Phosphorus-Sulfur Heterocycles Incorporating an O-P(S)-O or O-P(S)-S-S-P(S)-O Scaffold: One-Pot Synthesis and Crystal Structure Study


 and
and
Abstract
:
1. Introduction
2. Results and Discussion
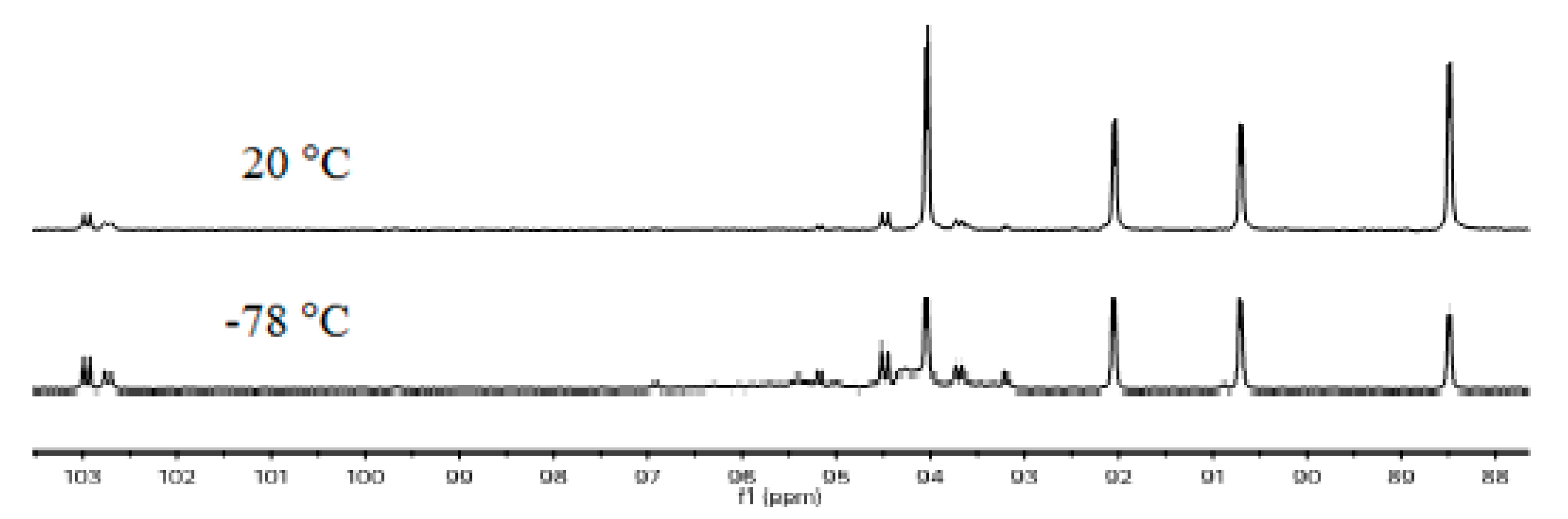
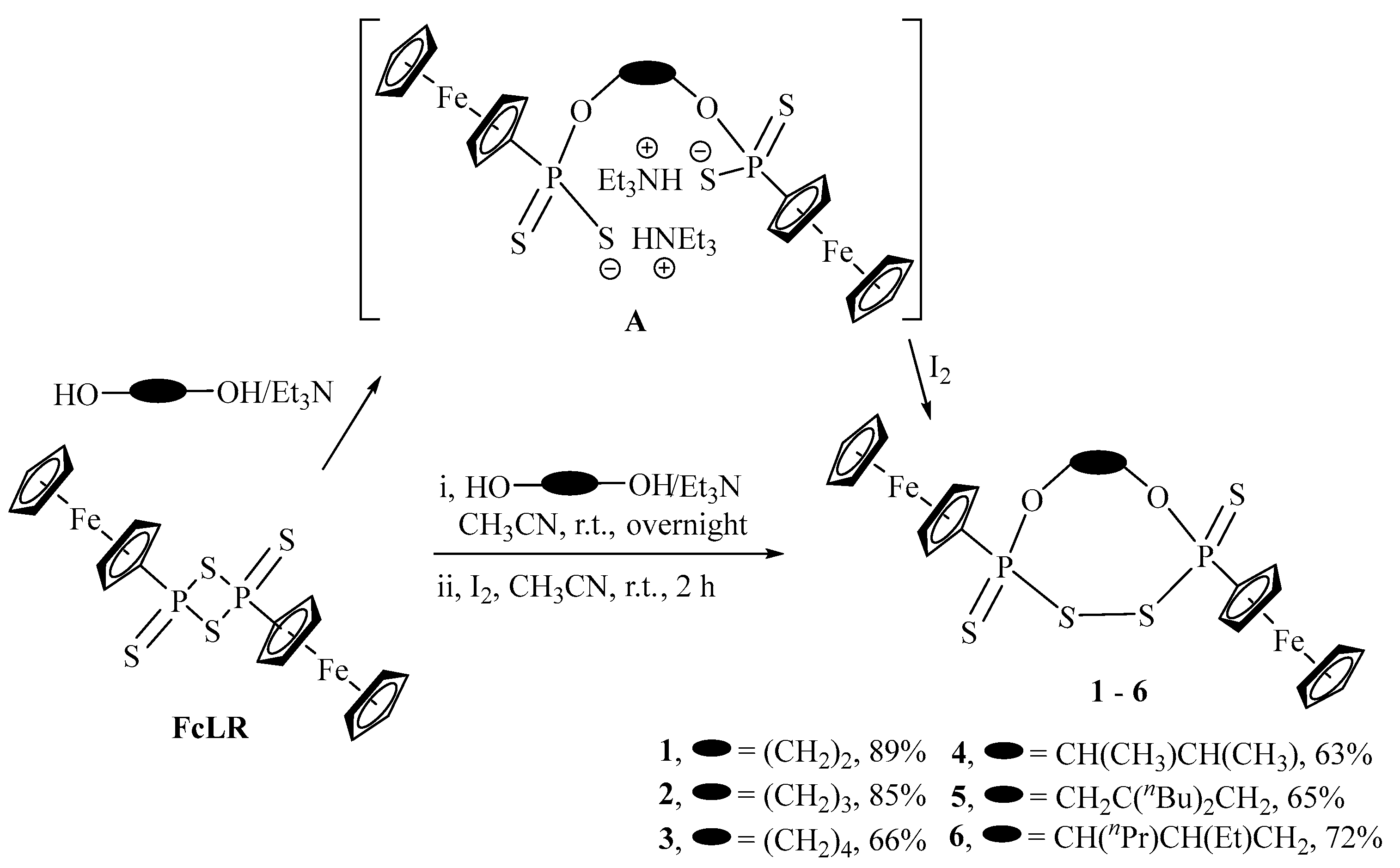
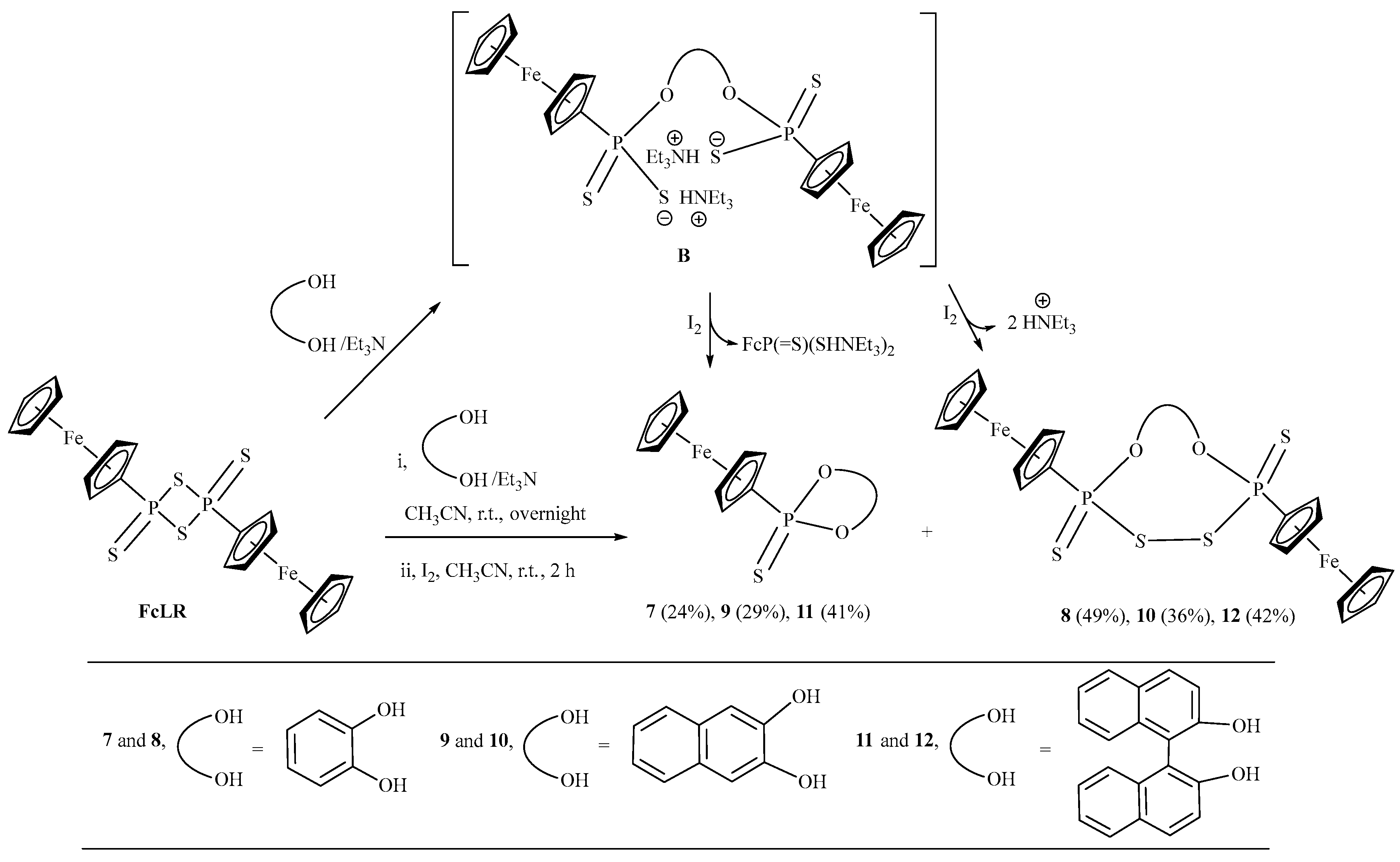
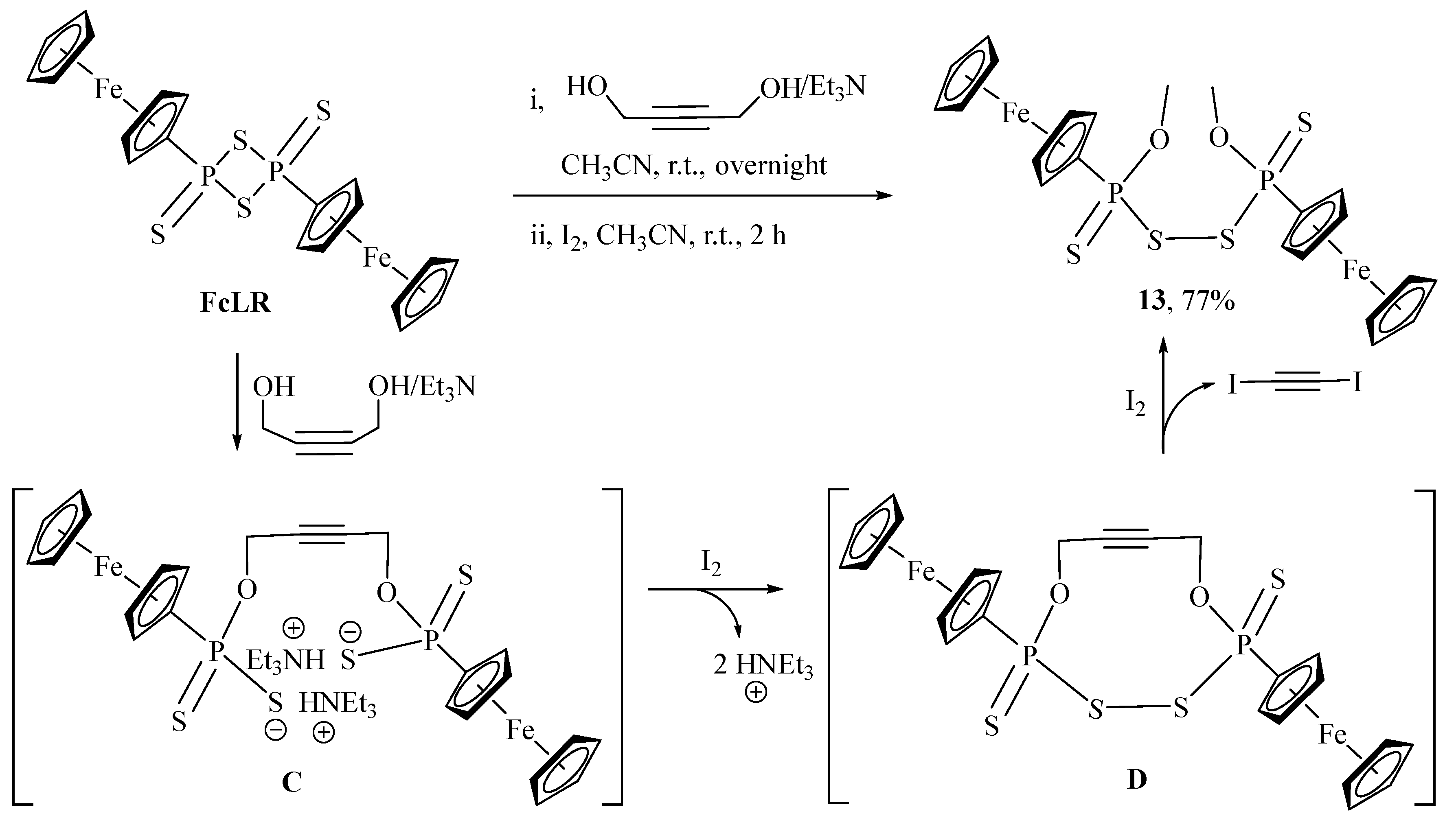
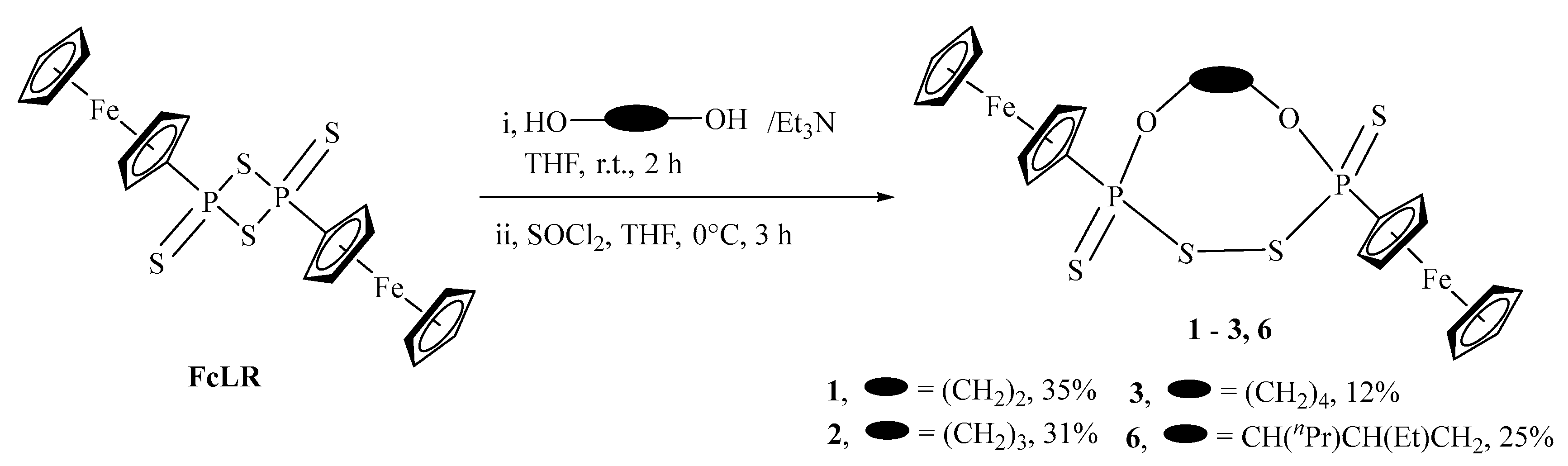
2.1. Synthesis and Characterization
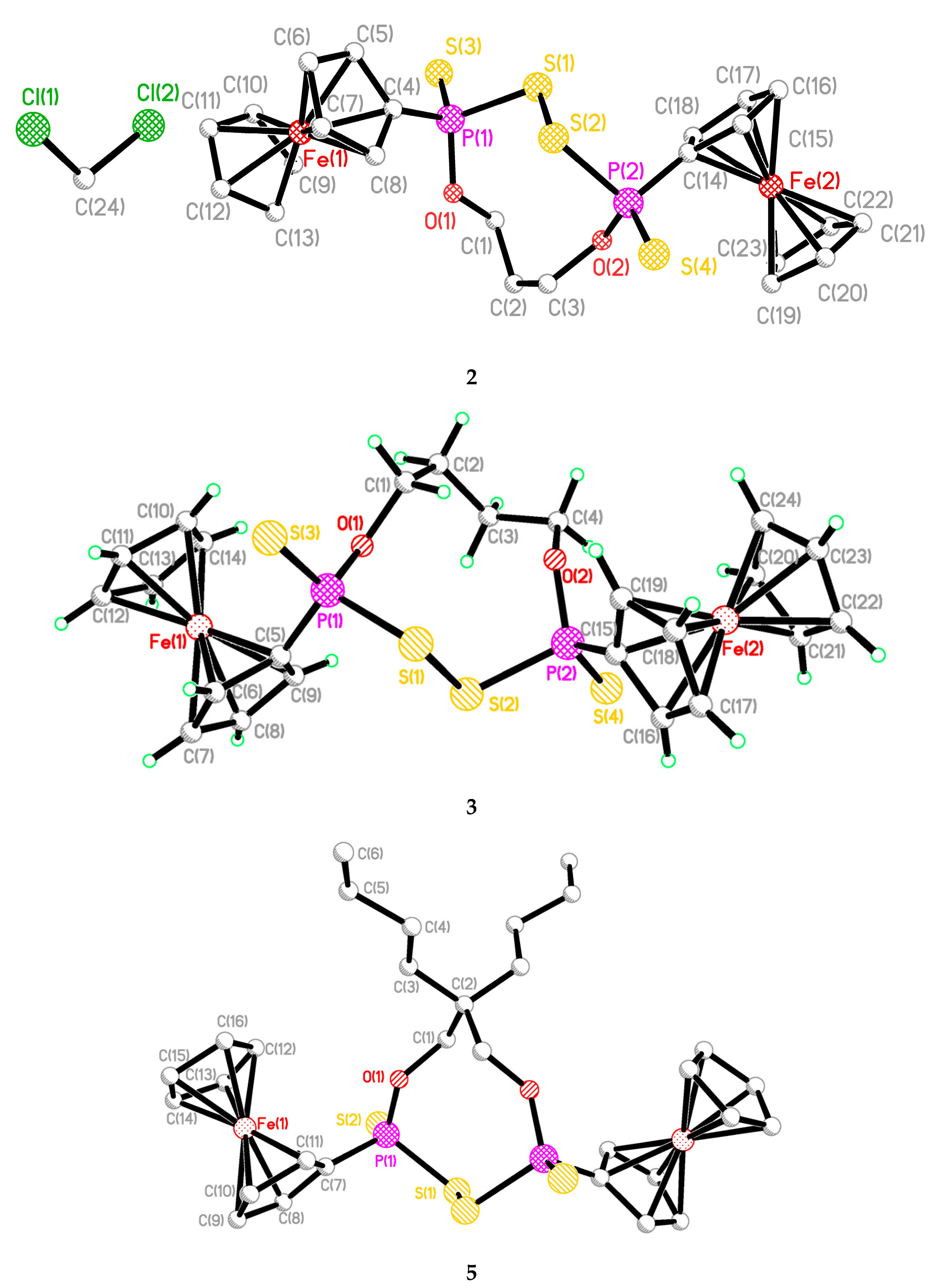
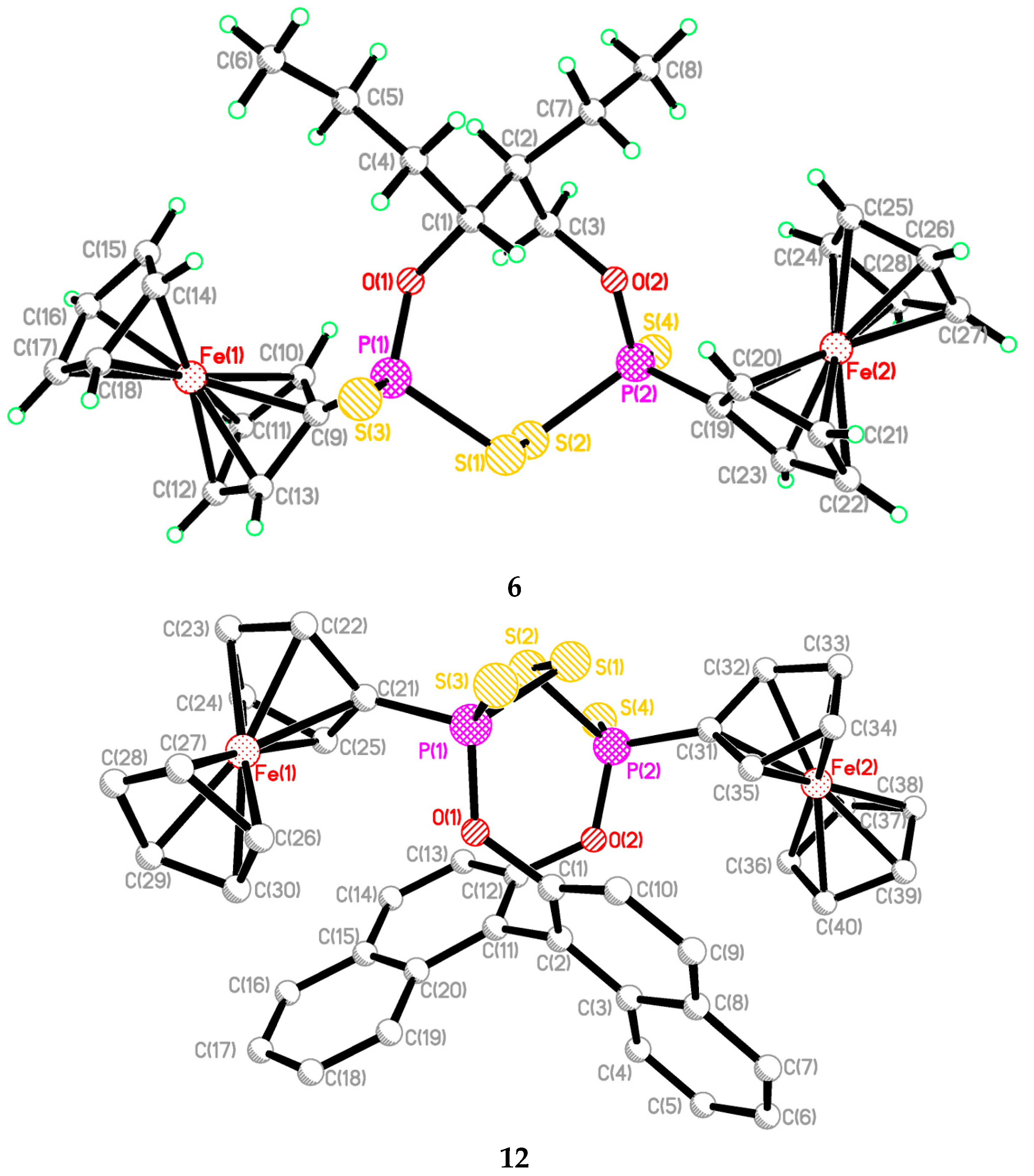
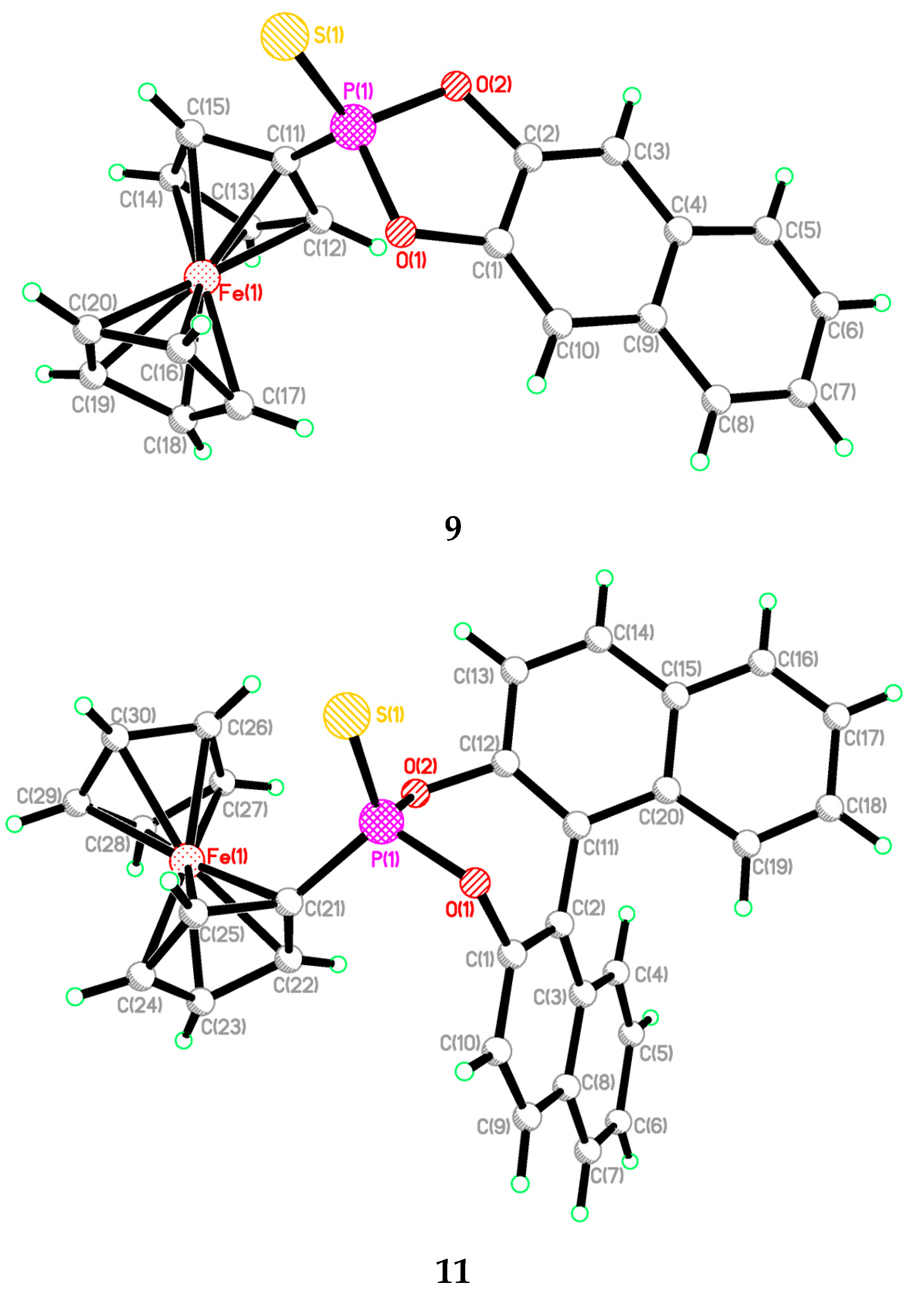
2.2. Single Crystal Structure Analysis
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.2.1. General Procedure for the Reaction of FcLR with Alkenyl-diols and I2 in the Presence of Triethylamine
3.2.2. General Procedure for the Reaction of FcLR with Aryl-diols (pyrocatechol, naphthalene-2,3-diol, [1,1′-binaphthalene]-2,2′-diol) and I2
3.2.3. Synthesis of Compound 13
3.2.4. General Procedure for the Reaction of FcLR with Alkenyl-diols and SOCl2
4. Conclusions
Supplementary Materials
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Bao, M.; Shimizu, M. N-Trifluoroacetyl arenesulfenamides, effective precursors for synthesis of unsymmetrical disulfides and sulfenamides. Tetrahedron 2003, 59, 9655–9659. [Google Scholar] [CrossRef]
- Cremlyn, R.; An, J. Introduction to Organosulfur Chemistry; Wiley & Sons: New York, NY, USA, 1996. [Google Scholar]
- Fletcher, J.M.; Hughes, R.A. A novel approach to the regioselective synthesis of a disulfide-linked heterodimeric bicyclic peptide mimetic of brain-derived neurotrophic factor. Tetrahedron Lett. 2004, 45, 6999–7001. [Google Scholar] [CrossRef]
- Vruhula, V.M.; MacMaster, J.F.; Li, Z.; Kerr, D.E.; Senter, P.D. Reductively activated disulfide prodrugs of paclitaxel. Bioorg. Med. Chem. Lett. 2002, 12, 3591–3594. [Google Scholar] [CrossRef]
- Roy, B.; Chambert, S.; Leoivre, M.; Aubertin, A.M.; Balzarini, J.; Decout, J.L. Deoxyribonucleoside-2′- or 3′-mixed disulfides: prodrugs to target ribonucleotide reductase and /or to inhibit HIV reverse transcription. J. Med. Chem. 2003, 46, 2565–2568. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Nodwell, M.; Pace, J.L.; Shaw, J.P.; Judice, J.K. Vancomycin disulfide derivatives as antibacterial agents. Bioorg. Med. Chem. Lett. 2004, 14, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Ulman, A. Formation and structure of self-assembled monolayers. Chem. Rev. 1996, 96, 1533–1554. [Google Scholar] [CrossRef] [PubMed]
- Ulman, A. Thin Films: Self-Assembled Monolayers of Thiols; Academic Press: Boston, MA, USA, 1998. [Google Scholar]
- Porter, L.A., Jr.; Ji, D.; Westcott, S.L.; Graupe, M.; Czernuszewicz, R.S.; Halas, N.J.; Lee, T.R. Gold and silver nanoparticles functionalized by the adsorption of dialkyl disulfides. Langmuir 1998, 14, 7378–7386. [Google Scholar] [CrossRef]
- Shon, Y.S.; Mazzitelli, C.; Murray, R.W. Unsymmetrical disulfides and thiol mixtures produce different mixed monolayer-protected gold clusters. Langmuir 2001, 17, 7735–7741. [Google Scholar] [CrossRef]
- Sato, R.; Ohyama, T.; Ogawa, S. Efficient synthesis and biological properties of new benzopentathiepins. Heterocycles 1995, 41, 893–896. [Google Scholar] [CrossRef]
- Toste, F.D.; Still, I.W.J. A new route to the synthesis of the naturally occurring benzopentathiepin varacin. J. Am. Chem. Soc. 1995, 117, 7261–7262. [Google Scholar] [CrossRef]
- Searle, P.A.; Molinski, T.F. Five new alkaloids from the tropical ascidian, Lissoclinum sp. lissoclinotoxin A is chiral. J. Org. Chem. 1994, 59, 6600–6605. [Google Scholar] [CrossRef]
- Davidson, B.S.; Ford, P.W.; Wahlman, M. Chirality in unsymmetrically substituted benzopentathiepins: The results of a high barrier to ring inversion. Tetrahedron Lett. 1994, 35, 7185–7188. [Google Scholar] [CrossRef]
- Behar, V.; Danishefsky, S.J. Total synthesis of the novel benzopentathiepin varacinium trifluoroacetate: The viability of “varacin-free base”. J. Am. Chem. Soc. 1993, 115, 7017–7018. [Google Scholar] [CrossRef]
- Ford, P.W.; Davidson, B.S. Synthesis of varacin, a cytotoxic naturally occurring benzopentathiepin isolated from a marine ascidian. J. Org. Chem. 1993, 58, 4522–4523. [Google Scholar] [CrossRef]
- Davidson, B.S.; Molinski, T.F.; Barrows, L.R.; Ireland, C.M. Varacin: A novel benzopentathiepin from lissoclinium vareau that is cytotoxic toward a human colon tumour. J. Am. Chem. Soc. 1991, 113, 4709–4710. [Google Scholar] [CrossRef]
- Janosik, T.; Bergman, J.; Stensland, B.; Stalhandske, C. Thionation of bisindole derivatives with P4S10 or elemental sulfur. J. Chem. Soc. Perkin Trans. I 2002, 330–334. [Google Scholar] [CrossRef]
- Macho, S.; Rees, C.W.; Rodriguez, T.; Torroba, T. A novel oxime to pentathiepin cascade reaction. Chem. Commun. 2001, 403–404. [Google Scholar] [CrossRef]
- Bergman, J.; Stalhandske, C. Transformation of isatin with P4S10 to pentathiepino[6,7-b]indole in one step. Tetrahedron Lett. 1994, 35, 5279–5282. [Google Scholar] [CrossRef]
- Grishina, O.N.; Kosova, L.M.; Lipatova, I.P.; Shagidullin, R.R. Sulfides of alkylthionophosphines. IX. Synthesis of pentaerythritol-O,O,O,O-tetrakis(alkyldithioophosphonates) and their derivatives. Zh. Obsch. KHim 1970, 40, 66–69. [Google Scholar]
- Przychodzen, W. New products of reaction of Lawesson’s ragent with diols. Phosphorus Sulfur Silicon Relat. Elem. 2004, 179, 1621–1633. [Google Scholar] [CrossRef]
- Pillay, M.N.; van der Wailt, H.; Staples, R.J.; Van Zyl, W.E. C/O/P/S cyclic derived from oxidative intramolecular disulfide (-S-S-) coupling of ferrocenyl dithiophopshonates. J. Organomet. Chem. 2015, 794, 33–39. [Google Scholar] [CrossRef]
- Hua, G.; Slawin, A.M.Z.; Randall, R.A.M.; Cordes, D.B.; Crawford, L.; Bühl, M.; Woollins, J.D. An efficient route for the synthesis of phosphorus-selenium macro-heterocycles. Chem. Commun. 2013, 49, 2619–2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, G.; Du, J.; Cordes, D.B.; Slawin, A.M.Z.; Woollins, J.D. One-pot three-component condensation synthesis and structural features of organophosphorus-sulfur macrocycles. J. Org. Chem. 2016, 81, 4210–4225. [Google Scholar] [CrossRef] [PubMed]
- Hua, G.; Du, J.; Slawin, A.M.Z.; Woollins, J.D. One-pot approach to organo-phosphorus-chalcogen macrocycles incorporating double OP(S)SCn or OP(Se)SeCn scaffolds: A synthetic and structural study. Chem. Eur. J. 2016, 22, 7782–7791. [Google Scholar] [CrossRef] [PubMed]
- Foreman, M.R., St.; Slawin, A.M.Z.; Woollins, J.D. 2,4-Diferrocenyl-1,3-dithiadiphosphetane 2,4-disulfide; structure and reactions with catechols and [PtCl2(PR3)2] (R = Et or Bu). J. Chem. Soc. Dalton Trans. 1996, 3653–3657. [Google Scholar] [CrossRef]
- Gray, I.P.; Slawin, A.M.Z.; Woollins, J.D. Synthesis and characterization of disulfides and esters derived from their sodium organodithiophosphonate salts. New J. Chem. 2004, 28, 1383–1389. [Google Scholar] [CrossRef]
- Grossmann, G.; Komber, H. NMR investigation on diastereomeric mixtures of bis(dialkoxythiophosphoryl) sulfides and polysulfides containing sec-butoxy groups. Assignment of phosphorus-31, carbon-13 and proton NMR signals in a mixture of seven diastereomers using shift-correlated 2D NMR spectra. Phosphorus Sulfur Silicon Relat. Elem. 1991, 61, 269–281. [Google Scholar]
- Ogawa, S.; Ohmiya, T.; Kikuchi, T.; Kawaguchi, A.; Saito, S.; Sai, A.; Kawai, Y.; Niizuma, S.; Nakajo, S.; Kimura, T.; et al. Synthesis, structure, and one-electron redox reactions of 4,7-disubstituted benzotrichalcogenoles containing sulfur and /or selenium atoms. J. Organoment. Chem. 2000, 611, 136–145. [Google Scholar] [CrossRef]
- Jesberger, M.; Davis, T.P.; Bamer, L. Applications of Lawesson’s reagent in organic and organometallic syntheses. Synthesis 2003, 1929–1958. [Google Scholar] [CrossRef]
- Fuller, A.L.; Scott-Hayward, L.A.S.; Li, Y.; Bühl, M.; Slawin, A.M.Z.; Woollins, J.D. Automated Chemical Crystallography. J. Am. Chem. Soc. 2010, 132, 5799–5802. [Google Scholar] [CrossRef] [PubMed]
- CrystalClear-SM Expert v3.1b27; Rigaku Corporation: The Woodlands, TX, USA; Tokyo, Japan, 2013.
- CrystalClear-SM Expert v2.1; Rigaku Corporation: The Woodlands, TX, USA; Tokyo, Japan, 2015.
- Palatinus, L.; Chapuis, G. SUPERFLIP–A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Cryst. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Polidori, G.; Spagna, R. SIR2004: An improved tool for crystal structure determination and refinement. J. Appl. Cryst. 2005, 38, 381–388. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G.; Spagna, R. SIR2011: A new package for crystal structure determination and refinement. J. Appl. Cryst. 2012, 45, 357–361. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar]
- CrystalStructure v4.2; Rigaku Corporation: The Woodlands, TX, USA; Tokyo, Japan, 2015.
Sample Availability: Samples of all compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 2 | 3 | 5 | 6 |
|---|---|---|---|---|
| Formula | C24H26Cl2Fe2O2P2S4 | C24H26Fe2O2P2S4 | C31H40Fe2O2P2S4 | C28H34Fe2O2P2S4 |
| M | 719.26 | 648.35 | 746.45 | 704.46 |
| Crystal system | triclinic | triclinic | monoclinic | triclinic |
| Space group | P-1 | P-1 | C2/c | P-1 |
| a/Å | 7.6366(5) | 7.6464(11) | 26.246(4) | 8.8513(18) |
| b/Å | 11.7048(8) | 13.1117(18) | 9.6717(15) | 13.173(3) |
| c/Å | 16.7834(11) | 14.453(2) | 13.720(2) | 15.034(3) |
| Α | 98.035(5) | 67.778(8) | 90 | 106.519(2) |
| Β | 100.985(6) | 83.624(11) | 102.900(4) | 105.439(3) |
| Γ | 99.739(17) | 73.660(10) | 90 | 104.131(3) |
| U/A3 | 1428.35(17) | 1287.2(3) | 3394.8(9) | 1519.8(6) |
| Z | 2 | 2 | 4 | 2 |
| µ/cm−1 | 16.286 | 15.968 | 12.214 | 13.591 |
| Reflections collected | 11190 | 16874 | 19978 | 25008 |
| Independent reflections | 4976 | 4632 | 3098 | 5552 |
| Rint | 0.0625 | 0.0405 | 0.0321 | 0.0355 |
| R1 | 0.0455 | 0.0289 | 0.0267 | 0.0244 |
| wR2 [I > 2σ(I)] | 0.1145 | 0.0713 | 0.0594 | 0.0575 |
| Compound | 9 | 11 | 12 |
|---|---|---|---|
| Formula | C20H15FeO2PS | C30H21FeO2PS | C40H30Fe2O2P2S8 |
| M | 406.22 | 532.38 | 972.80 |
| Crystal system | triclinic | monoclinic | Triclinic |
| Space group | P-1 | P21/n | P-1 |
| a/Å | 7.4940(13) | 7.8541(16) | 10.874(4) |
| b/Å | 10.6802(16) | 10.884(2) | 14.866(3) |
| c/Å | 10.8335(18) | 27.176(5) | 15.261(4) |
| α | 90.128(5) | 90 | 62.01(2) |
| β | 94.864(5) | 92.738(6) | 79.51(3) |
| γ | 101.765(5) | 90 | 77.78(3) |
| U/A3 | 845.6(2) | 2320.5(8) | 2119.3(11) |
| Z | 2 | 4 | 2 |
| µ/cm−1 | 11.192 | 8.362 | 11.882 |
| Reflections collected | 13750 | 27298 | 25606 |
| Independent reflections | 3066 | 4241 | 7692 |
| Rint | 0.0319 | 0.1309 | 0.1469 |
| R1 | 0.0253 | 0.0586 | 0.1261 |
| wR2 [I > 2σ(I)] | 0.1244 | 0.1236 | 0.3345 |
| 2 | 3 | 5 | 6 | 12 | |
|---|---|---|---|---|---|
| S(1)-P(1) | 2.099(2) | 2.1026(9) | 2.1034(8) | 2.1076(8) | 2.093(6) |
| S(2)-P(2) | 2.104(2) | 2.1087(10) | 2.1034(8) | 2.1031(8) | 2.107(5) |
| S(3)-P(1) | 1.930(2) | 1.9324(16) | 1.9210(9) | 1.9271(7) | 1.916(5) |
| S(4)-P(2) | 1.928(2) | 1.9255(12) | 1.9210(9) | 1.9286(7) | 1.921(5) |
| O(1)-P(1) | 1.584(3) | 1.587(2) | 1.5784(14) | 1.5846(14) | 1.613(7) |
| O(2)-P(2) | 1.581(3) | 1.5830(19) | 1.5784(14) | 1.5828(16) | 1.603(8) |
| S(1)-S(2) | 2.072(2) | 2.0789(11) | 2.0723(9) | 2.0739(7) | 2.064(4) |
| S(1)-P(1)-S(3) | 103.88(8) | 104.81(5) | 105.39(3) | 102.35(3) | 103.9(2) |
| S(2)-P(2)-S(4) | 105.07(7) | 103.75(4) | 105.39(3) | 104.07(4) | 105.70 (19) |
| S(1)-P(1)-O(1) | 107.63(13) | 107.44(7) | 106.92(6) | 108.93(6) | 107.3(4) |
| S(2)-P(2)-O(2) | 107.80(13) | 108.12(7) | 106.92(6) | 106.20(6) | 107.2(4) |
| S(3)-P(1)-O(1) | 118.95(14) | 118.77(6) | 119.24(8) | 119.55(6) | 118.7(3) |
| S(4)-P(2)-O(2) | 117.84(15) | 118.77(6) | 118.91(10) | 119.13(5) | 117.7(3) |
| P(1)-S(1)-S(2) | 103.96(8) | 102.06(4) | 103.72(3) | 105.11(3) | 106.9(2) |
| P(2)-S(2)-S(1) | 102.63(7) | 104.42(4) | 103.72(3) | 102.76(3) | 105.18(17) |
| 9 | 11 | |
|---|---|---|
| S(1)-P(1) | 1.9034(8) | 1.9103(17) |
| O(1)-P(1) | 1.6303(8) | 1.606(3) |
| O(2)-P(1) | 1.6328(17) | 1.608(3) |
| O(1)-C(1) | 1.390(3) | 1.409(5) |
| O(2)-C(2) | 1.390(3) | 1.393(5) |
| S(1)-P(1)-O(1) | 116.10(7) | 109.46(13) |
| S(1)-P(1)-O(2) | 115.32(7) | 118.65(13) |
| O(1)-P(1)-O(2) | 95.56(9) | 102.17(16) |
| P(1)-O(1)-C(1) | 110.60(15) | 118.4(3) |
| P(1)-O(2)-C(2) | 110.61(15) | 120.3(3) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hua, G.; Davidson, K.; Cordes, D.B.; Du, J.; Slawin, A.M.Z.; Woollins, J.D. Phosphorus-Sulfur Heterocycles Incorporating an O-P(S)-O or O-P(S)-S-S-P(S)-O Scaffold: One-Pot Synthesis and Crystal Structure Study. Molecules 2017, 22, 1687. https://doi.org/10.3390/molecules22101687
Hua G, Davidson K, Cordes DB, Du J, Slawin AMZ, Woollins JD. Phosphorus-Sulfur Heterocycles Incorporating an O-P(S)-O or O-P(S)-S-S-P(S)-O Scaffold: One-Pot Synthesis and Crystal Structure Study. Molecules. 2017; 22(10):1687. https://doi.org/10.3390/molecules22101687
Chicago/Turabian StyleHua, Guoxiong, Kate Davidson, David B. Cordes, Junyi Du, Alexandra M. Z. Slawin, and J. Derek Woollins. 2017. "Phosphorus-Sulfur Heterocycles Incorporating an O-P(S)-O or O-P(S)-S-S-P(S)-O Scaffold: One-Pot Synthesis and Crystal Structure Study" Molecules 22, no. 10: 1687. https://doi.org/10.3390/molecules22101687