Synthesis and Antitumor Activity of Triazole-Containing Sorafenib Analogs

Key Laboratory of Structure-Based Drug Design and Discovery (Shenyang Pharmaceutical University), Ministry of Education, 103 Wenhua Road, Shenyang 110016, China
*
Author to whom correspondence should be addressed.
Molecules 2017, 22(10), 1759; https://doi.org/10.3390/molecules22101759
Submission received: 20 September 2017
/
Revised: 13 October 2017
/
Accepted: 15 October 2017
/
Published: 24 October 2017
Abstract
:Using a highly effective binuclear Cu complex as the catalyst, the 1,3-dipolar cycloaddition reactions between 16 alkynes and two azides were successfully performed and resulted in the production of 25 new triazole-containing sorafenib analogs. Several compounds were evaluated as potent antitumor agents. Among them, 4-(4-(4-(3-fluorophenyl)-1H-1,2,3-triazol-1-yl)phenoxy)-N-methylpicolinamide (8f) potently suppressed the proliferation of HT-29 cancer cells by inducing apoptosis and almost completely inhibited colony formation at a low micromolar concentration.
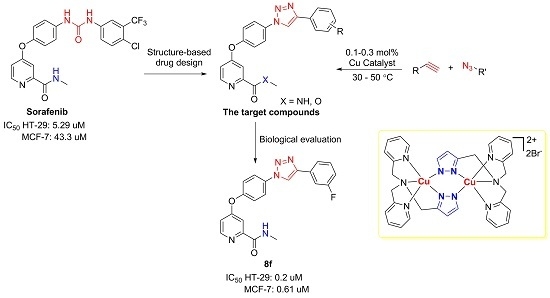
1. Introduction
Although significant progress has been achieved in the treatment of tumors, it is still a major health concern worldwide. Sorafenib is a well-known antitumor agent, which potently targets several kinases such as vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), stem cell factor receptor (c-Kit), and the downstream protein Raf [1]. It demonstrates excellent activity against a broad range of tumor types including renal, hepatocellular, and colorectal carcinomas [2,3,4]. However, the relative bioavailability of sorafenib is merely 38−49%, mainly due to its poor water solubility [5,6]. Thus, a feasible strategy for identifying antitumor agents could be the modification of sorafenib, paying equal attention to the potency and hydrophilicity of the derivatives.
The results of structure–activity relationship (SAR) analyses have indicated that the pyridine facilitates a suitable chemical interaction with Cys531 in a hinge region of B-Raf kinase, while the terminal phenyl ring occupies a hydrophobic pocket. The urea moiety connects both functional groups and forms important hydrogen-bond interactions with Glu500 and Asp593 [7,8,9,10]. Considerable studies were conducted on modification of the urea moiety, and potent compounds were identified by introducing an amide [11], rhodanine [12], or benzimidazole [13] group. From the viewpoint of medicinal chemistry, we considered that the urea moiety of sorafenib could be replaced by a 1,2,3-triazole fragment, which could act as a hydrogen-bond acceptor as well as a chemical linker between functional groups [14].
The constructing of 1,2,3-triazole cycles has been intensively studied in recent years. The Cu(I)-catalyzed Huisgen 1,3-dipolar cycloaddition reaction between alkynes and azides (CuAAC) is known as one of the most efficient methods [15,16]. Numerous efficient catalytic systems have been developed for accelerating the catalytic process and decreasing the amount of metal catalyst needed over the past decade [17,18,19]. Very recently, we explored Cu complexes 1–3 bearing an unsymmetrical bipyridine-pyrazole-amine ligand and demonstrated their applications in the CuAAC reaction (Scheme 1) [20,21]. To the best of our knowledge, the binuclear complex 2 is currently one of the most active catalyst systems used in the CuAAC reaction.
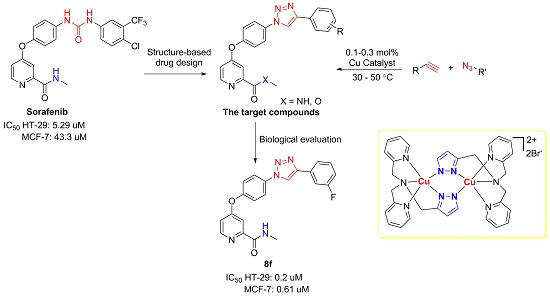
Herein, we reported the application of our highly active catalyst (2) in the synthesis of novel sorafenib analogs containing a 1,2,3-triazole moiety (Figure 1). The 3-CF3-4-Cl phenyl moiety in the chemical structure of sorafenib was also modified and evaluated to identify an optimal substitution pattern. The antiproliferative activity of these compounds was evaluated in HT-29 and MCF-7 cancer cell lines. The biological evaluation led to the discovery of compound 8f, which potently suppressed the proliferation of HT-29 cells by inducing apoptosis. Furthermore, 8f effectively inhibited colony formation of HT-29 cells.
2. Results and Discussion
2.1. Chemistry
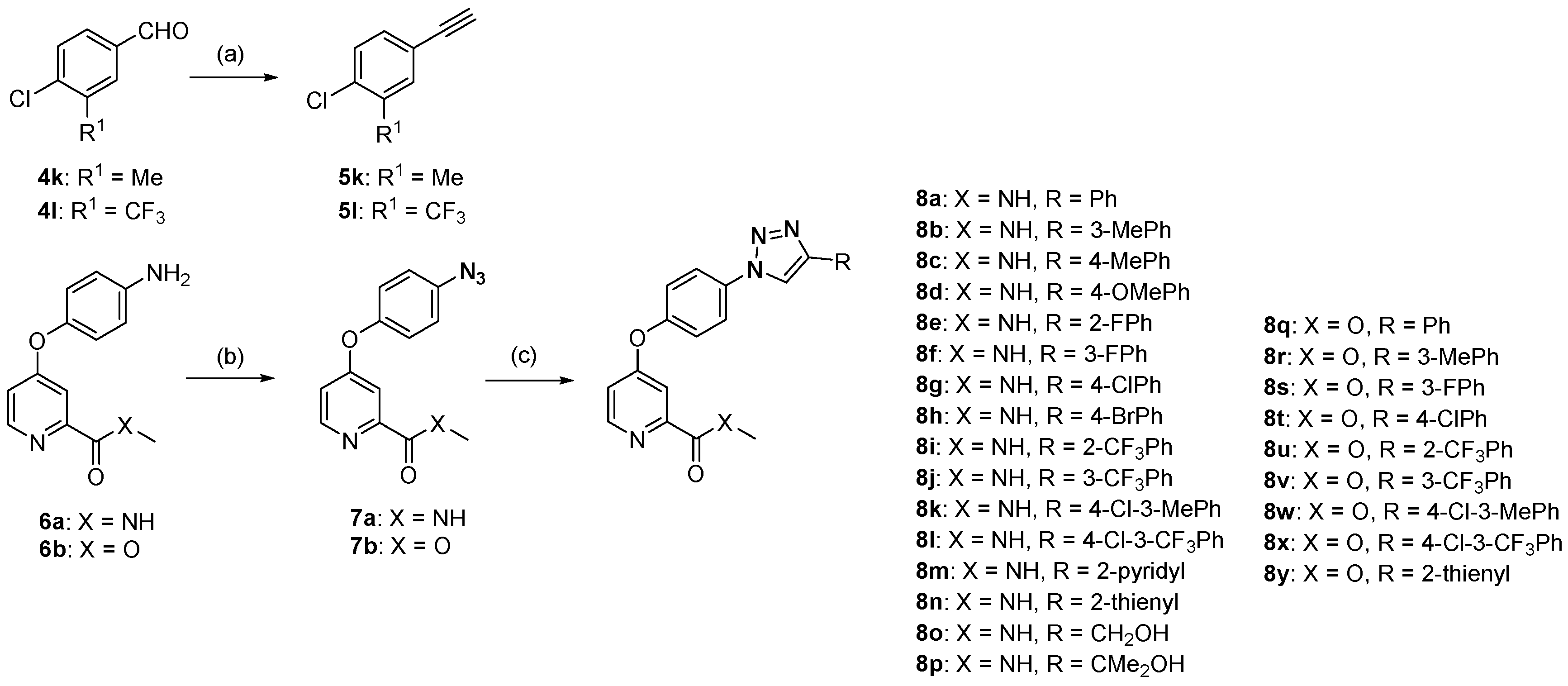
The preparation of compounds 8a–y is described in Scheme 2. Most alkynes were obtained from commercial sources and were used without further purification. Alkynes 5k and 5l were prepared using the Seyferth–Gilbert reaction from the corresponding aldehydes 4k and 4l, which were treated with 1.5 equivalents of the Bestmann–Ohira reagent in the presence of 2.0 equivalents of potassium carbonate in methanol (MeOH) at room temperature [22]. The azides 7a and 7b were synthesized using a previously reported method [20]. Next, a library of triazolic derivatives was synthesized by cycloaddition reactions between various alkynes and azides using the binuclear complex 2 as the catalyst with a 0.1–0.3 mol % catalyst loading and sodium l-ascorbate as the reductant in MeOH (Scheme 2). Twenty-five triazoles were obtained with excellent isolated yields between 91 and 99%. The chemical structures of the triazoles were confirmed using nuclear magnetic resonance (NMR) and high-resolution mass spectrometry (HRMS). The object products were 1,4-disubtituted triazoles, which were determined by comparing the NMR spectra of 8a–y with those of our previously reported triazoles [20].
2.2. Biological Evaluation
2.2.1. In Vitro Antiproliferative Activity and SARs
The newly synthesized sorafenib analogs were screened for antiproliferative activity in two cancer cell lines, namely the HT-29 (human colorectal cancer) and MCF-7 (human breast cancer) using a standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Sorafenib was used as the positive control. The biological data, expressed as half-maximal inhibitory concentration (IC50) values, are listed in Table 1. Interestingly, we discovered that most compounds exhibited significant antiproliferative activity, especially in the HT-29 cell line. This selectivity in cell lines was quite consistent with that of sorafenib. Seven compounds, 8a, 8e, 8f, 8i, 8j, 8m, and 8n, exhibited more potent activity than sorafenib did in inhibiting HT-29 cells. Notably, compounds 8e, 8f, and 8n potently inhibited the proliferation of both selected cells, with IC50 values less than 1 μM, which highlighted their potential for further investigations. Compound 8f was identified as the most promising compound, which exhibited a 26.5- and 80-fold increased potency compared with that of sorafenib in inhibiting the HT-29 and MCF-7 cell lines, respectively. In addition, compound 8f possessed a ClogP of 3.6, which is quite suitable for drug development.
In addition, relatively clear SAR results were obtained for these compounds. An aromatic ring at the terminal might be indispensable for antiproliferative activity. For example, both compounds 8o and 8p, which have aliphatic substituents connecting the triazole moiety were radically inactive against the tested cancer cells. In contrast, most compounds (8a, 8b, 8e, 8f, and 8i–8k) bearing a phenyl group exhibited obvious activity. Compounds bearing a pyridine (8m) or thiophene (8n) fragment exhibited promising antiproliferative activity, indicating that the heterocyclic ring was also critical for this region.
Further investigations of the phenyl ring showed close SARs. Notably, a substituent at the para-position was detrimental to the activity of amide series of compounds. None of the related compounds showed antiproliferative activity against the HT-29 or MCF-7 cell line, even when an electron-withdrawing (8g, R = 4-ClPh and 8h, R = 4-BrPh) or donating (8c, R = 4-MePh; 8d, R = 4-OMePh) group was incorporated. For compound 8c, shifting the methyl group to the meta-position produced compound 8b, which exhibited significantly improved activity against the HT-29 cancer cells (IC50 = 8.53 μM). The results suggest that substituents at the meta-position were better tolerated. A similar observation was reported when the activity of 8e (R = 2-FPh, IC50 = 0.84 μM) was compared with that of 8f (R = 3-FPh, IC50 = 0.20 μM). However, the introduction of substituents at both the meta- and para-positions failed to generate potent compounds (8k and 8l), which further confirmed that the para-position could not be substituted in this chemical series.
The analysis of the biological results showed that replacing the amide fragment at the 2-position of pyridine with an ester sharply decreased the activity. For example, compound 8a (X = NH, R = Ph; IC50: 0.74 μM) exhibited potent inhibitory activity in HT-29 cells, but a 20.6-fold decrease in potency was reported for compound 8q (X = O, R = Ph; IC50: 15.22 μM). Reduced potency was also observed when 8f (X = NH, R = 3-FPh; IC50: 0.20 μM) was compared with 8s (X = O, R = 3-FPh; IC50: 9.12 μM). These results indicate that a hydrogen-bond donor was necessary for activity, which was in line with the observation for sorafenib. In view of potent antiproliferative acitivity exhibited by 8f, it was evaluated for cytotoxicity against normal cells derived from human embryonic kidney (HEK293T). Attractively, compound 8f was less cytotoxic for HEK293T cells, with an IC50 value of 5.32 μM. These results indicated that compound 8f was a good candidate for subsequent studies.
2.2.2. Inhibition of Colony Formation by 8f
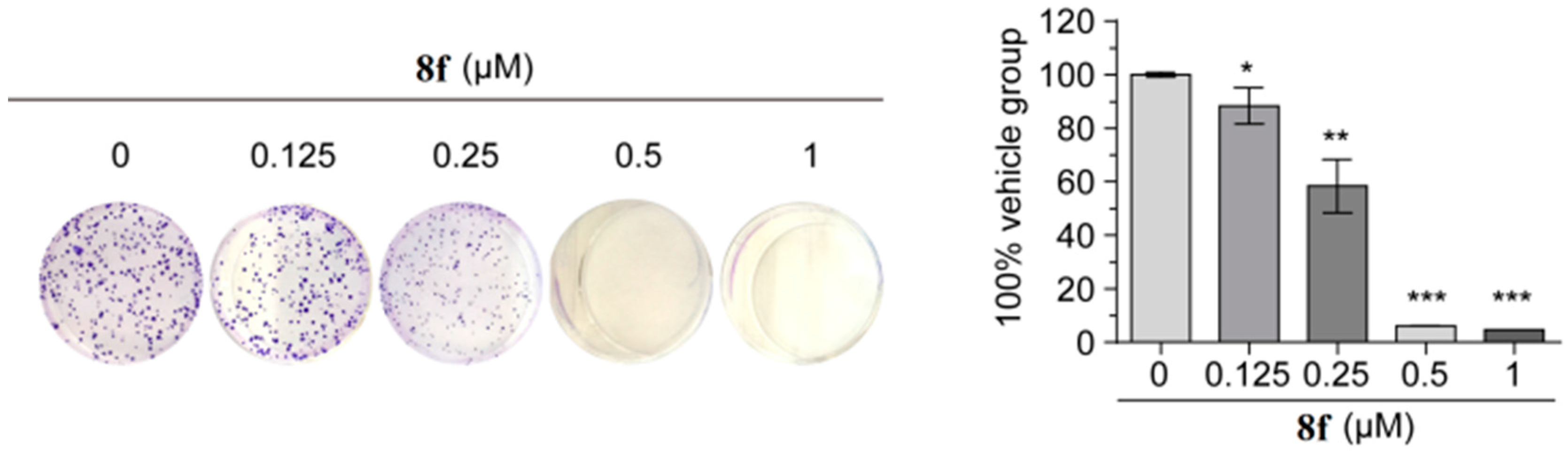
Considering the potent antiproliferative activity exhibited by 8f, a colony formation assay was conducted for a more visual result. HT-29 cells were treated with 8f at different concentrations (0, 0.125, 0.25, 0.5, and 1 μM) for 14 days. As shown in Figure 2, colony formation was significantly impaired in the 8f-treated group at a concentration of 0.25 μM. Furthermore, treatment with 0.5 μM 8f almost completely suppressed the colony formation.
2.2.3. Induction of Cell Apoptosis by 8f
The results of the MTT and colony formation assays clearly demonstrated that 8f potently inhibited the proliferation of HT-29 cells. To determine whether 8f acted by inducing apoptosis, a biparametric cytofluorimetric analysis using propidium iodide and annexin V-fluorescein isothiocyanate staining was performed. HT-29 cells were treated with 8f at various concentrations (0, 0.1, 0.3, and 1 μM) for 72 h before staining and flow cytometry. As shown in Figure 3, the apoptotic rate of the HT-29 cells obviously increased from 7.57% (control) to 61.7% following treatment with 1 μM 8f. The results suggest that 8f induced significant apoptosis of HT-29 cells in a dose-dependent manner.
3. Experimental Section
3.1. General Information
The 1H- and 13C-NMR spectra were recorded on a Bruker AV-400 spectrometer (Bruker Co., Ltd., Zurich, Switzerland), with TMS as an internal standard. Melting points were determined on a Büchi Melting Point B-540 apparatus (Büchi Labortechnik, Flawil, Switzerland). The HRMS analysis was obtained on an Agilent Technologies 6530 Accurate-Mass Q-TOF LC/MS (Version Q-TOF B.05.01, Agilent Technologies Inc., Santa Clara, CA, USA). All the alkynes except 5k and 5l were purchased from commercial sources. Intermediates 5k and 5l were prepared according to a literature procedure. The characterization of compounds 8a, 8h, and 8q have been reported in [20]. All of the solvents were purified according to the routine methods before being used.
3.2. General Procedure for the Synthesis of 5k and 5l
Dimethyl-1-diazo-2-oxopropylphosphonate (3.6 mmol) was added to a solution of aldehyde 4 (3.0 mmol) and K2CO3 (6.0 mmol) in MeOH (3 mL) and stirring was continued for 24 h. The reaction mixture was diluted with Et2O (60 mL), washed with an aq solution (5%) of NaHCO3 (85 mL) and water (25 mL) successively and dried over Na2SO4. After filtration and evaporation of the solvent in vacuo, the resulting residue was purified by flash column chromatography to afford the corresponding alkyne (petroleum ether (PE) as an elution solvent).
1-Chloro-4-ethynyl-2-methylbenzene (5k). Yield: 74%, white solid. m.p.: 29.4–31.2 °C. 1H-NMR (600 MHz, DMSO-d6) δ 7.36 (d, 1H, J = 1.1 Hz, ArH), 7.29–7.24 (m, 2H, ArH), 3.07 (s, 1H, C≡CH), 2.25 (s, 3H, CH3).
1-Chloro-4-ethynyl-2-(trifluoromethyl)benzene (5l). Yield: 84%, white solid. m.p.: 29.6–30.1 °C. 1H-NMR (600 MHz, DMSO-d6) δ 7.80 (d, 1H, J = 1.8 Hz, ArH), 7.58 (dd, 1H, J = 8.3, 1.9 Hz, ArH), 7.48 (d, 1H, J = 8.3 Hz, ArH), 3.19 (s, 1H, C≡CH).
3.3. General Procedure for 2-Catalyzed 1,3-Dipolar Cycloaddition Reactions
Under N2 atmosphere, the mixture of alkyne (0.3 mmol), azide (0.3 mmol), Na ascorbate (0.006 mmol, 2 mol %), and 0.3 × 10−3 M solution of catalyst 2 in MeOH (0.1–0.3 mol %) was stirred in a 10 mL Schlenk tube at 25 °C for 16 h. Evaporation of the solvents followed by purification by short column chromatography on silica gel provided the desired 1,4-disubstituted triazole product. The unreacted alkyne and azide were first eluted out with 3/1 petroleum ether (PE)/ethyl acetate, and the pure 1,4-disubstituted triazole product was then obtained by elution with pure ethyl acetate.
N-Methyl-4-(4-(4-(m-tolyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinamide (8b). Yield: 99%, white solid. m.p.: 185–187 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.33 (s, 1H, triazole-H), 8.82 (q, 1H, J = 4.8 Hz, NH), 8.57 (d, 1H, J = 5.6 Hz, ArH), 8.10 (dt, 2H, J = 8.9, 2.3 Hz, ArH), 7.79 (s, 1H, ArH), 7.75 (d, 1H, J = 7.7 Hz, ArH), 7.53 (dt, 2H, J = 8.9, 2.2 Hz, ArH), 7.49 (d, 1H, J = 2.6 Hz, ArH), 7.40 (t, 1H, J = 7.7 Hz, ArH), 7.27 (dd, 1H, J = 5.6, 2.6 Hz, ArH), 7.22 (d, 1H, J = 7.5 Hz, ArH), 2.80 (d, 3H, J = 4.9 Hz, NCH3), 2.39 (s, 3H, CCH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.18, 163.72, 153.28, 152.66, 150.67, 147.49, 138.21, 134.15, 130.13, 128.96, 125.91, 122.55, 122.37, 122.27, 119.80, 114.59, 109.34, 26.06, 21.12. HRMS Calcd for C22H19N5O2 [M]: 385.1539; found for C22H19N5O2 [M]: 385.1532.
N-Methyl-4-(4-(4-(p-tolyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinamide (8c). Yield: 99%, white solid. m.p.: 195–197 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.30 (s, 1H, triazole-H), 8.84 (q, 1H, J = 4.4 Hz, NH), 8.58 (d, 1H, J = 5.6 Hz, ArH), 8.10 (dt, 2H, J = 10.0, 3.2 Hz, ArH), 7.85 (d, 2H, J = 8.0 Hz, ArH), 7.54–7.49 (m, 3H, ArH), 7.33 (d, 2H, J = 7.9 Hz, ArH), 7.27 (dd, 1H, J = 5.6, 2.6 Hz, ArH), 2.80 (d, 3H, J = 4.9 Hz, NHCH3), 2.36 (s, 3H, CCH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.18, 163.70, 153.25, 152.65, 150.66, 147.45, 137.69, 134.17, 129.59, 127.44, 125.30, 122.36, 122.30, 119.46, 114.58, 109.32, 26.05, 20.90. HRMS Calcd for C22H19N5O2 [M]: 385.1539; found for C22H19N5O2 [M]: 385.1558.
4-(4-(4-(4-Methoxyphenyl)-1H-1,2,3-triazol-1-yl)phenoxy)-N-methylpicolinamide (8d). Yield: 97%, white solid. m.p.: 215–217 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.24 (s, 1H, triazole-H), 8.83 (d, 1H, J = 4.8 Hz, NH), 8.58 (d, 1H, J = 5.6 Hz, ArH), 8.10 (dt, 2H, J = 8.9, 3.2 Hz, ArH), 7.90 (dt, 2H, J = 8.8, 2.8 Hz, ArH), 7.54–7.49 (m, 3H, ArH), 7.27 (dd, 1H, J = 8.0, 2.6 Hz, ArH), 7.10 (dt, 2H, J = 8.8, 2.8 Hz, ArH), 3.82 (s, 3H, OCH3), 2.81(d, 3H, J = 4.9 Hz, NCH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.20, 163.72, 159.35, 153.21, 152.65, 150.68, 147.36, 134.21, 126.75, 122.75, 122.37, 122.26, 118.89, 114.58, 114.48, 109.33, 55.24, 26.07. HRMS Calcd for C22H19N5O3 [M]: 401.1488; found for C22H19N5O3 [M]: 401.1471.
4-(4-(4-(2-Fluorophenyl)-1H-1,2,3-triazol-1-yl)phenoxy)-N-methylpicolinamide (8e). Yield: 98%, white solid. m.p.: 182–183 °C. IR (KBr, cm−1): 3395.0, 1684.6, 1631.3, 1604.1, 1566.3, 1533.9, 1512.4, 901.8, 859.3, 837.4. 1H-NMR (600 MHz, DMSO-d6) δ 9.14 (d, 1H, J = 2.9 Hz, triazole-H), 8.83 (q, 1H, J = 4.4 Hz, NH), 8.57(d, 1H, J = 5.6 Hz, ArH), 8.21–8.14 (m, 3H, ArH), 7.52–7.46 (m, 4H, ArH), 7.42–7.37 (m, 2H, ArH), 7.27 (dd, 1H, J = 5.5, 2.6 Hz, ArH), 2.80 (d, 3H, J = 4.9 Hz, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.17, 163.72, 159.43, 157.79, 153.43, 152.66, 150.68, 141.01, 140.99, 134.01, 130.23, 130.18, 127.76, 127.74, 125.09, 125.07, 122.78, 122.29, 122.09, 122.02, 118.00, 117.91, 116.27, 116.13, 114.61, 109.34, 26.06. HRMS Calcd for C21H17N5O2 [M]: 389.1288; found for C21H17N5O2 [M]: 389.1304.
4-(4-(4-(3-Fluorophenyl)-1H-1,2,3-triazol-1-yl)phenoxy)-N-methylpicolinamide (8f). Yield: 99%, white solid. m.p.: 209–210 °C. IR (KBr, cm−1): 3403.3, 1676.7, 1630.7, 1610.4, 1589.8, 1568.9, 1538.8, 1514.7, 897.2, 862.4, 836.4. 1H-NMR (600 MHz, DMSO-d6) δ 9.42 (s, 1H, triazole-H), 8.83 (q, 1H, NH), 8.58 (d, 1H, J = 5.6 Hz, ArH), 8.09 (d, 2H, J = 8.9 Hz, ArH), 7.82 (d, 1H, J = 7.8 Hz, ArH), 7.76 (d, 1H, J = 10.1 Hz, ArH), 7.59–7.52 (m, 3H, ArH), 7.49 (d, 1H, J = 2.6 Hz, ArH), 7.27–7.22 (m, 2H, ArH), 2.80 (d, 3H, J = 4.9 Hz, CH3). 13C-NMR (150 MHz, DMSO-d6) δ 165.15, 163.72, 163.45, 161.83, 153.44, 152.67, 150.69, 146.29 (d, J = 2.7 Hz), 134.03, 132.61 (d, J = 8.6 Hz), 132.55 (d, J = 8.5 Hz), 131.30, 131.25, 122.44 (d, J = 2.8 Hz), 121.41 (d, J = 2.5 Hz), 120.71 (d, J = 2.6 Hz), 115.13, 114.99, 114.63, 112.03, 111.88, 109.36, 26.07. HRMS Calcd for C21H16FN5O2 [M]+: 389.1288; found for C21H16FN5O2 [M + H]+: 390.1355.
4-(4-(4-(4-Chlorophenyl)-1H-1,2,3-triazol-1-yl)phenoxy)-N-methylpicolinamide (8g). Yield: 94%, white solid. m.p.: 244–246 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.40 (s, 1H, triazole-H), 8.83 (d, 1H, J = 4.8 Hz, NH), 8.58 (d, 1H, J = 5.6 Hz, ArH), 8.09 (d, 2H, J = 8.9 Hz, ArH), 7.98 (d, 2H, J = 8.5 Hz, ArH), 7.60 (d, 2H, J = 8.5 Hz, ArH), 7.54 (d, 2H, J = 8.9 Hz, ArH), 7.49 (d, 1H, J = 2.5 Hz, ArH), 7.28 (dd, 1H, J = 5.6, 2.6 Hz, ArH), 2.81 (d, 3H, J = 4.9 Hz, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.16, 163.72, 153.39, 152.66, 150.69, 146.30, 134.06, 132.77, 129.17, 129.14, 127.05, 122.42, 122.40, 120.33, 120.31, 114.62, 109.35, 26.07. HRMS Calcd for C21H16ClN5O2 [M]: 405.0993; found for C21H16ClN5O2 [M]: 405.0991.
N-Methyl-4-(4-(4-(2-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinamide (8i). Yield: 99%, white solid. m.p.: 51–54 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.07 (s, 1H, triazole-H), 8.83 (q, 1H, J = 4.8 Hz, NH), 8.58 (d, 1H, J = 5.6 Hz, ArH), 8.14 (dt, 2H, J = 8.9, 3.2 Hz, ArH), 7.94 (d, 1H, J = 8.0 Hz, ArH), 7.84–7.83 (m, 2H, ArH), 7.73–7.71 (m, 1H, ArH), 7.53–7.50 (m, 3H, ArH), 7.27 (dd, 1H, J = 4.9, 2.5 Hz, ArH), 2.81 (d, 1H, J = 4.9 Hz, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.17, 163.71, 153.45, 152.67, 150.68, 144.84, 133.90, 132.76, 132.22, 129.36, 128.98, 127.08 (q, J = 30.2 Hz), 126.51 (q, J = 5.5 Hz), 124.89, 123.08, 122.59, 122.41, 114.59, 109.37, 26.06. HRMS Calcd for C21H16F3N5O2 [M]: 439.1256; found for C21H16F3N5O2 [M]: 439.1245.
N-Methyl-4-(4-(4-(3-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinamide (8j). Yield: 99%, white solid. m.p.: 216–218 °C. IR (KBr, cm−1): 3391.6, 1678.7, 1594.3, 1573.2, 1542.9, 835.8, 800.3. 1H-NMR (600 MHz, DMSO-d6) δ 9.56 (s, 1H, triazole-H), 8.83 (d, 1H, J = 4.9 Hz, NH), 8.59 (d, 1H, J = 5.6 Hz, ArH), 8.29 (d, 2H, J = 4.9 Hz, ArH), 8.11 (dt, 2H, J = 8.9, 2.0 Hz, ArH), 7.78–7.77 (m, 2H, ArH), 7.56 (dt, 2H, J = 8.9, 2.0 Hz, ArH), 7.50 (d, 1H, J = 2.5 Hz, ArH), 7.28 (dd, 1H, J = 5.6, 2.6 Hz, ArH), 2.81 (d, 3H, J = 4.9 Hz, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.14, 163.71, 153.47, 152.67, 150.69, 145.96, 134.00, 131.32, 130.36, 130.23 (q, JF = 31.8 Hz), 129.11, 126.87, 125.06, 124.84, 124.81, 123.26, 122.42, 122.38, 121.71, 121.68, 121.45, 120.96, 120.94, 114.64, 109.38, 26.06. HRMS Calcd for C21H16F3N5O2 [M]: 439.1256; found for C21H16F3N5O2 [M]: 439.1245.
4-(4-(4-(4-Chloro-3-methylphenyl)-1H-1,2,3-triazol-1-yl)phenoxy)-N-methylpicolinamide (8k). Yield: 99%, white solid. m.p.: 237–238 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.38 (s, 1H, triazole-H), 8.83 (d, 1H, J = 4.3 Hz, NH), 8.58 (d, 1H, J = 5.4 Hz, ArH), 8.09 (d, 1H, J = 8.5 Hz, ArH), 7.96 (s, 1H, ArH), 7.80 (d, 1H, J = 7.8 Hz, ArH), 7.57–7.49 (m, 4H, ArH), 7.28 (d, 1H, J = 2.9 Hz, ArH), 2.81 (d, 3H, J = 4.6 Hz, NCH3), 2.43 (s, 3H, CCH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.16, 163.71, 153.37, 152.66, 150.69, 146.42, 136.19, 134.07, 133.00, 129.59, 129.16, 127.93, 124.51, 122.39, 122.34, 120.20, 114.62, 109.35, 26.06, 19.75. HRMS Calcd for C22H18ClN5O2 [M]: 419.1149; found for C22H18ClN5O2 [M]: 419.1137.
4-(4-(4-(4-Chloro-3-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)phenoxy)-N-methylpicolinamide (8l). Yield: 98%, white solid. m.p.: 226–228 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.58 (s, 1H, triazole-H), 8.83 (d, 1H, J = 4.9 Hz, NH), 8.59 (d, 1H, J = 5.6 Hz, ArH), 8.38 (d, 1H, J = 1.9 Hz, ArH), 8.27 (dd, 1H, J = 8.3, 1.8 Hz, ArH), 8.10 (dt, 2H, J = 8.9, 3.2 Hz, ArH), 7.91 (d, 1H, J = 8.4 Hz, ArH), 7.56–7.50 (m, 3H, ArH), 7.28 (dd, 1H, J = 5.6, 2.6 Hz, ArH), 2.81 (d, 3H, J = 4.9 Hz, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.12, 163.71, 153.54, 152.67, 150.69, 145.06, 133.91, 132.62, 130.45, 130.09, 129.93, 127.52, 127.31, 124.27, 124.23, 123.70, 122.42, 122.41, 121.89, 121.28, 121.26, 114.65, 109.38, 26.06. HRMS Calcd for C22H15ClF3N5O2 [M]: 473.0866; found for C22H15ClF3N5O2 [M]: 473.0883.
N-Methyl-4-(4-(4-(pyridin-2-yl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinamide (8m). Yield: 94%, white solid. m.p.: 200–201 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.40 (s, 1H, triazole-H), 8.84 (q, 1H, J = 4.5 Hz, NH), 8.68 (d, 1H, J = 4.7 Hz, ArH), 8.58 (d, 1H, J = 5.6 Hz, ArH), 8.19–8.14 (m, 3H, ArH), 7.98 (dt, 1H, J = 7.7, 1.6 Hz, ArH), 7.52 (m, 3H, ArH), 7.43 (dd, 1H, J = 7.4, 4.9 Hz, ArH), 7.28 (dd, 1H, J = 5.5, 2.6 Hz, ArH), 2.81, 2.80 (d, 3H, J = 4.9 Hz, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.16, 163.72, 153.41, 152.65, 150.67, 149.73, 149.48, 148.29, 137.41, 134.05, 123.43, 122.54, 122.29, 121.52, 121.51, 119.86, 114.63, 109.36, 26.07. HRMS Calcd for C20H16N6O2 [M]: 372.1335; found for C20H16N6O2 [M]: 372.1340.
N-methyl-4-(4-(4-(thiophen-2-yl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinamide (8n). Yield: 92%, white solid. m.p.: 185–188 °C. IR (KBr, cm−1): 3381.7, 1677.0, 1631.4, 1587.3, 1571.9, 1530.8, 1511.8, 849.6, 838.6, 816.1. 1H-NMR (600 MHz, DMSO-d6) δ 9.26 (s, 1H, triazole-H), 8.83 (d, 1H, J = 4.8 Hz, NH), 8.58 (d, 1H, J = 5.6 Hz, ArH), 8.09 (d, 2H, J = 8.9 Hz, ArH), 7.63 (dd, 1H, J = 5.0, 0.8 Hz, ArH), 7.54–7.49 (m, 4H, ArH), 7.27 (dd, 1H, J = 5.5, 2.6 Hz, ArH), 7.21 (dd, 1H, J = 4.9, 3.6 Hz, ArH), 2.81 (d, 3H, J = 4.9 Hz, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.14, 163.69, 153.39, 152.65, 150.66, 142.79, 133.95, 132.30, 128.06, 126.01, 124.68, 122.43, 122.34, 119.14, 114.60, 109.35, 26.05. HRMS Calcd for C19H15N5O2S [M]: 377.0946; found for C19H15N5O2S [M]: 377.0938.
4-(4-(4-(Hydroxymethyl)-1H-1,2,3-triazol-1-yl)phenoxy)-N-methylpicolinamide (8o). Yield: 92%, white solid. m.p.: 191–192 °C. 1H-NMR (600 MHz, DMSO-d6) δ 8.82 (d, 1H, J = 4.9 Hz, NH), 8.74 (s, 1H, triazole-H), 8.57 (d, 2H, J = 5.6 Hz, ArH), 8.06 (dt, 2H, J = 8.9, 3.2 Hz, ArH), 7.49–7.46 (m, 3H, ArH), 7.26 (dd, 1H, J = 5.6, 2.6 Hz, ArH), 5.39 (t, 1H, J = 5.6 Hz, OH), 4.64 (d, 2H, J = 5.5 Hz, CH2), 2.80 (d, 3H, J = 4.9 Hz, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 165.18, 163.72, 153.11, 152.64, 150.66, 149.25, 134.27, 122.29, 122.26, 121.23, 114.58, 109.32, 54.98, 26.06. HRMS Calcd for C16H15N5O3 [M]: 325.1175; found for C16H15N5O3 [M]: 325.1191.
4-(4-(4-(2-Hydroxypropan-2-yl)-1H-1,2,3-triazol-1-yl)phenoxy)-N-methylpicolinamide (8p). Yield: 99%, white solid. m.p.: 150–152 °C. 1H-NMR (600 MHz, DMSO-d6) δ 8.83 (q, 1H, J = 4.5 Hz, NH), 8.65 (s, 1H, triazole-H), 8.57 (d, 1H, J = 5.6 Hz, ArH), 8.06 (dt, 2H, J = 8.9, 3.2 Hz, ArH), 7.48–7.45 (m, 3H, ArH), 7.25 (dd, 1H, J = 5.6, 2.6 Hz, ArH), 5.28 (s, 1H, OH), 2.81 (d, 3H, J = 4.9 Hz, NCH3), 1.55 (s, 6H, C(CH3)2). 13C-NMR (151 MHz, DMSO-d6) δ 165.19, 163.72, 157.03, 153.01, 152.64, 150.66, 134.34, 122.25, 122.17, 119.10, 114.57, 109.33, 67.02, 30.60, 26.06. HRMS Calcd for C18H19N5O3 [M]: 353.1488; found for C18H19N5O3 [M]: 353.1504.
Methyl 4-(4-(4-(m-tolyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinate (8r). Yield: 98%, white solid. m.p.: 147–149 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.34 (s, 1H, triazole-H), 8.65 (d, 1H, J = 5.5 Hz, ArH), 8.11 (d, 2H, J = 8.6 Hz, ArH), 7.79–7.74 (m, 2H, ArH), 7.56–7.52 (m, 3H, ArH), 7.41 (t, 1H, J = 7.6 Hz, ArH), 7.32 (dd, 1H, J = 5.2, 2.0 Hz, ArH), 7.22 (d, 1H, J = 7.44 Hz, ArH), 3.87 (s, 3H, OCH3), 2.40 (s, 3H, CCH3). 13C-NMR (151 MHz, DMSO-d6) δ164.82, 164.72, 153.22, 151.92, 149.74, 147.53, 138.24, 134.19, 130.13, 128.99, 125.93, 122.57, 122.28, 119.78, 115.51, 112.89, 52.67, 21.14. HRMS Calcd for C22H18N4O3 [M]: 386.1379; found for C22H18N4O3 [M]: 386.1370.
Methyl 4-(4-(4-(3-fluorophenyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinate (8s). Yield: 96%, white solid. m.p.: 154–157 °C. 1H-NMR (600 MHz, CDCl3) δ 9.42 (s, 1H, triazole-H), 8.65 (d, 1H, J = 5.5 Hz, ArH), 8.08 (d, 2H, J = 8.6 Hz, ArH), 7.81 (d, 1H, J = 7.6 Hz, ArH), 7.75 (d, 1H, J = 9.9 Hz, ArH), 7.58–7.53 (m, 4H, ArH), 7.32 (dd, 1H, J = 5.2, 2.0 Hz, ArH), 7.25 (t, 1H, J = 7.1 Hz, ArH), 3.86 (s, 3H, CH3). 13C-NMR (150 MHz, DMSO-d6) δ 164.84, 164.70, 163.47, 161.86, 153.39, 151.96, 149.76, 146.34, 134.07, 132.62, 132.56, 131.35, 131.29, 122.46, 122.34, 121.46, 120.70, 115.56, 115.17, 115.03, 112.93, 112.06, 111.91, 52.70. HRMS Calcd for C21H15FN4O3 [M]: 390.1128; found for C21H15FN4O3 [M]: 390.1142.
Methyl 4-(4-(4-(4-chlorophenyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinate (8t). Yield: 99%, white solid. m.p.: 173–175 °C. IR (KCl, cm−1): 3415.6, 1717.4, 1631.5, 1608.1, 1586.3, 884.0, 866.4, 855.6, 835.6. 1H-NMR (600 MHz, DMSO-d6) δ 9.40 (s, 1H, triazole-H), 8.65 (d, 1H, J = 5.6 Hz, ArH), 8.09 (d, 2H, J = 8.7 Hz, ArH), 7.98 (d, 2H, J = 8.3 Hz, ArH), 7.60 (d, 2H, J = 8.2 Hz, ArH), 7.56–7.53 (m, 3H, ArH), 7.32 (dd, 1H, J = 5.3, 1.7 Hz, ArH), 3.86 (s, 3H, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 164.82, 164.69, 153.34, 151.93, 149.75, 146.35, 134.09, 132.80, 129.18, 129.14, 127.07, 122.42, 122.30, 120.28, 115.53, 112.90, 52.67. HRMS Calcd for C21H15ClN4O3 [M]: 406.0833; found for C21H15ClN4O3 [M]: 406.0827.
Methyl 4-(4-(4-(2-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinate (8u). Yield: 99%, white solid. m.p.: 87–88 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.06 (s, 1H, triazole-H), 8.65 (d, 1H, J = 5.5 Hz, ArH), 8.14 (d, 2H, J = 8.8 Hz, ArH), 7.93 (d, 1H, J = 7.9 Hz, ArH), 7.83–7.82 (m, 2H, ArH), 7.72 (t, 1H, J = 6.8 Hz, ArH), 7.55–7.52 (m, 3H, ArH), 7.32 (dd, 1H, J = 5.5, 2.4 Hz, ArH), 3.86 (s, 3H, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 164.81, 164.71, 153.39, 151.92, 149.75, 144.88, 133.93, 132.78, 132.25, 129.39, 128.98, 127.11 (q, J = 30.0 Hz), 126.53 (q, J = 5.4 Hz), 124.91, 123.10, 122.58, 122.33, 115.51, 112.89, 52.67. HRMS Calcd for C22H15F3N4O3 [M]: 440.1096; found for C22H15F3N4O3 [M]: 440.1062.
Methyl 4-(4-(4-(3-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinate (8v). Yield: 93%, white solid. m.p.: 172–174 °C. 1H-NMR (600 MHz, CDCl3) δ 9.55 (s, 1H, triazole-H), 8.65 (d, 1H, J = 5.6 Hz, ArH), 8.28 (d, 2H, J = 5.0 Hz, ArH), 8.11 (dt, 2H, J = 8.9, 3.3 Hz, ArH), 7.77–7.76 (m, 2H, ArH), 7.55–7.53 (m, 3H, ArH), 7.32 (dd, 1H, J = 5.6, 2.6 Hz, ArH), 3.86 (s, 3H, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 164.78, 164.64, 153.39, 151.90, 149.73, 145.97, 134.00, 131.29, 130.33, 130.21 (q, J = 31.9 Hz), 129.08, 126.85, 125.05, 124.82, 124.80, 123.24, 122.34, 122.28, 121.69, 121.67, 121.44, 120.90, 120.88, 115.51, 112.90, 52.63. HRMS Calcd for C22H15F3N4O3 [M]: 440.1096; found for C22H15F3N4O3 [M]: 440.1102.
Methyl 4-(4-(4-(4-chloro-3-methylphenyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinate (8w). Yield: 99%, white solid. m.p.: 177–178 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.38 (s, 1H, triazole-H), 8.65 (d, 1H, J = 5.5 Hz, ArH), 8.10 (dt, 2H, J = 6.8, 3.3 Hz, ArH), 7.95 (d, 1H, J = 1.5 Hz, ArH), 7.80 (dd, 1H, J = 8.2, 1.8 Hz, ArH), 7.57–7.53 (m, 4H, ArH), 7.32 (dd, 1H, J = 5.6, 2.6 Hz, ArH), 3.86 (s, 3H, OCH3), 2.42 (s, 3H, CCH3). 13C-NMR (151 MHz, DMSO-d6) δ 164.84, 164.71, 153.33, 151.95, 149.76, 146.48, 136.23, 134.11, 133.06, 129.62, 129.16, 127.97, 124.55, 122.37, 122.32, 120.17, 115.55, 112.93, 52.70, 19.78. HRMS Calcd for C22H17ClN4O3 [M]: 420.0989; found for C22H17ClN4O3 [M]: 420.1007.
Methyl 4-(4-(4-(4-chloro-3-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinate (8x). Yield: 99%, white solid. m.p.: 175–177 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.59 (s, 1H, triazole-H), 8.66 (d, 1H, J = 5.6 Hz, ArH), 8.38 (d, 1H, J = 1.8 Hz, ArH), 8.27 (dd, 1H, J = 8.3, 1.9 Hz, ArH), 8.09 (dt, 2H, J = 8.9, 1.9 Hz, ArH), 7.92 (d, 1H, J = 8.4 Hz, ArH), 7.56–7.54 (m, 3H, ArH), 7.33 (dd, 1H, J = 5.5, 2.5 Hz, ArH), 3.87 (s, 3H, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 164.81, 164.64, 153.47, 151.93, 149.75, 145.09, 133.93, 132.62, 130.46, 130.12, 129.92, 127.52 (q, J = 30.8 Hz), 124.27, 123.71, 122.39, 122.31, 121.90, 121.20, 115.55, 112.94, 52.67. HRMS Calcd for C22H14ClF3N4O3 [M]: 474.0707; found for C22H14ClF3N4O3 [M]: 474.0730.
Methyl 4-(4-(4-(thiophen-2-yl)-1H-1,2,3-triazol-1-yl)phenoxy)picolinate (8y). Yield: 99%, white solid. m.p.: 152–153 °C. 1H-NMR (600 MHz, DMSO-d6) δ 9.26 (s, 1H, triazole-H), 8.65 (d, 1H, J = 5.5 Hz, ArH), 8.09 (d, 2H, J = 8.9 Hz, ArH), 7.63 (d, 1H, J = 5.0 Hz, ArH), 7.56–7.52 (m, 4H, ArH), 7.32 (dd, 1H, J = 5.5, 2.5 Hz, ArH), 7.21 (dt, 1H, J = 3.7, 1.2 Hz, ArH), 3.87 (s, 3H, CH3). 13C-NMR (151 MHz, DMSO-d6) δ 164.82, 164.69, 153.36, 151.93, 149.75, 142.85, 133.99, 132.29, 128.11, 126.06, 124.74, 122.46, 122.27, 119.12, 115.53, 112.91, 52.67. HRMS Calcd for C19H14N4O3S [M]: 378.0787; found for C19H14N4O3S [M]: 378.0797.
3.4. Cell Viability Assay
The viability of HT-29, MCF-7, and HEK293T cells was determined using an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. These cancer cell lines were cultured in minimum essential medium supplemented with 10% foetal bovine serum.
Approximately 4 × 103 cells were suspended in cell culture medium, plated in a 96-well plate and incubated at 37 °C in 5% CO2 for 24 h. The test compounds were added to the culture medium and incubated for a further 72 h. Fresh MTT was then added to each well at a final concentration of 5 μg/mL, and incubated with the cells at 37 °C for 4 h. The formazan crystals in each well were dissolved in 100 μL DMSO, and the absorbance at 492 nm (MTT formazan wavelength) and 630 nm (reference wavelength) was measured by a microplate reader. The reported IC50 results represented averages of at least three determinations and were calculated using the Bacus Laboratories Incorporated Slide Scanner (Bliss) software.
3.5. Colony Formation Assay
Logarithmic phase HT-29 cells were seeded into 6-well plates at a concentration of 500 cells per well. The next day, cells were treated with 8f at 0.125, 0.25, 0.5, and 1 μM. Colonies were fixed with methanol at room temperature for 15 min and then stained with a solution of 0.1% crystal violet diluted by phosphate buffered saline (PBS) from 0.5% crystal violet (dissolved in methanol) for 15 min and then washed by water. The stained colonies were then photographed and dissolved by 33% acetic acid. Optical density of absorbance was read at 570 nm.
3.6. Apoptosis Assay
Apoptosis of HT-29 cells was detected using a flow cytometric assay. Briefly, cells were seeded in 6-well plates and incubated overnight. The following day, cells were treated with different concentrations of compound 8f for 72 h. The cells and supernatants were harvested and washed twice with cold PBS and then resuspended in 100μL 1× Binding Buffer. 5 µL of FITC Annexin V and 5 µL PI were added in each tube and the cells were then gently vortexed incubated for 15 min at RT (25 °C) in the dark. In addition, 400 µL of 1× Binding Buffer was then added to each tube. The stained cells were analyzed by a flow cytometer (Thermo Fisher Scientific, San Jose, CA, USA).
4. Conclusions
In summary, we demonstrated the synthetic application of our highly active CuAAC reaction catalyst in preparing a novel series of sorafenib analogs. The binuclear Cu catalyst exhibited a satisfactory performance in the synthesis of the target compounds. At 0.1–0.3 mol % catalyst loading, all triazole products were obtained at 91–99% isolated yields. Using this catalytic system, 25 new compounds were prepared effectively and were evaluated biologically. The MTT and colony formation assays demonstrated that compound 8f potently inhibited the proliferation of HT-29 cells. The flow cytometric assay suggested that 8f caused significant apoptosis of HT-29 cells in a dose-dependent manner.
According to the chemical structural characteristics of 8f, we considered it to be a kinase inhibitor. Actually, we attempted to identify its biological target by screening it against a panel of kinases including B-Raf, VEGFR-3, PDGFRα, fibroblast growth factor receptor 1 (FGFR1), c-Kit, TRAF2 and NCK interacting kinase (TNIK), ret proto-oncogene (RET), cyclin-dependent 2 (CDK2), checkpoint kinase 1 (CHK1), and cyclin-dependent kinase 4 (CDK4). However, none of these kinases were significantly inhibited (Supplementary materials Table S1). These results indicated that 8f acted quite differently from sorafenib. More detailed investigations of the mechanisms of action of these compounds are in progress in our laboratory. Studies of the SARs indicated that a fluorine atom at the meta-position of the terminal phenyl ring was well tolerated, rather than the 3-CF3-4-Cl group in sorafenib. The hydrogen-bond donor at the 2-position of pyridine was indispensable for the activity of this chemical series. These findings all provide potentially valuable guidance for designing more powerful compounds.
Supplementary Materials
Supplementary materials are available online. Table S1: Enzymatic screening of selected compounds.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81502924 and 21502120) and the Innovation Training Project of Liaoning Province (201610163013).
Author Contributions
Mingze Qin and Wenjing Ye designed and wrote the paper; Qi Yao, Simiao Yu, and Ping Gong carried out the experiments. All authors have read and approved the final manuscript.
Conflicts of Interest
The authors have declared no conflict of interest.
References
- Guida, T.; Anaganti, S.; Provitera, L.; Gedrich, R.; Sullivan, E.; Wilhelm, S.; Santoro, M.; Carlomagno, F. Sorafenib inhibits imatinib-resistant KIT and platelet-derived growth factor receptor beta gatekeeper mutants. Clin. Cancer Res. 2007, 13, 3363–3369. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, F.; Radi, M.; Brullo, C.; Schenone, S. Vascular endothelial growth factor (VEGF) receptors: Drugs and new inhibitors. J. Med. Chem. 2012, 55, 10797–10822. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef] [PubMed]
- Drug Bank. Sorafenib Drug Card DB00398. Available online: https://www.drugbank.ca/drugs/DB00398 (accessed on 18 October 2017).
- Liu, C.; Chen, Z.; Chen, Y.; Lu, J.; Li, Y.; Wang, S.; Wu, G.; Qian, F. Improving oral bioavailability of sorafenib by optimizing the “spring” and “parachute” based on molecular interaction mechanisms. Mol. Pharm. 2016, 13, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Zhan, W.; Li, Y.; Huang, W.; Zhao, Y.; Yao, Z.; Yu, S.; Yuan, S.; Jiang, F.; Yao, S.; Li, S. Design, synthesis and antitumor activities of novel bis-aryl ureas derivatives as Raf kinase inhibitors. Bioorg. Med. Chem. 2012, 20, 4323–4329. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Cheng, H.; Tian, Z.; Zhao, J.; Li, G.; Chu, Y.; Sun, C.; Li, W. Rational drug design of indazole-based diarylurea derivatives as anticancer agents. Chem. Biol. Drug Des. 2017, 90, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, R.; Li, G.; Xue, X.; Sun, C.; Qu, X.; Li, W. Synthesis of indazole based diarylurea derivatives and their antiproliferative activity against tumor cell lines. Bioorg. Med. Chem. Lett. 2013, 23, 1989–1992. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; He, Z.; Chen, J.; Sun, W.; Fang, H.; Xu, W. Design, synthesis and biological activities of sorafenib derivatives as antitumor agents. Bioorg. Med. Chem. Lett. 2012, 22, 6549–6553. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wu, C.; Wang, J.; Luo, R.; Wang, C.; Liu, X.; Li, J.; Zhu, W.; Zheng, P. Synthesis, activity and docking studies of phenylpyrimidine-carboxamide sorafenib derivatives. Bioorg. Med. Chem. 2016, 24, 6166–6173. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhai, X.; Zhong, Z.; Li, G.; Pu, Y.; Gong, P. Design, synthesis and evaluation of novel rhodanine-containing sorafenib analogs as potential antitumor agents. Arch. Pharm. 2011, 11, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Ramurthy, S.; Subramanian, S.; Aikawa, M.; Amiri, P.; Costales, A.; Dove, J.; Fong, S.; Jansen, J.M.; Levine, B.; Ma, S.; et al. Design and synthesis of orally bioavailable benzimidazoles as Raf kinase inhibitors. J. Med. Chem. 2008, 51, 7049–7052. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, V.F.; Rocha, D.R.; Silva, F.C.; Ferreira, P.G.; Boechat, N.A.; Magalhães, J.L. Novel 1H-1,2,3-, 2H-1,2,3-, 1H-1,2,4- and 4H-1,2,4-triazole derivatives: A patent review (2008–2011). Expert Opin. Ther. Pat. 2013, 23, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. 2002, 41, 2596–2598. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Campbell-Verduyn, L.S.; Mirfeizi, L.; Dierckx, R.A.; Elsinga, P.H.; Feringa, B.L. ChemInform abstract: Phosphoramidite accelerated copper(I)-catalyzed [3 + 2] cycloadditions of azides and alkynes. Chem. Commun. 2009, 2139–2141. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, P.S.; Zanatta, S.D.; Zammit, S.C.; White, J.M.; Williams, S.J. ‘Click’ cycloaddition catalysts: Copper(I) and copper(II) tris(triazolylmethyl)amine complexes. Chem. Commun. 2008, 21, 2459–2461. [Google Scholar] [CrossRef] [PubMed]
- Ozkal, E.; Llanes, P.; Bravo, F.; Ferrali, A.; Pericàs, M.A. Fine-tunable tris(triazolyl)methane ligands for copper(I)-catalyzed azide-alkyne cycloaddition reactions. Adv. Synth. Catal. 2014, 356, 857–869. [Google Scholar] [CrossRef]
- Ye, W.J.; Xiao, X.; Wang, L.; Hou, S.C.; Hu, C. Synthesis of mono- and binuclear Cu(II) complexes bearing unsymmetrical bipyridine-pyrazole-amine ligand and their applications in azide-alkyne cycloaddition. Organometallics 2017, 36, 2116–2125. [Google Scholar] [CrossRef]
- Han, B.F.; Xiao, X.; Wang, L.; Ye, W.J.; Liu, X.P. Highly active binuclear Cu(II) catalyst bearing an unsymmetrical bipyridine-pyrazole-amine ligand for the azide-alkyne cycloaddition reaction. Chin. J. Catal. 2016, 37, 1446–1450. [Google Scholar] [CrossRef]
- Roth, G.J.; Liepold, B.; Müller, S.G.; Bestmann, H.J. Further improvements of the synthesis of alkynes from aldehydes. Synthesis 2004, 59–62. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 8a–8y are available from the authors. |
Scheme 1.
Cu complexes bearing an unsymmetrical bipyridine-pyrazole-amine ligand and application in CuAAC reaction.
Scheme 1.
Cu complexes bearing an unsymmetrical bipyridine-pyrazole-amine ligand and application in CuAAC reaction.

Figure 1.
Design of novel sorafenib analogs.

Scheme 2.
Synthesis route of the 1,2,3-triazole derivatives. Reagents and conditions: (a) K2CO3 2.0 eq., Bestman–Ohira reagent 1.5 eq., MeOH, rt, 48 h, 74% yield in 5k; 84% yield in 5l; (b) HCl/NaNO2, 1.2 eq. NaN3, EtOAc, rt, 3 h, 61% yield in 7a; 97% yield in 7b; (c) appropriate alkyne, 0.1–0.3 mol % Cat. 2, sodium l-ascorbate, MeOH, 25–50 °C, 16 h, 91–99% yields.
Scheme 2.
Synthesis route of the 1,2,3-triazole derivatives. Reagents and conditions: (a) K2CO3 2.0 eq., Bestman–Ohira reagent 1.5 eq., MeOH, rt, 48 h, 74% yield in 5k; 84% yield in 5l; (b) HCl/NaNO2, 1.2 eq. NaN3, EtOAc, rt, 3 h, 61% yield in 7a; 97% yield in 7b; (c) appropriate alkyne, 0.1–0.3 mol % Cat. 2, sodium l-ascorbate, MeOH, 25–50 °C, 16 h, 91–99% yields.

Figure 2.
Representative images of colony formation of HT-29 cells treated with 8f. Relative colony formation rate is shown as mean ± SEM. * p < 0.05, ** p < 0.01, and *** p < 0.001 versus vehicle group; p-values were calculated using an unpaired two-tailed Student’s t-test.
Figure 2.
Representative images of colony formation of HT-29 cells treated with 8f. Relative colony formation rate is shown as mean ± SEM. * p < 0.05, ** p < 0.01, and *** p < 0.001 versus vehicle group; p-values were calculated using an unpaired two-tailed Student’s t-test.

Figure 3.
Effect of compound 8f on cell apoptosis in HT-29 cells.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Antiproliferative activities and calculated logP-values of compounds 8a–8y.
| Products | IC50 (μM) a | ClogP b | |
|---|---|---|---|
| HT-29 | MCF-7 | ||
| 8a | 0.74 ± 0.12 | 7.53 ± 0.65 | 3.45 |
| 8b | 8.53 ± 1.09 | 10.17 ± 0.77 | 3.97 |
| 8c | >100 | >100 | 3.97 |
| 8d | 89.83 ± 3.28 | >100 | 3.30 |
| 8e | 0.84 ± 0.17 | 0.33 ± 0.02 | 3.60 |
| 8f | 0.20 ± 0.01 | 0.61 ± 0.12 | 3.60 |
| 8g | >100 | >100 | 4.06 |
| 8h | >100 | >100 | 4.22 |
| 8i | 0.55 ± 0.08 | 10.99 ± 1.66 | 4.33 |
| 8j | 0.76 ± 0.13 | 26.70 ± 3.37 | 4.33 |
| 8k | 13.51 ± 1.28 | 50.96 ± 2.76 | 4.57 |
| 8l | 49.20 ± 2.39 | 99.19 ± 3.58 | 4.94 |
| 8m | 0.60 ± 0.05 | 7.21± 0.96 | 2.62 |
| 8n | 0.58 ± 0.13 | 0.73 ± 0.32 | 3.23 |
| 8o | >100 | >100 | 0.74 |
| 8p | >100 | >100 | 1.74 |
| 8q | 15.22 ± 2.68 | 71.92 ± 3.66 | 4.18 |
| 8r | 5.33 ± 0.31 | 48.33 ± 2.56 | 4.70 |
| 8s | 9.12 ± 0.60 | 17.06 ± 2.17 | 4.33 |
| 8t | 11.81 ± 2.31 | 36.23 ± 1.70 | 4.79 |
| 8u | 44.54 ± 2.28 | 43.93 ± 2.67 | 5.06 |
| 8v | >100 | >100 | 5.06 |
| 8w | 9.57 ± 1.05 | 44.08 ± 2.69 | 5.30 |
| 8x | 54.98 ± 2.70 | 40.84 ± 3.56 | 5.67 |
| 8y | 8.79 ± 0.97 | 47.36 ± 1.38 | 3.96 |
| sorafenib | 5.29 ± 0.32 | 43.30 ± 1.36 | 4.34 |
a Biological data are generated from at least three independent experiments; b ClogP-values calculated using MarvinSketch 6.1.0.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ye, W.; Yao, Q.; Yu, S.; Gong, P.; Qin, M. Synthesis and Antitumor Activity of Triazole-Containing Sorafenib Analogs. Molecules 2017, 22, 1759. https://doi.org/10.3390/molecules22101759
AMA Style
Ye W, Yao Q, Yu S, Gong P, Qin M. Synthesis and Antitumor Activity of Triazole-Containing Sorafenib Analogs. Molecules. 2017; 22(10):1759. https://doi.org/10.3390/molecules22101759
Chicago/Turabian StyleYe, Wenjing, Qi Yao, Simiao Yu, Ping Gong, and Mingze Qin. 2017. "Synthesis and Antitumor Activity of Triazole-Containing Sorafenib Analogs" Molecules 22, no. 10: 1759. https://doi.org/10.3390/molecules22101759