
Preparative Scale Resolution of Enantiomers Enables Accelerated Drug Discovery and Development


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Identifying the Optimal Method for Preparative-Scale Chiral Separation
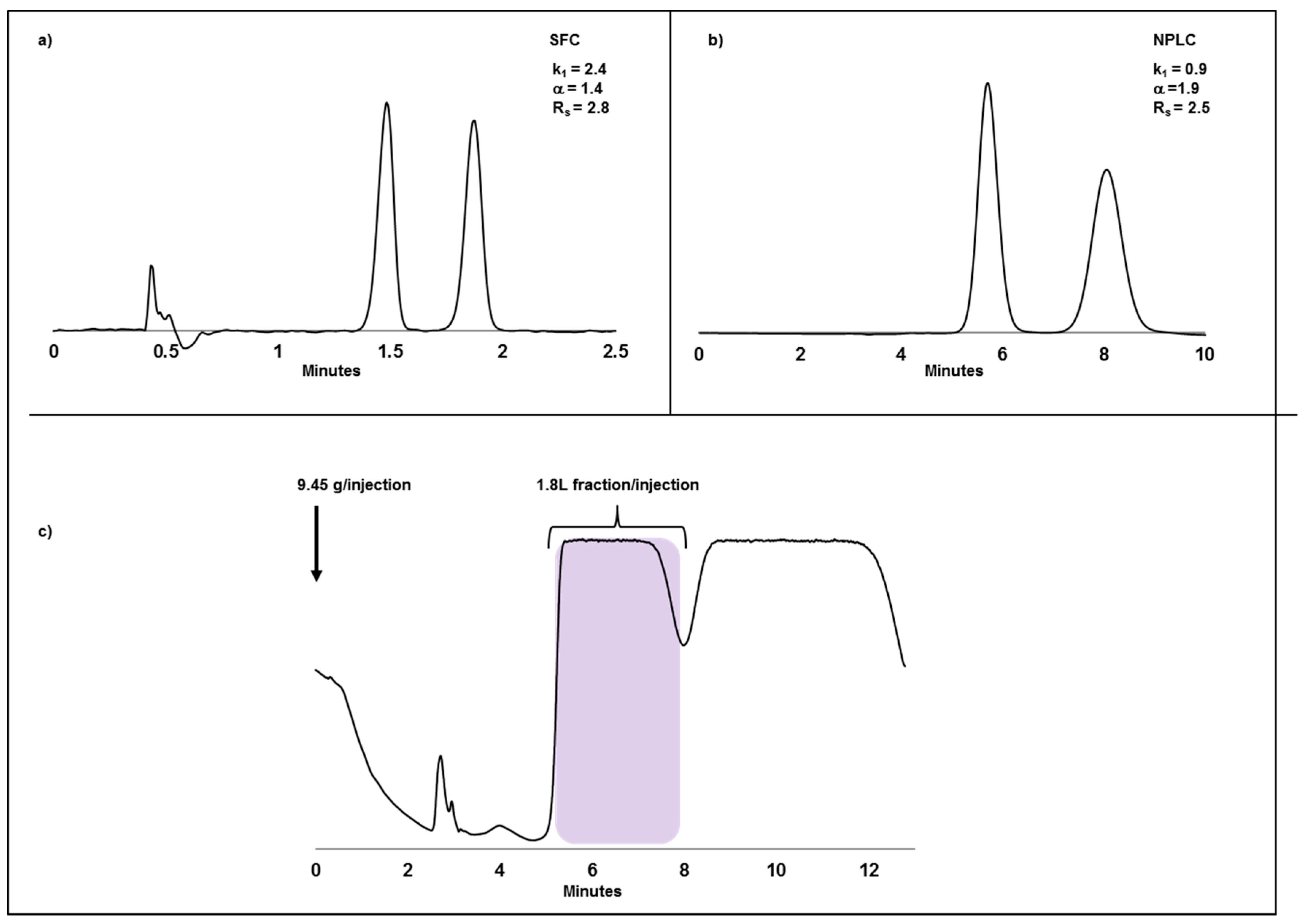
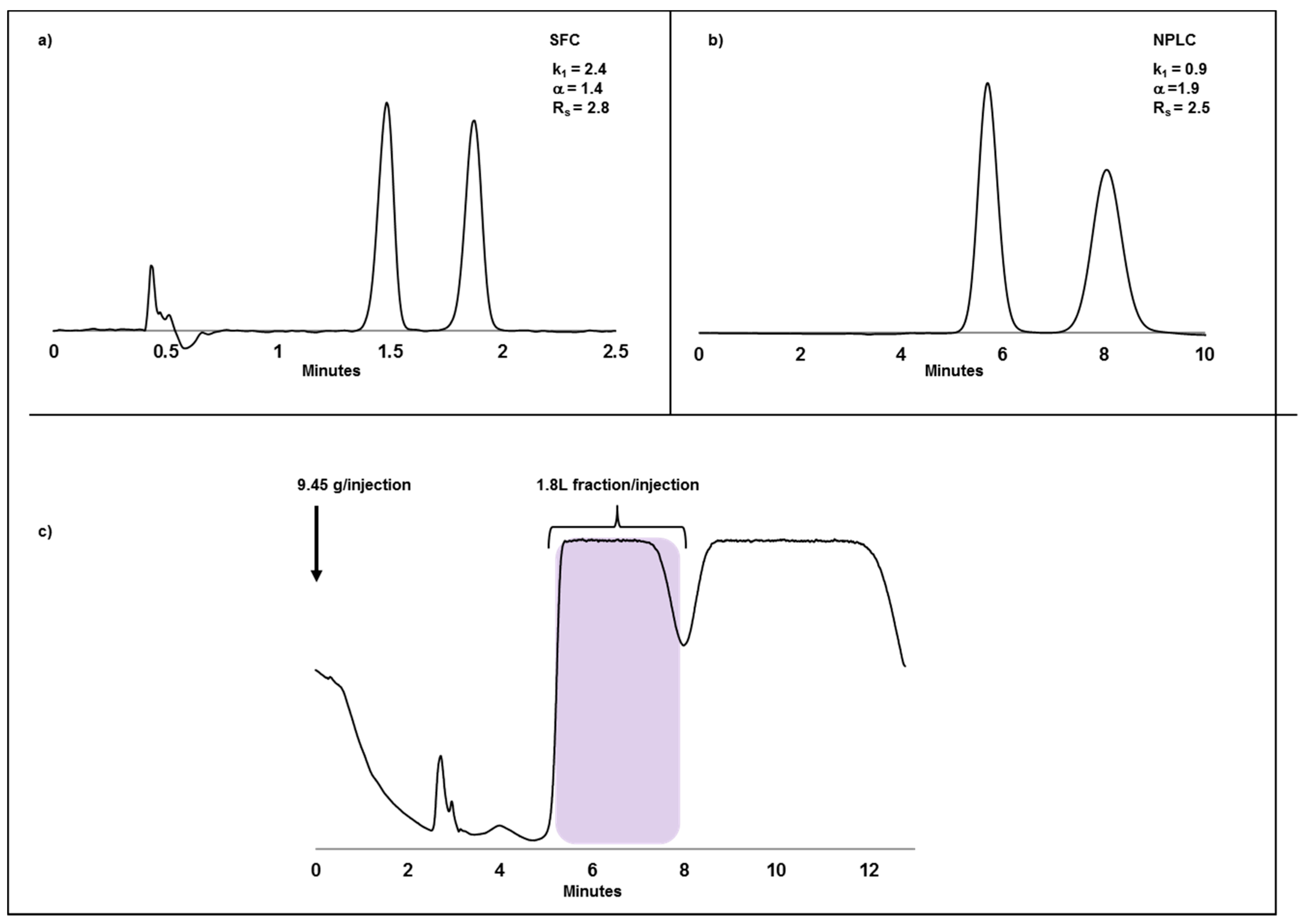
2.2. Case Study I: Application of SFC or NPLC in Enantiomeric Separations and Impact of Compound Solubility
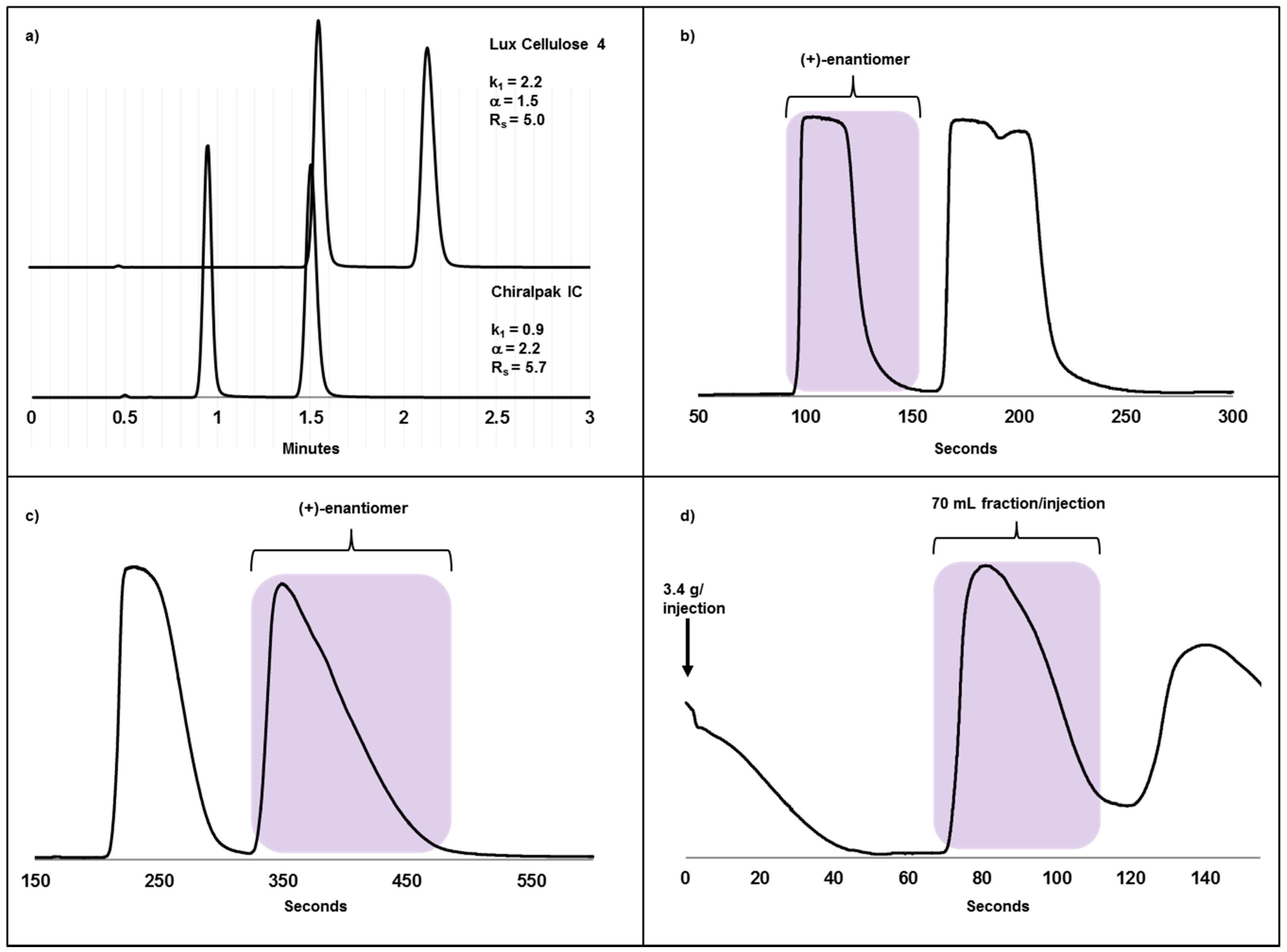
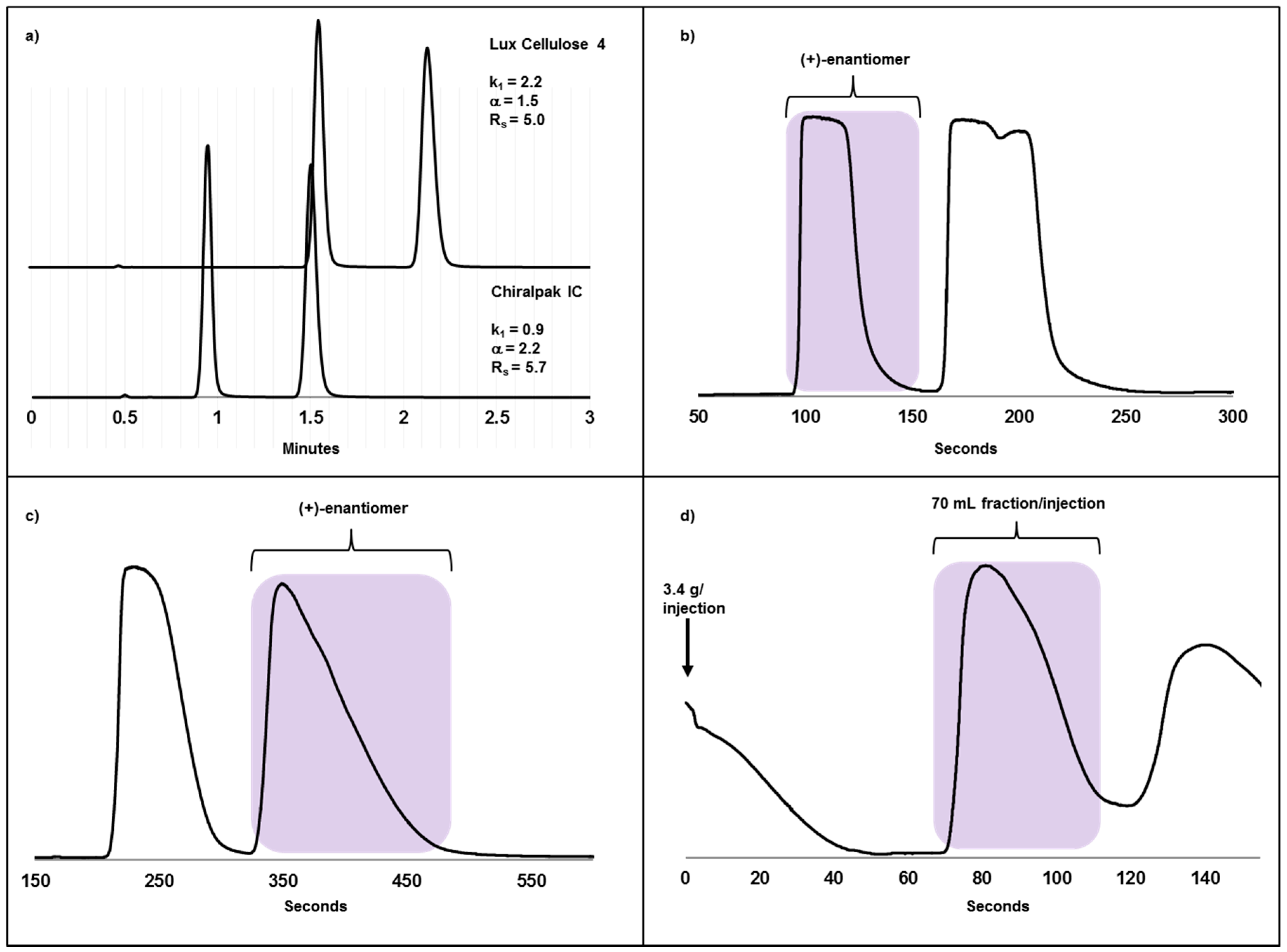
2.3. Case Study II: A Highly Efficient Chiral Separation of Kilogram Amounts of a Racemic Drug Compound on Chiralpak IC
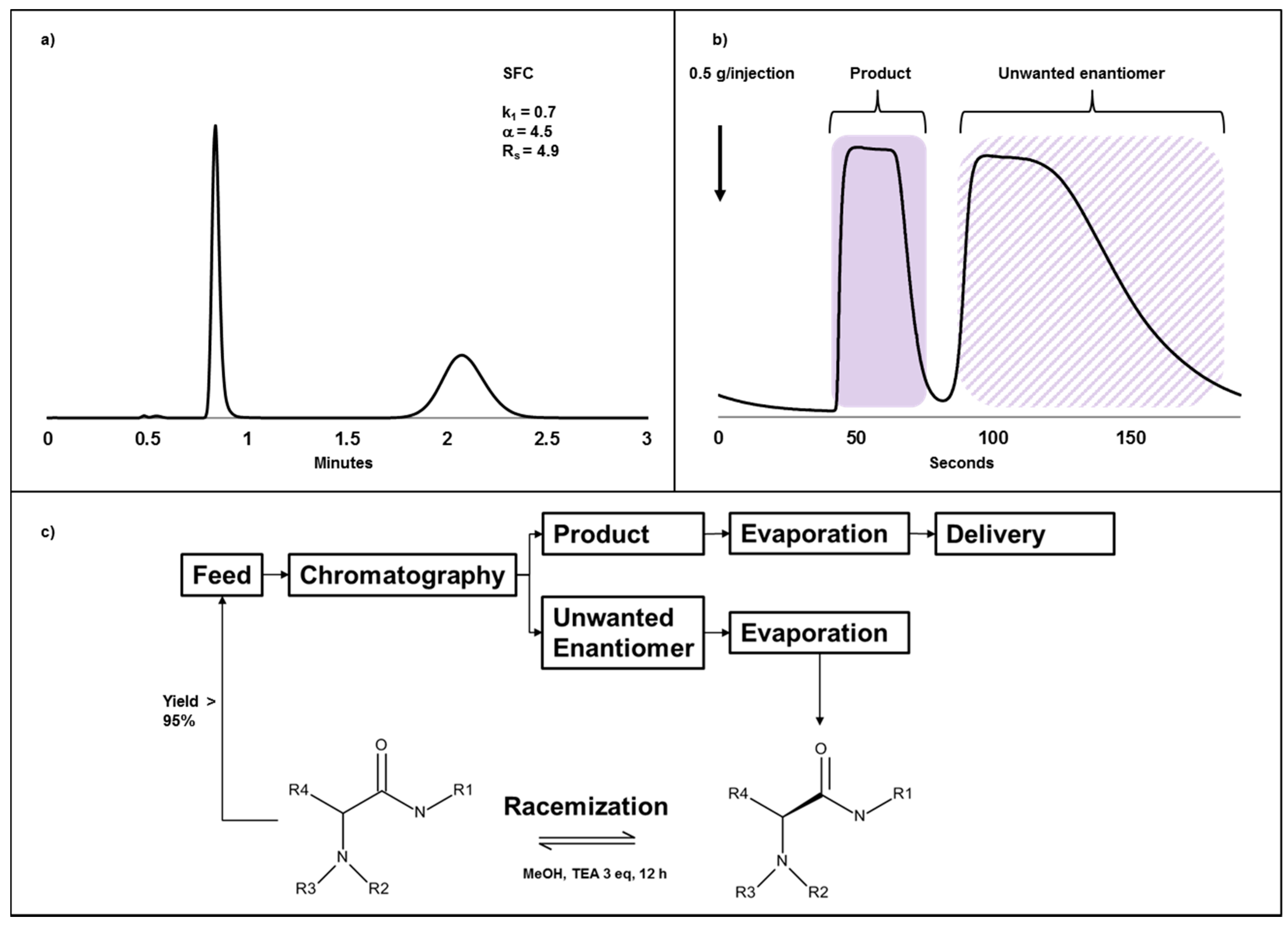
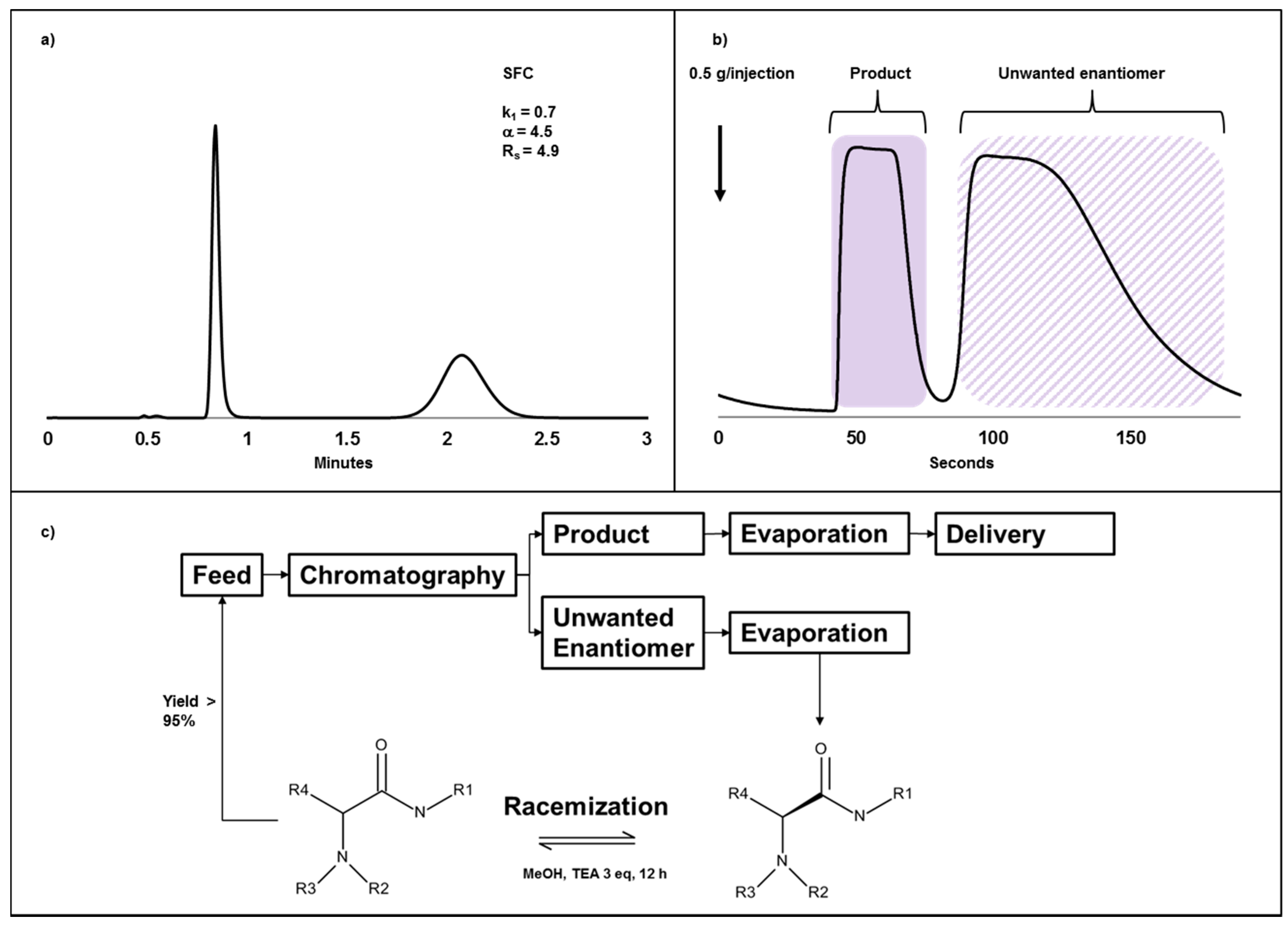
2.4. Case Study III: A Combination of Chiral Chromatography and Racemization of the Undesired Isomer Gives a Highly Efficient Process for Obtaining a Single Enantiomer
3. Materials and Methods
3.1. Chemicals
3.2. Instrumentation
3.3. Chiral Stationary Phases (CSPs)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Testa, B.; Vistoli, G.; Pedretti, A.; Caldwell, J. Organic Stereochemistry. Part 5 Stereoselectivity in Molecular and Clinical Pharmacology. Helv. Chim. Acta 2013, 96, 747–798. [Google Scholar] [CrossRef]
- Leach, A.G.; Pilling, E.A.; Rabow, A.A.; Tomasi, S.; Asaad, N.; Buurma, N.J.; Ballard, A.; Narduolo, S. Enantiomeric pairs reveal that key medicinal chemistry parameters vary more than simple physical property based models can explain. Med. Chem. Commun. 2012, 3, 528–540. [Google Scholar] [CrossRef]
- Triggle, D.J. Stereoselectivity of Drug Action. Drug Discov. Today 1997, 2, 138–147. [Google Scholar] [CrossRef]
- Andersson, S. Preparative Chiral Chromatography—A powerful and efficient tool in drug discovery. In Chiral Separation Techniques, a Practical Approach, 3rd ed.; Subramanian, G., Ed.; Wiley-VCH: Weinheim, Germany, 2007; pp. 585–599. [Google Scholar]
- Von Langermann, J.; Kaspereit, M.; Shakeri, M.; Lorenz, H.; Hedberg, M.; Jones, M.J.; Larson, K.; Herschend, B.; Arnell, R.; Temmel, E.; et al. Design of an Integrated Process of Chromatography, Crystallization and Racemization for the Resolution of 2′,6′-Pipecoloxylidide (PPX). Org. Process Res. Dev. 2012, 16, 343–352. [Google Scholar] [CrossRef]
- Lorenz, H.; Seidel-Morgenstern, A. Processes to Separate Enantiomers. Angew. Chem. Int. Ed. Engl. 2014, 53, 1218–1250. [Google Scholar] [CrossRef] [PubMed]
- Andersson, S.; Nelander, H.; Ohlen, K. Preparative Chiral Chromatography and Chiroptical Characterization of Enantiomers of Omeprazole and Related Benzimidazoles. Chirality 2007, 19, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.C.; Kim, H.; Yang, X.; Ross, D. Large Scale Chiral Chromatography for the Separation of an Enantiomer to Accelerate Drug Development. Chirality 2011, 23, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Leonard, W.R., Jr.; Henderson, D.W.; Miller, R.A.; Spencer, G.A.; Sudah, O.S.; Biba, M.; Welch, C.J. Strategic use of Preparative Chiral Chromatography for the Synthesis of a Preclinical Pharmaceutical Candidate. Chirality 2007, 19, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Okamoto, Y. Efficient Separation of Enantiomers Using Stereoregular Chiral Polymers. Chem. Rev. 2016, 116, 1094–1138. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Ikai, T. Chiral HPLC for efficient resolution of enantiomers. Chem. Soc. Rev. 2008, 37, 2593–2608. [Google Scholar] [CrossRef] [PubMed]
- Allenmark, S.G.; Andersson, S.; Möller, P.; Sanchez, D. A New Class of Network-Polymeric Chiral Stationary Phases. Chirality 1995, 7, 248–256. [Google Scholar] [CrossRef]
- Pirkle, W.H.; Lee, W.; Welch, C.J. Chromatographic separation of the enantiomers of 2-aryloxypropionic acids, esters and amides. Enantiomer 1997, 2, 423–431. [Google Scholar]
- Lämmerhofer, M.; Lindner, W. Quinine and Quinidine Derivatives as Chiral Selectors I. Brush Type Chiral Stationary Phases for High-Performance Liquid Chromatography Based on Cinchonan Carbamates and Their Application as Chiral Anion Exchangers. J. Chromatogr. A 1996, 741, 33–48. [Google Scholar] [CrossRef]
- Blehaut, J.; Franco, P.; Zhang, T.; Lang, E.; Valery, E.; Marcoux, J.F. Industrial applications of chiral chromatography. In Comprehensive Chirality; Carreira, E.M., Yamamoto, H., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 9, pp. 400–456. [Google Scholar]
- Andersson, S.; Allenmark, S.G. Preparative Chiral Chromatographic Resolution of Enantiomers in Drug Discovery. J. Biochem. Biophys. Methods 2002, 54, 11–23. [Google Scholar] [CrossRef]
- Schulte, M.; Joehnck, M.; Skudas, R.; Unger, K.K.; von Hohenesche, C.; Wewers, W.; Dingenen, J.; Kinkel, J. Stationary phases and chromatographic systems. In Preparative Chromatography, 2nd ed.; Schmidt-Traub, H., Schulte, M., Seidel-Morgenstern, A., Eds.; Wiley-VCH: Weinheim, Germany, 2012; pp. 47–198. [Google Scholar]
- De Mas, N.; Natalie, J.K.; Quiroz, F.; Rosso, W.V.; Chen, C.D.; Conlon, A.D. A Partial Classical Resolution/Preparative Chiral Supercritical Fluid Chromatography Method for the Rapid Preparation of the Pivotal Intermediate in the Synthesis of Two Nonsteroidal Glucocorticoid Receptor Modulators. Org. Process Res. Dev. 2016, 20, 934–939. [Google Scholar] [CrossRef]
- Speybrouck, D.; Lipka, E. Preparative Supercritical Fluid Chromatography: A Powerful Tool for Chiral Separations. J. Chromatogr. A 2016, 1467, 33–55. [Google Scholar] [CrossRef] [PubMed]
- Desfontaine, V.; Guillarme, D.; Francotte, E.; Novakova, L. Supercritical Fluid Chromatography in Pharmaceutical Analysis. J. Pharm. Biomed. Anal. 2015, 113, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Lesellier, E.; West, C. The Many Faces of Packed Column Supercritical Fluid Chromatography—A Critical Review. J. Chromatogr. A 2015, 1382, 2–46. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.; Potter, M. Preparative chromatographic resolution of racemates using HPLC and SFC in a pharmaceutical discovery environment. J. Chromatogr. B 2008, 875, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Lindskog, M.A.; Nelander, H.; Jonson, A.C.; Halvarsson, T. Delivering the Promise of SFC: A Case Study. Drug Discov. Today 2014, 19, 1607–1612. [Google Scholar] [CrossRef] [PubMed]
- Dispas, A.; Lebrun, P.; Sacré, P.; Hubert, P. Screening Study of SFC Critical Method Parameters for the Determination of Pharmaceutical Compounds. J. Pharm. Biomed. Anal. 2016, 125, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Franco, P.; Zhang, T. Common Screening Approaches for Efficient Analytical Method Development in LC and SFC on Columns Packed with Immobilized Polysaccharide-Derived Chiral Stationary Phases. Methods Mol. Biol. 2013, 970, 113–126. [Google Scholar] [PubMed]
- Speybrouck, D.; Corens, D.; Argoullon, J.M. Screening Strategy for Chiral and Achiral Separations in Supercritical Fluid Chromatography Mode. Curr. Top. Med. Chem. 2012, 12, 1250–1263. [Google Scholar] [CrossRef] [PubMed]
- Miller, L. Evaluation of Non-Traditional Modifiers for Analytical and Preparative Enantioseparations using Supercritical Fluid Chromatography. J. Chromatogr. A 2012, 1256, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Thunberg, L.; Hashemi, J.; Andersson, S. Comparative Study of Coated and Immobilized Polysaccharide-Based Chiral Stationary Phases and their Applicability in the Resolution of Enantiomers. J. Chromatogr. B 2008, 875, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Francotte, E.; Zhang, T. Preparation and evaluation of immobilized 4-methylbenzoylcellulosestationary phases for enantioselective separations. J. Chromatogr. A 2016, 1467, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Ikai, T.; Okamoto, Y. Synthesis and application of immobilized polysaccharide-based chiral stationary phases for enantioseparation by high-performance liquid chromatography. J. Chromatogr. A 2014, 1363, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Szekely, G.; Amores de Sousa, M.C.; Gil, M.; Ferreira, F.C.; Heggie, W. Genotoxic Impurities in Pharmaceutical Manufacturing: Sources, Regulations, and Mitigation. Chem. Rev. 2015, 115, 8182–8229. [Google Scholar] [CrossRef] [PubMed]
- Leek, H.; Thunberg, L.; Jonson, A.C.; Öhlén, K.; Klarqvist, M. Strategy for Large-Scale Isolation of Enantiomers in Drug Discovery. Drug Discov. Today 2017, 22, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leek, H.; Andersson, S. Preparative Scale Resolution of Enantiomers Enables Accelerated Drug Discovery and Development. Molecules 2017, 22, 158. https://doi.org/10.3390/molecules22010158
Leek H, Andersson S. Preparative Scale Resolution of Enantiomers Enables Accelerated Drug Discovery and Development. Molecules. 2017; 22(1):158. https://doi.org/10.3390/molecules22010158
Chicago/Turabian StyleLeek, Hanna, and Shalini Andersson. 2017. "Preparative Scale Resolution of Enantiomers Enables Accelerated Drug Discovery and Development" Molecules 22, no. 1: 158. https://doi.org/10.3390/molecules22010158