
ent-Pimarane and ent-Kaurane Diterpenes from Aldama discolor (Asteraceae) and Their Antiprotozoal Activity †

 ,
, 
Abstract
:
1. Introduction
2. Results
2.1. Antiprotozoal Activity of A. discolor Crude Extracts
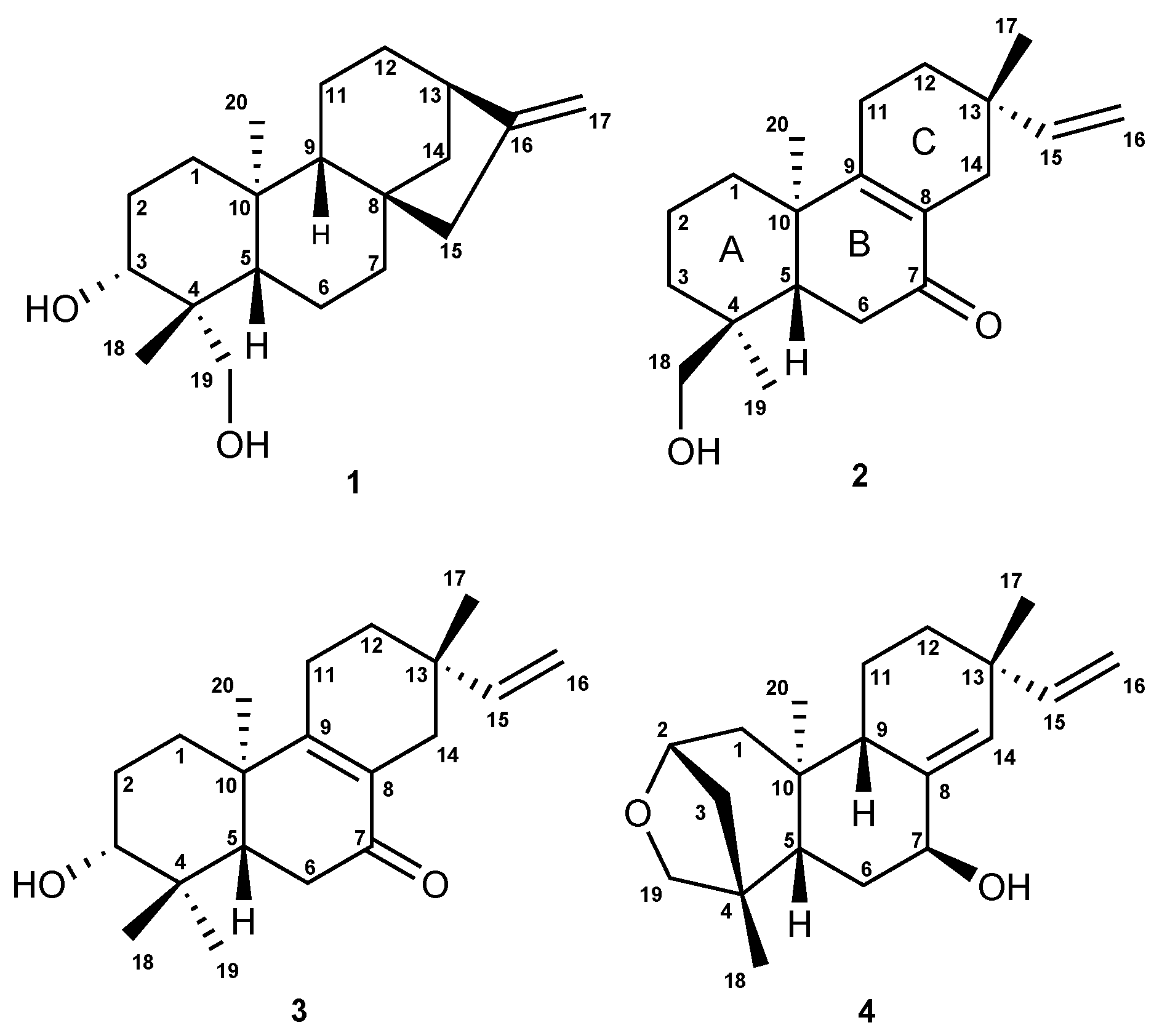
2.2. Isolation and Characterization of the Compounds
2.3. In Vitro Antiprotozoal Activities
3. Discussion
4. Materials and Methods
General Procedures
5. Plant Material
Chromatographic Separation of the Dichloromethane Extract of Aldama discolor Leaves: Isolation of Compounds 1–4
6. Chromatographic Analysis of the Plant Extract and Pure Compounds by UHPLC-MS
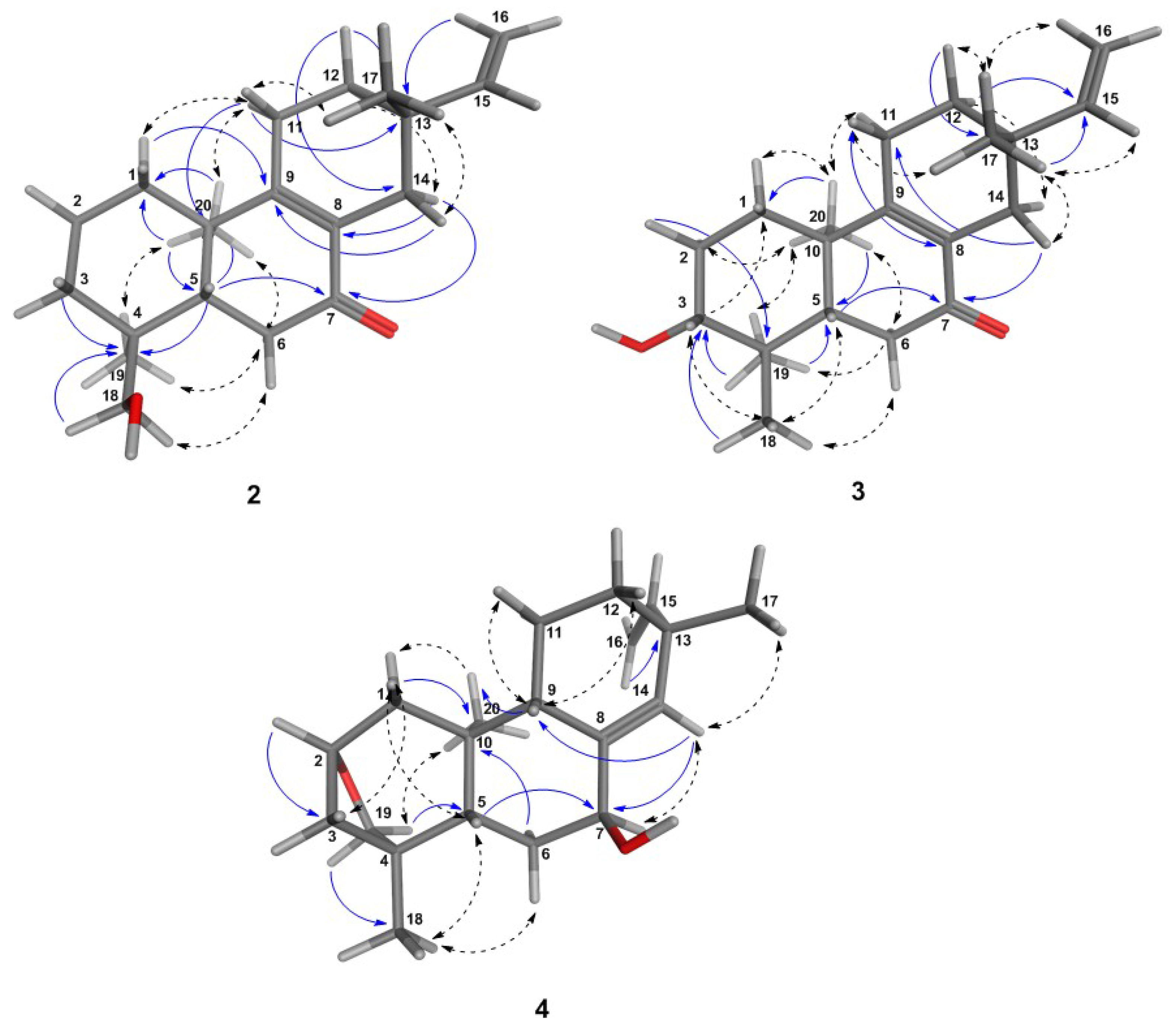
6.1. Structural Determination of the Isolated Compounds
6.1.1. General
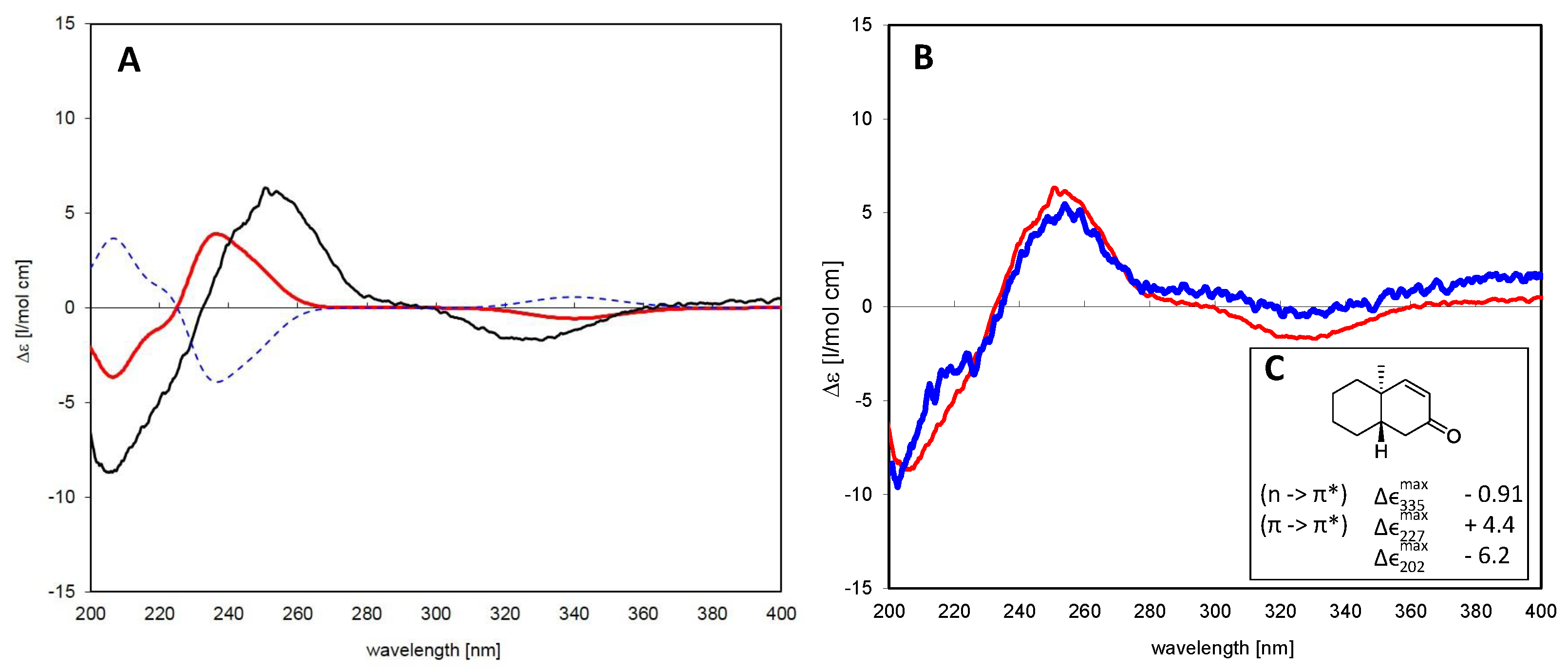
6.1.2. Assignment of Absolute Configuration for ent-Pimaranes
6.2. Analytical Data for Compounds 1–5
6.3. In Vitro Assays for the Antiprotozoal Activity of the Crude Extract and Pure Compounds
7. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schilling, E.E.; Panero, J.L. A revised classificationcation of subtribe Helianthinae (Asteraceae: Heliantheae). I. Basal lineages. Bot. J. Linn. Soc. 2011, 140, 65–76. [Google Scholar] [CrossRef]
- Blake, S.F. A Revision of the Genus Viguiera; Contrib. Gray Herb. Harvard Univ.: Cambridge, MA, USA, 1918; Volume 54. [Google Scholar]
- Meragelman, K.M.; Espinar, L.A.; Sosa, V.E.; Uriburu, M.L.; de la Fuente, J.R. Terpenoid constituents of Viguiera tucumanensis. Phytochemistry 1996, 41, 499–502. [Google Scholar] [CrossRef]
- Da Costa, F.B.; Schorr, K.; Arakawa, N.S.; Schilling, E.E.; Spring, O. Infraspecific variation in the chemistry of glandular trichomes of two Brazilian Viguiera species (Heliantheae; Asteraceae). J. Braz. Chem. Soc. 2001, 12, 403–407. [Google Scholar] [CrossRef]
- Delgado, G.; Vivar, A.R.D.E.; Herz, W.; Blake, V. Sesquiterpene lactones from Viguiera species. Phytochemistry 1982, 21, 1305–1308. [Google Scholar] [CrossRef]
- Delgado, G.; Cirdenas, H.; Pelez, G.; Vivar, A.R.D.; Pereda-Miranda, R. Terpenoids from Viguiera excelsa and Vigueira oaxacana. J. Nat. Prod. 1984, 4, 1042–1045. [Google Scholar] [CrossRef]
- Spring, O.; Zipper, R.; Klaiber, I.; Reeb, S.; Vogler, B. Sesquiterpene lactones in Viguiera eriophora and Viguiera puruana (Heliantheae; Asteraceae). Phytochemistry 2000, 55, 255–261. [Google Scholar] [CrossRef]
- Gao, F.; Miski, M.; Gage, D.A.; Norris, J.A.; Mabry, T.J. Terpenoids from Viguiera potosina. J. Nat. Prod. 1991, 30, 4172–4174. [Google Scholar] [CrossRef]
- Gao, F.; Miski, M.; Gage, D.A.; Mabry, T.J. Terpenoid constituents of Viguiera dentata. J. Nat. Prod. 1985, 48, 316–318. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, F.B.; Albuquerque, S.; Vichnewski, W. Diterpenes and synthetic derivatives from Viguiera aspillioides with trypanomicidal activity. Planta Med. 1996, 62, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Delgado, G.; Vivar, A.R.D.E.; Ortega, A.; Cardenas, J. Diterpenoids from Viguiera insignis. Phytochemistry 1983, 22, 1227–1230. [Google Scholar] [CrossRef]
- Delgado, G.; De Vivar, A.E.; Cardenas, J.; Pereda-Miranda, R.; Huerta, E. ent-Beyerene and ent-atisene diterpenes from Viguiera insignis. Phytochemistry 1984, 23, 2285–2288. [Google Scholar] [CrossRef]
- Zamilpa, A.; Tortoriello, J.; Navarro, V.; Delgado, G.; Alvarez, L. Antispasmodic and antimicrobial diterpenic acids from Viguiera hypargyrea roots. Planta Med. 2002, 68, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Bohlmann, F.; Zdero, C.; Schmeda-Hirschmann, G.; Jakupovic, J.; Castro, V.; King, R.M.; Robinson, H. Heliangolide, trachyloban- und villanovan-derivate aus Viguiera arten. Liebigs Annal. Chem. 1984, 3, 495–502. [Google Scholar] [CrossRef]
- Guerrero, C.; Nava, A.L.; Quevedo, F.; Toscano, R.A.; Soriano-Garcia, M. Further constituents of Viguiera stenoloba and Viguiera pinnatilobata. Rev. Latinoam. Quim. 1986, 16, 126–128. [Google Scholar]
- Ambrosio, S.R.; Schorr, K.; da Costa, F.B. Terpenoids of Viguiera arenaria (Asteraceae). Biochem. Syst. Ecol. 2004, 32, 221–224. [Google Scholar] [CrossRef]
- Ambrosio, S.R.; Arakawa, N.S.; Esperandim, V.R.; de Albuquerque, S.; da Costa, F.B. Trypanocidal activity of pimarane diterpenes from Viguiera arenaria (Asteraceae). Phytother. Res. 2008, 22, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, M.L.; Alvarezb, L.; Alonsoa, D.; Navarroa, V.; Garciab, P.; Delgadoc, G. Cytotoxic and antimicrobial screening of selected terpenoids from Asteraceae species. J. Ethnopharmacol. 1994, 42, 25–29. [Google Scholar] [CrossRef]
- Carvalho, T.C.; Furtado, N.A.J.C.; Veneziani, R.C.S.; Heleno, V.C.G.; Souza, M.G.M.; Martins, C.H.G.; Franca, U.D.; Campinas, D. Antimicrobial activity of diterpenes from Viguiera arenaria against Endodontic Bacteria. Molecules 2011, 8, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Souza, T.; Furtado, N.A.J.C.; Heleno, V.C.G.; Martins, C.H.G.; Da, F.B.; Severiano, M.E.; Silva, A.N.; Veneziani, R.C.S.; Ambrósio, S.R. Antimicrobial ent-pimarane diterpenes from Viguiera arenaria against gram-positive bacteria. Fitoterapia 2009, 80, 432–436. [Google Scholar]
- Chagas-Paula, D.A.; Oliveira, T.B.; Faleiro, D.P.V.; Oliveira, R.B.; Costa, F.B.D. Outstanding anti-inflammatory potential of selected Asteraceae species through the potent dual inhibition of Cyclooxygenase-1 and 5-Lipoxygenase. Planta Med. 2015, 81, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Marquina, S.; Maldonado, N.; Gardun, M.L.; Aranda, E.; Villarreal, M.L.; Navarro, V.; Bye, R.; Delgado, G.; Alvarez, L. Bioactive oleanolic acid saponins and other constituents from the roots of Viguiera decurrens. Phytochemistry 2001, 56, 93–97. [Google Scholar] [CrossRef]
- Guillet, G.; Chauret, D.; Arnason, J.T. Phototoxic polyacetylenes from Viguiera annua and adaptations of a Chrysomelide Beetle, Zygogramma continua, feeding on this plant. Phytochemistry 1997, 45, 695–699. [Google Scholar] [CrossRef]
- Ambrosio, S.R.; Tirapelli, C.R.; Fernando, B.; Oliveira, A.M.D. Kaurane and pimarane-type diterpenes from the Viguiera species inhibit vascular smooth muscle contractility. Life Sci. 2006, 79, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.J.; Khalid, S.A.; Romanha, A.J.; Alves, T.M.; Biavatti, M.W.; Brun, R.; da Costa, F.B.; de Castro, S.L.; Ferreira, V.F.; de Lacerda, M.V. The potential of secondary metabolites from plants as drugs or leads against protozoan neglected diseases—Part I. Curr. Med. Chem. 2012, 19, 2128–2175. [Google Scholar] [PubMed]
- Schmidt, T.J.; da Costa, F.B.; Lopes, N.P.; Kaiser, M.; Brun, R. Silico prediction and experimental evaluation of furanoheliangolide sesquiterpene lactones as potent agents against Trypanosoma brucei rhodesiense. Antimicrob. Agents Chemother. 2014, 58, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, M.S. The Use of Chemometric and Chemoinformatic Tools for Identification and Targeted Isolation of Compounds from Asteraceae with Antiprotozoal Activity. Ph.D. Thesis, IPBP, University of Münster, Münster, Germany, 2016; p. 253. [Google Scholar]
- Nogueira, M.S.; da Costa, F.B.; Magenta, M.A.; Kaiser, M.; Brun, R.; Schmidt, T.J. Screening of some Asteraceae to discover new active compounds against Trypanosoma brucei by metabolite profiling and PLS analysis. Planta Med. 2013, 79, SL4. [Google Scholar] [CrossRef]
- Lloyd, H.A.; Fallis, A. Terpene alcohols of Helichrysum dendroideum. Tetr. Lett. 1967, 48, 4891–4895. [Google Scholar] [CrossRef]
- Dutra, L.M.; Bomfim, L.M.; Rocha, S.L.A.; Nepel, A.; Soares, M.B.P.; Barison, A.; Costa, E.V.; Bezerra, D.P. ent-Kaurane diterpenes from the stem bark of Annona vepretorum (Annonaceae) and cytotoxic evaluation. Bioorgan. Med. Chem. Lett. 2014, 24, 3315–3320. [Google Scholar] [CrossRef] [PubMed]
- Seca, A.M.L.; Pinto, D.C.G.A.; Silva, A.M.S. Structural elucidation of pimarane and isopimarane diterpenoids: The 13C-NMR contribution. Nat. Prod. Commun. 2008, 3, 399–412. [Google Scholar]
- Lightner, D.A.; Gurst, E.J. Organic Analysis and Stereochemistry from Circular Dichroism Spectroscopy; Wiley-VCH: New York, NY, USA, 2000; p. 487. [Google Scholar]
- Schmidt, T.J.; Nour, A.M.M.; Khalid, S.A.; Kaiser, M.; Brun, R. Quantitative structure—Antiprotozoal activity relationships of sesquiterpene lactones. Molecules 2009, 14, 2062–2076. [Google Scholar] [CrossRef] [PubMed]
- Asili, J.; Lambert, M.; Ziegler, H.L.; Stærk, D.; Sairafianpour, M.; Witt, M.; Asghari, G.; Ibrahimi, I.S.; Jaroszewski, J.W. Erythrocyte membrane and on Plasmodium falciparum growth in the erythrocyte host cells. J. Nat. Prod. 2004, 67, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Thongnest, S.; Mahidol, C.; Sutthivaiyakit, S.; Ruchirawat, S. Oxygenated pimarane diterpenes from Kaempferia marginata. J. Nat. Prod. 2005, 68, 1632–1636. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, H.L.; Franzyk, H.; Sairafianpour, M.; Tabatabai, M.; Tehrani, M.D.; Bagherzadeh, K.; Hägerstrand, H.; Stærk, D.; Jaroszewski, J.W. Erythrocyte membrane modifying agents and the inhibition of Plasmodium falciparum growth: Structure-activity relationships for betulinic acid analogues. Bioorgan. Med. Chem. 2004, 12, 119–127. [Google Scholar] [CrossRef]
- Ziegler, H.L.; Hansen, H.S.; Staerk, D.; Christensen, S.B.; Hägerstrand, H.; Jaroszewski, J.W. The antiparasitic compound licochalcone A is a potent echinocytogenic agent that modifies the erythrocyte membrane in the concentration range where antiplasmodial activity is observed. Antimicrob. Agents Chemother. 2004, 48, 4067–4071. [Google Scholar] [CrossRef] [PubMed]
- Otoguro, K.; Iwatsuki, M.; Ishiyama, A.; Namatame, M.; Nishihara-Tukashima, A.; Kiyohara, H.; Hashimoto, T.; Asakawa, Y.; Mura, S.; Yamada, H. In vitro antitrypanosomal activity of plant terpenes against Trypanosoma brucei. Phytochemistry 2011, 72, 2024–2030. [Google Scholar] [CrossRef] [PubMed]
- Wagner, H.; Bladt, S. Plant Drug Analysis: A Thin Layer Chromatography Atlas, 2nd ed.; Springer: Berlin, Germany, 2001; p. 383. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision C.02, 2004; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Schmidt, T.; Vößing, S.; Klaes, M.; Grimme, S. An aryldihydronaphthalene lignan with a novel type of ring system and further new lignans from Linum perenne L. Planta Med. 2007, 73, 1574–1580. [Google Scholar] [CrossRef] [PubMed]
- Barbic, M.; Schmidt, T.J.; Jügenliemk, G. Novel phenyl-1-benzoxepinols from butcher’s broom (Rusci rhizoma). Chem. Biodivers. 2012, 9, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of compounds 1–4 are available from the corresponding author on request.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extract | T. b. rhodesiense | T. cruzi | L. donovani | P. falciparum | Yield (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| 2 µg/mL | 10 µg/mL | 2 µg/mL | 10 µg/mL | 2 µg/mL | 10 µg/mL | 2 µg/mL | 10 µg/mL | ||
| Hex | 31.2 | 100.0 | 0.0 | 6.8 | 25.0 | 86.9 | - | 99.8 | 3.0 |
| DCM | 100.0 | 100.0 | 0.0 | 0.0 | 21.1 | 86.4 | 5.7 | 99.7 | 28.0 |
| EtOAc | 100.0 | 99.8 | 6.9 | 14.8 | 14.2 | 87.8 | 17.8 | 100.0 | 25.2 |
| MeOH | 100.0 | 100.0 | 0.0 | 5.4 | 34.1 | 69.0 | 45.2 | 87.4 | 9.4 |
| 1 | 2 | 3 | 4 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δ C | δ H | J (Hz) | δ C | δ H | J (Hz) | δ C | δ H | J (Hz) | δ C | δ H | J (Hz) | |
| 1 | 38.6 | β 0.89 ddd | 35.5 | a 1.84 ddd | 16.6, 12.8, 3.8 | 34.3 | α 1.88 dt | 13, 3.5 | 36.8 | α 1.79 m | 13, 11, 3.6 | |
| α 1.87 ddd | b 1.28 ddd | 16.6, 12.8, 4.1 | β 1.42 ddd | 13, 4.2, 3.5 | β 1.21 m | 13, 6, 2.7 | ||||||
| 2 | 27.9 | β 1.84 ddd | 13.4, 11.8, 3.6 | 18.3 | a 1.65 dt | 13.8, 3.8 | 27.4 | α 1.69 ddd | 13.3, 11.7, 3.5 | 81 | α 3.53 ddd | 11.5, 4.6, 1.3 |
| α 1.71 ddd | 13.4, 8, 4.4 | b 1.71 dt | 13.8, 3.5 | β 1.77 dq | 13.3, 3.9 | |||||||
| 3 | 81.1 | 3.42 ddd | 11.8, 4.4, 1.3 | 34.6 | a 1.33 ddt | 12.8, 3.8, 1.3 | 78.1 | 3.29 dd | 11.7, 4.4 | 28.1 | a 1.74 m | |
| b 1.52 ddd | 13.8, 12.8, 4.2 | b 1.78 ddd | 13.2, 3.6, 1.8 | |||||||||
| 4 | 43.1 | 37.7 | 38.9 | 42.4 | ||||||||
| 5 | 55.9 | 0.87 ddd | 11.8, 6, 1.7 | 43.2 | 2.08 dd | 13.4, 4.5 | 49.1 | 1.69 dd | 11.7, 3.9 | 47.5 | 1.71 m | |
| 6 | 20.2 | β 1.30 ddd | 16, 11.8, 3.8 | 35.3 | α 2.37 m | 35.1 | α 2.42 dd | 17.7, 11.7 | 28.9 | α 1.55 m | ||
| α 1.64 m | β 2.42 m | β 2.51 dd | 17.7,3.9 | β 1.88 dt | 13.9, 3.8 | |||||||
| 7 | 41.5 | β 1.47 m | 199.8 | 199.8 | 72.8 | α 4.25 dd | 6.8, 3.8 | |||||
| α 1.51 m | 175.2 | 176.2 | ||||||||||
| 8 | 44.1 | 129.1 | 129.5 | 140.1 | ||||||||
| 9 | 56 | 1.03 d | 6.9 | 165.8 | 164.7 | 46.7 | 2.05 ddd | 10.2, 6.4, 2.1 | ||||
| 10 | 38.9 | 39.5 | 39.5 | 38.1 | ||||||||
| 11 | 18.5 | β 1.62 m | 22.8 | α 2.20 m | 22.8 | α 2.19 m | 18.9 | α 1.54 m | ||||
| α 1.52 ddd | 13.4, 6.9, 3.6 | β 2.31 m | β 2.27 m | β 1.30 m | ||||||||
| 12 | 39.7 | β 1.91 d | 11.5 | 33.1 | α 1.47 m | 33 | α 1.43 m | 35.4 | α 1.57 m | |||
| α 1.09 ddt | 11.5, 5, 1.8 | β 1.44 m | β 1.46 m | β 1.23 m | ||||||||
| 13 | 44 | 2.64 t | 5 | 34.3 | 34.2 | 38.8 | ||||||
| 14 | 33.2 | pax 1.47 m | 33.6 | β 1.99 dq | 17, 2.3 | 33.6 | α 2.30 dt | 17, 2 | 133.9 | 5.47 t | 1.7 | |
| peq 1.63 m | α 2.31 dq | 17, 2.3 | β 1.99 dq | 17, 1.4 | ||||||||
| 15 | 49 | 2.05 t | 2.5 | 147.4 | 5.75 dd | 17.7, 10.6 | 147.2 | 5.74 dd | 17.2, 10.7 | 146.5 | 5.68 dd | 17.3, 10.4 |
| 16 | 155.7 | 110.8 | 4.89 dd | 17.7, 1 | 110.9 | 4.88 dd | 17.2, 1.4 | 113.5 | 4.82 dd | 17.3, 1.7 | ||
| 4.91 dd | 10.6, 1 | 4.9 | 10.7, 1.4 | 4.97 dd | 10.4, 1.7 | |||||||
| 17 | 103.3 | 4.73 ddt | 2.5, 1.8, 1.3 | 24.8 | 0.95 s | 24.8 | 0.94 s | 29.2 | 1.02 s | |||
| 4.79 m | 119 | 119 | ||||||||||
| 18 | 22.8 | 1.22 s | 70.9 | a 3.13 d | 10.9 | 27.5 | 1.0 s | 22.4 | 1.27 s | |||
| b 3.41 d | 10.9 | |||||||||||
| 19 | 64.5 | a 4.20 d | 11.2 | 17.5 | 0.87 s | 15.3 | 0.89 s | 64.4 | a 4.25 d | 11.1 | ||
| b 3.31 dd | 11.2, 1.3 | b 3.34 dd | 11.1, 1.5 | |||||||||
| 20 | 18.3 | 0.98 s | 18.8 | 1.10 s | 18.3 | 1.06 s | 15.2 | 0.65 s | ||||
| Comp. | T. b. rhodesiense | SI | T. cruzi | SI | L. donovani | SI | P. falciparum | SI | L6 Cells |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 20.1 ± 0.5 | 2 | 55.6 | 1 | 2.5 ± 1.5 | 16 | 3.5 ± 0.2 | 11 | 40.2 ± 5.5 |
| 2 | 24.3 | 2 | 15.4 | 3 | 18.2 | 3 | 3.8 | 13 | 48.3 ± 5.9 |
| 3 | NA | 19.4 | 4 | 13.8 | 5 | 16.5 ± 4.9 | 4 | 69.3 ± 10.9 | |
| 4 | 47.3 ± 4.1 | 2 | 58.9 | 2 | 21.9 | 5 | 16.1 ± 6.1 | 6 | 101.0 ± 45.7 |
| Pos. controls | 0.025 ± 0.0005 | 0.653 ± 0.135 | 0.049 ± 0.001 | 0.003 ± 0.001 | 0.008 + 0.001 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nogueira, M.S.; Da Costa, F.B.; Brun, R.; Kaiser, M.; Schmidt, T.J. ent-Pimarane and ent-Kaurane Diterpenes from Aldama discolor (Asteraceae) and Their Antiprotozoal Activity. Molecules 2016, 21, 1237. https://doi.org/10.3390/molecules21091237
Nogueira MS, Da Costa FB, Brun R, Kaiser M, Schmidt TJ. ent-Pimarane and ent-Kaurane Diterpenes from Aldama discolor (Asteraceae) and Their Antiprotozoal Activity. Molecules. 2016; 21(9):1237. https://doi.org/10.3390/molecules21091237
Chicago/Turabian StyleNogueira, Mauro S., Fernando B. Da Costa, Reto Brun, Marcel Kaiser, and Thomas J. Schmidt. 2016. "ent-Pimarane and ent-Kaurane Diterpenes from Aldama discolor (Asteraceae) and Their Antiprotozoal Activity" Molecules 21, no. 9: 1237. https://doi.org/10.3390/molecules21091237