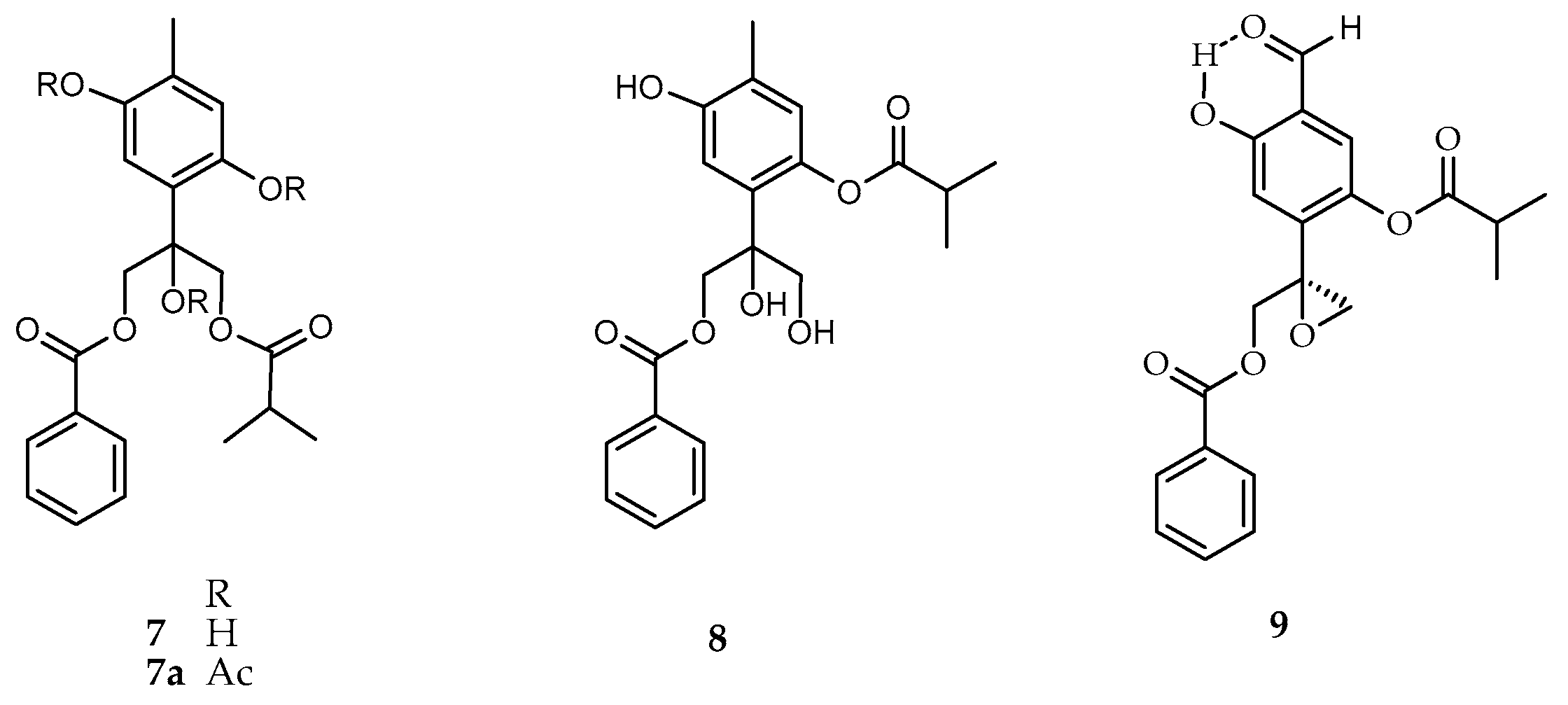
2. Results and Discussion
3. Materials and Methods
3.1. General Procedures
3.2. Plant Material
3.3. Extraction, Isolation and Characterization
3.4. X-ray Crystallography of Compound 1
3.5. Antiprotozoal Assays
3.6. Animals
3.7. Effect on Charcoal-Gum Acacia-Induced Hyperperistalsis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Vivar, A.R.; Cuevas, L.A.; Guerrero, C. Eupaglabrina, un nuevo terpeno aislado de Eupatorium glabratum. Rev. Latinoam. Chem. 1971, 2, 32–34. [Google Scholar]
- Guerrero, C.; Silva, M.; Maldonado, E.; Martinez, M. Acido eupaglabrico un Nuevo compuesto aislado de Eupatorium glabratum H.B.K. Rev. Latinoam. Chem. 1978, 9, 71–75. [Google Scholar]
- Bohlmann, F.; Jakupovic, J.; Lonitz, M. Naturally Ocurring Terpene Derivatives, 76, On Constituents of the Eupatorium group. Chem. Ber. 1977, 110, 301–314. [Google Scholar] [CrossRef]
- Pimienta-Rámirez, L.; García-Rodriguez, Y.M.; Rios-Ramirez, E.M.; Linding-Cisneros, R.; Espinoza-Garcia, F.J. Chemical composition and evaluation of the essential oil from Eupatorium glabratum as biopesticide against Sitophilus zeamais and several stored maize fungi. J. Essent. Oil Res. 2016, 28, 113–120. [Google Scholar] [CrossRef]
- Garcia-Sanchez, E.; Ramirez-Lopez, C.B.; Martinez-Muñoz, R.E.; Flores-Garcia, A.; del Rio, R.E.; Martinez-Pacheco, M.M. Antibacterial activity of some medicinal Eupatorium species against antibiotic resistant pathogenic bacteria. Polibotánica 2015, 39, 91–101. [Google Scholar] [CrossRef]
- García, P.G.; del Río, R.E.; Guzmán, M.R.; Martinez, G.M.I.; Scior, T.R.F. Preliminary studies on the analgesic effect of leaf extract from Ageratina glabrata in two thermal models of acute pain. Rev. Mex. Cienc. Farm. 2011, 42, 45–51. [Google Scholar]
- Bustos-Brito, C.; Sánchez-Castellanos, M.; Esquivel, B.; Calderón, J.S.; Calzada, F.; Yépez-Mulia, L.; Hernández, A.B.; Joseph-Nathan, P.; Cuevas, G.; Quijano, L. Structure, absolute configuration, and antidiarrheal activity of a thymol derivative from Ageratina cylindrica. J. Nat. Prod. 2014, 77, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Bustos-Brito, C.; Sánchez-Castellanos, M.; Esquivel, B.; Calderón, J.S.; Calzada, F.; Yépez-Mulia, L.; Joseph-Nathan, P.; Cuevas, G.; Quijano, L. ent-Kaurene Glycosides from Ageratina cylindrica. J. Nat. Prod. 2015, 78, 2580–2587. [Google Scholar] [PubMed]
- Deng, Y.R.; Song, A.X.; Wang, H.Q. Chemical Components of Seriphidium santolium Poljak. J. Chin. Chem. Soc. 2004, 51, 629–636. [Google Scholar] [CrossRef]
- Flack, H.D.; Bernardinelli, G. The use of X-ray chrystallography to determine absolute configuration. Chirality 2008, 20, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Tamayo-Castillo, G.; Jakupovic, J.; Bohlmann, F.; Rojas, A.; Castro, V.; King, R.M. Germacranolides and other constituents from Ageratina species. Phytochemistry 1988, 27, 2893–2897. [Google Scholar] [CrossRef]
- Maldonado, E.; Marquez, L.C.; Ortega, A. A thymol derivative from Calea nelsonii. Phytochemistry 1992, 31, 2527–2528. [Google Scholar] [CrossRef]
- Bohlmann, F.; Nuedballa, U.; Schulz, J. Über einige thymolderivate aus Gaillardia-und Helenium-arten. Chem. Ber. 1969, 102, 864–871. [Google Scholar] [CrossRef]
- García, P.G.; García, S.E.; Martínez, G.I.; Scior, T.R.F.; Salvador, J.L.; Martínez, P.M.M.; del Río, R.E. Analgesic effect of leaf extract from Ageratina glabrata in the hot plate test. Braz. J. Pharmacog. 2011, 21, 928–935. [Google Scholar]
- Bruker. SAINT & APEX-2 Programs; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, C71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Calzada, F.; Meckes, M.; Cedillo-Rivera, R.; Tapia-Contreras, A.; Mata, R. Screening of Mexican medicinal plants from antiprotozoal activity. Pharm. Biol. 1998, 36, 305–309. [Google Scholar] [CrossRef]
- Norma Oficial Mexicana NOM-062-ZOO-1999, Especificaciones Técnicas Para la Producción, Cuidado y Uso de Los Animales de Laboratorio; Diario Oficial de la Federación: México City, México, 2001.
- Williamson, E.M.; Okpako, D.T.; Evans, F.J. Pharmacological Methods in Phytotherapy Research: Selection, Preparation and Pharmacological Evaluation of Plant Material, 1st ed.; John Wiley & Sons: Chichester, UK, 1996; Volume 1, p. 28. [Google Scholar]
- Sample Availability: Samples of the compounds 1, 1a,2, 3 and 7 are available from the authors.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 1a | |||
|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | HMBC | δC, Type | δH (J in Hz) | |
| 1 | 126.2, C | 132.2, C | |||
| 2 | 124.8, CH | 6.79, d (0.8) | 1, 3, 4, 6, 7 | 125.0, CH | 6.96, brs |
| 3 | 141.7, C | 146.2, C | |||
| 4 | 127.2, C | 127.9, C | |||
| 5 | 114.8, CH | 6.94, s | 1, 3, 6, 8 | 122.7, CH | 7.23. s |
| 6 | 152.1, C | 147.0, C | |||
| 7 | 15.8, CH3 | 2.18, s | 1, 2, 6 | 16.3, CH3 | 2.17, s |
| 8 | 57.3, C | 56.9, C | |||
| 9a | 51.3, CH2 | 3.12, d (5.2) | 4, 8, 10 | 51.2, CH2 | 3.11, d (5.2) |
| 9b | 2.86, d (5.2) | 4, 8, 10 | 2.87, d (5.2) | ||
| 10a | 65.6, CH2 | 4.77, d (12.3) | 1′, 4, 8, 9 | 65.9, CH2 | 4.76, d (12.2) |
| 10b | 4.48, d (12.3) | 1′, 4, 8, 9 | 4.47, d (12.2) | ||
| 1′ | 166.3, C | 166.1, C | |||
| 2′ | 129.7, C | 129.0, C | |||
| 3′ | 129.8, CH | 7.97, m | 1′, 5′ | 129.9, CH | 7.97, m |
| 4′ | 128.5, CH | 7.40, m | 3′, 5′, 7′ | 128.5, CH | 7.42, m |
| 5′ | 133.4, CH | 7.55, tt (7.5, 1.3) | 3′, 7′ | 133.3, CH | 7.55, tt (7.4, 1.2) |
| 6′ | 128.5, CH | 7.40, m | 3′, 5′, 7′ | 128.5, CH | 7.42, m |
| 7′ | 129.8, CH | 7.97, m | 1′, 5′ | 129.9, CH | 7.97, m |
| 1″ | 176.2, C | 175.3, C | |||
| 2″ | 34.3, CH | 2.83, hep (7.2) | 1″, 3″, 4″ | 34.3, CH | 2.84, hep (7.2) |
| 3″, 4″ | 19.1, 19.2, CH3 | 1.312, 1.307 d (7.2) | 1″, 2″ | 19.0, 19.1, CH3 | 1.31, 1.32, d(7.2) |
| 6-OH | 5.78, brs | ||||
| COCH3 | 20.8, CH3 | 2.31 | |||
| COCH3 | 168.9, C | ||||
| Position | 2 | 3 | ||
|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 1 | 128.6, C | 140.0, C | ||
| 2 | 124.6, CH | 6.83, brs | 123.1, CH | 6.88, brd (0.7) |
| 3 | 141.6, C | 148.7, C | ||
| 4 | 126.9, C | 126.1, C | ||
| 5 | 109.9, CH | 6.97, s | 128.9, CH | 7.43, d (7.8) |
| 6 | 155.6, C | 126.8, CH | 7.07, ddd (7.8, 1.6, 0.7) | |
| 7 | 16.2, CH3 | 2.19, brd (0.4) | 21.1, CH3 | 2.35, s |
| 8 | 57.3, C | 57.0, C | ||
| 9a | 51.3, CH2 | 3.13, d (5.3) | 50.9, CH2 | 3.12, d (5.3) |
| 9b | 2.87, d (5.3) | 2.85, d (5.3) | ||
| 10a | 65.8, CH2 | 4.79, d (12.2) | 65.9, CH2 | 4.76, d (12.2.) |
| 10b | 4.48, d (12.2) | 4.47, d (12.2) | ||
| 1′ | 166.1, C | 165.9, C | ||
| 2′ | 129.9, C | 129.72, C | ||
| 3′ | 129.8, CH | 7.99, m | 129.67, CH | 7.98, m |
| 4′ | 128.5, CH | 7.43, m | 128.4, CH | 7.42, m |
| 5′ | 133.3, CH | 7.55, m | 133.1, CH | 7.55, tt (7.6, 1.3) |
| 6′ | 128.5, CH | 7.42, m | 128.4, CH | 7.42, m |
| 7′ | 129.8, CH | 7.99, m | 129.67, CH | 7.98, m |
| 1″ | 175.9, C | 175.3, C | ||
| 2″ | 34.3, CH | 2.83, hep (7.2) | 34.2, CH | 2.85, hep (7.0) |
| 3″, 4″ | 19.1, 19.2, CH3 | 1.31, 1.32 d(7.2) | 18.9, 19.0, CH3 | 1.32, 1.33, d(7.0) |
| OCH3 | 55.9, CH3 | 3.82, s | ||
| Position | 4 | 5 | 6 | |||
|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 1 | 125.0, C | 140.2, C | 138.0, C | |||
| 2 | 124.9, CH | 6.80, brs | 116.9, CH | 6.78, d (0.7) | 116.8, CH | 6.68, d (0.8) |
| 3 | 141.4, C | 153.5, C | 154.0, C | |||
| 4 | 130.9, C | 122.6, C | 125.3, C | |||
| 5 | 115.9, CH | 6.74, s | 129.3, CH | 7.03, d (7.7) | 127.2, CH | 7.10, d (7.8) |
| 6 | 151.6, C | 121.2, CH | 6.71, ddd (7.7, 1.7, 0.7) | 121.6, C | 6.74, ddd (7.6, 0.8, 0.4) | |
| 7 | 15.8, CH3 | 2.22, brs | 21.4, CH3 | 2.32, s | 21.1, CH3 | 2.28, s |
| 8 | 140.2, C | - | 141.9, C | 32.4, C | 3.50, dqd (8.0, 6.8, 4.8) | |
| 9a | 117.3, CH2 | 5.48, q (1.6) | 116.7, CH2 | 5.56, q (1.6) | 16.7, CH3 | 1.42, d (6.8) |
| 9b | 5.25, q (1.2) | 5.31, q ( 1.2) | ||||
| 10a | 66.7, CH2 | 5.0, dd (1.6, 1.2) | 66.6, CH2 | 4.99, dd (1.6, 1.2) | 70.2, CH2 | 4.53, d (10.9, 4.8) |
| 10b | 4.19 dd (10.9, 8.0) | |||||
| 1′ | 166.3, C | 167.2, C | 167.5, C | |||
| 2’ | 130.2, C | 129.8, C | 130.3, C | |||
| 3′ | 129.8, CH | 8.03, m | 129.9, CH | 8.09, m | 129.8, CH | 8.04, m |
| 4′ | 128.6, CH | 7.43, m | 128.7, CH | 7.47, m | 128.5, CH | 7.56, tt (7.2, 1.6) |
| 5′ | 133.2, CH | 7.55, tt (7.4, 1.4) | 133.6, CH | 7.60, tt (7.2, 1.3) | 133.2, CH | 7.44, m |
| 6′ | 128.6, CH | 7.43, m | 128.7, CH | 7.47, m | 128.5, CH | 7.56, tt (7.2, 1.6) |
| 7′ | 129.8, CH | 8.03, m | 129.9, CH | 8.09, m | 129.8, CH | 8.04, m |
| 1″ | 176.1, C | |||||
| 2″ | 34.2, CH | 2.75, hep (7.0) | ||||
| 3″, 4″ | 19.1, CH3 | 1.27, d (7.0) | ||||
| Position | 7 | 7a | |||
|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | HMBC | δC, Type | δH (J in Hz) | |
| 1 | 126.2, C | 132.3, C | |||
| 2 | 120.2, CH | 6.66, brs | 3, 4, 6, 7 | 126.7, CH | 6.96, d (0.7) |
| 3 | 149.8, C | 145.3, C | |||
| 4 | 120.3, C | 127.7, C | |||
| 5 | 113.0, CH | 6.60, s | 1, 3, 6, 8 | 121.8, CH | 7.16, s |
| 6 | 147.2, C | 147.1, C | |||
| 7 | 15.7, CH3 | 2.17, brs | 1, 2 | 16.1, CH3 | 1.63, brs |
| 8 | 78.3, C | 80.8, C | |||
| 9a | 67.5, CH2 | 4.58, d(11.9) | 4, 8, 10, 1″ | 62.9, CH2 | 4.96, d (11.4) |
| 9b | 4.51, d(11.9) | 4, 8, 10, 1″ | 4.81, d (11.4) | ||
| 10a | 68.1, CH2 | 4.68 d(12.0) | 1′, 4, 8, 9 | 63.6, CH2 | 5.12, d (11.4) |
| 10b | 4.65, d(12.0) | 1’, 4, 8, 9 | 4.91, d (11.4) | ||
| 1′ | 167.0, C | 165.8, C | |||
| 2′ | 129.1, C | 129.6, C | |||
| 3′ | 129.9, CH | 7.98, m | 1′, 5′, 7′ | 129.8, CH | 7.95, m |
| 4′ | 128.7, CH | 7.41, m | 3′, 5′, 7′ | 128.6, CH | 7.43, m |
| 5′ | 133.6, CH | 7.55, tt (7.4,1.3) | 3′, 7′ | 133.4, CH | 7.55, tt (7.5, 1.3) |
| 6′ | 128.7, CH | 7.41, m | 3′, 5′, 7′ | 128.6, CH | 7.43, m |
| 7′ | 129.9, CH | 7.98, m | 1′, 5′, 3′ | 129.8, CH | 7.95, m |
| 1″ | 177.9, C | 176.3, C | |||
| 2″ | 34.1, CH | 2.53, hep (7.0) | 1″, 3″, 4″ | 34.0, CH | 2.52, hep (7.0) |
| 3″, 4″ | 18.9. 19.0, CH3 | 1.05, 1.08, d (7.0) | 1″, 2″ | 18.8, 18.9, CH3 | 1.08, 1.10, d (7.0) |
| 3-OCOCH3 | 21.3, 169.4, CH3, CO | 2.38, s | |||
| 6-OCOCH3 | 20.9, 168.8, CH3, CO | 2.31, s | |||
| 8-OCOCH3 | 21.4, 168.8, CH3, CO | 2.00, s | |||
| Compound | IC50 μM (CI) a | ID50 μmol/kg ± SD b | |
|---|---|---|---|
| Entamoeba histolytica | Giardia lamblia | Inhibition of Hyperperistalsis | |
| 1 | 1.6 (1.8–1.57) | 36.9 (38.3–5.9) * | 2.000 ± 0.003 ** |
| 1a | 0.84 (0.87–0.80) | 24.2 (24.7–24.0) * | 0.810 ± 0.021 ** |
| 2 | 169.6 (171.3–168.9) * | 191.2 (192.1–190.8) * | 0.457 ± 0.004 ** |
| 3 | 25.9 (26.2–25.7) * | 48.3 (48.4–45.4) * | 0.740 ± 0.003 ** |
| 4 | 61.2 (62.3–59.8) * | 68.0 69.7–67.4) * | 1.430 ± 0.006 ** |
| 7 | 45.6 (46.9–44.3) * | 60.7 (62.3–56.9) * | 0.380 ± 0.003 ** |
| Pectolinaringenin | 43.6 (44.9–41.9) * | 68.7 (70.3–67.4) * | 0.598 ± 0.001 ** |
| 9 c | 184.9 (186.9–180.7) * | 167.4 (168.7–165.8) * | 0.85 ± 0.005 |
| Emetine d | 2.18 (2.2–2.14) | 0.83 (0.87–0.82) | |
| Metronidazole d | 0.23 (0.58–0.17) | 1.22 (1.57–0.81) | |
| Quercetin | - | - | 1.1 ± 0.001 |
| Loperamide hydrochloride d | - | - | 0.2 ± 0.001 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bustos-Brito, C.; Vázquez-Heredia, V.J.; Calzada, F.; Yépez-Mulia, L.; Calderón, J.S.; Hernández-Ortega, S.; Esquivel, B.; García-Hernández, N.; Quijano, L. Antidiarrheal Thymol Derivatives from Ageratina glabrata. Structure and Absolute Configuration of 10-Benzoyloxy-8,9-epoxy-6-hydroxythymol Isobutyrate. Molecules 2016, 21, 1132. https://doi.org/10.3390/molecules21091132
Bustos-Brito C, Vázquez-Heredia VJ, Calzada F, Yépez-Mulia L, Calderón JS, Hernández-Ortega S, Esquivel B, García-Hernández N, Quijano L. Antidiarrheal Thymol Derivatives from Ageratina glabrata. Structure and Absolute Configuration of 10-Benzoyloxy-8,9-epoxy-6-hydroxythymol Isobutyrate. Molecules. 2016; 21(9):1132. https://doi.org/10.3390/molecules21091132
Chicago/Turabian StyleBustos-Brito, Celia, Valeria J. Vázquez-Heredia, Fernando Calzada, Lilian Yépez-Mulia, José S. Calderón, Simón Hernández-Ortega, Baldomero Esquivel, Normand García-Hernández, and Leovigildo Quijano. 2016. "Antidiarrheal Thymol Derivatives from Ageratina glabrata. Structure and Absolute Configuration of 10-Benzoyloxy-8,9-epoxy-6-hydroxythymol Isobutyrate" Molecules 21, no. 9: 1132. https://doi.org/10.3390/molecules21091132