
Literature Survey and Further Studies on the 3-Alkylation of N-Unprotected 3-Monosubstituted Oxindoles. Practical Synthesis of N-Unprotected 3,3-Disubstituted Oxindoles and Subsequent Transformations on the Aromatic Ring



Abstract
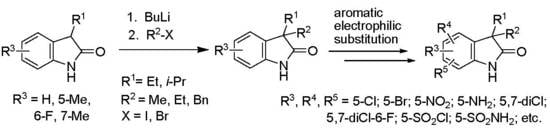
:
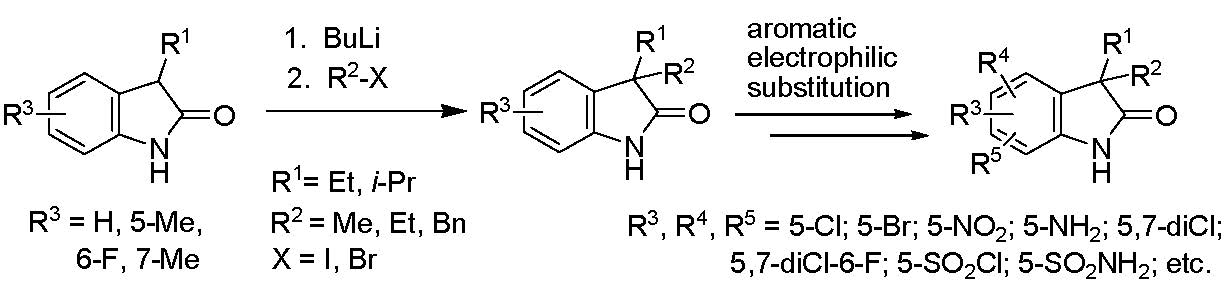
1. Introduction
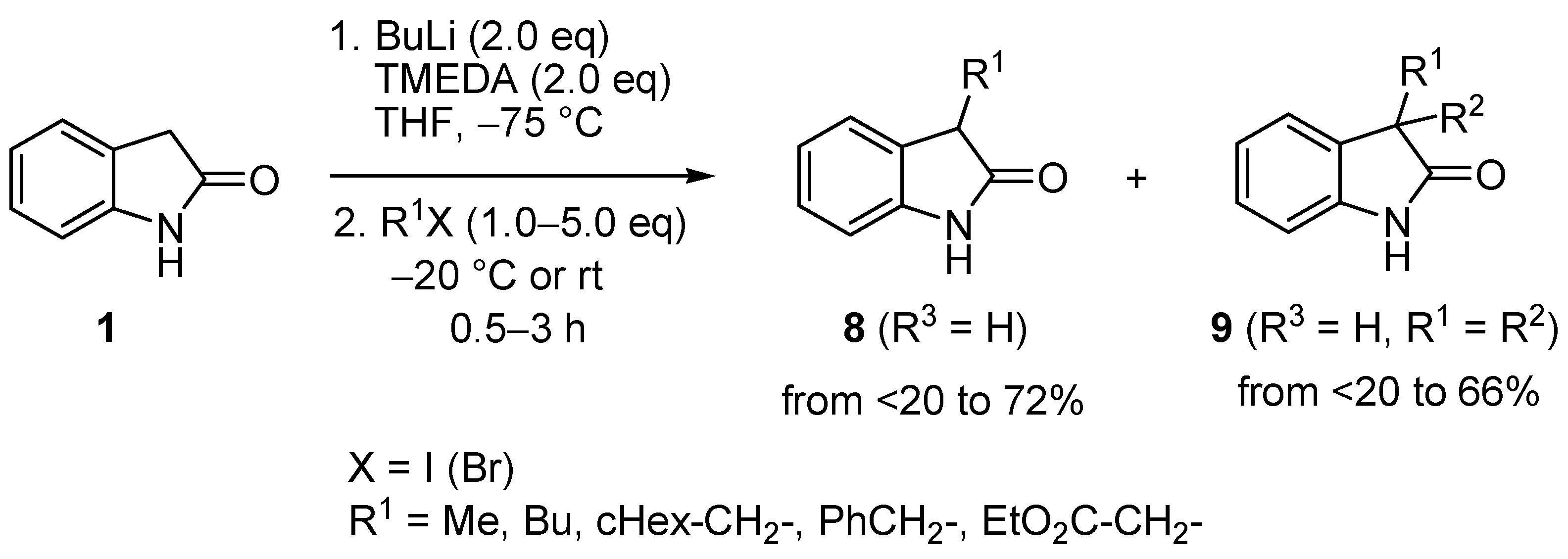
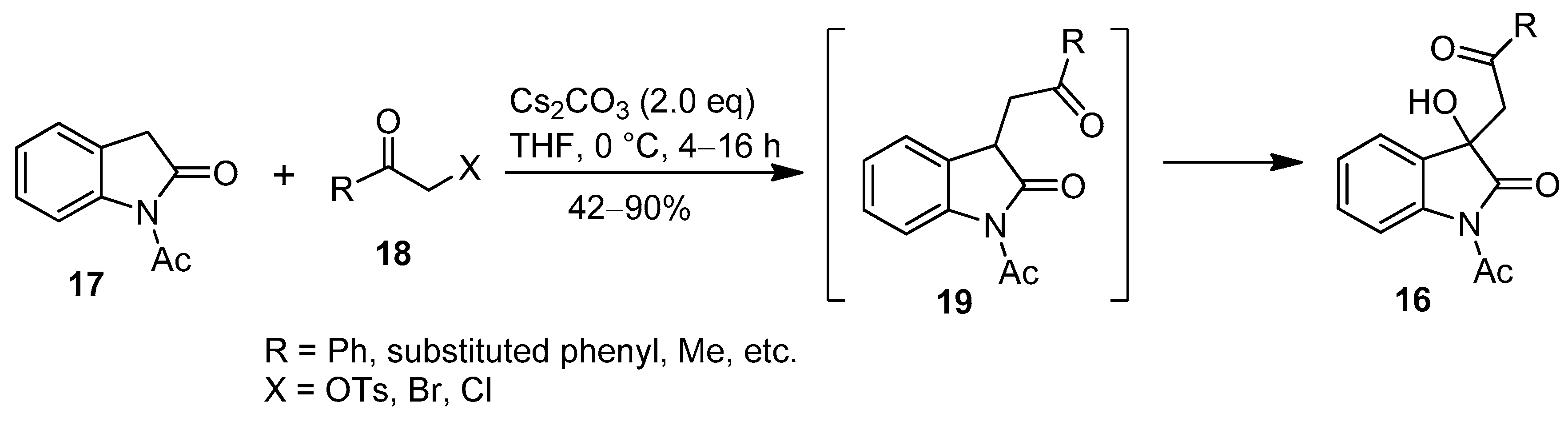
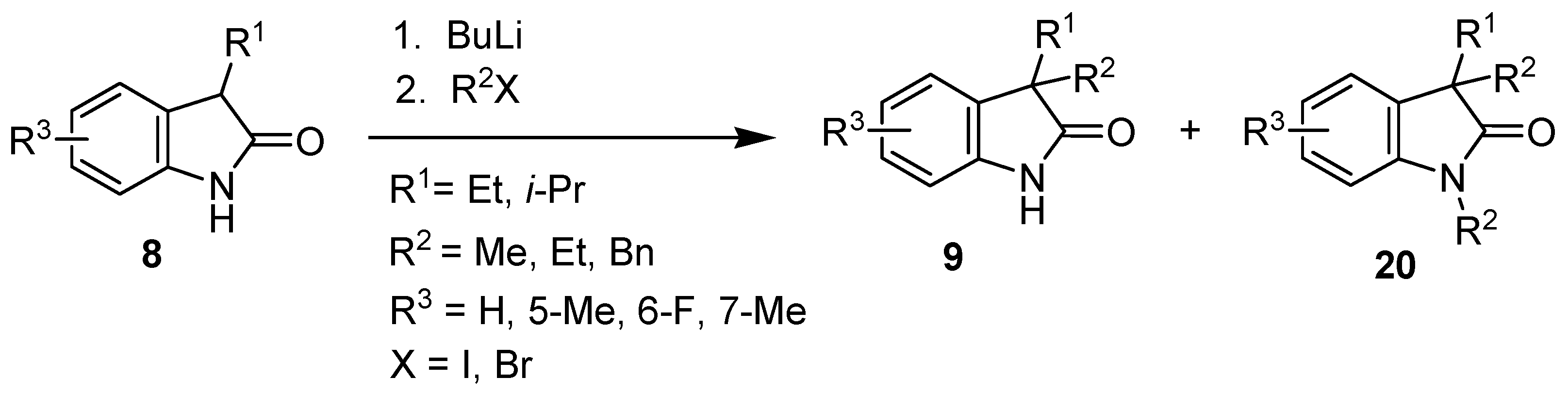
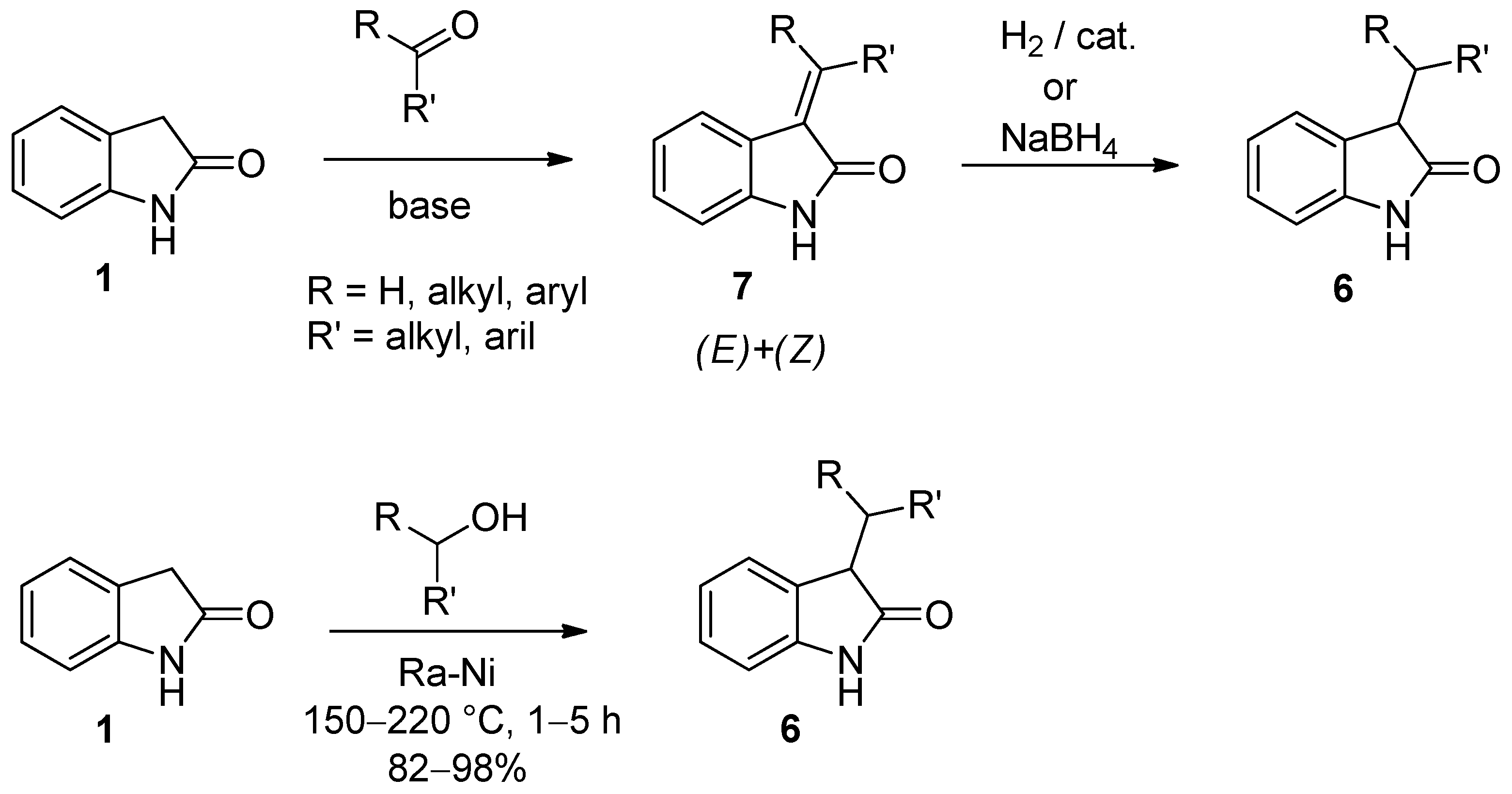
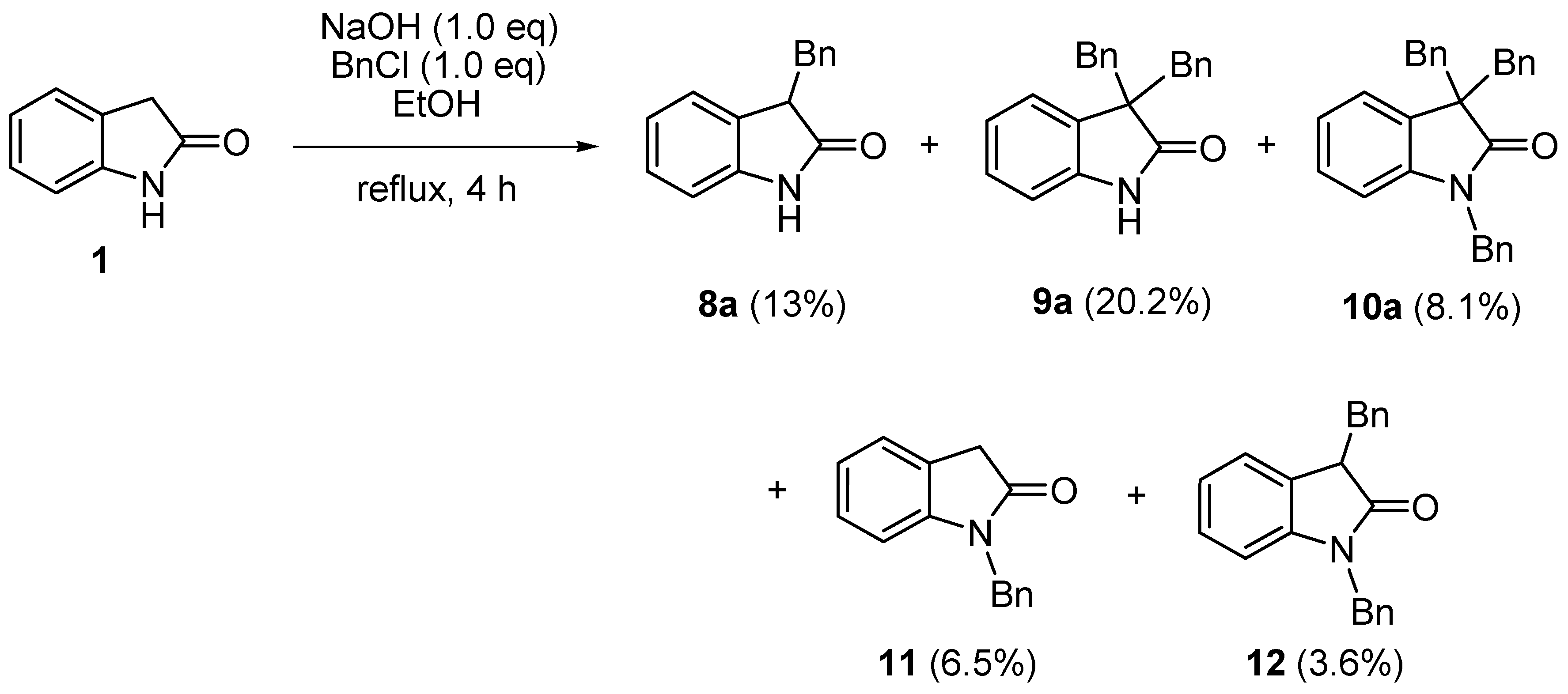
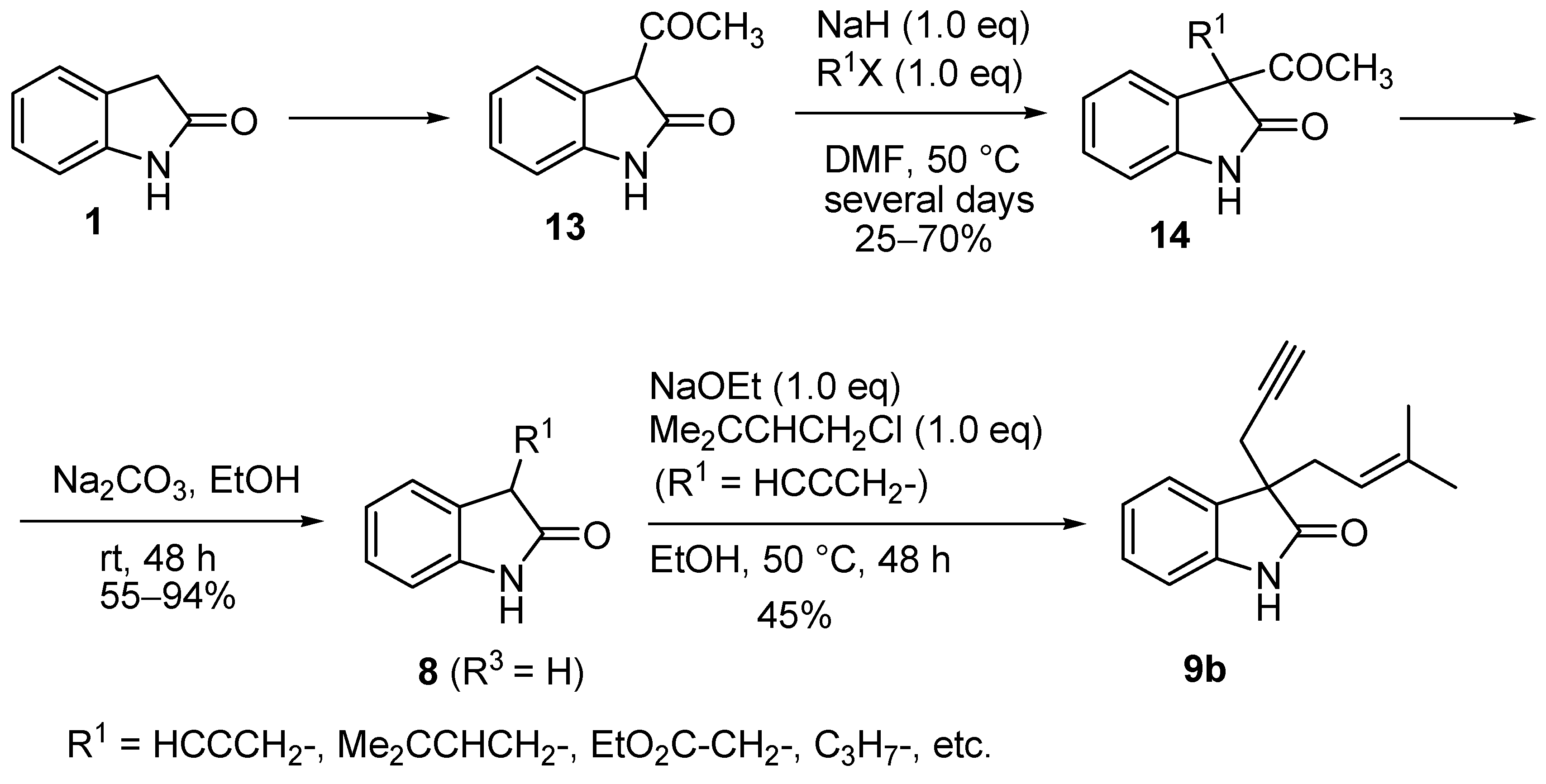
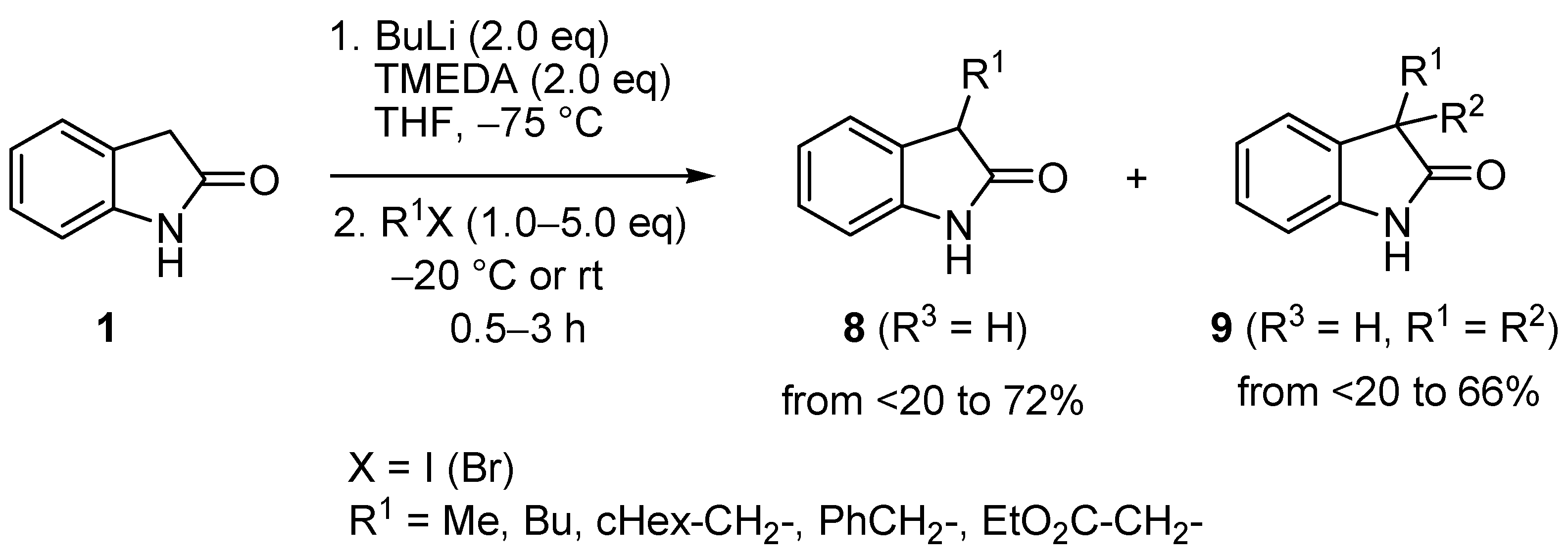
2. Results and Discussion
3. Experimental Section
3.1. General Information
3.2. General Procedure I for the Synthesis of Compounds 9c–h (and By-Product 20a)
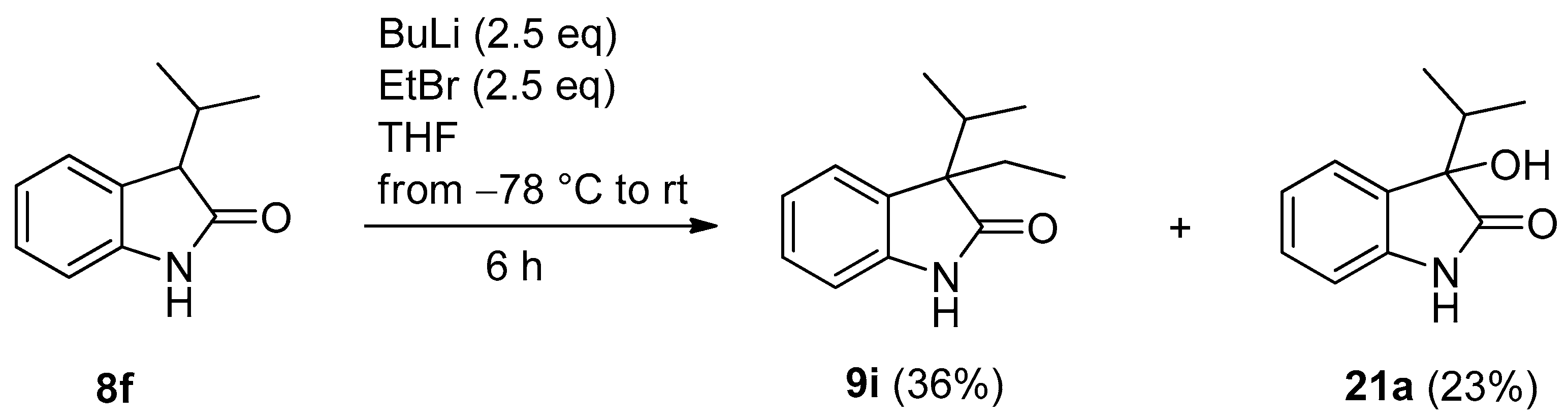
3.3. General Procedure II for the Synthesis of Compounds 9i–k (and By-Product 20b)
3.4. General Procedure III for the Synthesis of Compounds 21
4. Conclusions
Author Contributions
Conflicts of Interest
References and Notes
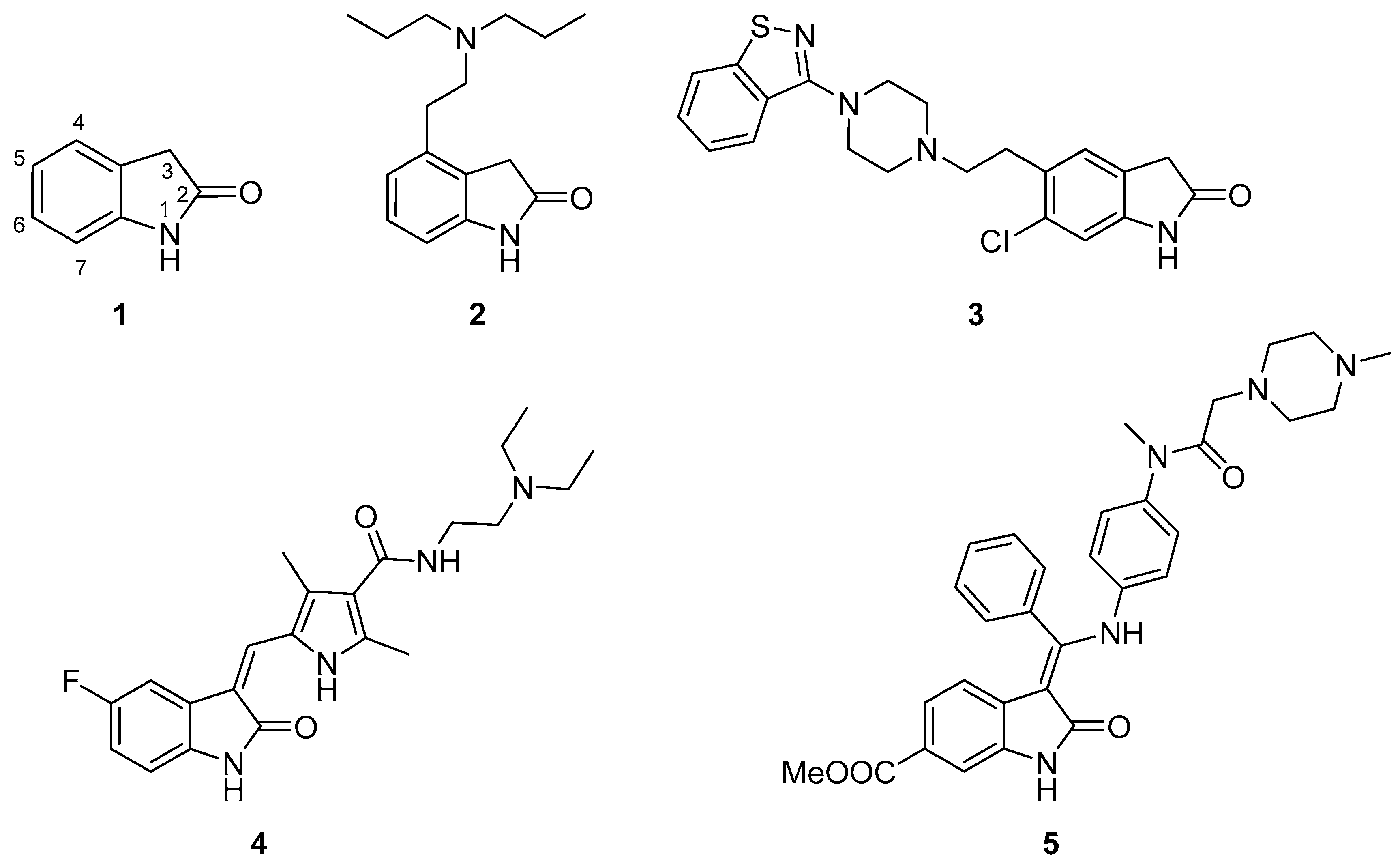
- Oxindole derivatives reaching human Phase III clinical trials are as follows: Tenidap, famitinib, linopirdine, flindokalner, satavaptan.
- Oxindole derivatives reaching human Phase II clinical trials are as follows: Aplindore, cipargamin, icopezil maleate, amcasertib, adibendan, funapide, NS-1209, semaxanib, indolidan.
- Oxindole derivatives reaching human Phase I clinical trials are as follows: SNAP-37889, PF-03382792, CP-126998, henatinib, BI-847325, CFI-400945, tafetinib, XEN-401, SSR-126768A, T-0632, DS-3032b.
- Nozoye, T.; Nakai, T.; Kubo, A. (E)- and (Z)-2-Oxoindolin-3-ethylidenes. Chem. Pharm. Bull. 1977, 25, 196–198. [Google Scholar] [CrossRef]
- Mertens, A.; Müller-Beckmann, B.; Kampe, W.; Hölck, J.-P.; von der Saal, W. Nonsteroidal cardiotonics. 1. 2-Pyridyl-6,7-dihydro-3H,5H-pyrrolo[2,3-f]benzimidazol-6-ones, a novel class of cardiotonic agents. J. Med. Chem. 1987, 30, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Tacconi, G.; Maggi, L.D.; Righetti, P.; Desimoni, G.; Azzolina, O.; Ghislandi, V. (Z)- and (E)-3-Alkylidene-1,3-dihydroindol-2-ones. Influence of configuration on the transmission of the inductive effect to the carbonyl group. J. Chem. Soc., Perkin Trans. 2 1976, 150–154. [Google Scholar] [CrossRef]
- Wenkert, E.; Bringi, N.V. A new procedure for forming carbon-carbon bonds. J. Am. Chem. Soc. 1958, 80, 5575–5576. [Google Scholar] [CrossRef]
- Wenkert, E.; Bringi, N.V.; Choulett, H.E. Raney nickel-induced alkylation reactions. Acta Chem. Scand. Ser. B 1982, 36, 348–350. [Google Scholar] [CrossRef]
- Volk, B.; Mezei, T.; Simig, G. Raney nickel-induced 3-alkylation of oxindole with alcohols and diols. Synthesis 2002, 595–597. [Google Scholar] [CrossRef]
- Gruda, I. Formation of N- and C-substituted derivatives during alkylation of indol-2(3H)-one. Can. J. Chem. 1972, 50, 18–23. [Google Scholar] [CrossRef]
- Reisch, J.; Müller, M.; Labitzke, H. Synthetic pathways for 3-substituted indol-2-ones. Arch. Pharm. 1984, 317, 639–646. [Google Scholar] [CrossRef]
- Horner, L. Syntheses in the oxindole series. Liebigs Ann. Chem. 1941, 548, 117–146. [Google Scholar] [CrossRef]
- Wenkert, E.; Bernstein, B.S.; Udelhofen, J.H. A novel conversion of derivatives of oxindoles to indoles. J. Am. Chem. Soc. 1958, 80, 4899–4903. [Google Scholar] [CrossRef]
- Shaun, J.; Tetsuro, K.; Katsura, T.; Tsuyoshi, H.; Yasufumi, U. Carbostyril derivative 5-HT1a receptor subtype agonist for treatment of central nervous system disorders. US 2011195954 U.S. Pat. Appl., Nov 21, 2002. Chem. Abstr. 2002, 137, 363096. [Google Scholar]
- Karig, G.; Ford, M.J.; Siegel, K.; Schnatterer, S. Method for producing N-sulfonyl-substituted oxindoles. US 2015133660 PCT Intern. Pat. Appl., May 14, 2015. Chem. Abstr. 2012, 157, 165628. [Google Scholar]
- Cook, B.N.; Huber, J.D.; Hughes, R.O.; Kirrane, T.M., Jr.; Lasota, C.; Li, X.; Liang, S.; Mugge, I.A.; Zhang, Q. Preparation of bicyclic compounds as modulators of retinoid-related orphan receptor γt (RORγt or RORc). WO 2015035032 PCT Intern. Pat. Appl., Mar 12, 2015. Chem. Abstr. 2015, 162, 425159. [Google Scholar]
- Alkylation of a 3-(substituted phenyl)oxindole and related compounds with α,ω-iodochloroalkanes with t-BuOK in DMF is described with undisclosed yields in: Foulon, L.; Goullieux, L.; Pouzet, B.; Le Serradeil, G.; Claudine, V.G. Novel 3-aminoalkyl-1,3-dihydro-2H-indol-2-one derivatives, preparation thereof and therapeutic use thereof. US 2011059955 U.S. Pat. Appl., Mar 10, 2011, also published as WO 2009115685 PCT Intern. Pat. Appl., Sep 24, 2009. Chem. Abstr. 2009, 151, 403108. [Google Scholar]
- Alkylation of 3-phenyloxindole with t-BuOK and 1,2-dibromoethane in THF is described in 53% yield in: Kato, K.; Doi, T.; Sugiura, Y.; Kawada, M. Preparation of 3-[2-(4-arylazino)ethyl]-2-indolones and analogs as antiincontinence agents. WO 9802432 PCT Intern. Pat. Appl., Jan 22, 1998. Chem. Abstr. 1998, 128, 140729. [Google Scholar]
- 3-Methyl-4-methoxyoxindole was 3-methylated with potassium bis(trimethylsilyl)amide and methyliodide in THF to give the product in 58% yield after laborious work-up, see: Jesudason, C.D.; Sall, D.J.; Stevens, F.C.; Werner, J.A. Preparation of oxindolyloxypropanolamines as β3 adrenergic receptor agonists. WO 0238543 PCT Intern. Pat. Appl., May 16, 2002. Chem. Abstr. 2002, 136, 369605. [Google Scholar]
- Reaction of 5-chloro-3-(4-methoxyphenyl)oxindole with benzyl bromide in the presence of K2CO3 and KI in acetone, subsequent work-up by flash chromatography, and preparation of several analogous derivatives in low to moderate yields in a similar manner are described in: Kin-Chun, L.; Sung-Sau, S.; Jing, Z.; Zhuming, Z. Preparation of oxindoles as inhibitors of MDM2-p53 interaction for the treatment of cancer. WO 2006136606 PCT Intern. Pat. Appl., Dec 28, 2006. Chem. Abstr. 2006, 146, 100555. [Google Scholar]
- Reaction of 5-(3,5-dimethylisoxazol-4-yl)-3-phenyloxindole with ethyl bromoacetate in the presence of K2CO3 and KI in acetone, and reaction of 5-(3,5-dimethylisoxazol-4-yl)-3-phenyloxindole with 3-bromopropan-1-ol in the presence of K2CO3 and KI in THF, and subsequent work-up by flash chromatography are disclosed in 60% and 41% yields, respectively, in: Bo, R.; Changyou, Z.; Hexiang, W. 5-(3,5-Dimethylisoxazol-4-yl)indolin-2-ones as BRD4 inhibitors and their preparation and use for the treatment of cancer. WO 2014173241 PCT Intern. Pat. Appl., Oct 30, 2014. Chem. Abstr. 2014, 161, 664198. [Google Scholar]
- 3-(Substituted phenyl) or 3-methyloxindoles, eventually substituted on the aromatic ring of the oxindole skeleton, are alkylated with K2CO3 and variously substituted benzyl bromides in DMA. Chromatographic purification is necessary, yields are not disclosed, see: Wilson, J.E.; Kurukulasuriya, R.; Reibarkh, M.; Reiter, M.; Zwicker, A.; Zhao, K.; Zhang, F.; Anand, R.; Colandrea, V.J.; Cumiskey, A.-M.; Crespo, A.; Duffy, R.A.; Murphy, B.A.; Mitra, K.; Johns, D.G.; Duffy, J.L.; Vachal, P. Discovery of novel indoline cholesterol ester transfer protein inhibitors (CETP) through a structure-guided approach. ACS Med. Chem. Lett. 2016, 7, 261–265. [Google Scholar] [CrossRef] [PubMed]
- 3-Arlyoxindoles are alkylated with K2CO3 and substituted benzyl bromides in DMA. The products are obtained after chromatography in unknown yields, see: Wilson, J.E.; Vachal, P.; Kurukulasuriya, R. 3,3'-Disubstituted indolines as inhibitors of cholesterol ester transfer protein. WO 2015054088 PCT Intern. Pat. Appl., Apr 16, 2015. Chem. Abstr. 2015, 162, 544611. [Google Scholar]
- Reaction of 6-bromo-3-methyloxindole with methyl bromoacetate and allyl bromide in the presence of Cs2CO3 in DMF is described, after purification by flash chromatography, in 71% and 51% yields, respectively, in: Chen, G.; Cushing, T.D.; Fisher, B.; He, X.; Li, K.; Li, Z.; McGee, L.R.; Pattaropong, V.; Faulder, P.; Seganish, J.L.; et al. Alkynyl alcohols as kinase inhibitors and their preparation, pharmaceutical compositions and use in the treatment of inflammation and inflammatory disorders. WO 2009158011 PCT Intern. Pat. Appl., Dec 30, 2009. Chem. Abstr. 2009, 152, 119631. [Google Scholar]
- Kende, A.S.; Hodges, J.C. Regioselective C-3 alkylations of oxindole dianion. Synth. Commun. 1982, 12, 1–10. [Google Scholar] [CrossRef]
- Fensome, A.; Mccomas, C.C.; Melenski, E.G.; Marella, M.A.; Wrobel, J.E. Progesterone receptor modulators comprising pyrrole-oxindole derivatives and uses thereof. US 2006030615 U.S. Pat. Appl., Feb 9, 2006. Chem. Abstr. 2006, 144, 192104. [Google Scholar]
- Brittain, W.D.G.; Buckley, B.R.; Fossey, J.S. Kinetic resolution of alkyne-substituted quaternary oxindoles via copper catalysed azide-alkyne cycloadditions. Chem. Commun. 2015, 51, 17217–17220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Woods, K.W.; Zhu, G.-D.; Fischer, J.P.; Gong, J.; Li, T.; Gandhi, V.; Thomas, S.A.; Packard, G.; Song, X.; et al. Preparation of pyridine derivatives as protein kinase inhibitors. WO 03051366 PCT Intern. Pat. Appl., Oct 2, 2003. Chem. Abstr. 2003, 139, 69152. [Google Scholar]
- For the introduction of identical alkyl groups (Me, Et, Bn) in position 3 of 4-methoxyoxindole (2 eq BuLi, TMEDA, THF), see: Stevens, F.C.; Bloomquist, W.E.; Borel, A.G.; Cohen, M.L.; Droste, C.A.; Heiman, M.L.; Kriauciunas, A.; Sall, D.J.; Tinsley, F.C.; Jesudason, C.D. Potent oxindole based human β3 adrenergic receptor agonists. Bioorg. Med. Chem. Lett. 2007, 17, 6270–6273. [Google Scholar] [CrossRef] [PubMed]
- 3-Alkylation (benzylation) of 3-(substituted phenyl)oxindoles with BuLi (2.2 eq), TMEDA (2.2 eq) and alkylating agent (2.2 eq) with chromatographic work-up is described in: Christensen, M.K.; Bjoerkling, F. Preparation of 3-(4-hydroxyphenyl)-indolin-2-ones for the treatment of cancer. WO 2008129075 PCT Intern. Pat. Appl., Oct 30, 2008. Chem. Abstr. 2008, 149, 513691. [Google Scholar]
- 3-Ethylation of 3-ethyloxindole with BuLi (2.0 eq), TMEDA (2.0 eq) and EtI (1.0 eq) in 45% yield is described in: Grubb, G.S.; Zhi, L.; Jones, T.K.; Tegley, C.M.; Fensome, A.; Miller, L.L.; Ullrich, J.W.; Bender, R.H.W.; Zhang, P.; Wrobel, J.E.; et al. Preparation of oxospiro[cycloalkane-1,3′-indoline]derivatives and analogs as progesterone receptor antagonists. WO 0066167 PCT Intern. Pat. Appl., Nov 9, 2000. Chem. Abstr. 2000, 133, 350135. [Google Scholar]
- Fensome, A.; Zhang, P.; Koko, M.C.; Zhi, L.; Jones, T.K.; Marschke, K.B.; Tegley, C.M. Preparation of thioxospiro[cycloalkane-1,3′-indole]derivatives and analogs as progesterone receptor agonists. WO 0066555 PCT Intern. Pat. Appl., Nov 9, 2000. Chem. Abstr. 2000, 133, 350136. [Google Scholar]
- Fensome, A.; Miller, L.L.; Ullrich, J.W.; Bender, R.H.W.; Zhang, P.; Wrobel, J.E.; Zhi, L.; Jones, T.K.; Marschke, K.B.; Tegley, C.M. Preparation of indolinones as progesterone antagonists. WO 0066556 PCT Intern. Pat. Appl., Nov 9, 2000. Chem. Abstr. 2000, 133, 350137. [Google Scholar]
- 3-Methylation of 3-methyl-6-methoxyoxindole (2.0 eq BuLi, 2.0 eq TMEDA, 1.14 eq MeI, THF, −78 °C, flash chromatography, 82%) is described in: Jayyosi, Z.; McGeehan, G.M.; Kelley, M.F.; Labaudiniere, R.F.; Zhang, L.; Groneberg, R.D.; McGarry, D.G.; Caulfield, T.J.; Minnich, A.; Bobko, M. Preparation of diaryl carboxylic acids and derivatives as peroxisome proliferator-activated receptor ligands. WO 0064888 PCT Intern. Pat. Appl., Nov 2, 2000. Chem. Abstr. 2000, 133, 335167. [Google Scholar]
- Reaction (2.0 eq BuLi, 2.0 eq TMEDA, THF, 25 °C, 60%) of 5-bromo-3-methyloxindole with BrCH2CO2Et (1.0 eq) is described in: Jiang, T.; Kuhen, K.L.; Wolff, K.; Yin, H.; Bieza, K.; Caldwell, J.; Bursulaya, B.; Wu, T.Y.-H.; He, Y. Design, synthesis and biological evaluations of novel oxindoles as HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 2105–2108. [Google Scholar] [CrossRef] [PubMed]
- Volk, B.; Barkóczy, J.; Hegedűs, E.; Udvari, S.; Gacsályi, I.; Mezei, T.; Pallagi, K.; Kompagne, H.; Lévay, G.; Egyed, A.; et al. (Phenylpiperazinyl-butyl)oxindoles as selective 5-HT7 receptor antagonists. J. Med. Chem. 2008, 51, 2522–2532. [Google Scholar] [CrossRef] [PubMed]
- Volk, B.; Gacsályi, I.; Pallagi, K.; Poszávácz, L.; Gyönös, I.; Szabó, É.; Bakó, T.; Spedding, M.; Simig, G.; Szénási, G. Optimization of (arylpiperazinylbutyl)oxindoles exhibiting selective 5-HT7 receptor antagonist activity. J. Med. Chem. 2011, 54, 6657–6669. [Google Scholar] [CrossRef] [PubMed]
- For methylation using trimethyloxonium tetrafluoroborate, see: Zhou, H.-J.; Wang, J.; Yao, B.; Wong, S.; Djakovic, S.; Kumar, B.; Rice, J.; Valle, E.; Soriano, F.; Menon, M.-K.; et al. Discovery of a first-in-class, potent, selective, and orally bioavailable inhibitor of the p97 AAA ATPase (CB-5083). J. Med. Chem. 2015, 58, 9480–9497. [Google Scholar] [CrossRef] [PubMed]
- For ethylation of unsubstituted oxindole using triethyloxonium tetrafluoroborate, see: Harley-Mason, J.; Leeney, T.J. 2-Ethoxyindoles and -indolenines. A case of indole-indolenine tautomerism. Proc. Chem. Soc. Lond. 1964, 368–369. [Google Scholar]
- Tóth, F.; Fráter, G.; Ondi, L. Compounds and the use thereof in metathesis reactions. WO 2015155593 PCT Intern. Pat. Appl., Oct 15, 2015. Chem. Abstr. 2015, 163, 585851. [Google Scholar]
- Borovik, V.P.; Gatilov, Y.V.; Shkurko, O.P. Reaction of 3-(R-methylidene)-2-ethoxylindolenines with N,N'-binucleophiles. Russ. J. Org. Chem. 2016, 52, 228–234. [Google Scholar] [CrossRef]
- For ethylation of 5-chlorooxindole using triethyloxonium tetrafluoroborate, see: Esses-Reiter, K.; Reiter, J. Attempted synthesis of a tenidap isomer and formation of an unexpected stable water adduct. J. Het. Chem. 2000, 37, 927–933. [Google Scholar] [CrossRef]
- For ethylation of 3-methyloxindole using triethyloxonium tetrafluoroborate, see: Nakagawa, M.; Hino, T. 2-Ethoxy and 2-ethylthioindoles. Autoxidation and nucleophilic substitutions. Tetrahedron 1970, 26, 4491–4503. [Google Scholar] [CrossRef]
- Hino, T.; Nakagawa, M.; Akaboshi, S. 2-Ethoxyindoles and 2-ethylthioindoles: their autoxidation and reactions with piperidine. Chem. Commun. 1967, 656–658. [Google Scholar] [CrossRef]
- For O-alkylation with ethyl diazoacetate in the presence of Rh2(OCOCF3)4, see: Busch-Petersen, J.; Corey, E.J. Rhodium(II) catalytic approach to the synthesis of ethers of a minor component in a tautomeric set. Org. Lett. 2000, 2, 1641–1643. [Google Scholar] [CrossRef] [PubMed]
- For the introduction of a monofluoromethyl moiety to the oxygen atom using N,N-(dimethylamino)-S-phenyl-S-monofluoromethyloxosulfonium hexafluorophosphate in the presence of tert-butylimino-tris(dimethylamino) phosphorane, see: Nomura, Y.; Tokunaga, E.; Shibata, N. Inherent oxygen preference in enolate monofluoromethylation and a synthetic entry to monofluoromethyl ethers. Angew. Chem. Int. Ed. 2011, 50, 1885–1889. [Google Scholar] [CrossRef] [PubMed]
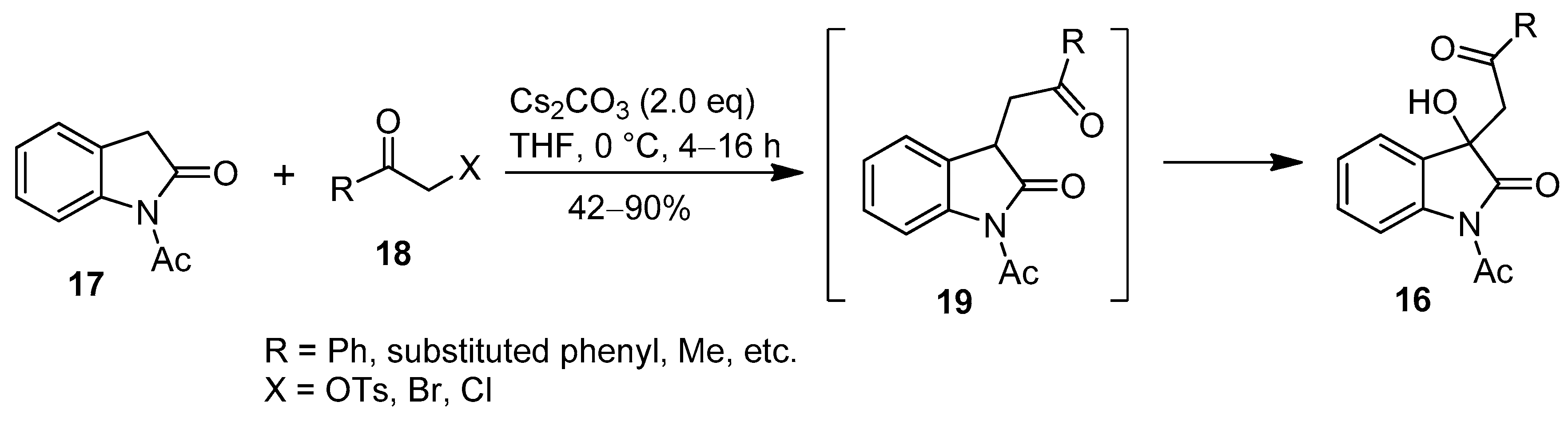
- Bai, M.; You, Y.; Chen, Y.-Z.; Xiang, G.-Y.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. An unprecedented protocol for the synthesis of 3-hydroxy-3-phenacyloxindole derivatives with indolin-2-ones and α-substituted ketones. Org. Biomol. Chem. 2016, 14, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Green, J.R. Science of Synthesis; Majewski, M., Snieckus, V., Eds.; Georg Thieme Verlag: Stuttgart-New York, Germany, 2006; Volume 8a, pp. 427–486. [Google Scholar]
- Clugston, M.; Fleming, R. Advanced Chemistry; Oxford University Press: Oxford, United Kingdom, 2000. [Google Scholar]
- Volk, B.; Simig, G. New one-pot synthesis of 3-alkyl- and 3-(ω-hydroxyalkyl)oxindoles from isatins. Eur. J. Org. Chem. 2003, 3991–3996. [Google Scholar] [CrossRef]
- Drug candidates with a 5-substituted oxindole skeleton are as follows: Tenidap, famitinib, and satavaptan (Phase III); cipargamin, amcasertib, adibendan, NS-1209 and indolidan (Phase II).
- Takayma, K.; Isobe, M.; Harano, K.; Taguchi, T. A general synthetic route of 3,3-disubstituted 3H-indoles and rearrangement of their acyl chloride adducts. Tetrahedron Lett. 1973, 14, 365–368. [Google Scholar] [CrossRef]
- Fan, J.-H.; Zhou, M.-B.; Liu, Y.; Wei, W.-T.; Ouyang, X.-H.; Song, R.-J.; Li, J.-H. Iron-catalyzed oxidative arylmethylation of activated alkenes using a peroxide as the methyl source. Synlett 2014, 25, 657–660. [Google Scholar] [CrossRef]
- Hseih, J.-C.; Cheng, A.-Y.; Fu, J.-H.; Kang, T.-W. Copper-catalyzed domino coupling reaction: an efficient method to synthesize oxindoles. Org. Biomol. Chem. 2012, 10, 6404–6409. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Altman, R.A.; Buchwald, S.L. Palladium-catalyzed enantioselective α-arylation and α-vinylation of oxindoles facilitated by an axially chiral P-stereogenic ligand. J. Am. Chem. Soc. 2009, 131, 9900–9901. [Google Scholar] [CrossRef] [PubMed]
- Braudeau, E.; David, S.; Fischer, J.-C. 2-Hydroxyindoxyls. General and novel preparation, properties, and their role in the perphthalic acid oxidation of indoles. Tetrahedron 1974, 30, 1445–1455. [Google Scholar] [CrossRef]
- Nakazaki, A.; Mori, A.; Kobayashi, S.; Nishikawa, T. Diastereoselective synthesis of 3,3-disubstituted oxindoles from atropisomeric N-aryl oxindole derivatives. Tetrahedron Lett. 2012, 53, 7131–7134. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 9c–u are available from the authors.
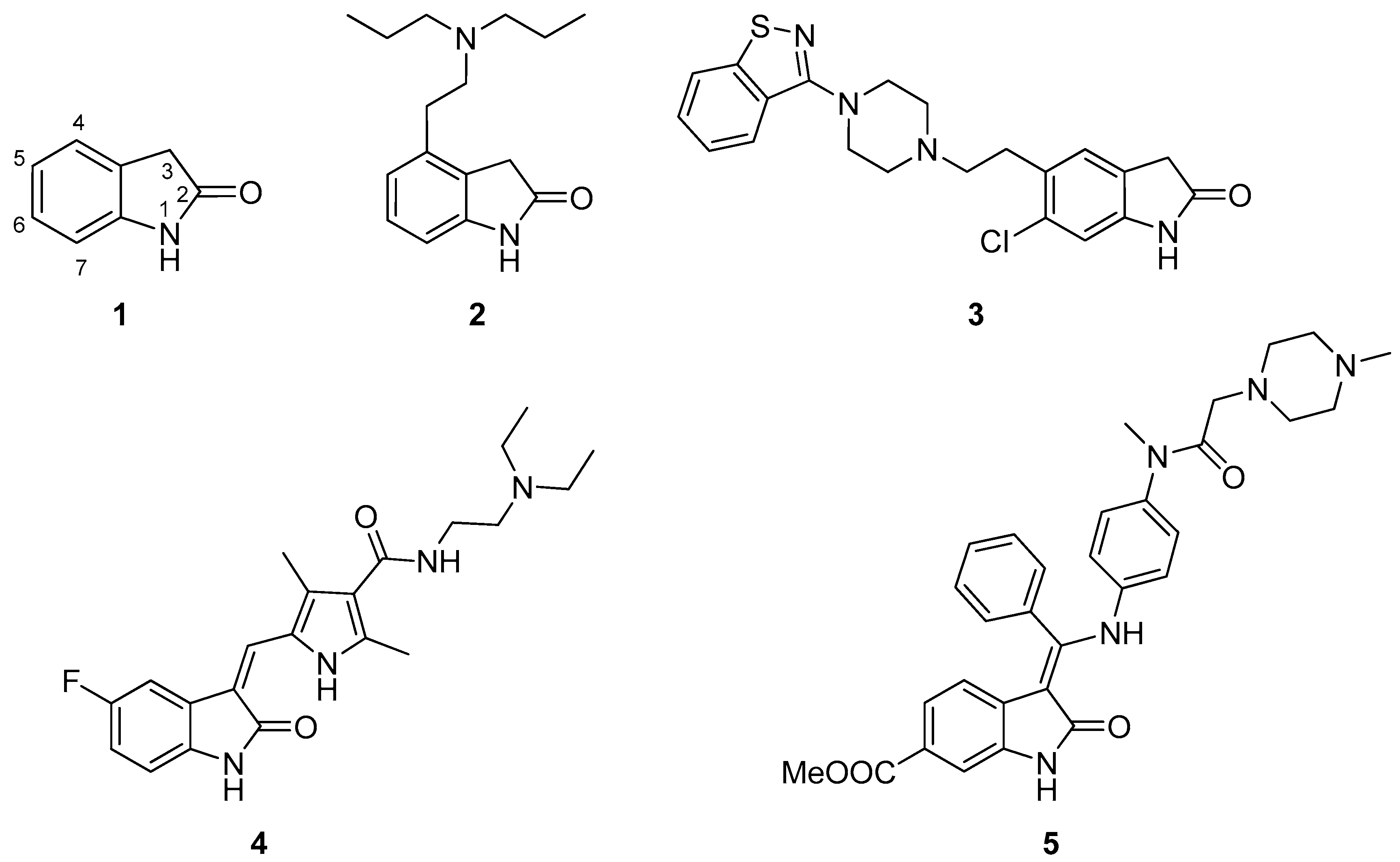
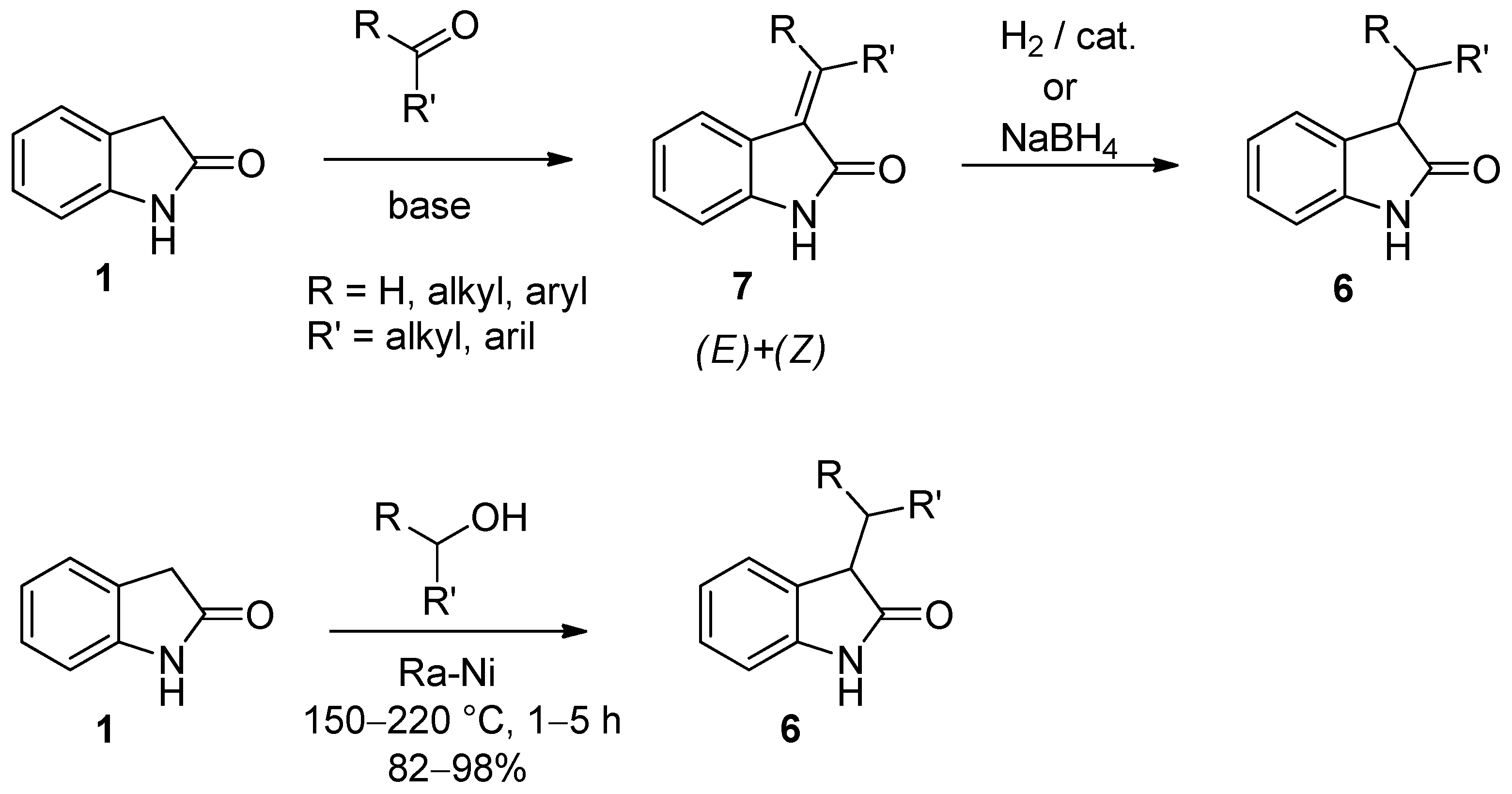
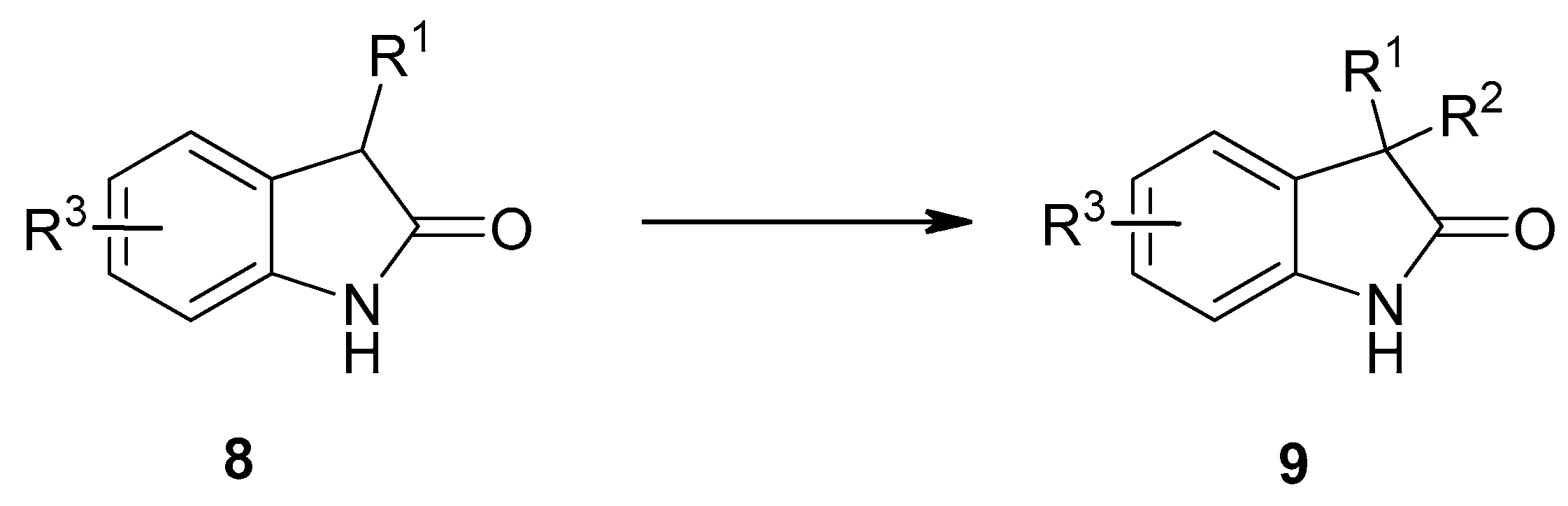


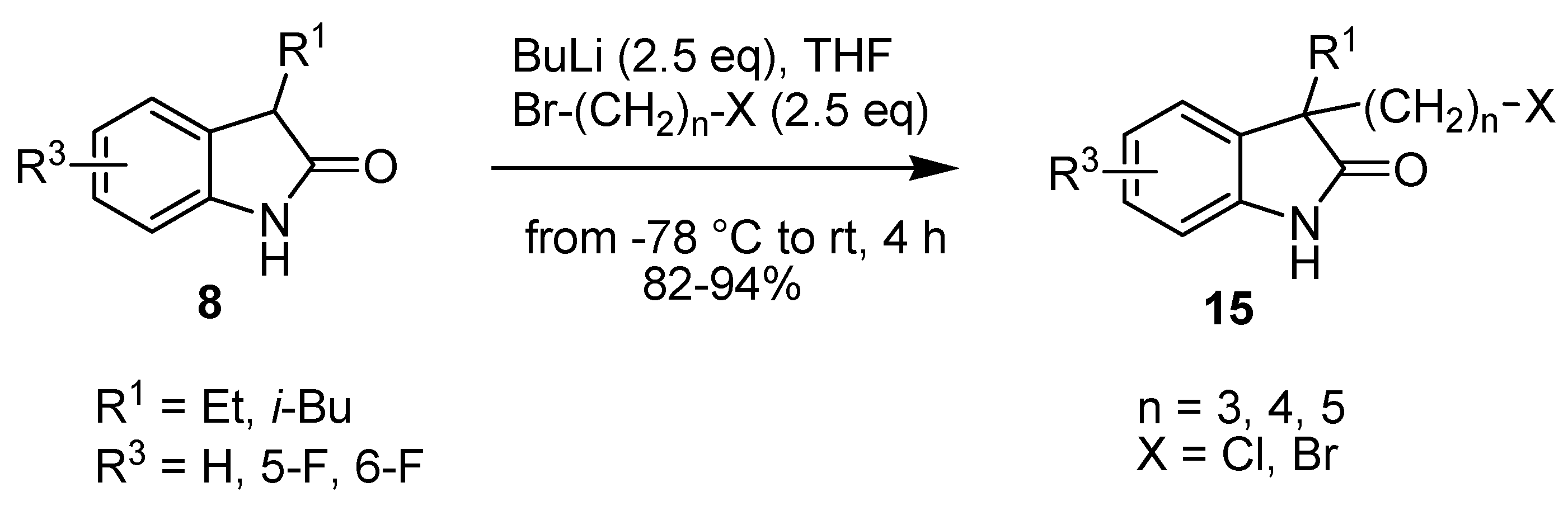
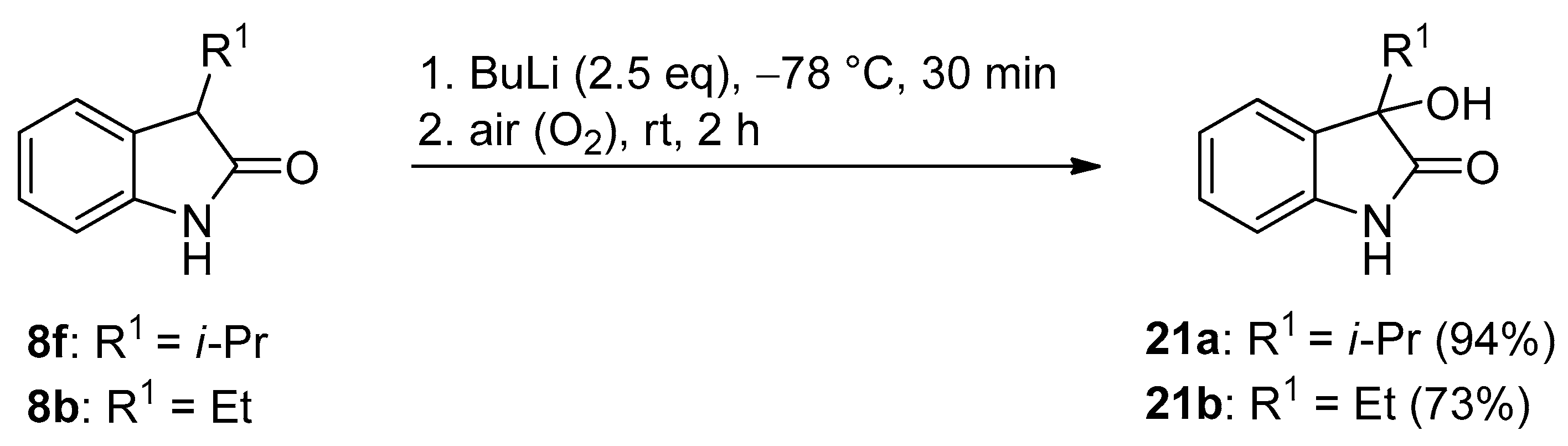
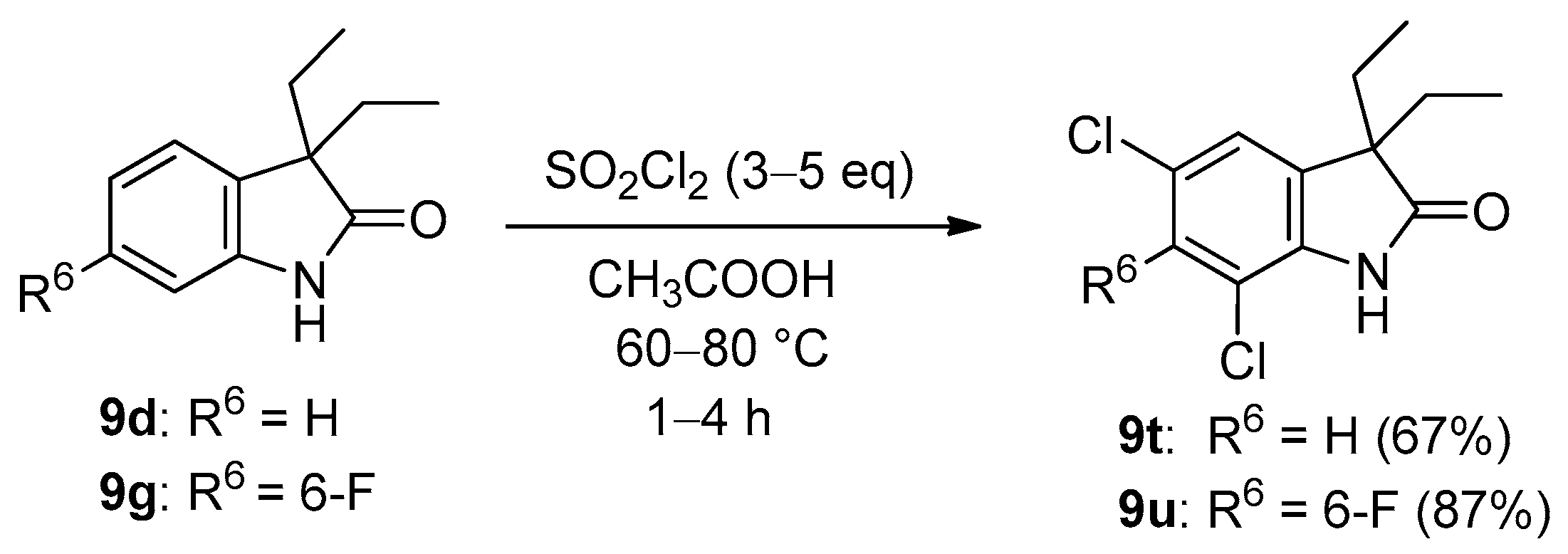

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
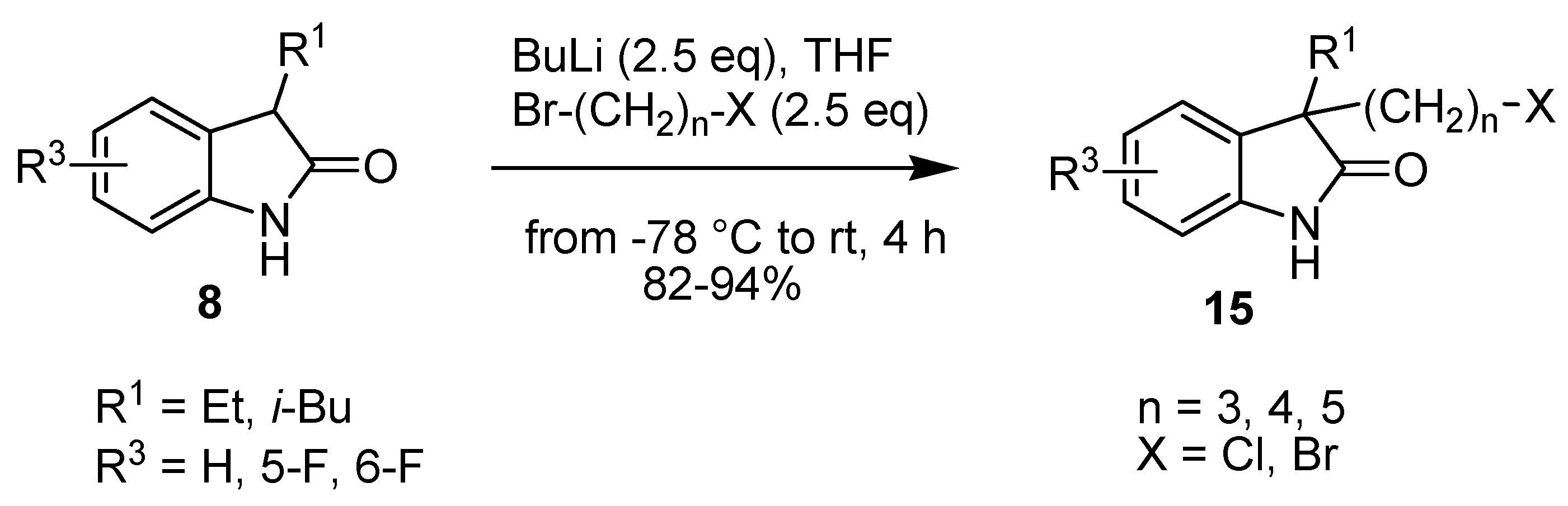{kind=link}
{kind=link}
{kind=link}
{kind=link}
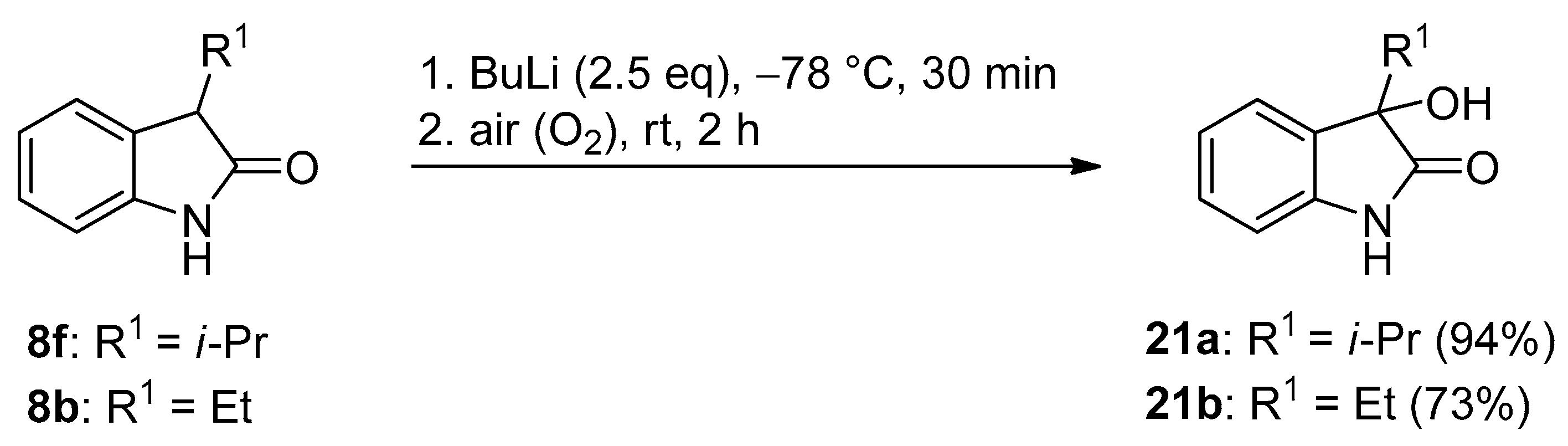{kind=link}
{kind=link}
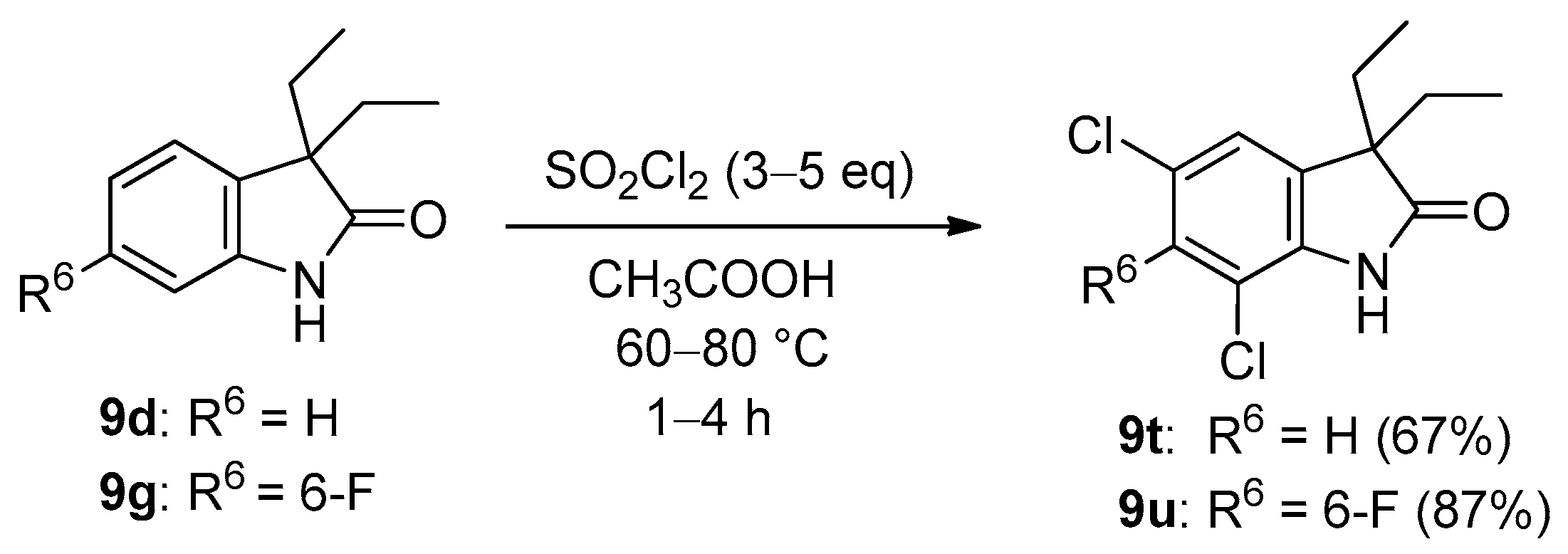{kind=link}
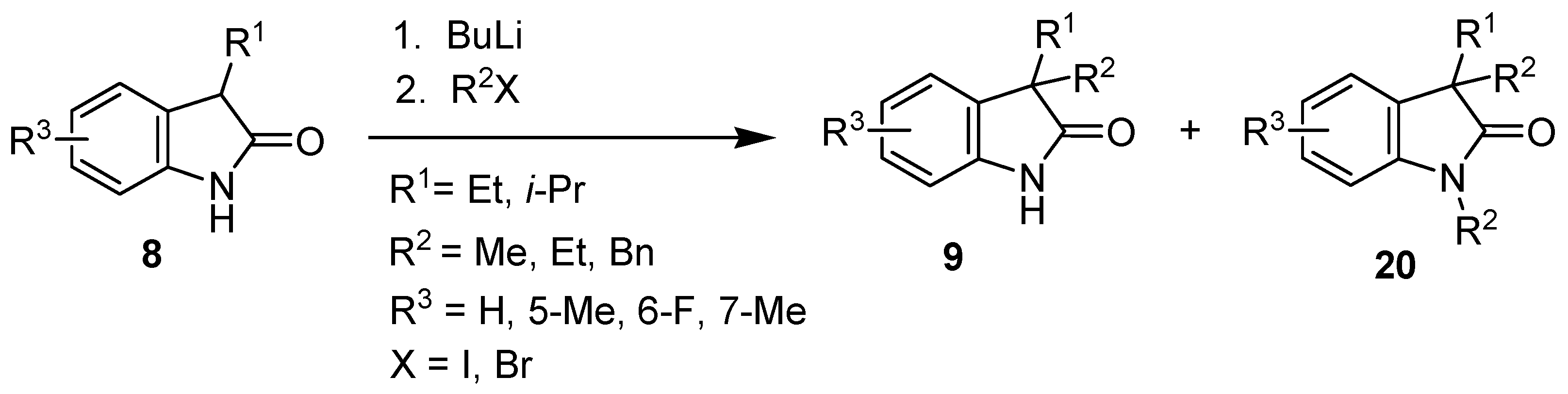
| Entry | 8 | R1 | R3 | BuLi (eq) | R2X (eq) | 9 | 9 Yield (%) | 20 Yield (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | b | Et | H | 2.5 | MeI (2.5) | c | 28 a | 55 a (20a) |
| 2 | b | Et | H | 2.2 | MeI (1.2) | c | 71 | 0 |
| 3 | b | Et | H | 2.5 | EtI (2.5) | d | 73 | 0 |
| 4 | b | Et | H | 2.2 | EtBr (1.2) | d | 90 | 0 |
| 5 | b | Et | H | 2.2 | BnBr (1.2) | e | 80 | 0 |
| 6 | c | Et | 5-Me | 2.2 | EtBr (1.2) | f | 76 | 0 |
| 7 | d | Et | 6-F | 2.2 | EtBr (1.2) | g | 77 | 0 |
| 8 | e | Et | 7-Me | 2.2 | EtBr (1.2) | h | 66 | 0 |
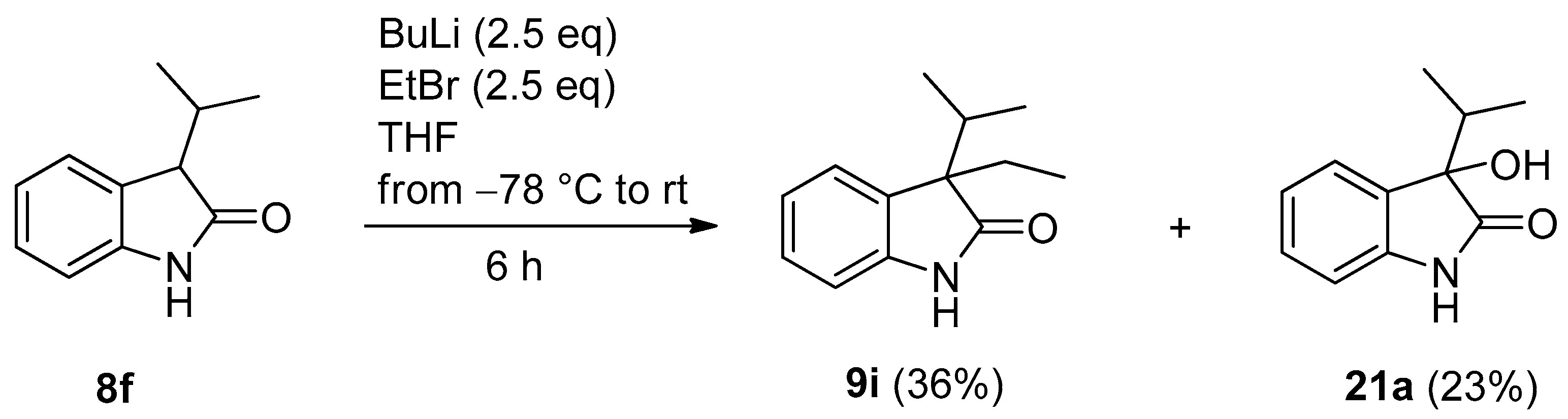
| 9 | f | i-Pr | H | 2.5 | MeI (2.5) | i | 40 a | 35 a (20b) |
| 10 | f | i-Pr | H | 2.5 | MeI (1.2) | i | 56 | 0 |
| 11 | f | i-Pr | H | 2.2 | EtBr (1.2) | j | 65 | 0 |
| 12 | f | i-Pr | H | 2.2 | BnBr (1.2) | k | 63 | 0 |
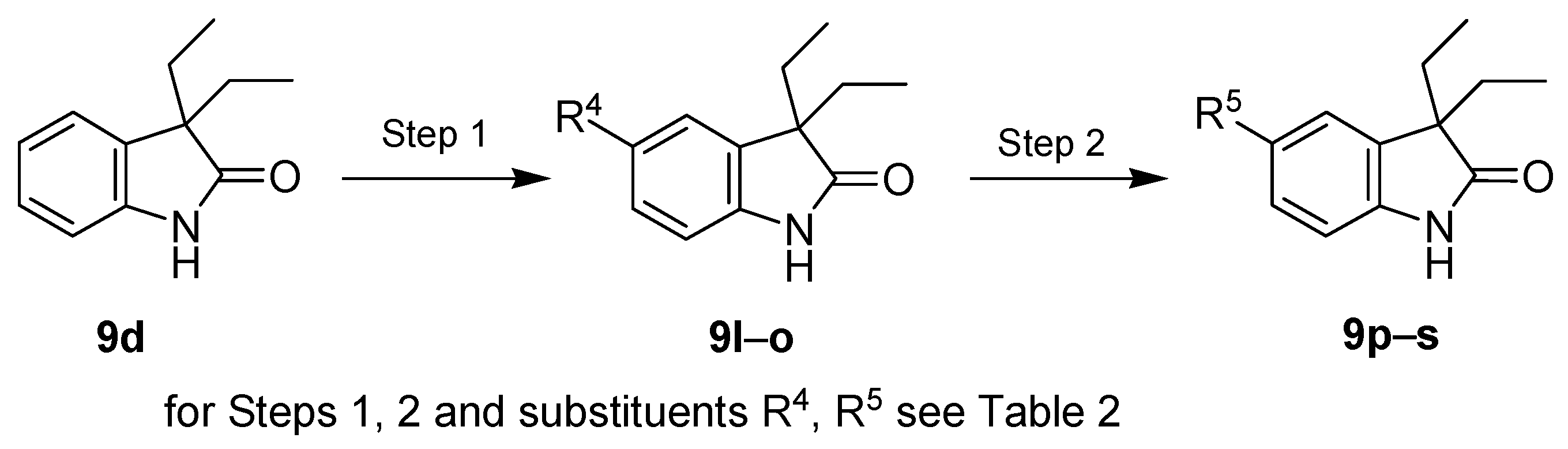
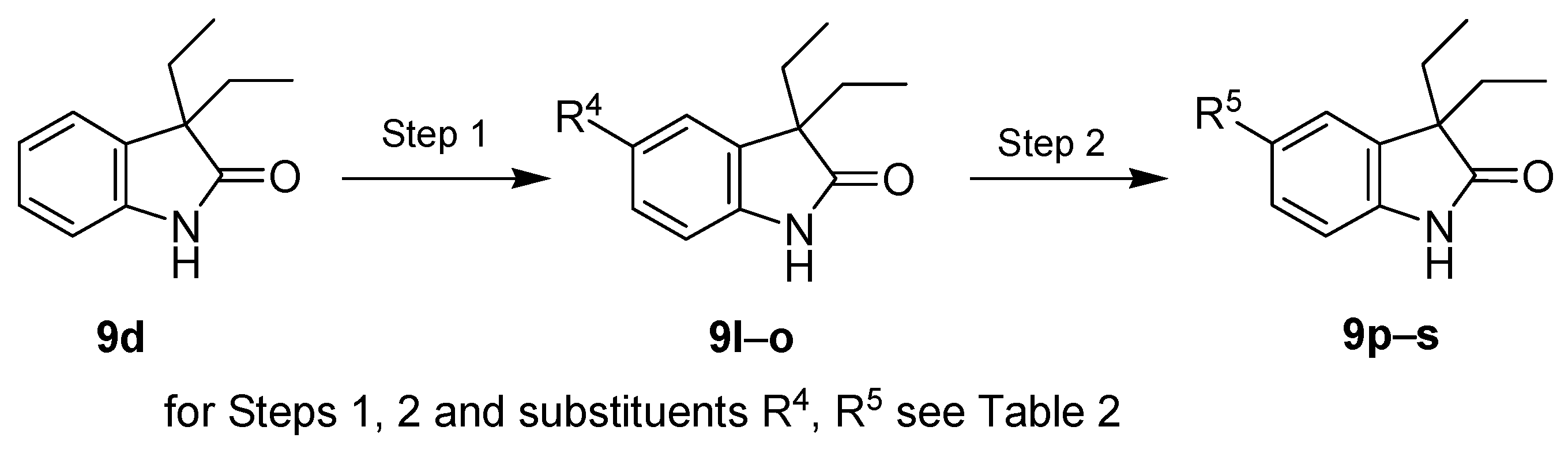
| Entry | Reagents of Step 1 a | Product of Step 1 | R4 | Yield of Step 1 (%) | Reagents of Step 2 | Product of Step 2 | R5 | Yield of Step 2 (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | SO2Cl2 | 9l | Cl | 73 | - | - | - | - |
| 2 | Br2, KBr | 9m | Br | 94 | - | - | - | - |
| 3 | HNO3, H2SO4 | 9n | NO2 | 85 | H2/Pd/C | 9p | NH2 | 87 |
| 4 | ClSO3H | 9o | SO2Cl | 98 | NH3 | 9q | NH2SO2 | 60 |
| 5 | t-BuNH2 | 9r | t-BuNHSO2 | 61 | ||||
| 6 | morpholine | 9s | (morph)SO2 | 76 |
© 2016 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kókai, E.; Simig, G.; Volk, B. Literature Survey and Further Studies on the 3-Alkylation of N-Unprotected 3-Monosubstituted Oxindoles. Practical Synthesis of N-Unprotected 3,3-Disubstituted Oxindoles and Subsequent Transformations on the Aromatic Ring. Molecules 2017, 22, 24. https://doi.org/10.3390/molecules22010024
Kókai E, Simig G, Volk B. Literature Survey and Further Studies on the 3-Alkylation of N-Unprotected 3-Monosubstituted Oxindoles. Practical Synthesis of N-Unprotected 3,3-Disubstituted Oxindoles and Subsequent Transformations on the Aromatic Ring. Molecules. 2017; 22(1):24. https://doi.org/10.3390/molecules22010024
Chicago/Turabian StyleKókai, Eszter, Gyula Simig, and Balázs Volk. 2017. "Literature Survey and Further Studies on the 3-Alkylation of N-Unprotected 3-Monosubstituted Oxindoles. Practical Synthesis of N-Unprotected 3,3-Disubstituted Oxindoles and Subsequent Transformations on the Aromatic Ring" Molecules 22, no. 1: 24. https://doi.org/10.3390/molecules22010024