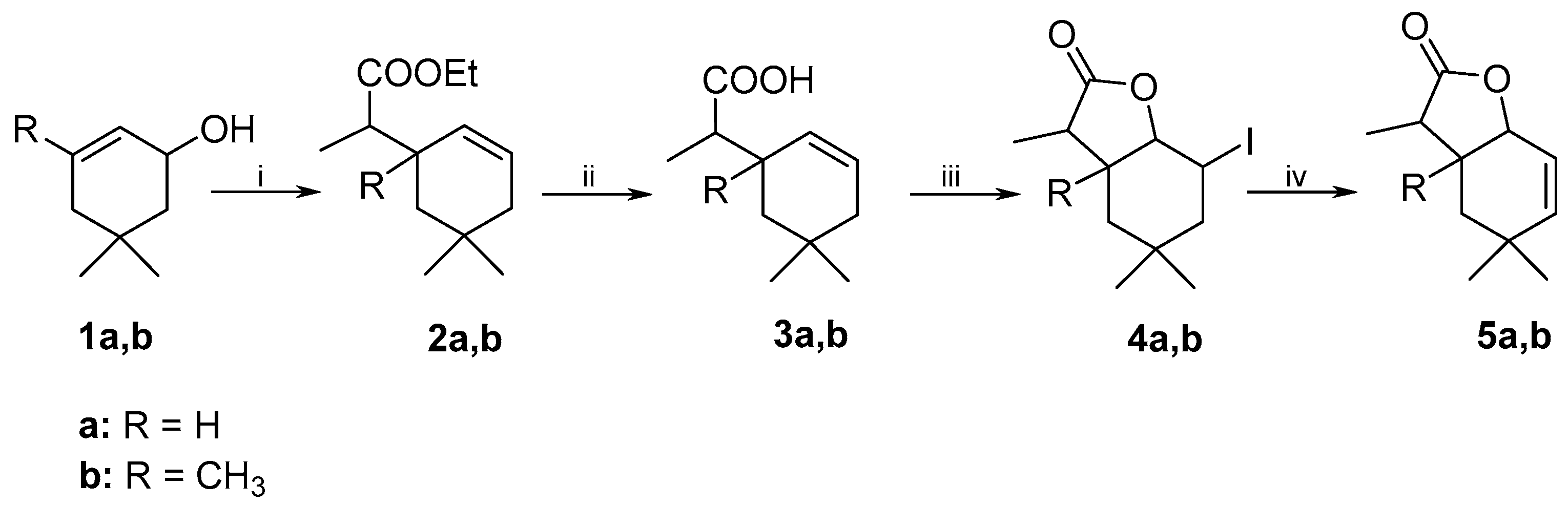
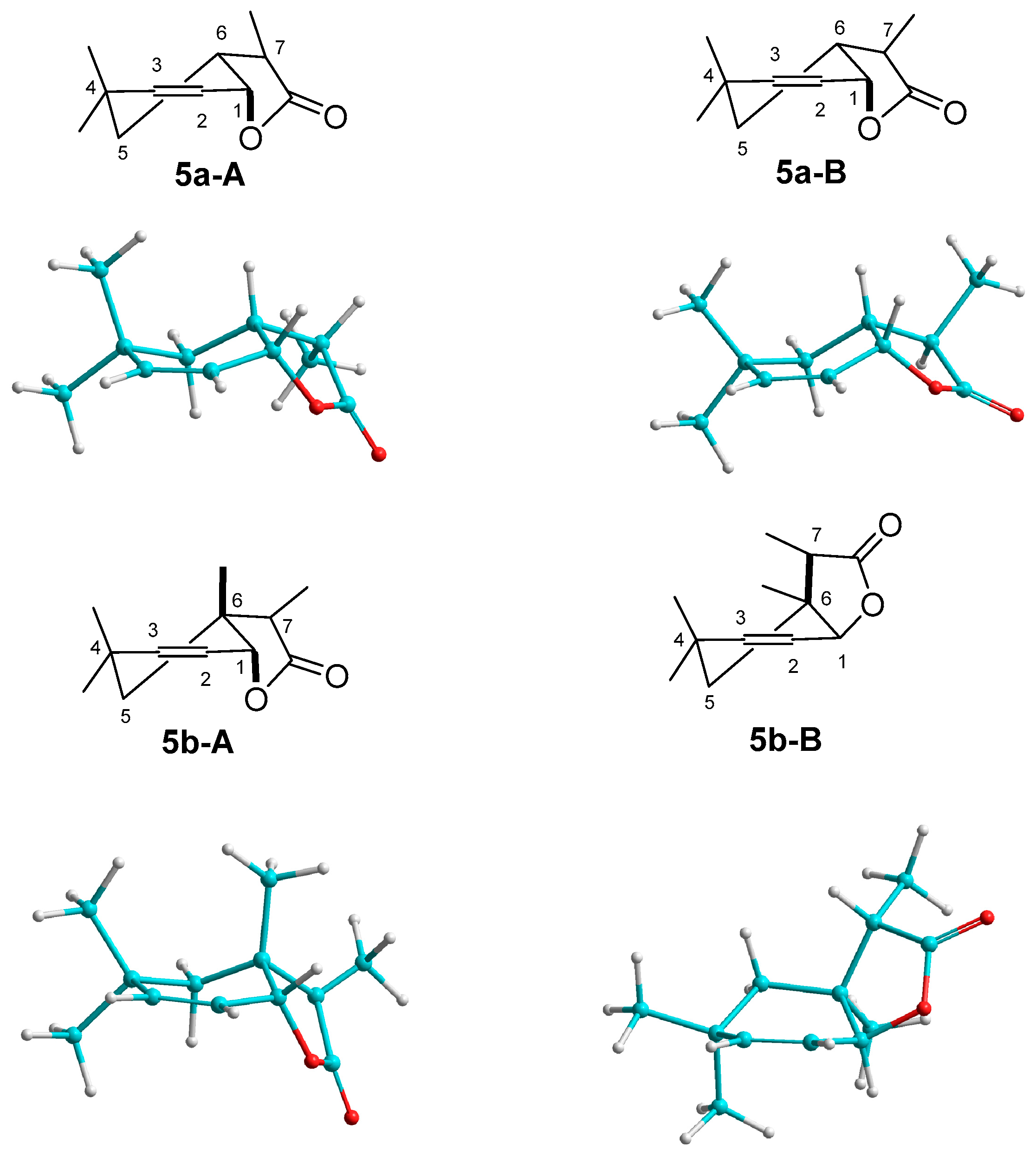
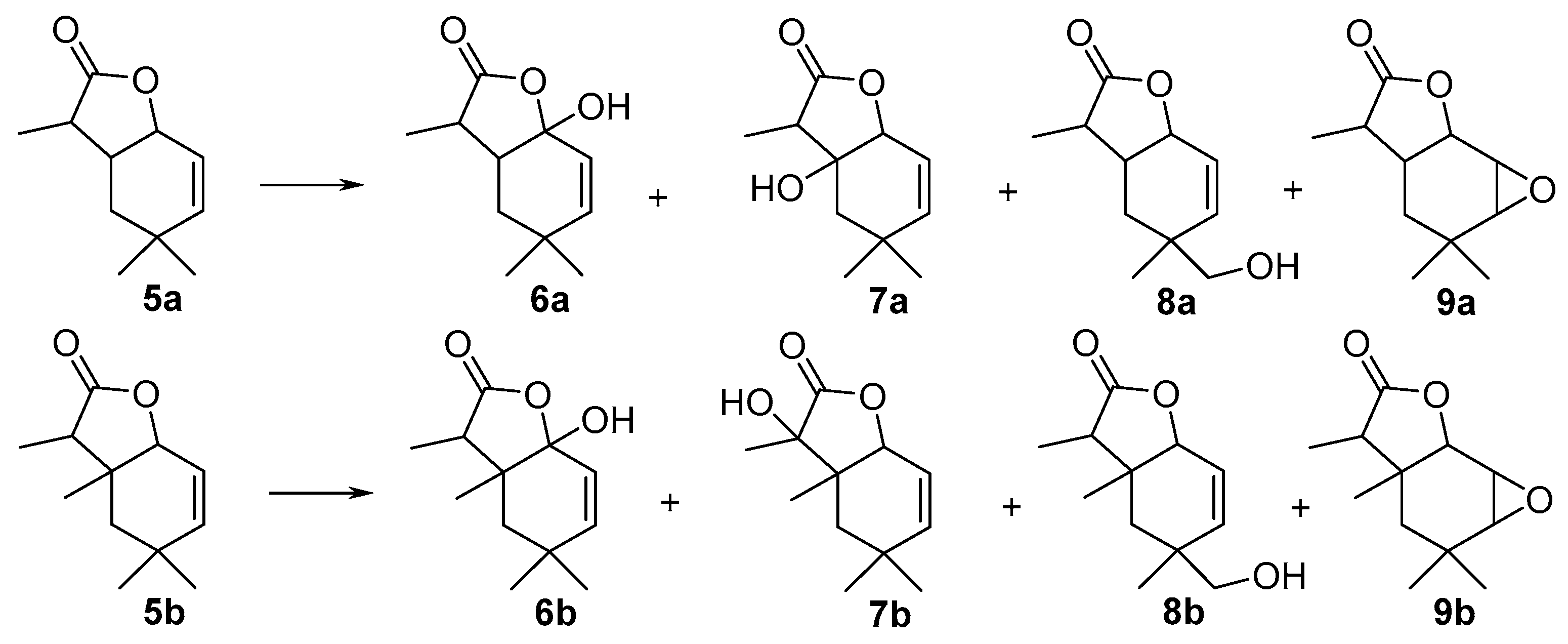
3.3.3. Preparative Biotransformation
Preparative biotransformation was carried out in ten 300 mL Erlenmeyer flasks containing cultures of 3-day fungal strains (prepared in a similar manner as described in the screening procedure). Substrate (100 mg) was dissolved in 10 mL of acetone and added to ten bottles. After 14 days the reaction mixture was extracted with dichloromethane (3 × 40 mL). The combined organic fractions were dried (MgSO4) and evaporated under reduced pressure. The pure product was purified by column chromatography (silica gel, hexane−acetone 3:1). As a result of these reactions two products were obtained.
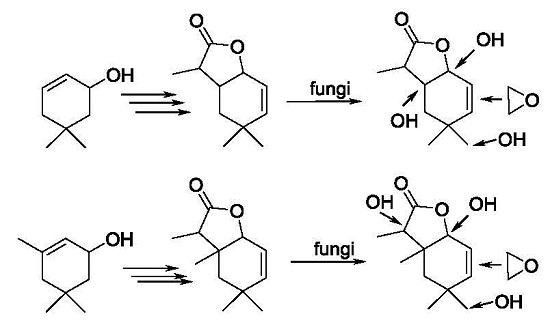
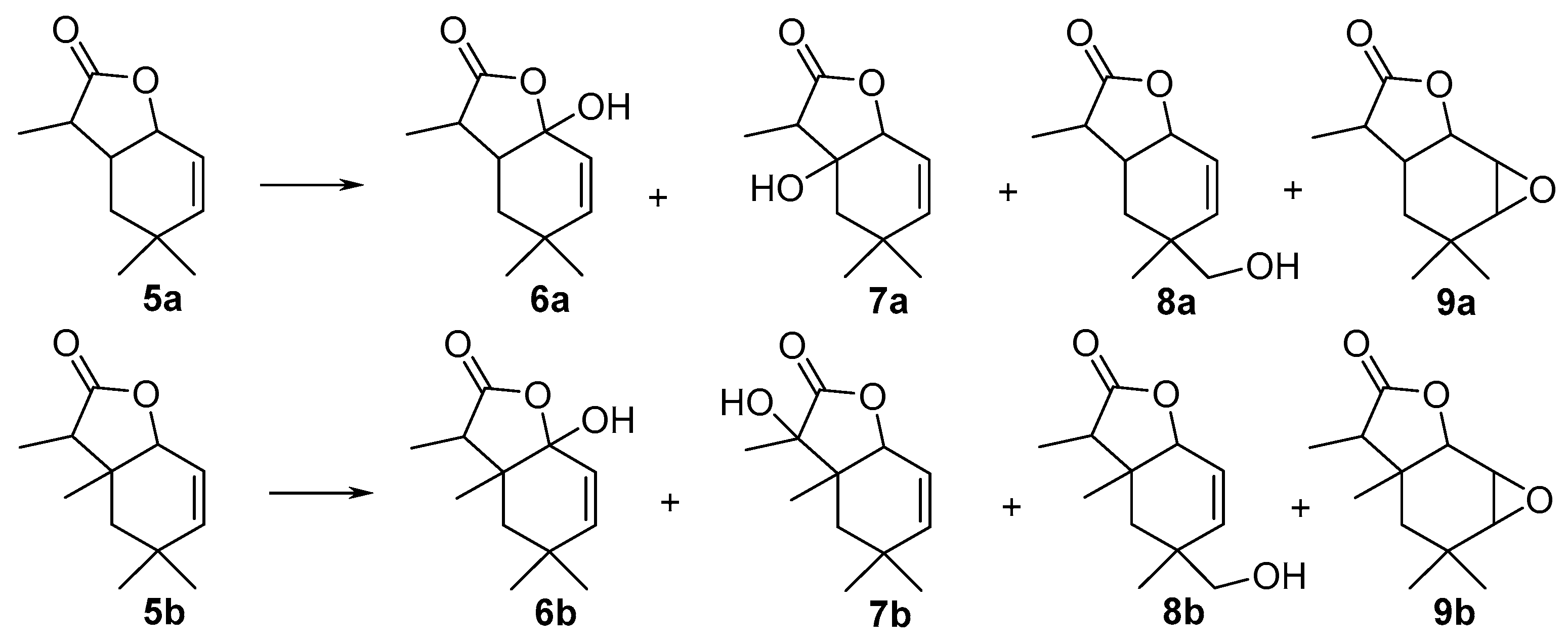
1-Hydroxy-4,4,7-trimethyl-9-oxabicyclo[4.3.0]non-2-en-8-one (
6a) was characterized by the following physical and spectral properties: m.p. = 77–78 °C;
1H-NMR: 1.14 (d,
J = 7.2 Hz, 3H, CH
3-11A), 1.19 (s, 3H, CH
3-9A), 1.20 (s, 3H, CH
3-9B), 1.23 (s, 3H, CH
3-10B), 1.24 (s, 3H, CH
3-10A), 1.25 (d,
J = 7.2 Hz, 3H, CH
3-11B), 1.78–1.79 (m, 2H, CH
2-5A), 1.80–1.81 (m, 1H, CH
2-5B), 1.95–2.00 (m, 1H, one of CH
2-5B), 2.91–2.95 (m, 1H, one of H-6B), 3.02–3.04 (m, 1H, H-7B), 3.06–3.09 (m, 1H, H-6A), 3.16–3.18 (m, 1H, H-7A), 5.86 (d,
J = 10.0 Hz, 1H, H-3A), 5.87 (d,
J = 10.0 Hz, 1H, H-3B), 6.63–6.66 (m, 2H, H-2A, H-2B);
13C-NMR: 12.0 (C-11A), 12.2 (C-11B), 24.3 (C-10A), 24.8 (C-10B), 30.1 (C-9B), 30.2 (C-9A), 33.2 (C-4A), 33.2 (C-4B), 37.4 (C-7B), 37.4 (C-7A), 38.2 (C-5B), 38.3 (C-5A), 44.3 (C-6A), 44.7 (C-6B), 125.9 (C-3A), 126.1 (C-3B), 158.6 (C-2B), 158.7 (C-2A), 179.4 (C-1B), 181.3 (C-1A), 198.6 (C-8A), 198.7 (C-8B); ESIHRMS: calcd. for C
11H
16O
3,
m/
z 197.1178 [M + H]
+, found 197.1173. (
Supplementary Materials, Figures S25–S29).
6-Hydroxy-4,4,7-trimethyl-9-oxabicyclo[4.3.0]non-2-en-8-one (
7a) was characterized by the following physical and spectral properties: m.p. = 76–77 °C;
1H-NMR: 1.04 (s, 3H, CH
3-9), 1.09 (s, 3H, CH
3-10), 1.43 (d,
J = 5.0 Hz, 3H, CH
3-11), 1.45 (dd,
J = 14.9 and 4.5 Hz, 2H, CH
2-5), 2.50 (dt,
J = 14.4 and 4.8 Hz, 1H, H-7), 5.03 (dd,
J = 8.8 and 4.4 Hz, 1H, H-1), 5.83 (dd,
J = 9.9 and 4.4 Hz, 1H, H-2), 5.94 (d,
J = 9.9 Hz, 1H, H-3);
13C-NMR: 19.1 (C-11), 26.7 (C-9), 30.3 (C-10), 32.1 (C-4), 34.0 (C-5), 42.0 (C-7), 73.0 (C-1), 77.7 (C-6), 119.6 (C-2), 145.1 (C-3), 177.1 (C-8); ESIHRMS: calcd. for C
11H
16O
3,
m/
z 197.1178 [M + H]
+, found 197.1176. (
Supplementary Materials, Figures S30–S34).
10-Hydroxy-4,4,7-trimethyl-9-oxabicyclo[4.3.0]non-2-en-8-one (
8a) was characterized by the following physical and spectral properties: colourless oil;
1H-NMR: 1.06 (s, 3H, CH
3-9), 1.20–1.22 (m, 1H, one of CH
2-5), 1.35 (d,
J = 7.6 Hz, 3H, CH
3-11), 1.88 (dd,
J = 13.6 and 4.9 Hz, 1H, one of CH
2-5), 2.40–2.44 (m, 1H, H-7), 2.45–2.49 (m, 1H, H-6), 3.40 (d,
J = 2.4 Hz, 2H, CH
2-10), 4.84 (dd,
J = 5.0 and 4.8 Hz, 1H, H-1), 5.82 (d,
J = 10.0 Hz, 1H, H-3), 5.94 (dd,
J = 10.0 and 3.9 Hz, 1H, H-2);
13C-NMR: 15.3 (C-11), 24.8 (C-9), 33.5 (C-5), 37.3 (C-4), 38.6 (C-7), 42.9 (C-6), 69.2 (C-10), 73.12 (C-1), 123.01 (C-2), 139. 9 (C-3), 179.8 (C-8); IR (KBr, cm
−1): 3468, 2964, 1785, 1456, 1170, 1024; ESIHRMS: calcd. for C
11H
16O
3,
m/
z 197.1178 [M + H]
+, found 197.1181. (
Supplementary Materials, Figures S35–S40).
2,3-Epoxy-4,4,7-trimethyl-9-oxabicyclo[4.3.0]nonan-8-one (
9a) was characterized by the following physical and spectral properties: colourless oil;
1H-NMR: 0.85 (dd,
J = 13.7 and 13.7 Hz, 1H, one of CH
2-5B), 1.05 (s, 3H, CH
3-9B), 1.08 (s, 3H, CH
3-9A), 1.15 (d,
J = 7.1 Hz, 3H, CH
3-11B), 1.17 (s, 6H, CH
3-10A, CH
3-10B), 1.18–1.19 (m, 1H, one of CH
2-5B), 1.20–1.21 (m, 2H, CH
2-5A), 1.32 (d,
J = 7.5 Hz, 3H, CH
3-11A), 12.15–2.20 (m, 1H, H-6A), 2.41–2.47 (m, 2H, H-6B and H-7A), 2.84 (quintet,
J = 7.1 Hz, 1H, H-7B), 3.02 (d,
J = 3.2 Hz, 1H, H-3A), 3.04 (d,
J = 3.3 Hz, 1H, H-3B), 3.46 (d,
J = 3.2 Hz, 1H, H-2A), 3.52 (dd,
J = 3.3 and 1.3 Hz, 1H, H-2B), 4.67 (d,
J = 4.2 Hz, 1H, H-1B), 4.82 (d,
J = 6.1 Hz, 1H, H-1A);
13C-NMR: 9.2 (C-11B), 14.4 (C-11A), 23.4 (C-9B), 25.4 (C-9A), 28.3 (C-4B), 28.8 (C-10A), 28. 8 (C-4A), 30.0 (C-10B), 33.2 (C-5B), 34.4 (C-6B), 35.7 (C-5A), 27.6 (C-6A), 39.9 (C-7B), 41.4 (C-7A), 52.3 (C-2B), 52.4 (C-2A), 61.1 (C-3A), 61.2 (C-3B), 73.5 (C-A), 74.1 (C-1B), 178.2 (C-8A), 178.2 (C-8B); IR (KBr, cm
−1): 2952, 1785, 1476, 1457, 1160, 1024; ESIHRMS: calcd. for C
11H
16O
3,
m/
z 197.1178 [M + H]
+, found 197.1179. (
Supplementary Materials, Figures S41–S46)
1-Hydroxy-4,4,6,7-tetramethyl-9-oxabicyclo[4.3.0]non-2-en-8-one (
6b) was characterized by the following physical and spectral properties: colourless oil;
1H-NMR: 1.13 (d,
J = 7.0 Hz, 3H, CH
3-11A), 1.20 (s, 3H, CH
3-9A), 1.21 (s, 3H, CH
3-9B), 1.23 (d,
J = 7.5 Hz, 3H, CH
3-11B), 1.28 (s, 6H, CH
3-10B, CH
3-12B), 1.30 (s, 3H, CH
3-10A), 1.34 (s, 3H, CH
3-12A), 1.65 (d,
J = 14.1 Hz, 1H, one of CH
2-5B), 1.86–1.89 (m, 1H, CH
2-5A), 2.02–2.05 (m, 1H, one of CH
2-5A), 2.19–2.22 (m, 1H, one of CH
2-5B), 3.01–3.02 (m, 1H, H-7B), 3.12–3.13 (m, 1H, H-7A), 5.86 (d,
J = 9.9 Hz, 1H, H-3A), 5.89 (d,
J = 10.1 Hz, 1H, H-3A), 6.60–6.62 (m, 2H, H-2A, H-2B);
13C-NMR: 10.9 (C-11B), 12.5 (C-11A), 24.5 (C-12A), 24.3 (C-10A), 28.8 (C-10B), 29.2 (C-12B), 32.2 (C-9B), 32.4 (C-9A), 40.8 (C-5A), 42.1 (C-5B), 44.7 (C-7B), 45.0 (C-7A), 44.7 (C-4A), 45.0 (C-4B), 44.4 (C-6A), 44.7 (C-6B), 124.4 (C-3A), 124.7 (C-3B), 157.3 (C-2B), 157.3 (C-2A), 178.1 (C-1B), 178.2 (C-1A), 178.2 (C-8A), 179.6 (C-8B); IR (KBr, cm
−1): 3480, 2962, 1751, 1675, 1461, 1382, 1242, 1154, 1065; ESIHRMS: calcd. for C
11H
18O
3,
m/
z 211.1334 [M + H]
+, found 211.1330. (
Supplementary Materials, Figures S83–S88).
7-Hydroxy-4,4,6,7-tetramethyl-9-oxabicyclo[4.3.0]non-2-en-8-one (
7b) was characterized by the following physical and spectral properties: colourless oil;
1H-NMR: 1.05 (s, 3H, CH
3-9), 1.15–1.16 (m, 1H, one of CH
2-5), 1.17 (s, 3H, CH
3-10), 1.19 (s, 3H, CH
3-11), 1.32 (s, 3H, CH
3-12), 1.33–1.36 (m, 1H, one of CH
2-5), 4.67 (d,
J = 5.3 Hz, 1H, H-1), 5.80 (dd,
J = 10.0 and 5.3 Hz, 1H, H-2), 5.90 (d,
J = 10.0 Hz, 1H, H-3);
13C-NMR: 14.7 (C-11), 16.9 (C-12), 17.0 (C-4), 28.8 (C-9), 32.3 (C-6), 33.2 (C-10), 40.0 (C-5), 43.0 (C-7), 77.3 (C-1), 117.4 (C-2), 144.4 (C-3), 177.7 (C-8B); IR (KBr, cm
−1): 2464, 2927, 1757, 1464, 1382, 1141, 974; ESIHRMS: calcd. for C
11H
18O
3,
m/
z 211.1334 [M + H]
+, found 211.1331, calcd. for C
11H
18O
3Na,
m/
z 233.1154 [M + H]
+, found 233.1166. (
Supplementary Materials, Figures S89–S94).
10-Hydroxy-4,4,6,7-tetramethyl-9-oxabicyclo[4.3.0]non-2-en-8-one (
8b) was characterized by the following physical and spectral properties: colourless oil;
1H-NMR: 1.06 (s, 3H, CH
3-9), 1.09 (s, 3H, CH
3-12), 1.12 (d,
J = 7.3 Hz, 3H, CH
3-11), 1.57 (d,
J = 15.0 Hz, 1H, one of CH
2-5), 1.80 (d,
J = 15.0 Hz, 1H, one of CH
2-5), 2.80 (q,
J = 7.3 Hz, 1H, H-7), 3.29 (d,
J = 10.5 Hz, 1H, one of CH
2-10), 3.38 (d,
J = 10.5 Hz, 1H, one of CH
2-10), 4.53 (s, 1H, H-1), 5.64 (dd,
J = 10.4 and 5.3 Hz, 1H, H-2), 5.79 (d,
J = 10.4 Hz, 1H, H-3);
13C-NMR: 7.2 (C-11), 23.2 (C-9), 24.1 (C-12), 36.03 (C-5), 37.2 (C-4), 40.4 (C-6), 40.8 (C-7), 72.0 (C-10), 80.9 (C-1), 125.2 (C-3), 135.8 (C-2), 178.7 (C-8B); IR (KBr, cm
−1): 3409, 2933, 1757, 1451, 1207, 1175, 1041; ESIHRMS: calcd. for C
11H
18O
3,
m/
z 211.1334 [M + H]
+, found 211.1331, calcd. for C
11H
18O
3Na,
m/
z 233.1154 [M + H]
+, found 233.1019. (
Supplementary Materials, Figures S95–S100).
2,3-Epoxy-4,4,6,7-tetramethyl-9-oxabicyclo[4.3.0]nonan-8-one (
9b) was characterized by the following physical and spectral properties: colourless oil;
1H-NMR: 0.98 (s, 6H, CH
3-9A, CH
3-9B), 1.08 (d,
J = 7.2 Hz, 3H, CH
3-11A), 1.10 (d,
J = 7.2 Hz, 3H, CH
3-11B), 1.12 (s, 3H, CH
3-12A), 1.16 (s, 3H, CH
3-12B), 1.21 (s, 6H, CH
3-10A, CH
3-10B), 1.26–1.30 (dd,
J = 15.1 and 9.9 Hz, 2H, CH
2-5A), 1.37 (d,
J = 15.1 Hz, 2H, CH
2-5B), 2.37 (q,
J = 7.2 Hz, 1H, H-7A), 2.80 (q,
J = 7.2 Hz, 1H, H-7B), 2.98 (d,
J = 3.3 Hz, 1H, H-3B), 3.02 (d,
J = 3.5 Hz, 1H, H-3A), 3.33 (d,
J = 3.3 Hz, 1H, H-2B), 3.60 (dd,
J = 3.5 and 3.3 Hz, 1H, H-2A), 4.29 (s, 1H, H-1B), 4.42 (m, 1H, H-1A);
13C-NMR: 6.9 (C-11A), 7.4 (C-11B), 23.5 (C-9A), 23.6 (C-9B), 26.5 (C-5B), 26.6 (C-5A), 29.5 (C-6B), 30.1 (C-12B), 31.1 (C-12A), 33.1 (C-6A), 38.0 (C-10A), 38.2 (C-10B), 39.6 (C-4B), 39.9 (C-7B), 41.1 (C-4A), 49.9 (C-7A), 53.4 (C-2A), 53.8 (C-2B), 60.7 (C-3B), 60.8 (C-3A), 78.3 (C-1A), 79.3 (C-1B), 178.2 (C-8A), 178.2 (C-8B); IR (KBr, cm
−1): 2963, 1707, 1673, 1459, 1383, 1241, 1096; ESIHRMS: calcd. for C
11H
18O
3,
m/
z 211.1334 [M + H]
+, found 211.1331, calcd. for C
11H
18O
3Na,
m/
z 233.1154 [M + H]
+, found 233.1019. (
Supplementary Materials, Figures S101–S106).


 and
and 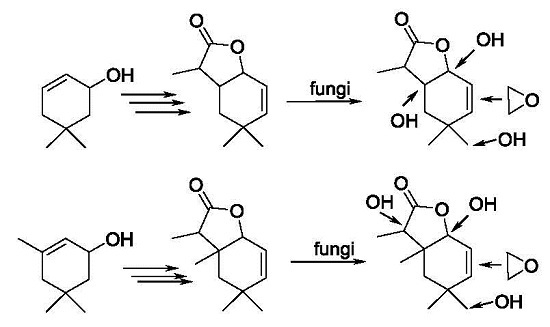


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}