
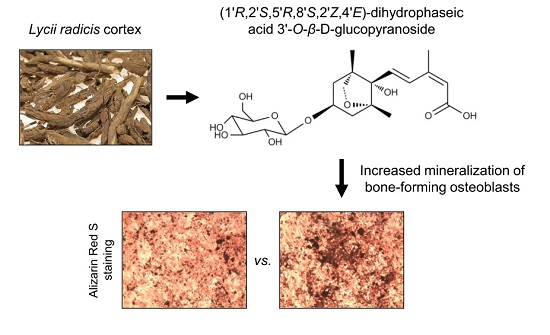
Effects of Dihydrophaseic Acid 3′-O-β-d-Glucopyranoside Isolated from Lycii radicis Cortex on Osteoblast Differentiation

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
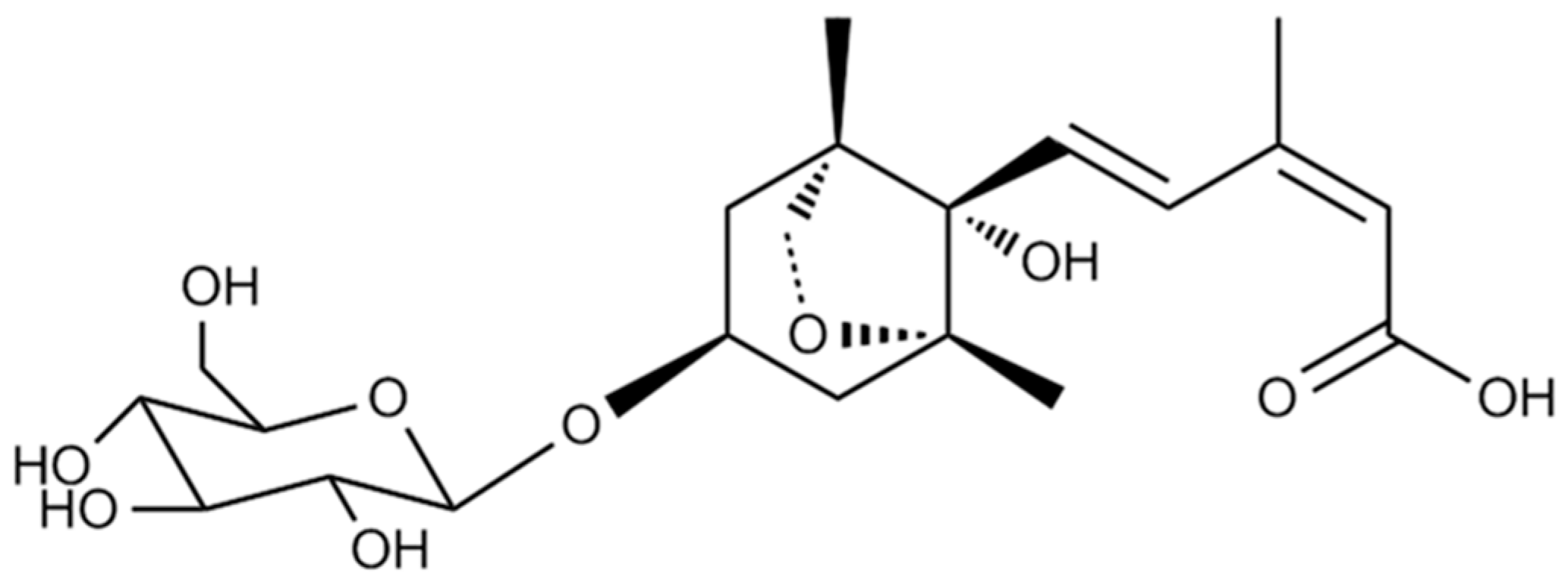
2.1. Isolation and Identification of DPA3G as a Bioactive Component of the LRC Extract for Enhancing Osteoblast Differentiation
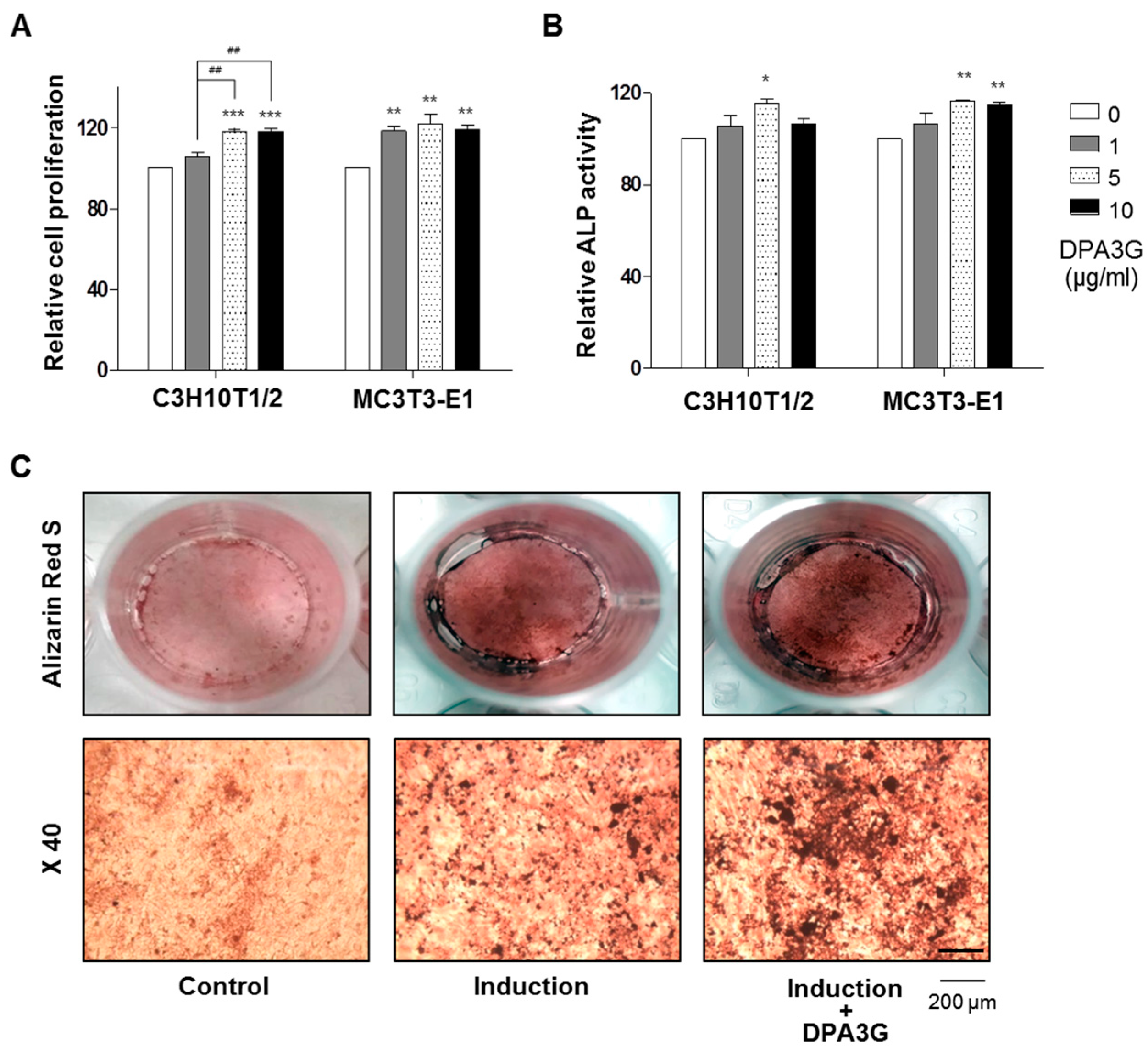
2.2. DPA3G Increased the Cellular Proliferation, Differentiation, and Mineralized Nodule Formation of Osteoblasts
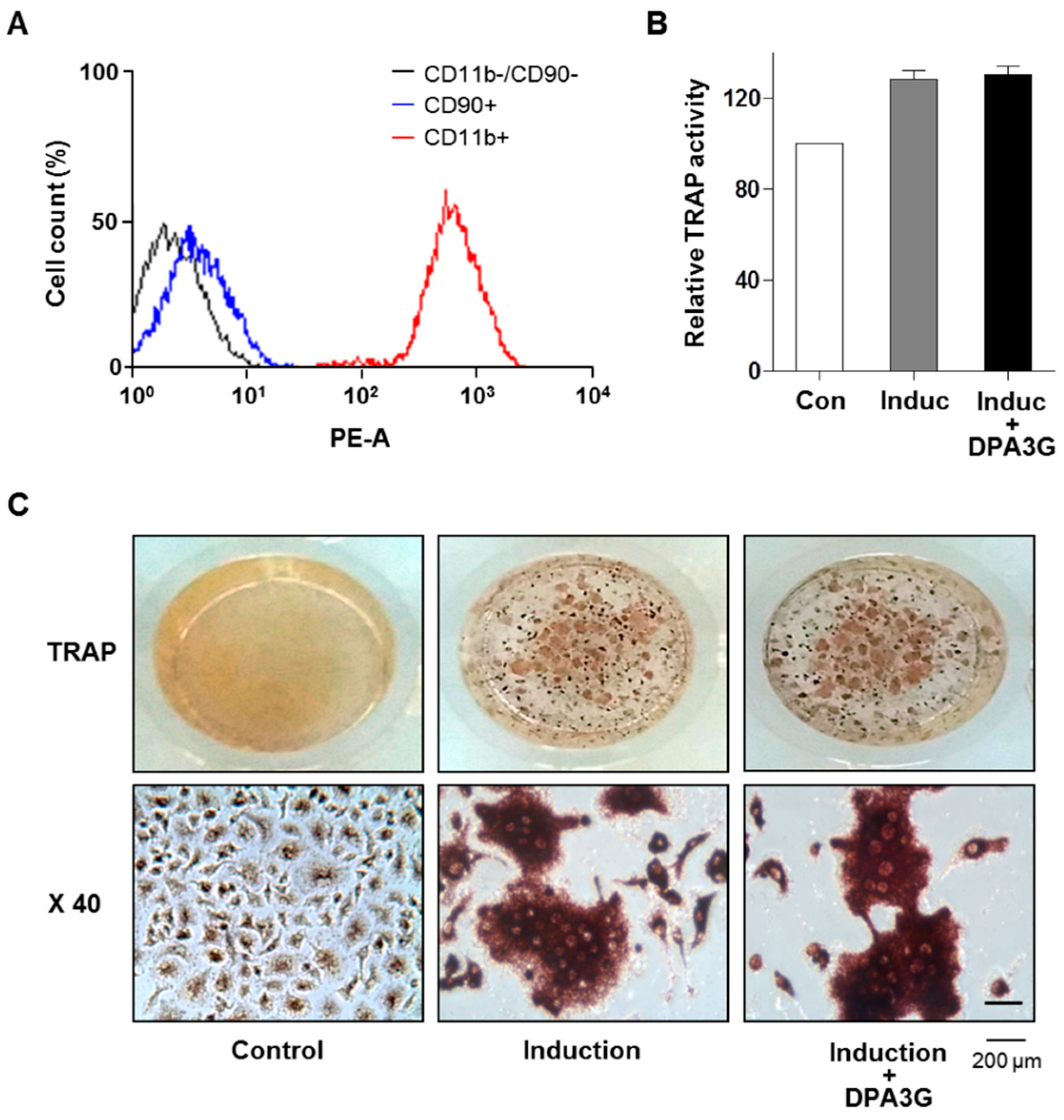
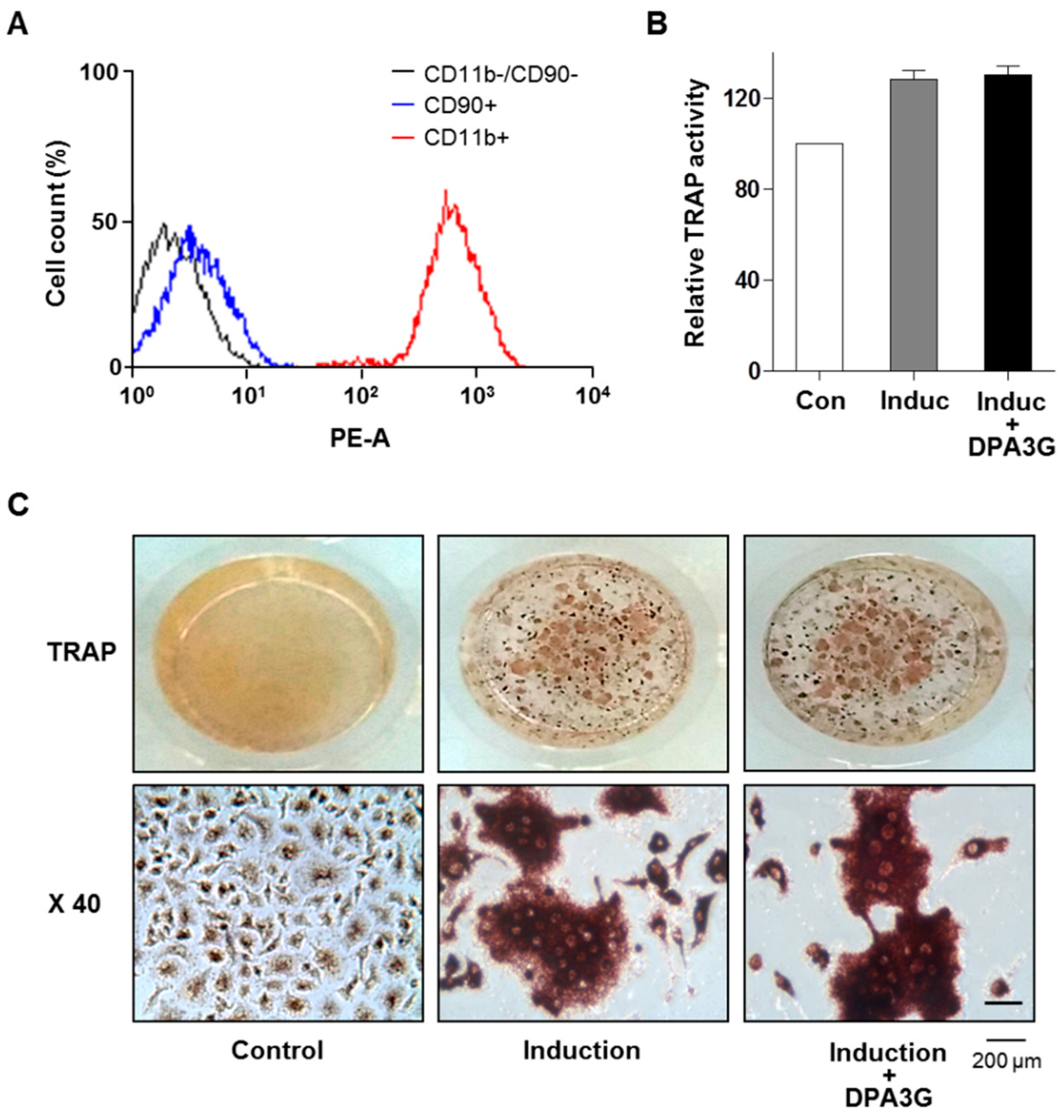
2.3. DPA3G Did Not Influence Differentiation of Osteoclasts
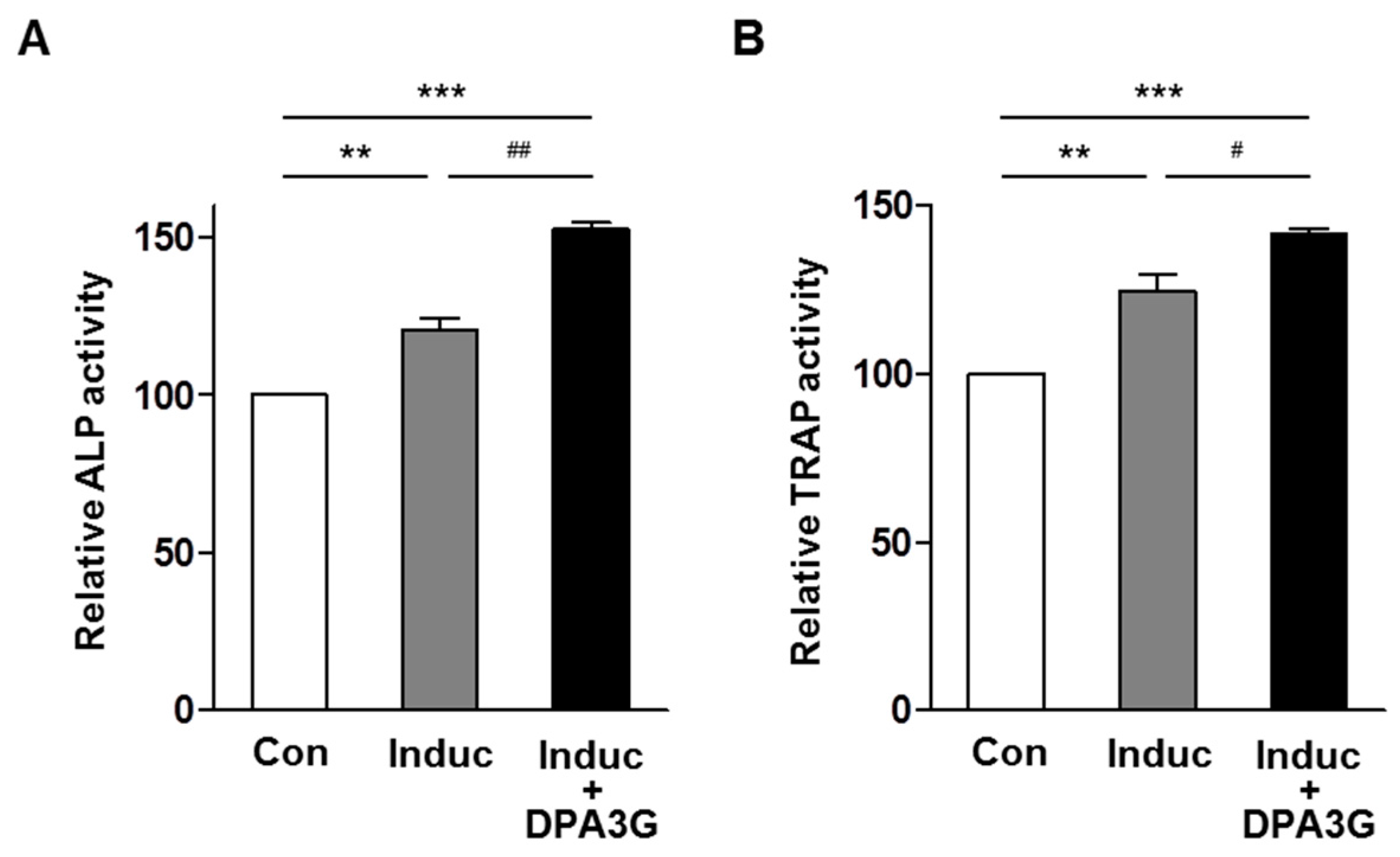
2.4. DPA3G Enhanced both Osteoblast and Osteoclast Differentiation in the MC3T3-E1 and Primary Monocyte Co-Culture System
3. Experimental Section
3.1. Fractionation, Isolation, and Structure Elucidation of the Bioactive Component
3.2. Cell Culture
3.3. Water-soluble Tetrazolium Salt (WST) Assay in Osteoblast Cells
3.4. Alkaline Phosphatase (ALP) Activity Assay in Osteoblast Cells
3.5. Mineralized Nodule Formation in Osteoblast Cells
3.6. Quantitative Reverse-Transcription PCR (qRT-PCR)
3.7. Osteoclastogenesis of Primary Monocytes and Tartrate-Resistant Acid Phosphatase (TRAP) Activity Assay and Staining
3.8. Co-Culture of MC3T3-E1 Cells and Primary Monocytes
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contribution
Conflict of Interest
References
- Feng, X.; McDonald, J.M. Disorders of bone remodeling. Annu. Rev. Pathol. 2011, 6, 121–145. [Google Scholar] [CrossRef] [PubMed]
- Raggatt, L.J.; Partridge, N.C. Cellular and molecular mechanisms of bone remodeling. J. Biol. Chem. 2010, 285, 25103–25108. [Google Scholar] [CrossRef] [PubMed]
- Fakhry, M.; Hamade, E.; Badran, B.; Buchet, R.; Magne, D. Molecular mechanisms of mesenchymal stem cell differentiation towards osteoblasts. World J. Stem Cells 2013, 5, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Rosenberg, E.; de Papp, A.E.; Duong, L.T. The osteoclast, bone remodelling and treatment of metabolic bone disease. Eur. J. Clin. Investig. 2012, 42, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Kikuta, J.; Ishii, M. Osteoclast migration, differentiation and function: novel therapeutic targets for rheumatic diseases. Rheumatology (Oxford) 2013, 52, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Irie, N. Osteoclast-osteoblast communication. Arch. Biochem. Biophys. 2008, 473, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.A.; Martin, T.J. Coupling the activities of bone formation and resorption: A multitude of signals within the basic multicellular unit. Bonekey Rep. 2014, 3, 481. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, N. Signaling Pathways in Osteoclast Differentiation. Chonnam Med. J. 2016, 52, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Nicolaidou, V.; Wong, M.M.; Redpath, A.N.; Ersek, A.; Baban, D.F.; Williams, L.M.; Cope, A.P.; Horwood, N.J. Monocytes induce STAT3 activation in human mesenchymal stem cells to promote osteoblast formation. PLoS ONE 2012, 7, e39871. [Google Scholar] [CrossRef] [PubMed]
- Rachner, T.D.; Khosla, S.; Hofbauer, L.C. Osteoporosis: now and the future. Lancet 2011, 377, 1276–1287. [Google Scholar] [CrossRef]
- Sambrook, P.; Cooper, C. Osteoporosis. Lancet 2006, 367, 2010–2018. [Google Scholar] [CrossRef]
- Yoshida, K.; Oida, H.; Kobayashi, T.; Maruyama, T.; Tanaka, M.; Katayama, T.; Yamaguchi, K.; Segi, E.; Tsuboyama, T.; Matsushita, M.; et al. Stimulation of bone formation and prevention of bone loss by prostaglandin E EP4 receptor activation. Proc. Natl. Acad. Sci. USA 2002, 99, 4580–4585. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Crockett, J.C. Osteoporosis-A current view of pharmacological prevention and treatment. Drug Des. Devel. Ther. 2013, 7, 435–448. [Google Scholar] [PubMed]
- Gennari, L.; Rotatori, S.; Bianciardi, S.; Nuti, R.; Merlotti, D. Treatment needs and current options for postmenopausal osteoporosis. Expert Opin. Pharmacother. 2016, 17, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Harslof, T.; Langdahl, B.L. New horizons in osteoporosis therapies. Curr. Opin. Pharmacol. 2016, 28, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Ma, Q.; Ye, L.; Piao, G. The Traditional Medicine and Modern Medicine from Natural Products. Molecules 2016, 21, 559. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.; McLachlan, A.J.; Sherwin, C.M.; Enioutina, E.Y. Herbal medicines: Challenges in the modern world. Part 1. Australia and New Zealand. Expert Rev. Clin. Pharmacol. 2016, 9, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Sammons, H.M.; Gubarev, M.I.; Krepkova, L.V.; Bortnikova, V.V.; Corrick, F.; Job, K.M.; Sherwin, C.M.; Enioutina, E.Y. Herbal medicines: challenges in the modern world. Part 2. European Union and Russia. Expert Rev. Clin. Pharmacol. 2016, 9, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Mukwaya, E.; Xu, F.; Wong, M.S.; Zhang, Y. Chinese herbal medicine for bone health. Pharm Biol. 2014, 52, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.D.; Han, T.; Huang, B.K.; Rahman, K.; Jiang, Y.P.; Xu, H.T.; Qin, L.P.; Xin, H.L.; Zhang, Q.Y. Traditional Chinese Medicine formulas for the treatment of osteoporosis: Implication for antiosteoporotic drug discovery. J. Ethnopharmacol. 2016, 189, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Che, C.T.; Wong, M.S.; Lam, C.W. Natural Products from Chinese Medicines with Potential Benefits to Bone Health. Molecules 2016, 21, 239. [Google Scholar] [CrossRef] [PubMed]
- Li, T.M.; Huang, H.C.; Su, C.M.; Ho, T.Y.; Wu, C.M.; Chen, W.C.; Fong, Y.C.; Tang, C.H. Cistanche deserticola extract increases bone formation in osteoblasts. J. Pharm. Pharmacol. 2012, 64, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Jin, H.S.; Cho, D.Y.; Kim, J.; Kim, M.C.; Chio, C.W.; Lee, J.W.; Park, J.H.; Chung, Y.S.; Huh, D.; et al. The effect of Lycii Radicis Cortex extract on bone formation in vitro and in vivo. Molecules 2014, 19, 19594–19609. [Google Scholar] [CrossRef] [PubMed]
- Potterat, O. Goji (Lycium barbarum and L. chinense): Phytochemistry, pharmacology and safety in the perspective of traditional uses and recent popularity. Planta Med. 2010, 76, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; Guan, S.H.; Feng, R.H.; Wang, Y.; Wu, Z.Y.; Zhang, Y.B.; Chen, X.H.; Bi, K.S.; Guo, D.A. Neolignanamides, lignanamides, and other phenolic compounds from the root bark of Lycium chinense. J. Nat. Prod. 2013, 76, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, E.Y.; Lee, B.; Min, J.H.; Song, D.U.; Lim, J.M.; Eom, J.W.; Yeom, M.; Jung, H.S.; Sohn, Y. The effects of Lycii Radicis Cortex on RANKL-induced osteoclast differentiation and activation in RAW 264.7 cells. Int. J. Mol. Med. 2016, 37, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, J.S.; Lee, J.H.; Kim, Y.S.; Kang, S.S. A New Saponin from the Seeds of Zizyphus jujuba var. spinosa. Bull. Korean Chem. Soc. 2013, 34, 657–660. [Google Scholar] [CrossRef]
- Youn, U.J.; Lee, J.; Nam, J.W.; Lee, Y.J.; Seo, E.K. Identification of a New Isomer of Dihydrophaseic Acid 3'-O-β-d-Glucopyranoside from Nelumbo nucifera. Bull. Korean Chem. Soc. 2011, 32, 4083–4085. [Google Scholar] [CrossRef]
- Ngan, N.T.; Quang, T.H.; Tai, B.H.; Song, S.B.; Lee, D.; Kim, Y.H. Anti-inflammatory and PPAR transactivational effects of components from the stem bark of Ginkgo biloba. J. Agric. Food Chem. 2012, 60, 2815–2824. [Google Scholar] [CrossRef] [PubMed]
- Tarapore, R.S.; Lim, J.; Tian, C.; Pacios, S.; Xiao, W.; Reid, D.; Guan, H.; Mattos, M.; Yu, B.; Wang, C.Y.; et al. NF-kappaB Has a Direct Role in Inhibiting Bmp- and Wnt-Induced Matrix Protein Expression. J. Bone Miner. Res. 2016, 31, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Moussa, F.M.; Mankoci, S.; Ustriyana, P.; Zhang, N.; Abdelmagid, S.; Molenda, J.; Murphy, W.L.; Safadi, F.F.; Sahai, N. Orthosilicic acid, Si(OH)4, stimulates osteoblast differentiation in vitro by upregulating miR-146a to antagonize NF-kappaB activation. Acta Biomater. 2016, 39, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Scholtysek, C.; Katzenbeisser, J.; Fu, H.; Uderhardt, S.; Ipseiz, N.; Stoll, C.; Zaiss, M.M.; Stock, M.; Donhauser, L.; Bohm, C.; et al. PPARbeta/delta governs Wnt signaling and bone turnover. Nat. Med. 2013, 19, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.B.; Stein, G.S. Development of the osteoblast phenotype: molecular mechanisms mediating osteoblast growth and differentiation. Iowa Orthop. J. 1995, 15, 118–140. [Google Scholar] [PubMed]
- Komori, T. Signaling networks in RUNX2-dependent bone development. J. Cell Biochem. 2011, 112, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Neve, A.; Corrado, A.; Cantatore, F.P. Osteocalcin: Skeletal and extra-skeletal effects. J. Cell. Physiol. 2013, 228, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Kida, K.; Yamaguchi, A.; Hata, K.; Ichida, F.; Meguro, H.; Aburatani, H.; Nishimura, R.; Yoneda, T. BMP2 regulates Osterix through Msx2 and Runx2 during osteoblast differentiation. J. Biol. Chem. 2008, 283, 29119–29125. [Google Scholar] [CrossRef] [PubMed]
- Balint, E.; Szabo, P.; Marshall, C.F.; Sprague, S.M. Glucose-induced inhibition of in vitro bone mineralization. Bone 2001, 28, 21–28. [Google Scholar] [CrossRef]
- Watts, N.B. Clinical utility of biochemical markers of bone remodeling. Clin. Chem. 1999, 45, 1359–1368. [Google Scholar] [PubMed]
- Liu, J.; Nam, H.K.; Campbell, C.; Gasque, K.C.; Millan, J.L.; Hatch, N.E. Tissue-nonspecific alkaline phosphatase deficiency causes abnormal craniofacial bone development in the Alpl(−/−) mouse model of infantile hypophosphatasia. Bone 2014, 67, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B. Normal bone anatomy and physiology. Clin. J. Am. Soc. Nephrol. 2008, 3 (Suppl. 3), S131–S139. [Google Scholar] [CrossRef] [PubMed]
- Gough, J.E.; Jones, J.R.; Hench, L.L. Nodule formation and mineralisation of human primary osteoblasts cultured on a porous bioactive glass scaffold. Biomaterials 2004, 25, 2039–2046. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Harruff, R.; Ambrosius, W.; Burr, D.B. Trabecular bone volume and microdamage accumulation in the femoral heads of women with and without femoral neck fractures. Bone 1997, 21, 521–526. [Google Scholar] [CrossRef]
- Komori, T. Regulation of osteoblast differentiation by transcription factors. J. Cell Biochem. 2006, 99, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Atalay, S.; Elci, A.; Kayadibi, H.; Onder, C.B.; Aka, N. Diagnostic utility of osteocalcin, undercarboxylated osteocalcin, and alkaline phosphatase for osteoporosis in premenopausal and postmenopausal women. Ann. Lab. Med. 2012, 32, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Shieh, A.; Han, W.; Ishii, S.; Greendale, G.A.; Crandall, C.J.; Karlamangla, A.S. Quantifying the Balance Between Total Bone Formation and Total Bone Resorption: An Index of Net Bone Formation. J. Clin. Endocrinol. Metab. 2016, 101, 2802–2809. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Ye, X.; Yu, X.; Xu, Q.; Pan, K.; Lu, S.; Yang, P. Co-culture with periodontal ligament stem cells enhanced osteoblastic differentiation of MC3T3-E1 cells and osteoclastic differentiation of RAW264.7 cells. Int. J. Clin. Exp. Pathol. 2015, 8, 14596–14607. [Google Scholar] [PubMed]
- Wu, L.; Feyerabend, F.; Schilling, A.F.; Willumeit-Romer, R.; Luthringer, B.J. Effects of extracellular magnesium extract on the proliferation and differentiation of human osteoblasts and osteoclasts in coculture. Acta. Biomater. 2015, 27, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, A.; Thieme, S.; Domaschke, H.; Springer, A.; Rosen-Wolff, A.; Gelinsky, M. Crosstalk of osteoblast and osteoclast precursors on mineralized collagen—Towards an in vitro model for bone remodeling. J. Biomed. Mater. Res. A 2010, 95, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Yang, X.G.; Wang, F.; Ma, X.Y. IL-1alpha induces apoptosis and inhibits the osteoblast differentiation of MC3T3-E1 cells through the JNK and p38 MAPK pathways. Int. J. Mol. Med. 2016, 38, 319–327. [Google Scholar] [PubMed]
- Zeng, Q.; Guo, Y.; Liu, Y.; Li, R.; Zhang, X.; Liu, L.; Wang, Y.; Zhang, X.; Zou, X. Integrin-beta1, not integrin-beta5, mediates osteoblastic differentiation and ECM formation promoted by mechanical tensile strain. Biol. Res. 2015, 48, 25. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not Available.

© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, E.; Kim, M.-C.; Choi, C.W.; Kim, J.; Jin, H.-S.; Lee, R.; Lee, J.-W.; Park, J.-H.; Huh, D.; Jeong, S.-Y. Effects of Dihydrophaseic Acid 3′-O-β-d-Glucopyranoside Isolated from Lycii radicis Cortex on Osteoblast Differentiation. Molecules 2016, 21, 1260. https://doi.org/10.3390/molecules21091260
Park E, Kim M-C, Choi CW, Kim J, Jin H-S, Lee R, Lee J-W, Park J-H, Huh D, Jeong S-Y. Effects of Dihydrophaseic Acid 3′-O-β-d-Glucopyranoside Isolated from Lycii radicis Cortex on Osteoblast Differentiation. Molecules. 2016; 21(9):1260. https://doi.org/10.3390/molecules21091260
Chicago/Turabian StylePark, Eunkuk, Mun-Chang Kim, Chun Whan Choi, Jeonghyun Kim, Hyun-Seok Jin, Ryunjin Lee, Ji-Won Lee, Jin-Hyok Park, Dam Huh, and Seon-Yong Jeong. 2016. "Effects of Dihydrophaseic Acid 3′-O-β-d-Glucopyranoside Isolated from Lycii radicis Cortex on Osteoblast Differentiation" Molecules 21, no. 9: 1260. https://doi.org/10.3390/molecules21091260