
Fenofibrate Therapy Restores Antioxidant Protection and Improves Myocardial Insulin Resistance in a Rat Model of Metabolic Syndrome and Myocardial Ischemia: The Role of Angiotensin II

 ,
,  ,
,  and
and
Abstract
:1. Introduction
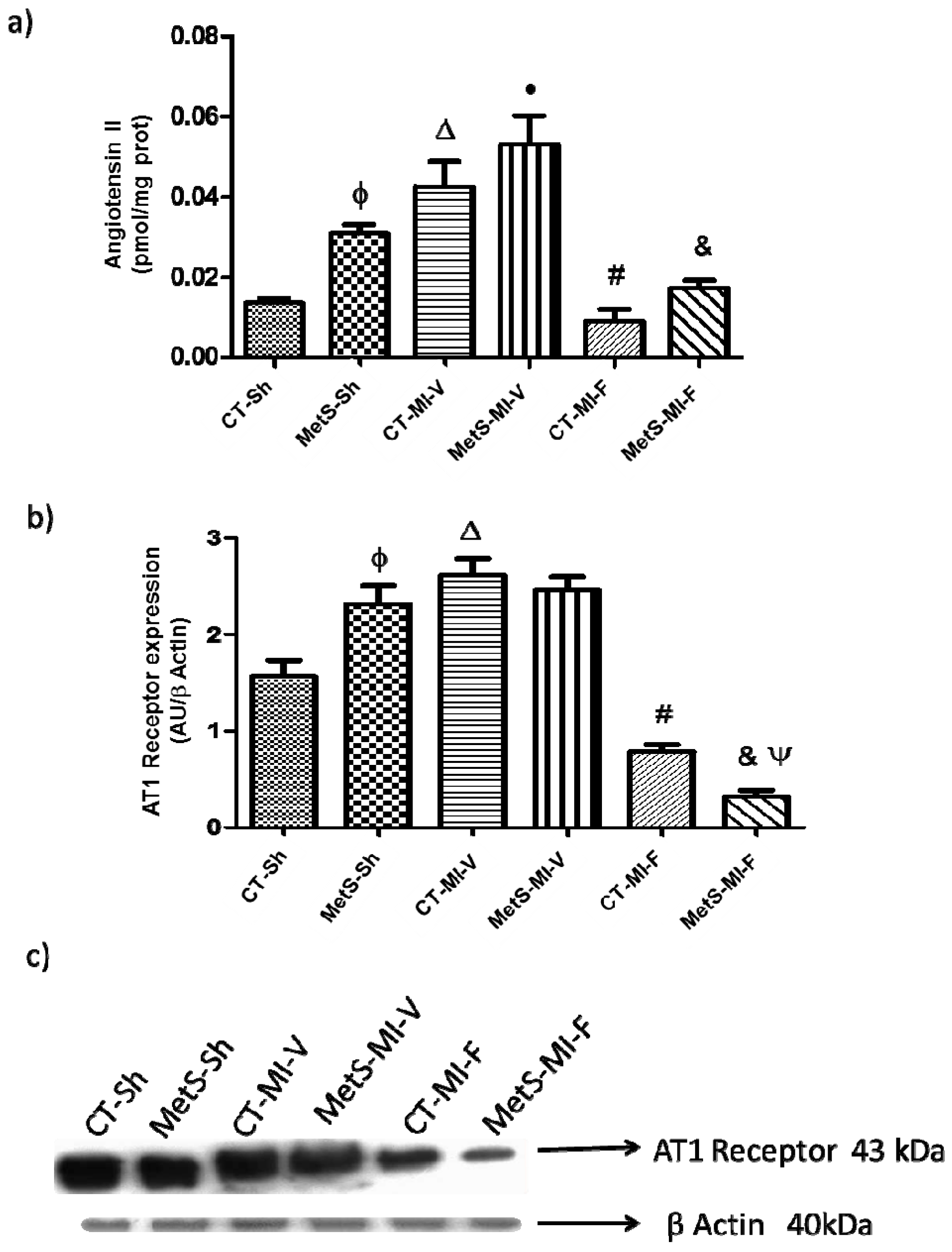
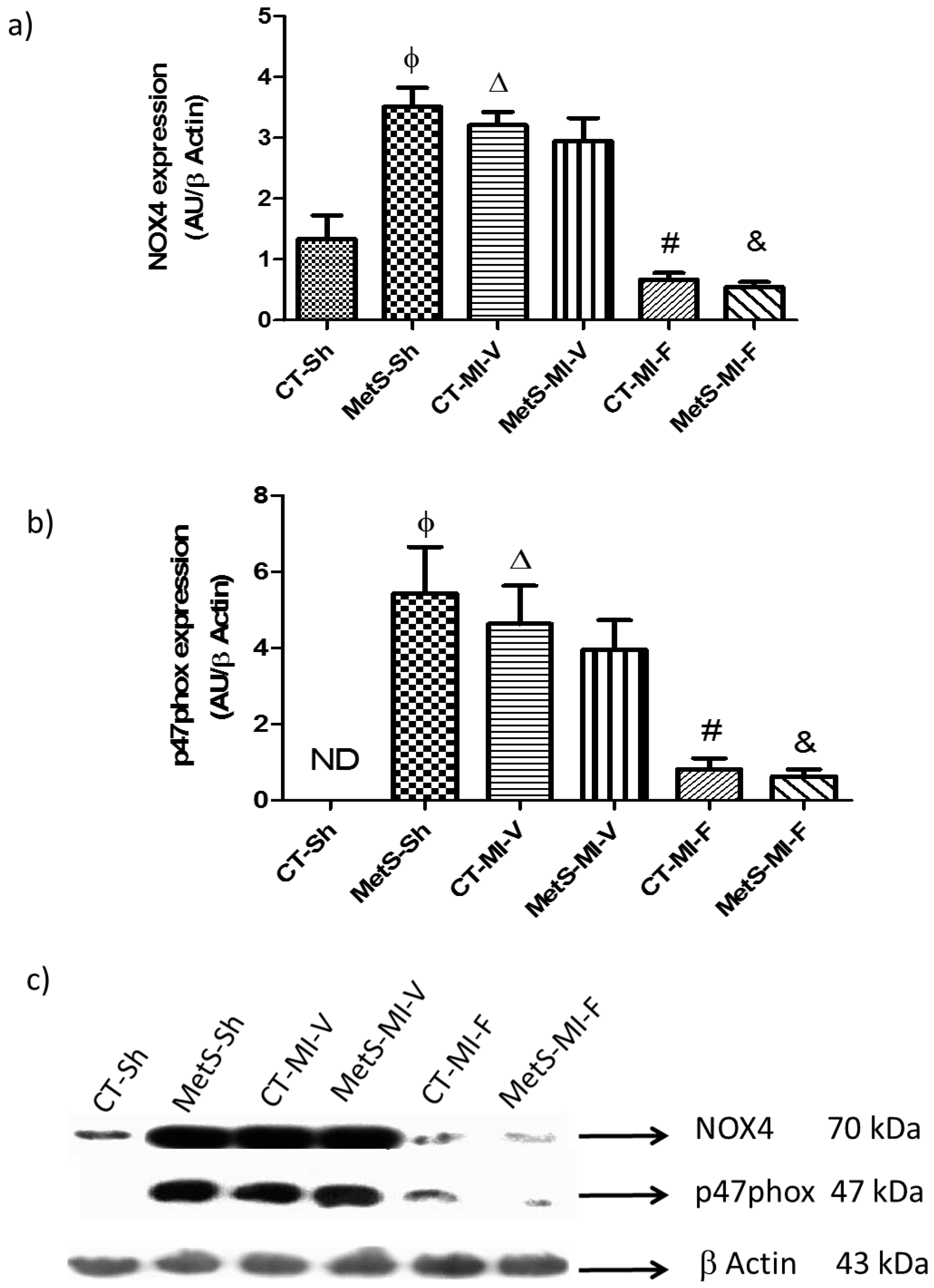
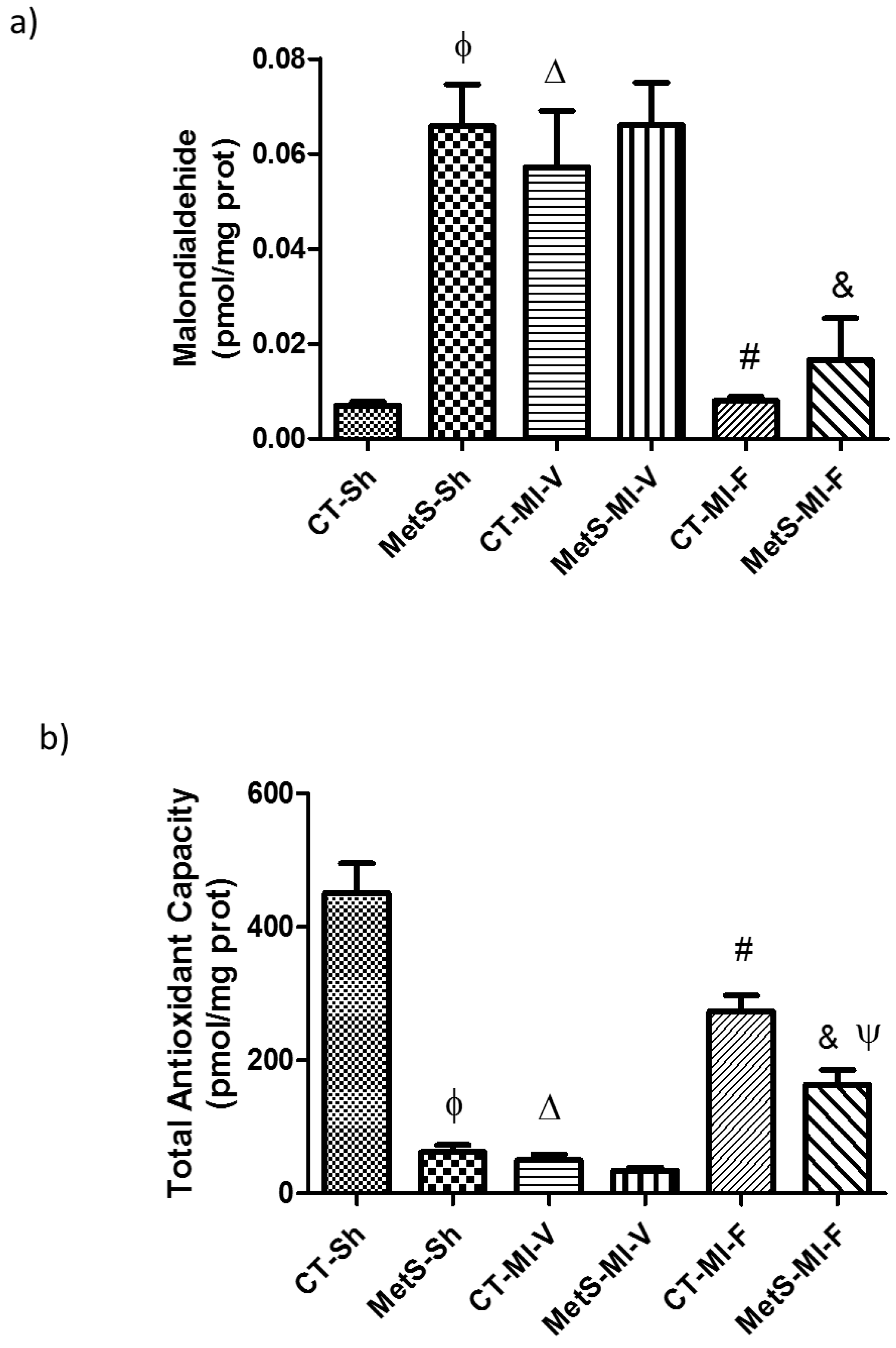
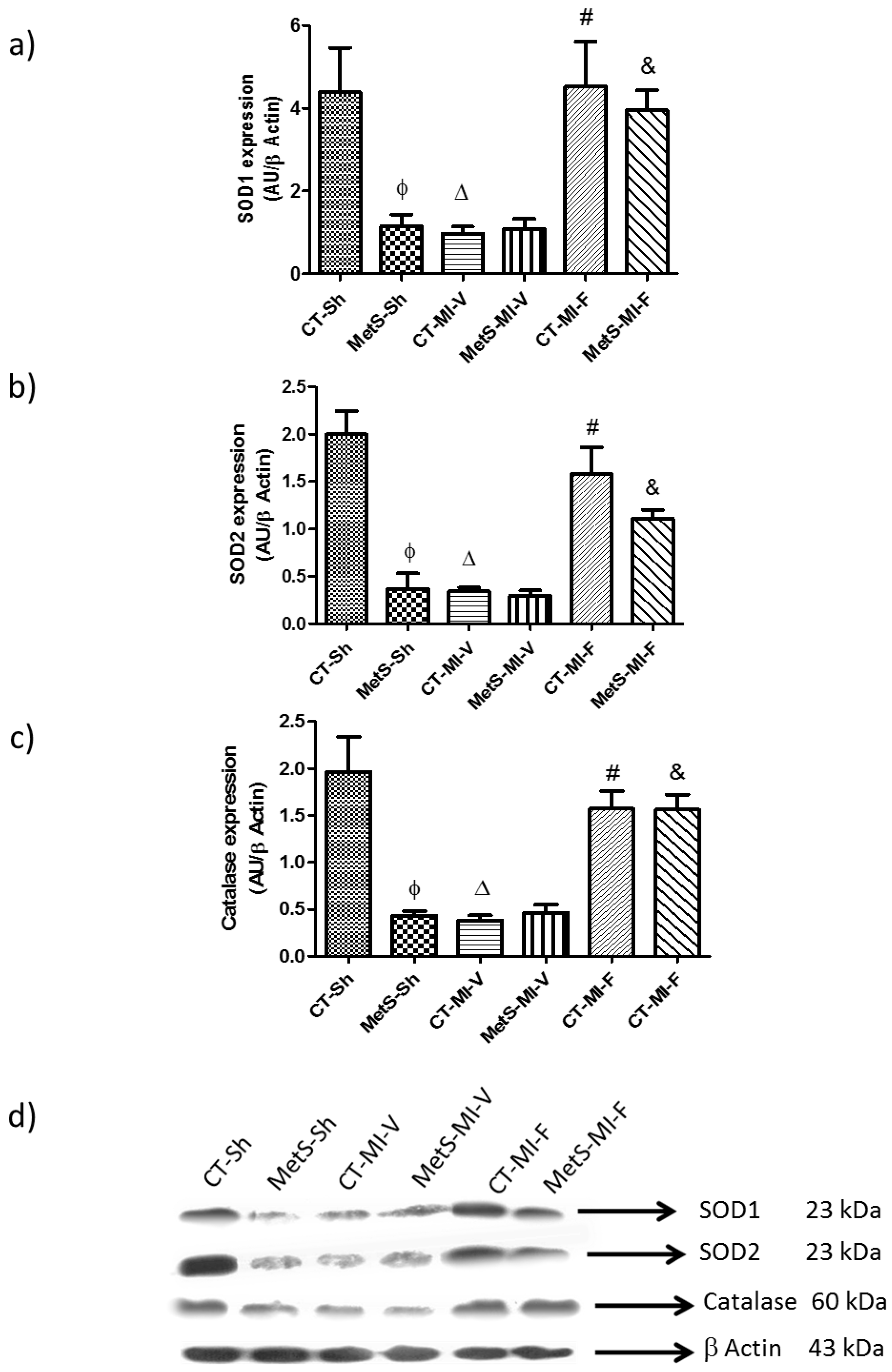
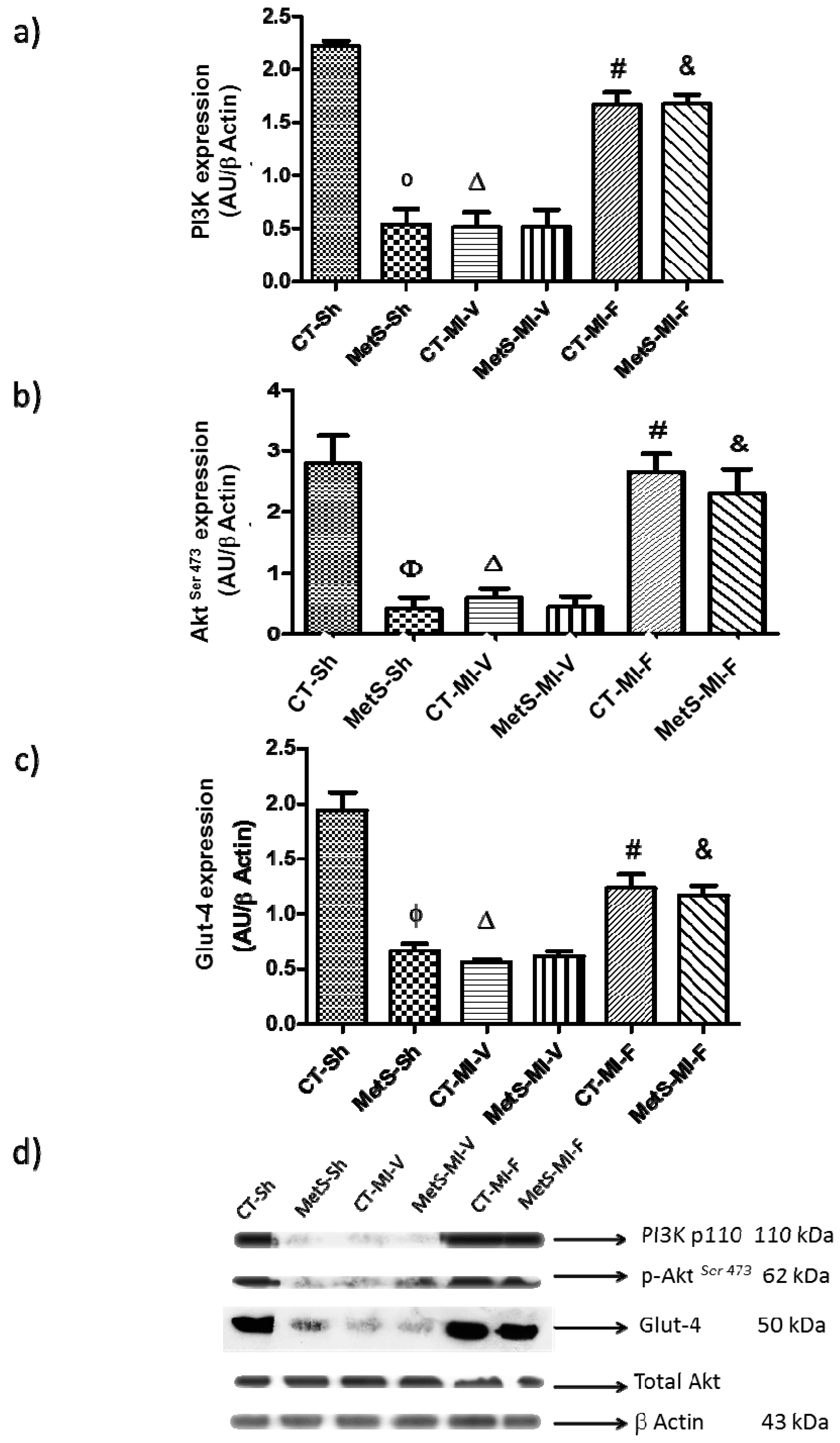
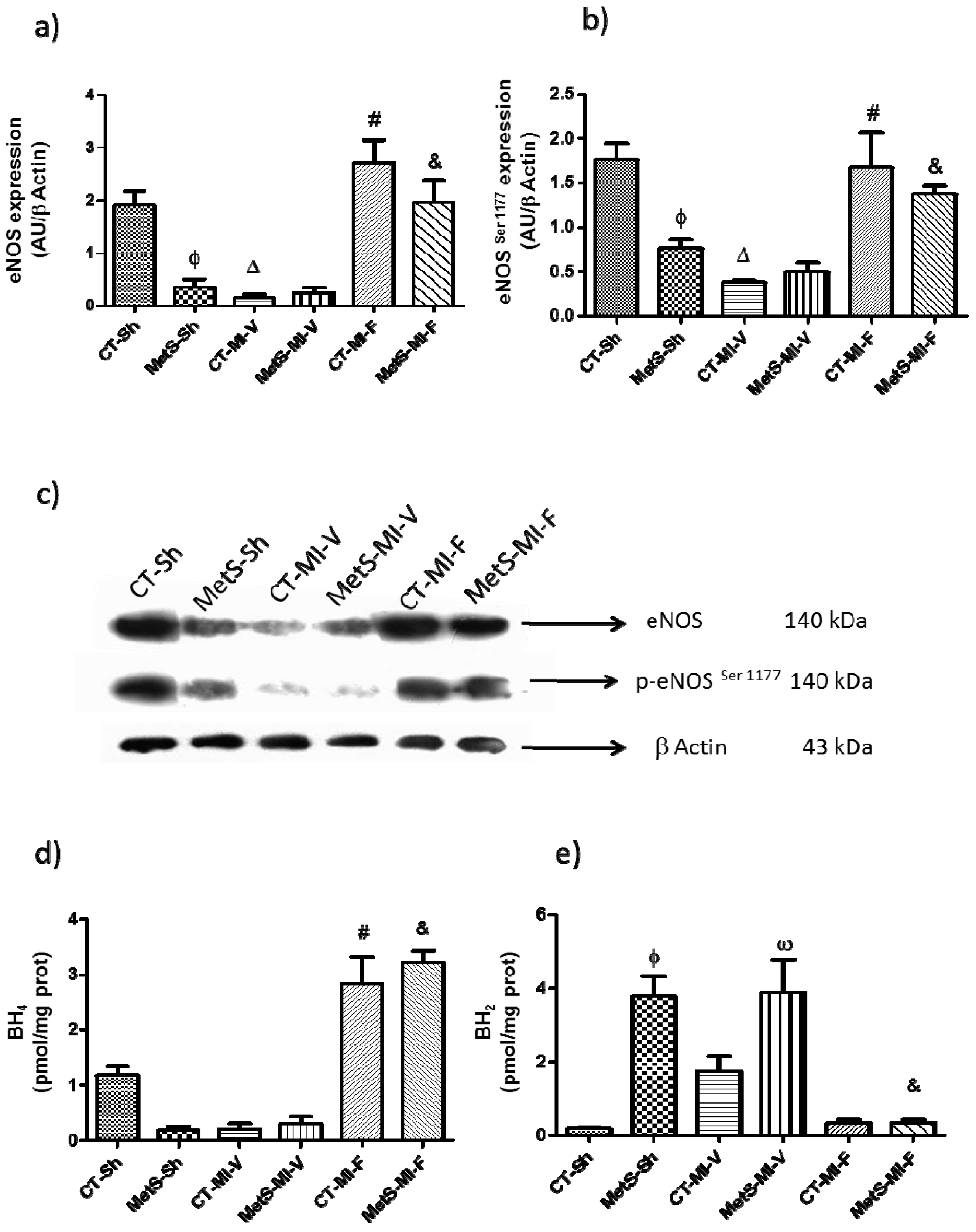
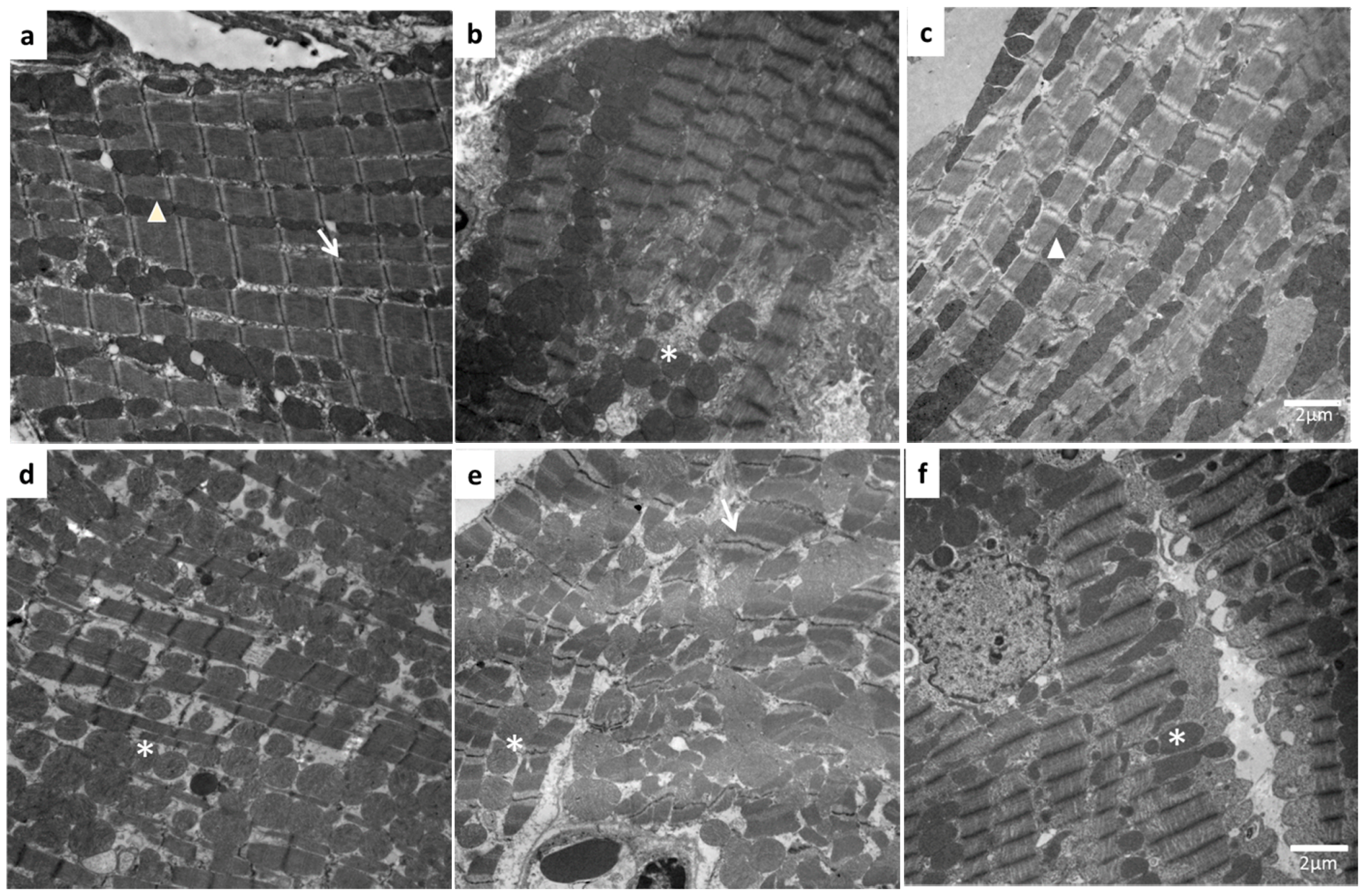
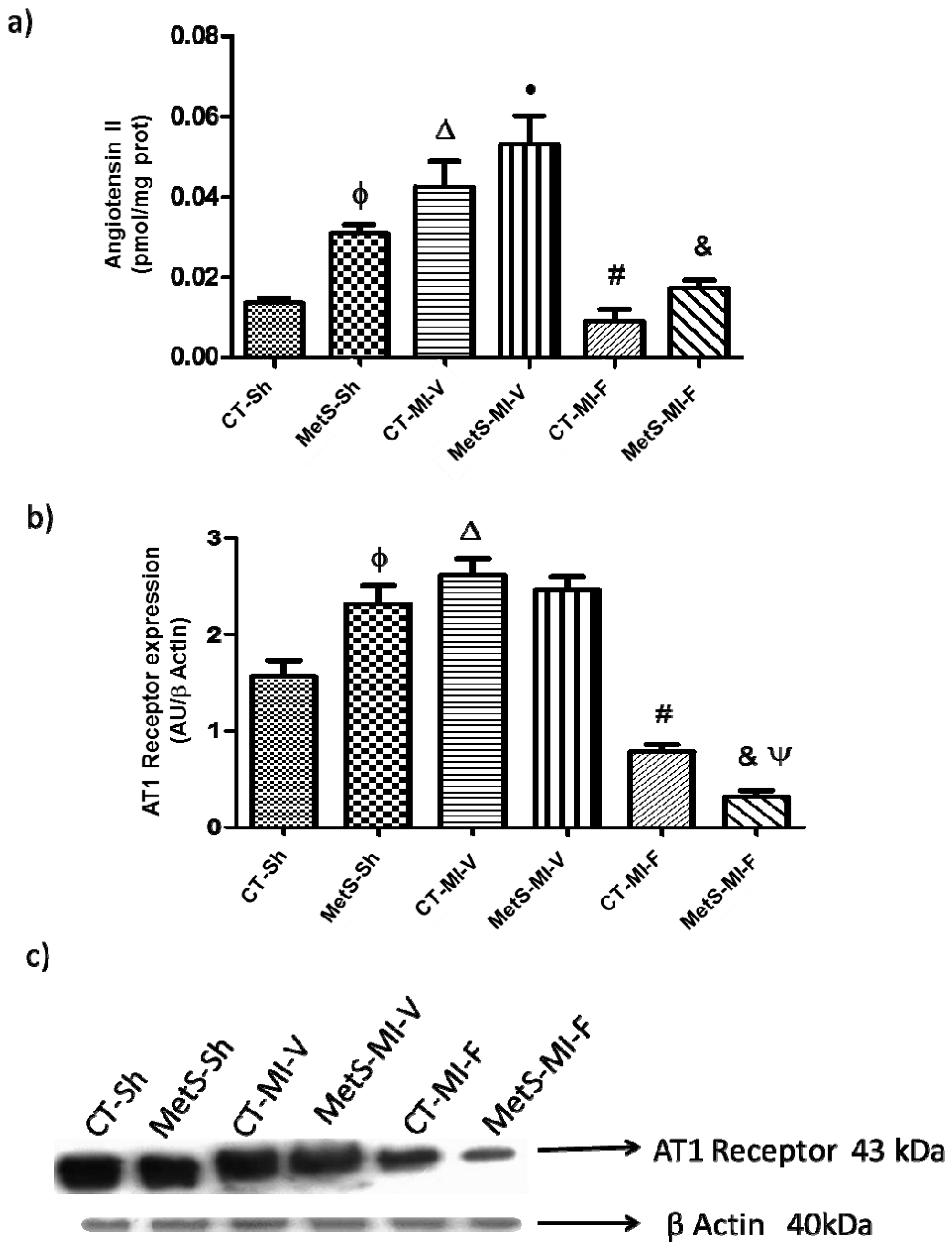
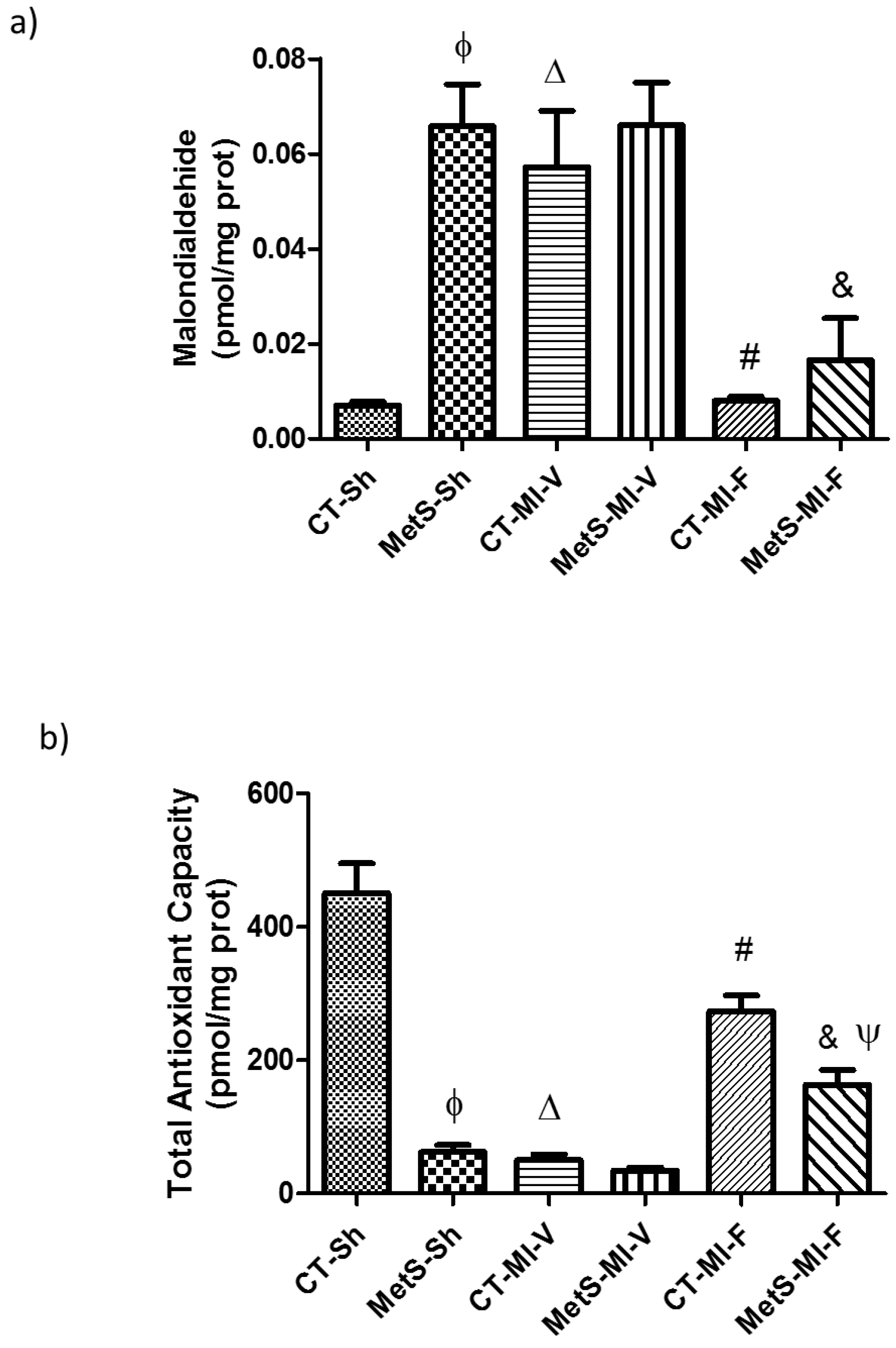
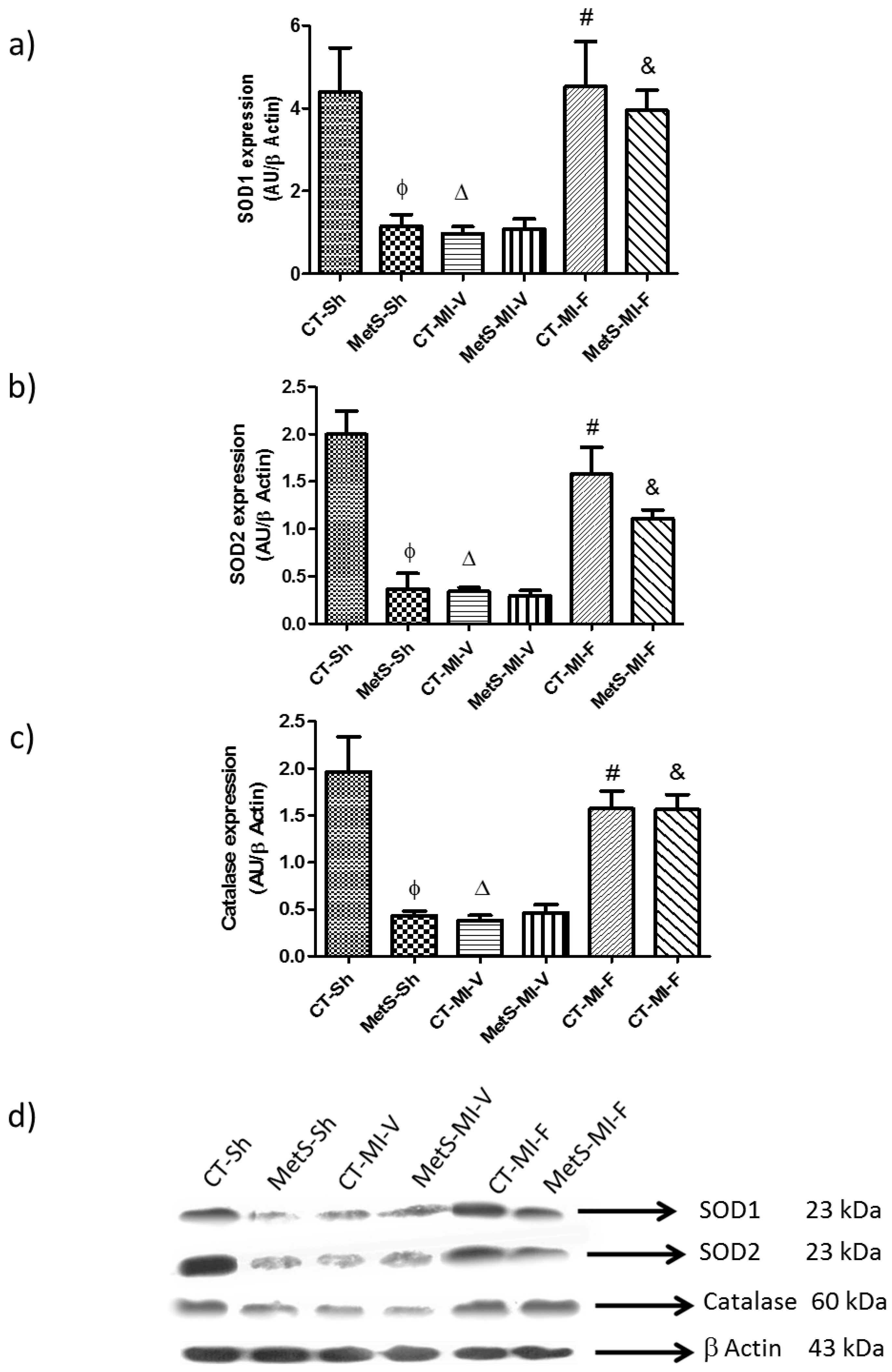
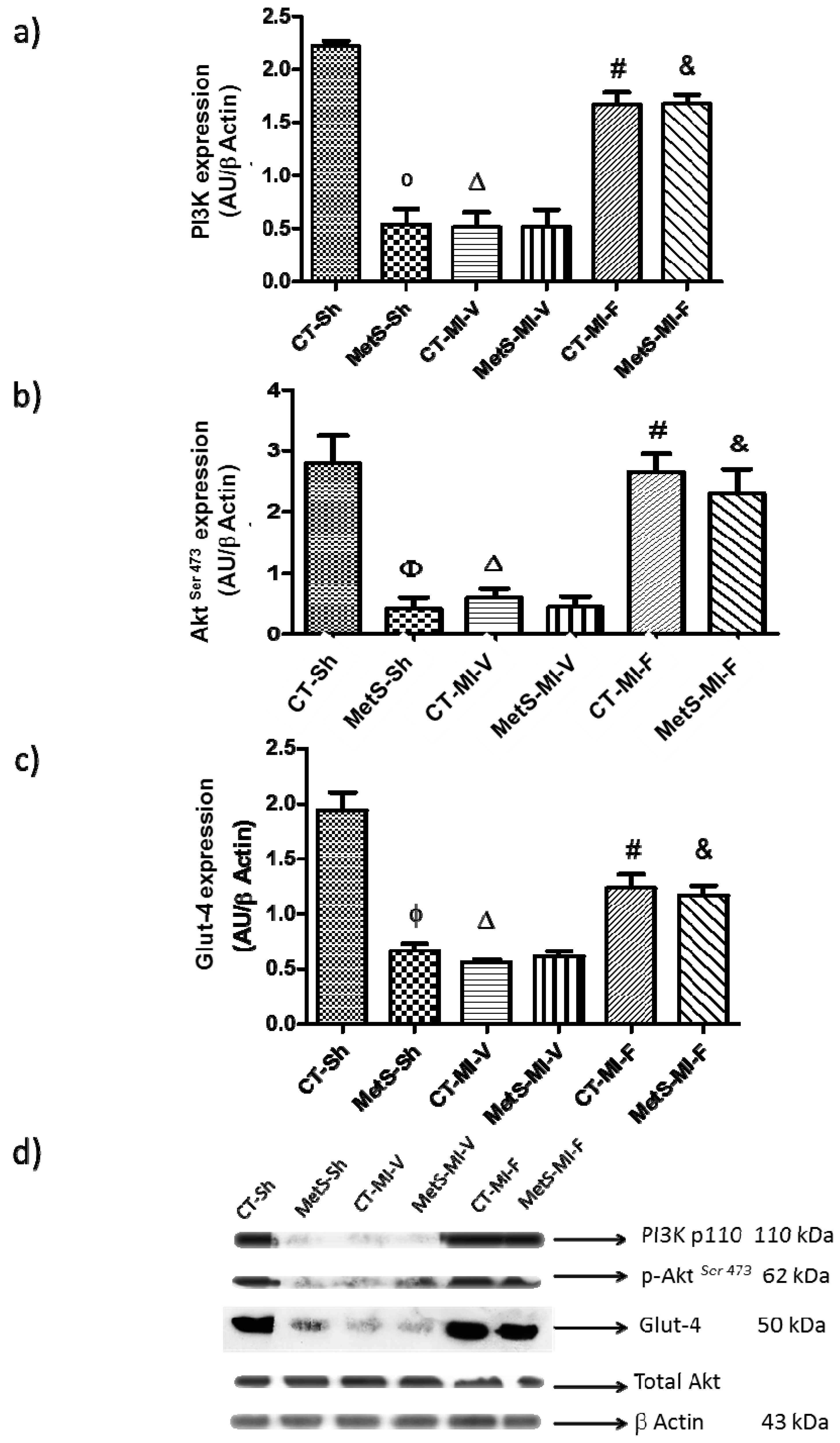
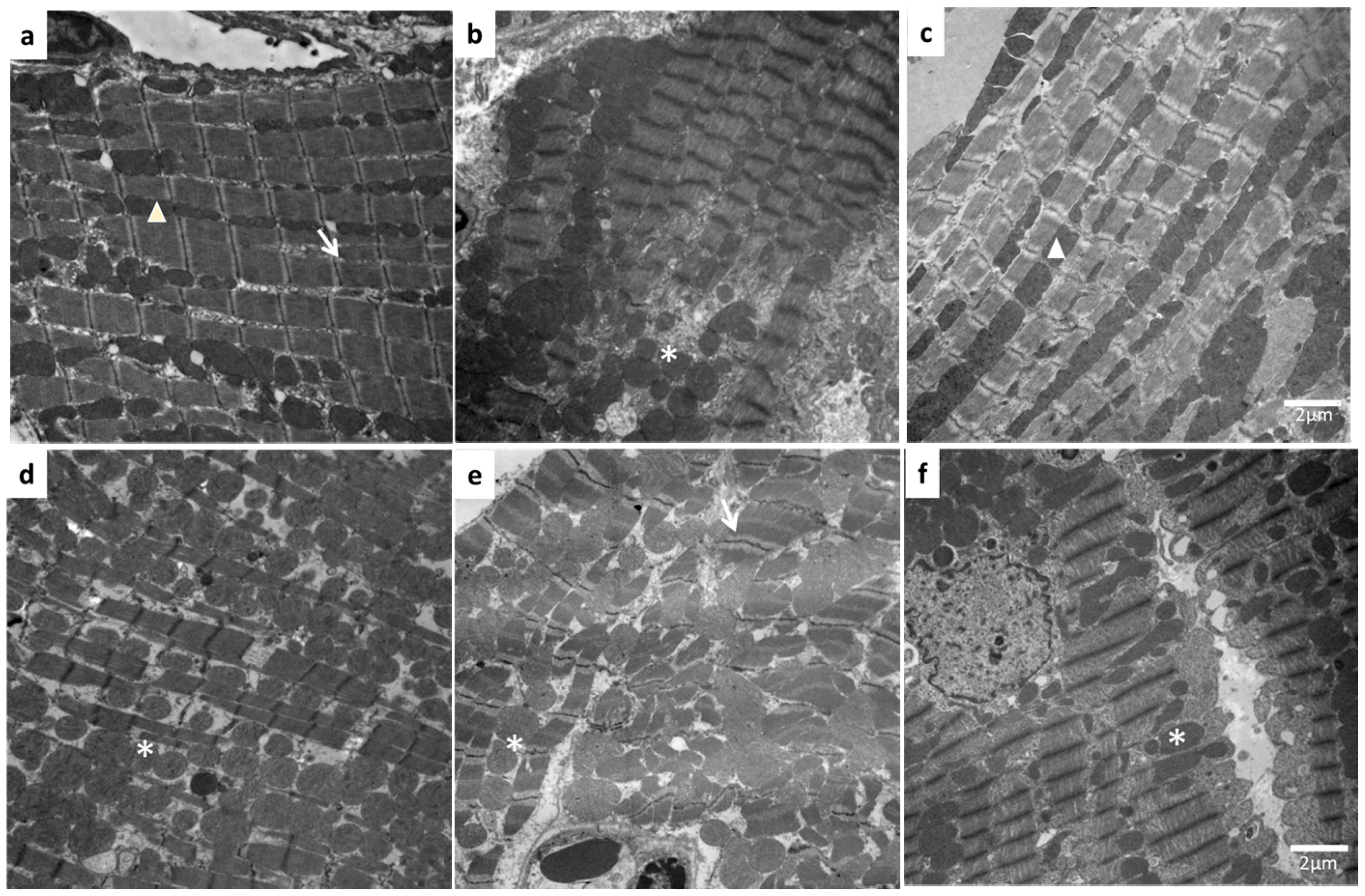
2. Results
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. Myocardial Infarction
4.3. Biochemical Serum Measurement
4.4. Measurement of Total Antioxidant Capacity
4.5. Quantification of Ang II, Malondialdehyde, Tetrahydrobiopterin and Dihydrobiopterin
4.6. Western Blotting
4.7. Structural Analysis by Electron Microscopy
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Abel, E.D.; O’Shea, K.M.; Ramasamy, R. Insulin resistance: Metabolic mechanisms and consequences in the heart. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2068–2076. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, J.; Ago, T.; Matsushima, S.; Zhai, P.; Schneider, M.D.; Sadoshima, J. NADPH oxidase 4 (Nox4) is a major source stress in the of oxidative failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 15565–15570. [Google Scholar] [CrossRef] [PubMed]
- Beauloye, C.; Bertrand, L.; Krause, U.; Marsin, A.S.; Dresselaers, T.; Vanstapel, F.; Vanoverschelde, J.L.; Hue, L. No-flow ischemia inhibits insulin signaling in heart by decreasing intracellular pH. Circ. Res. 2001, 88, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.; Zhao, K.; Li, J.; Xu, J.; Zhang, Y.; Liu, C.; Yang, W.; Gao, C.; Li, J.; Zhang, H.; et al. Direct evidence that myocardial insulin resistance following myocardial ischemia contributes to post-ischemic heart failure. Sci. Rep. 2015, 2015, 17927. [Google Scholar] [CrossRef] [PubMed]
- Amorim, P.A.; Nguyen, T.D.; Shingu, Y.; Schwarzer, M.; Mohr, F.W.; Schrepper, A.; Doenst, T. Myocardial infarction in rats causes partial impairment in insulin response associated with reduced fatty acid oxidation and mitochondrial gene expression. J. Thorac. Cardiovasc. Surg. 2010, 140, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Guarner, V.; Rubio-Ruiz, M.E. Aging, metabolic syndrome and the heart. Aging Dis. 2012, 3, 269–279. [Google Scholar]
- Rubio-Ruíz, M.E.; Del Valle-Mondragón, L.; Castrejón-Tellez, V.; Carreón-Torres, E.; Díaz-Díaz, E.; Guarner-Lans, V. Angiotensin II and 1–7 during aging in metabolic syndrome rats. Expression of AT1, AT2 and Mas receptors in abdominal white adipose tissue. Peptides 2014, 57, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Ibarra-Lara, L.; Sánchez-Aguilar, M.; Hong, E.; del Valle-Mondragón, L.; Soria-Castro, E.; Pérez-Severiano, F.; Torres-Narváez, J.C.; Ramírez-Ortega, M.; Pastelín-Hernández, G.S.; Cervantes-Pérez, L.G.; et al. PPARα stimulation modulates myocardial ischemia-induced activation of renin-angiotensin system. J. Cardiovasc. Pharmacol. 2015, 65, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Ayajiki, K.; Nishio, Y.; Sugaya, T.; Kashiwagi, A.; Okamura, T. Evidence for a causal role of the renin-angiotensin system in vascular dysfunction associated with insulin resistance. Hypertension 2004, 43, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Kobayashi, N.; Ohno, T.; Fukushima, H.; Matsuoka, H. Cardioprotective effect of angiotensin II type 1 receptor antagonist associated with bradykinin-endothelial nitric oxide synthase and oxidative stress in Dahl salt-sensitive hypertensive rats. J. Hypertens. 2007, 25, 1633–1642. [Google Scholar] [CrossRef] [PubMed]
- Oidor-Chan, V.H.; Hong, E.; Pérez-Severiano, F.; Montes, S.; Torres-Narváez, J.C.; Del Valle-Mondragón, L.; Pastelín-Hernández, G.; Sánchez-Mendoza, A. Fenofibrate plus Metformin Produces Cardioprotection in a Type 2 Diabetes and Acute Myocardial Infarction Model. PPAR Res. 2016, 2016, 8237264. [Google Scholar] [CrossRef] [PubMed]
- Sowers, J.R. Insulin resistance and hypertension. Am. J. Physiol. Heart. Circ. Physiol. 2004, 286, H1597–H1602. [Google Scholar] [CrossRef] [PubMed]
- Tuck, M.L.; Bounoua, F.; Eslami, P.; Nyby, M.D.; Eggena, P.; Corry, D.B. Insulin stimulates endogenous angiotensin II production via a mitogen-activated protein kinase pathway in vascular smooth muscle cells. J. Hypertens 2004, 22, 1779–1785. [Google Scholar] [CrossRef] [PubMed]
- Ibarra-Lara, L.; Hong, E.; Soria-Castro, E.; Torres-Narváez, J.C.; Pérez-Severiano, F.; Del Valle-Mondragón, L.; Cervantes-Pérez, L.G.; Ramírez-Ortega, M.; Pastelín-Hernández, G.S.; Sánchez-Mendoza, A. Clofibrate PPARα activation reduces oxidative stress and improves ultrastructure and ventricular hemodynamics in no-flow myocardial ischemia. J. Cardiovasc. Pharmacol. 2012, 60, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Ibarra-Lara, L.; Del Valle-Mondragón, L.; Soria-Castro, E.; Torres-Narváez, J.C.; Pérez-Severiano, F.; Sánchez-Aguilar, M.; Ramírez-Ortega, M.; Cervantes-Pérez, L.G.; Pastelín-Hernández, G.S.; Oidor-Chan, V.H.; et al. Peroxisome proliferator-activated receptor-α stimulation by clofibrate favors an antioxidant and vasodilator environment in a stressed left ventricle. Pharmacol. Rep. 2016, 68, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Ilkun, O.; Boudina, S. Cardiac dysfunction and oxidative stress in the metabolic syndrome: An update on antioxidant therapies. Curr. Pharm. Des. 2013, 19, 4806–4817. [Google Scholar] [CrossRef] [PubMed]
- Hiuge, A.; Tenenbaum, A.; Maeda, N.; Benderly, M.; Kumada, M.; Fisman, E.Z.; Tanne, D.; Matas, Z.; Hibuse, T.; Fujita, K.; et al. Effects of peroxisome proliferator-activated receptor ligands, bezafibrate and fenofibrate, on adiponectin level. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Nita, C.; Bala, C.; Porojan, M.; Hancu, N. Fenofibrate improves endothelial function and plasma myeloperoxidase in patients with type 2 diabetes mellitus: An open-label interventional study. Diabetol. Metab. Syndr. 2014, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.; Djouadi, F.; Nelly, D.P. Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via de peroxisome-activaded receptor alpha. J. Biol. Chem. 1998, 273, 23786–23792. [Google Scholar] [CrossRef] [PubMed]
- Ibarra-Lara, L.; Cervantes-Pérez, L.G.; Pérez-Severiano, F.; Del Valle, L.; Rubio-Ruíz, E.; Soria-Castro, E.; Pastelín-Hernández, G.S.; Sánchez-Aguilar, M.; Martínez-Lazcano, J.C.; Sánchez-Mendoza, A. PPARalpha stimulation exerts a blood pressure lowering effect through different mechanisms in a time-dependent manner. Eur. J. Pharmacol. 2010, 627, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.; O’Brien, R.; Fulcher, G.; Pardy, C.; D’Emden, M.; Tse, D.; Taskinen, M.R.; Ehnholm, C.; Keech, A. Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) Study Investigators. Effects of fenofibrate treatment on cardiovascular disease risk in 9795 individuals with type 2 diabetes and various components of the metabolic syndrome: The Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study. Diabetes Care 2009, 32, 493–498. [Google Scholar] [PubMed]
- Flores-Castillo, C.; Zamora-Pérez, J.Á.; Carreón-Torres, E.; Arzola-Paniagua, A.; Aguilar-Salinas, C.; López-Olmos, V.; Fragoso, J.M.; Luna-Luna, M.; Rodríguez-Pérez, J.M.; Franco, M.; et al. Atorvastatin and fenofibrate combination induces the predominance of the large HDL subclasses and increased apo AI fractional catabolic rates in New Zealand white rabbits with exogenous hypercholesterolemia. Fundam. Clin. Pharmacol. 2015, 29, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Xie, Y.; Jiang, J.; Wang, Y.; Xu, X.; Zhao, C.; Huang, F. Pleiotropic effects of fenofibrate therapy on rats with hypertriglycemia. Lipids Health Dis. 2015, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M.; Ura, N.; Murakami, H.; Hyakukoku, M.; Yamaguchi, K.; Higashiura, K.; Shimamoto, K. Fenofibrate improves insulin sensitivity in connection with intramuscular lipid content, muscle fatty acid-binding protein, and β-oxidation in skeletal muscle. J. Endocrinol. 2002, 174, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Ruiz, M.E.; Guarner-Lans, V. Inflammation and the Use of Anti-Inflammatory Agents in Signs and Cardiovascular Consequences of Metabolic Syndrome. In Handbook on Metabolic Syndrome. Classification, Risk Factors and Health Impact, 1st ed.; Nova Biomedical: New York, NY, USA, 2012; pp. 169–188. [Google Scholar]
- Schuchard, J.; Winkler, M.; Stölting, I.; Schuster, F.; Vogt, F.M.; Barkhausen, J.; Thorns, C.; Santos, R.A.; Bader, M.; Raasch, W. Lack of weight gain after angiotensin AT1 receptor blockade in diet-induced obesity is partly mediated by an angiotensin-(1–7)/Mas-dependent pathway. Br. J. Pharmacol. 2015, 172, 3764–3778. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.L.; Duan, R.; El-Marakby, A.; Alhashim, A.; Lee, D.L. Peroxisome Proliferator Activated Receptor-α Agonist Slows the Progression of Hypertension, Attenuates Plasma Interleukin-6 Levels and Renal Inflammatory Markers in Angiotensin II Infused Mice. PPAR Res. 2012, 2012, 645969. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Baker, K.M.; Kumar, R. Activation of the intracellular renin angiotensin system in cardiac fibroblasts by high glucose: Role in extracellular matrix production. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1675–H1684. [Google Scholar] [CrossRef] [PubMed]
- Varagic, J.; Ahmad, S.; Nagata, S.; Ferrario, C.M. ACE2: Angiotensin II/angiotensin-(1–7) balance in cardiac and renal injury. Curr. Hypertens. Rep. 2014, 16, 420. [Google Scholar] [CrossRef] [PubMed]
- Toba, H.; Miki, S.; Shimizu, T.; Yoshimura, A.; Inoue, R.; Sawai, N.; Tsukamoto, R.; Murakami, M.; Morita, Y.; Nakayama, Y.; et al. The direct antioxidative and anti-inflammatory effects of peroxisome proliferator-activated receptors ligands are associated with the inhibition of angiotensin converting enzyme expression in streptozotocin-induced diabetic rat aorta. Eur. J. Pharm. 2006, 549, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Ibarra-Lara, M.L.; Sánchez-Aguilar, M.; Soria, E.; Torres-Narváez, J.C.; Del Valle-Mondragón, L.; Cervantes-Pérez, L.G.; Pérez-Severiano, F.; Ramírez-Ortega, M.C.; Pastelín-Hernández, G.; Oidor-Chan, V.H.; et al. Peroxisome proliferator-activated receptors (PPAR) downregulate the expression of pro-inflammatory molecules in an experimental model of myocardial infarction. Can. J. Phys. Pharmacol. 2016, 94, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Oudot, A.; Vargely, C.; Rochette, L. Angiotensin II activates NADPH oxidase in isolated rat hearts subject to ischemia-reperfusion. Eur. J. Pharmacol. 2003, 462, 145–154. [Google Scholar] [CrossRef]
- Manrique, C.; Lastra, G.; Gardner, M.; Sowers, J. The renin Angiotensin Aldosterone System in hypertension: Role of insulin resistance and oxidative stress. Med. Clin. N. Am. 2009, 93, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Calkin, A.C.; Cooper, M.E.; Jandeleit-Dahm, K.A.; Allen, T.J. Gemfibrozil decreases atherosclerosis in experimental diabetes in association with a reduction in oxidative stress and inflammation. Diabetologia 2006, 49, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Newaz, M.; Blanton, A.; Fidelis, P.; Oyekan, A. NAD(P)H oxidase/nitric oxide interactions in peroxisome proliferator activated receptor (PPAR)alpha mediated cardiovascular effects. Mutat. Res. 2005, 579, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Inoue, I.; Goto, S.; Matsunaga, T.; Nakajima, T.; Awata, T.; Hokari, S.; Komoda, T.; Katayama, S. The ligands/activators for peroxisomal proliferator-activated receptor alpha (PPARalpha) and PPARgamma increase Cu2+,Zn2+-superoxide dismutase and decrease p22phox message expression in primary endothelial cells. Metabolism 2001, 50, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Umemoto, S.; Kawahara, S.; Kubo, M.; Itoh, S.; Umeji, K.; Matsuzaki, M. Angiotensin II type 1 receptor antagonist and angiotensin-converting enzyme inhibitor altered the activation of Cu/Zn-containing superoxide dismutase in the heart of stroke-prone spontaneously hypertensive rats. Hypertens. Res. 2005, 28, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; Pastorello, M.; Arzola, J.; Zavala, L.E.; De Jesús, S.; Varela, M.; Matos, M.G.; del Rosario Garrido, M.; Israel, A. AT1 receptor and NAD(P)H oxidase mediate angiotensin II-stimulated antioxidant enzymes and mitogen-activated protein kinase activity in the rat hypothalamus. J. RAS Syst. 2010, 11, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Guellich, A.; Damy, T.; Lecarpentier, Y.; Conti, M.; Claes, V.; Samuel, J.L.; Quillard, J.; Hébert, J.L.; Pineau, T.; Coirault, C. Role of oxidative stress in cardiac dysfunction of PPARα-/- mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H93–H102. [Google Scholar] [CrossRef] [PubMed]
- Mehendale, H.M. PPAR-alpha: A key to the mechanism of hepatoprotection by clofibrate. Toxicol. Sci. 2000, 57, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Peredo-Escárcega, A.E.; Guarner-Lans, V.; Pérez-Torres, I.; Ortega-Ocampo, S.; Carreón-Torres, E.; Castrejón-Tellez, V.; Díaz-Díaz, E.; Rubio-Ruiz, M.E. The Combination of Resveratrol and Quercetin Attenuates Metabolic Syndrome in Rats by Modifying the Serum Fatty Acid Composition and by Upregulating SIRT 1 and SIRT 2 Expression in White Adipose Tissue. Evid. Based Complement. Altern. Med. 2015, 2015, 474032. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; DeMarco, V.G.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2016, 12, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, E.; Diamond-Stanic, M.K.; Marchionne, E. Oxidative stress and the etiology of insulin resistance and Type 2 Diabetes. Free Radic. Biol. Med. 2011, 51, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Mori, J.; Alrob, O.A.; Wagg, C.S.; Harris, R.A.; Lopaschuk, G.D.; Oudit, G.Y. ANG II causes insulin resistance and induces cardiac metabolic switch and inefficiency: A critical role of PDK4. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1103–H1113. [Google Scholar] [CrossRef] [PubMed]
- Ide, T.; Tsunoda, M.; Mochizuki, T.; Murakami, K. Enhancement of insulin signaling through inhibition of tissue lipid accumulation by activation of peroxisome proliferator-activated receptor (PPAR) alpha in obese mice. Med. Sci. Monit. 2004, 10, BR388–395. [Google Scholar] [PubMed]
- Yang, F.J.; He, Y.H.; Zhou, J.H. Fenofibrate pre-treatment suppressed inflammation by activating phosphoinositide 3 kinase/protein kinase B (PI3K/Akt) signaling in renal ischemia-reperfusion injury. J. Huazhong. Univ. Sci. Technol. Med. Sci. 2015, 35, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Bulhak, A.A.; Jung, C.; Ostenson, C.G.; Lundberg, J.O.; Sjöquist, P.O.; Pernow, J. PPAR-alpha activation protects the type 2 diabetic myocardium against ischemia-reperfusion injury: Involvement of the PI3-Kinase/Akt and NO pathway. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H719–H727. [Google Scholar] [CrossRef] [PubMed]
- Vásquez-Vivar, J.; Kalyanaraman, B.; Martásek, P.; Hogg, N.; Masters, B.S.; Karoui, H.; Tordo, P.; Pritchard, K.A., Jr. Superoxide generation by endothelial nitric oxide synthase: The influence of cofactors. Proc. Natl. Acad. Sci. USA 1998, 95, 9220–9225. [Google Scholar] [CrossRef] [PubMed]
- Dumitrescu, C.; Biondi, R.; Xia, Y.; Cardounel, A.J.; Druhan, L.J.; Ambrosio, G.; Zweier, J.L. Myocardial ischemia results in tetrahydrobiopterin (BH4) oxidation with impaired endothelial function ameliorated by BH4. Proc. Natl. Acad. Sci. USA 2007, 104, 15081–15086. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CT-V | CT-F | MetS-V | MetS-F | |
|---|---|---|---|---|
| Body weight (g) | 522.0 ± 12.4 | 463.5 ± 28.9 | 524.9 ± 23.9 | 490.4 ± 17.1 |
| Visceral fat (g) | 6.2 ± 0.8 | 4.4 ± 0.9 | 12.0 ± 0.6 a | 12.1 ± 1.4 |
| Blood pressure (mmHg) | 97.3 ± 7.2 | 90.7 ± 2.6 | 142.1 ± 1.8 a | 142.7 ± 16.8 a |
| Glucose (mg/dL) | 105.2 ± 9.8 | 97.8 ± 6.1 | 115.2 ± 17.8 | 102.6 ± 6.3 |
| Insulin (ng/mL) | 0.08 ± 0.04 | 0.09 ± 0.03 | 0.29 ± 0.04 a | 0.10 ± 0.05 b |
| HOMA-IR | 0.81 ± 0.32 | 1.15 ± 0.38 | 3.52 ± 0.6 a | 1.2 ± 0.53 b |
| Total cholesterol (mg/dL) | 62.4 ± 7.5 | 48.8 ± 1.9 | 57.6 ± 1.7 | 40.6 ± 5.3 |
| HDL-C (mg/dL) | 36.9 ± 5.8 | 36.7 ± 1.6 | 23.7 ± 4.3 a | 25.6 ± 6.4 |
| Non-HDL-C (mg/dL) | 21.7 ± 3.2 | 12.1 ± 2.2 b | 30.4 ± 1.6a | 11.2 ± 3.1 b |
| Triglycerides (mg/dL) | 51.20 ± 21.3 | 20.8 ± 2.3 | 140.6 ± 25.2 a | 41.30 ± 13.2 b |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibarra-Lara, L.; Sánchez-Aguilar, M.; Sánchez-Mendoza, A.; Del Valle-Mondragón, L.; Soria-Castro, E.; Carreón-Torres, E.; Díaz-Díaz, E.; Vázquez-Meza, H.; Guarner-Lans, V.; Rubio-Ruiz, M.E. Fenofibrate Therapy Restores Antioxidant Protection and Improves Myocardial Insulin Resistance in a Rat Model of Metabolic Syndrome and Myocardial Ischemia: The Role of Angiotensin II. Molecules 2017, 22, 31. https://doi.org/10.3390/molecules22010031
Ibarra-Lara L, Sánchez-Aguilar M, Sánchez-Mendoza A, Del Valle-Mondragón L, Soria-Castro E, Carreón-Torres E, Díaz-Díaz E, Vázquez-Meza H, Guarner-Lans V, Rubio-Ruiz ME. Fenofibrate Therapy Restores Antioxidant Protection and Improves Myocardial Insulin Resistance in a Rat Model of Metabolic Syndrome and Myocardial Ischemia: The Role of Angiotensin II. Molecules. 2017; 22(1):31. https://doi.org/10.3390/molecules22010031
Chicago/Turabian StyleIbarra-Lara, Luz, María Sánchez-Aguilar, Alicia Sánchez-Mendoza, Leonardo Del Valle-Mondragón, Elizabeth Soria-Castro, Elizabeth Carreón-Torres, Eulises Díaz-Díaz, Héctor Vázquez-Meza, Verónica Guarner-Lans, and María Esther Rubio-Ruiz. 2017. "Fenofibrate Therapy Restores Antioxidant Protection and Improves Myocardial Insulin Resistance in a Rat Model of Metabolic Syndrome and Myocardial Ischemia: The Role of Angiotensin II" Molecules 22, no. 1: 31. https://doi.org/10.3390/molecules22010031