Ureidopyrazine Derivatives: Synthesis and Biological Evaluation as Anti-Infectives and Abiotic Elicitors

 , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
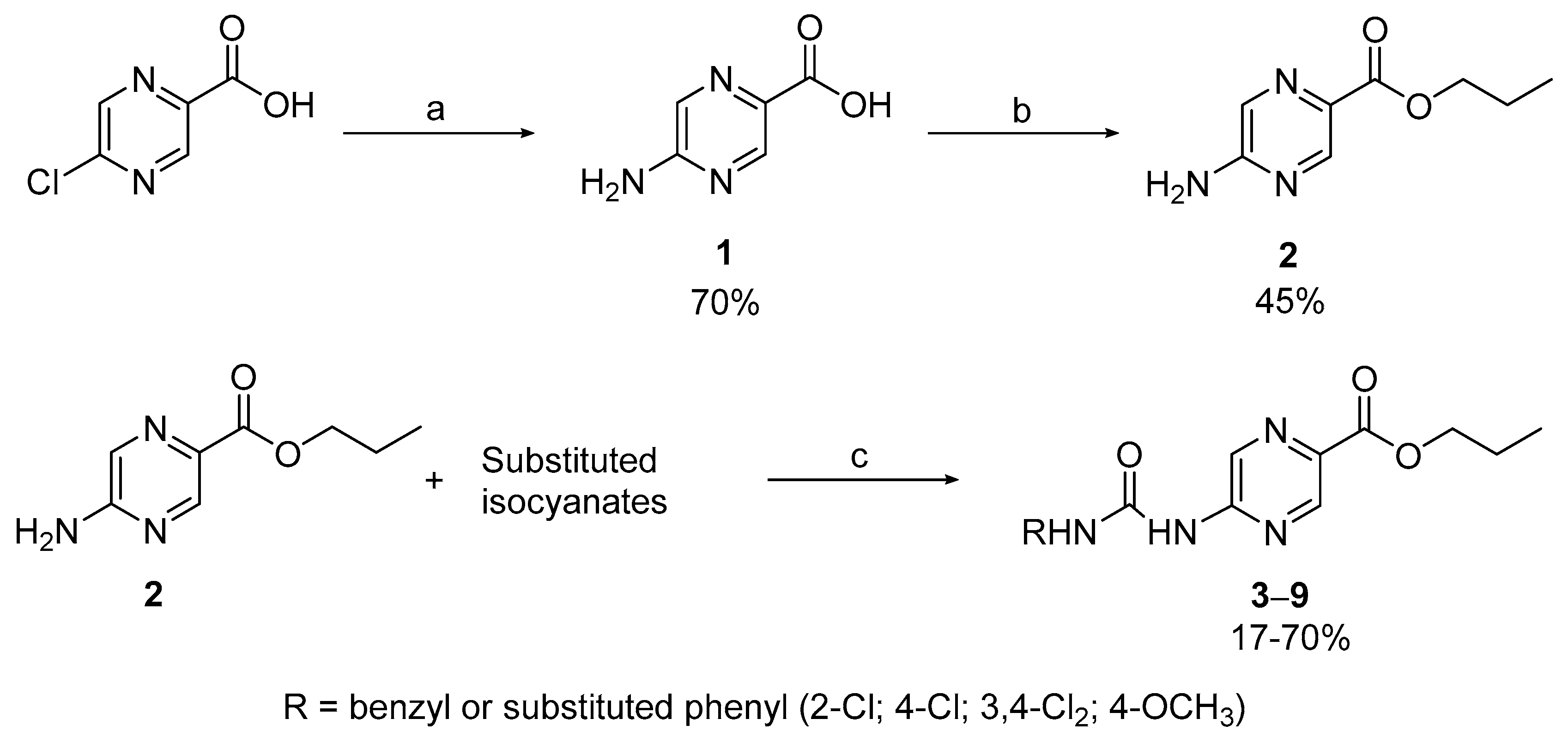
2.1.1. Compounds 1–9
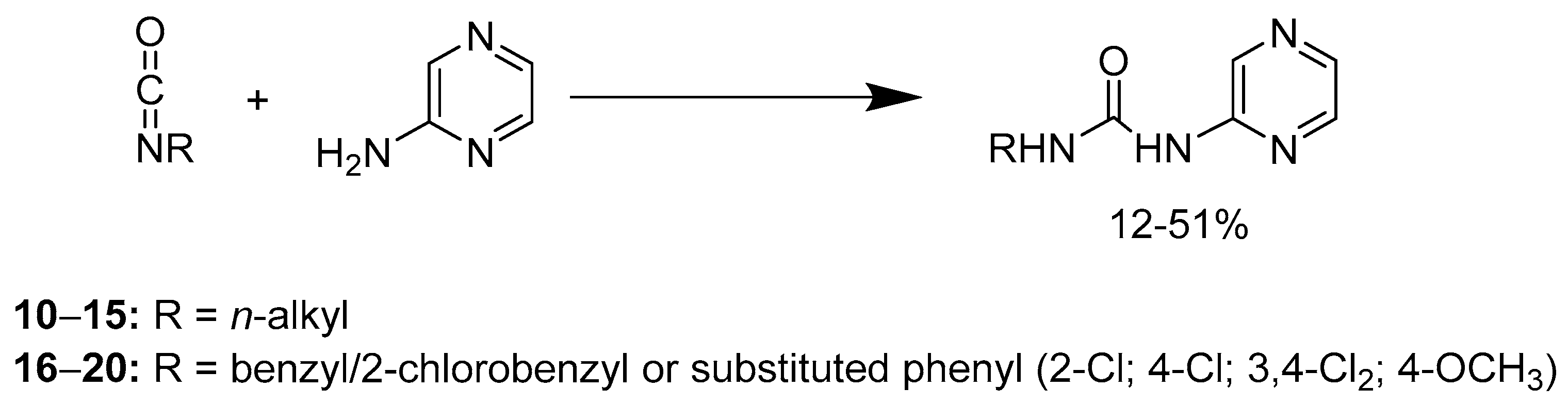
2.1.2. Compounds 10–20
2.2. Biological Activity
2.2.1. Antimycobacterial Activity Evaluation against Mycobacterium tuberculosis, Mycobacterium kansasii, and Mycobacterium avium
2.2.2. Antimycobacterial Activity Evaluation against Mycobacterium smegmatis and Mycobacterium aurum
2.2.3. Antibacterial and Antifungal Activity Evaluation
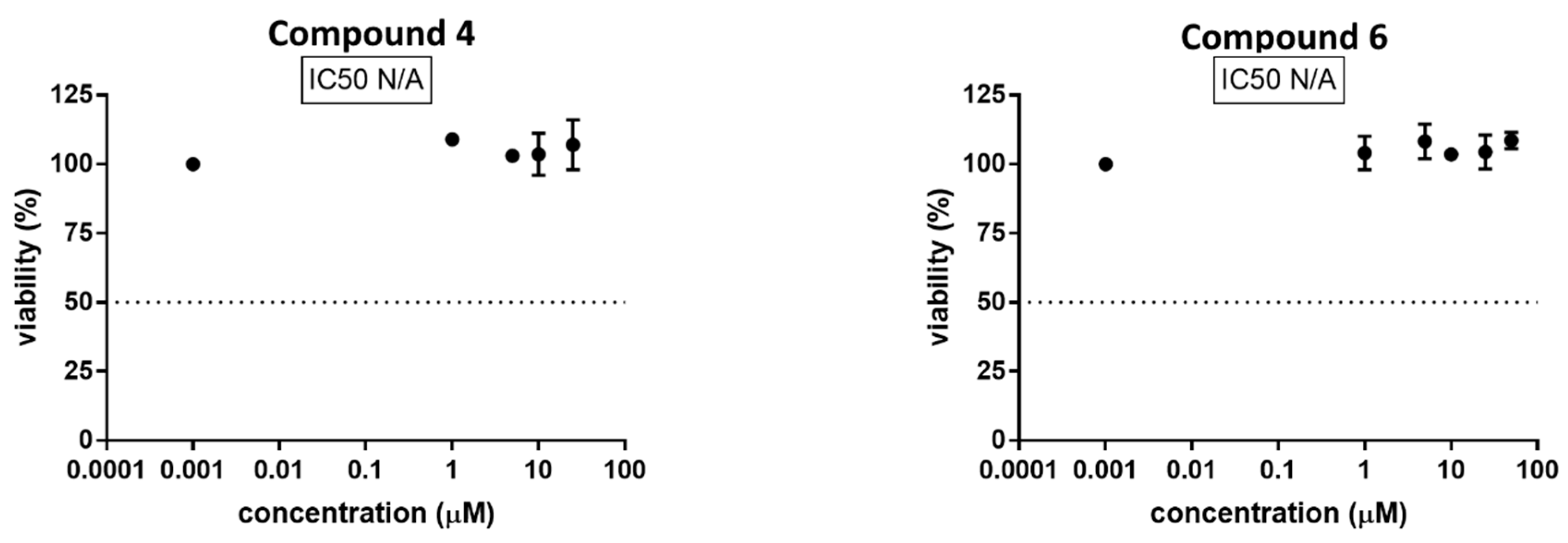
2.2.4. In Vitro Cytotoxicty Assays
2.2.5. Plant Growth Regulation Activity Evaluation
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.2.1. Compounds 1 and 2
3.2.2. Compounds 3–9
3.2.3. Compounds 10–20
3.3. Analytical Data of the Prepared Compounds
3.4. Biological Assays
3.4.1. In Vitro Activity Evaluation against Mycobacterium tuberculosis, Mycobacterium kansasii, and Mycobacterium avium
3.4.2. In Vitro Activity Evaluation against Mycobacterium smegmatis and Mycobacterium aurum
3.4.3. In Vitro Antibacterial Activity Evaluation
3.4.4. In Vitro Antifungal Activity Evaluation
3.4.5. Cytotoxicity Determination
3.4.6. Plant Growth Regulation Activity Evaluation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sia, I.G.; Wieland, M.L. Current Concepts in the Management of Tuberculosis. Mayo Clin. Proc. 2011, 86, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Shenoi, S.; Heysell, S.; Moll, A.; Friedland, G. Multidrug-resistant and extensively drug-resistant tuberculosis: Consequences for the global HIV community. Curr. Opin. Infect. Dis. 2009, 22, 11–17. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Tuberculosis Report 2016. WHO/HTM/TB/2016.13. Available online: http://www.who.int/tb/publications/global_report/en/ (accessed on 2 February 2017).
- Review on Antimicrobial Resistance. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. Available online: http://www.his.org.uk/files/4514/1829/6668/AMR_Review_Paper_-_Tackling_a_crisis_for_the_health_and_wealth_of_nations_1.pdf (accessed on 27 November 2016).
- Chaluvaraju, K.C.; Bhat, K.I. Synthesis and antimicrobial activities of amino benzylated mannich bases of pyrazinamide. Int. J. ChemTech Res. 2010, 2, 1368–1371. [Google Scholar]
- Jandourek, O.; Tauchman, M.; Paterova, P.; Konecna, K.; Navratilova, L.; Kubicek, V.; Holas, O.; Zitko, J.; Dolezal, M. Synthesis of Novel Pyrazinamide Derivatives Based on 3-Chloropyrazine-2-carboxamide and Their Antimicrobial Evaluation. Molecules 2017, 22, 223. [Google Scholar] [CrossRef] [PubMed]
- Kucerova-Chlupacova, M.; Vyskovska-Tyllova, V.; Richterova-Finkova, L.; Kunes, J.; Buchta, V.; Vejsova, M.; Paterova, P.; Semelkova, L.; Jandourek, O.; Opletalova, V. Novel Halogenated Pyrazine-Based Chalcones as Potential Antimicrobial Drugs. Molecules 2016, 21, 1421. [Google Scholar] [CrossRef] [PubMed]
- Semelkova, L.; Janoscova, P.; Fernandes, C.; Bouz, G.; Jandourek, O.; Konecna, K.; Paterova, P.; Navratilova, L.; Kunes, J.; Dolezal, M.; Zitko, J. Design, Synthesis, Antimycobacterial Evaluation, and In Silico Studies of 3-(Phenylcarbamoyl)-pyrazine-2-carboxylic Acids. Molecules 2017, 22, 1491. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Yang, S.; Huang, G. Design, synthesis and biological activity of pyrazinamide derivatives for anti-Mycobacterium tuberculosis. J. Enzym. Inhibit. Med. Chem. 2017, 32, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Zitko, J.; Barbora, S.-V.; Paterová, P.; Navrátilová, L.; Trejtnar, F.; Kuneš, J.; Doležal, M. Design, synthesis and anti-mycobacterial evaluation of some new iV-phenylpyrazine-2-carboxamides. Chem. Pap. 2016, 70, 649. [Google Scholar]
- Conde, M.B.; Lapa, E.S.J.R. New regimens for reducing the duration of the treatment of drug-susceptible pulmonary tuberculosis. Drug Dev. Res. 2011, 72, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Zhang, X.; Jiang, X.; Yuan, H.; Lee, J.S.; Barry, C.E., 3rd; Wang, H.; Zhang, W.; Zhang, Y. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 2011, 333, 1630–1632. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Shibayama, K.; Rimbara, E.; Mori, S. Biochemical Characterization of Quinolinic Acid Phosphoribosyltransferase from Mycobacterium tuberculosis H37Rv and Inhibition of Its Activity by Pyrazinamide. PLoS ONE 2014, 9, e100062. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.L.; Chen, J.Z.; Feng, J.; Cui, P.; Zhang, S.; Weng, X.H.; Zhang, W.H.; Zhang, Y. Aspartate decarboxylase (PanD) as a new target of pyrazinamide in Mycobacterium tuberculosis. Emerg. Microbes Infect. 2014, 3, e58. [Google Scholar] [CrossRef] [PubMed]
- Boehringer, D.; Ban, N.; Leibundgut, M. 7.5-Å Cryo-EM Structure of the Mycobacterial Fatty Acid Synthase. J. Mol. Biol. 2013, 425, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, L.; Connell, S.R.; Enderle, M.; Mills, D.J.; Vonck, J.; Grininger, M. Structure and Conformational Variability of the Mycobacterium tuberculosis Fatty Acid Synthase Multienzyme Complex. Structure 2013, 21, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, T.; Nakagawa, H.; Hori, R.; Watanabe, Y.; Soga, S.; Iida, K.; Onodera, H. The synthesis and evaluation of the antiproliferative activity of deacidified GEX1A analogues. J. Antibiot. 2017, 70, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Hackbarth, C.J.; Chen, D.Z.; Lewis, J.G.; Clark, K.; Mangold, J.B.; Cramer, J.A.; Margolis, P.S.; Wang, W.; Koehn, J.; Wu, C.; et al. N-Alkyl Urea Hydroxamic Acids as a New Class of Peptide Deformylase Inhibitors with Antibacterial Activity. Antimicrob. Agents Chemother. 2002, 46, 2752–2764. [Google Scholar] [CrossRef] [PubMed]
- Boyle, R.G.; Imogai, H.J.; Cherry, M. Preparation of Diarylureas as Chk-1 Kinase Inhibitors for the Treatment of Cancer. Patent WO200,310,144,4A1, 11 August 2005. [Google Scholar]
- Sorenson, W.R. Reaction of an Isocyanate and a Carboxylic Acid in Dimethyl Sulfoxide. J. Org. Chem. 1959, 24, 978–980. [Google Scholar] [CrossRef]
- Franzblau, S.G.; Witzig, R.S.; McLaughlin, J.C.; Torres, P.; Madico, G.; Hernandez, A.; Degnan, M.T.; Cook, M.B.; Quenzer, V.K.; Ferguson, R.M.; et al. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J. Clin. Microbiol. 1998, 36, 362–366. [Google Scholar] [PubMed]
- Zitko, J.; Servusová, B.; Paterová, P.; Mandíková, J.; Kubíček, V.; Kučera, R.; Hrabcová, V.; Kuneš, J.; Soukup, O.; Doležal, M. Synthesis, Antimycobacterial Activity and In Vitro Cytotoxicity of 5-Chloro-N-phenylpyrazine-2-carboxamides. Molecules 2013, 18, 14807. [Google Scholar] [CrossRef] [PubMed]
- Semelkova, L.; Konecna, K.; Paterova, P.; Kubicek, V.; Kunes, J.; Novakova, L.; Marek, J.; Naesens, L.; Pesko, M.; Kralova, K.; et al. Synthesis and Biological Evaluation of N-Alkyl-3-(alkylamino)-pyrazine-2-carboxamides. Molecules 2015, 20, 8687–8711. [Google Scholar] [CrossRef] [PubMed]
- Nguta, J.M.; Appiah-Opong, R.; Nyarko, A.K.; Yeboah-Manu, D.; Addo, P.G. Current perspectives in drug discovery against tuberculosis from natural products. Int. J. Mycobacteriol. 2015, 4, 165–183. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, C.H.; Tsai, T.F.; Hsueh, P.R. Characteristics of skin and soft tissue infection caused by non-tuberculous mycobacteria in Taiwan. Int. J. Tuberc. Lung Dis. 2011, 15, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Katoch, V.M. Infections due to non-tuberculous mycobacteria (NTM). Indian J. Med. Res. 2004, 120, 290–304. [Google Scholar] [PubMed]
- Shimizu, F.; Hatano, Y.; Okamoto, O.; Katagiri, K.; Fujiwara, S.; Sato, S.; Kato, A.; Uezato, H.; Asato, Y.; Takahashi, K. Mycobacterium smegmatis soft tissue infection. Int. J. Dermatol. 2012, 51, 1518–1520. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Bhakta, S. An integrated surrogate model for screening of drugs against Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2012, 67, 1380–1391. [Google Scholar] [CrossRef] [PubMed]
- Espinel-Ingroff, A.; Fothergill, A.; Ghannoum, M.; Manavathu, E.; Ostrosky-Zeichner, L.; Pfaller, M.; Rinaldi, M.; Schell, W.; Walsh, T. Quality control and reference guidelines for CLSI broth microdilution susceptibility method (M 38-A document) for amphotericin B, itraconazole, posaconazole, and voriconazole. J. Clin. Microbiol. 2005, 43, 5243–5246. [Google Scholar] [CrossRef] [PubMed]
- Yew, W.W.; Leung, C.C. Antituberculosis drugs and hepatotoxicity. Respirology 2006, 11, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Tostmann, A.; Boeree, M.J.; Peters, W.H.M.; Roelofs, H.M.J.; Aarnoutse, R.E.; van der Ven, A.J.A.M.; Dekhuijzen, P.N.R. Isoniazid and its toxic metabolite hydrazine induce in vitro pyrazinamide toxicity. Int. J. Antimicrob. Agents 2008, 31, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Guo, K.; Bai, Y.; Dong, J.; Gao, Z.; Yuan, Y.; Wang, Y.; Liu, L.; Yue, T. Identification, Synthesis, and Safety Assessment of Forchlorfenuron (1-(2-Chloro-4-pyridyl)-3-phenylurea) and Its Metabolites in Kiwifruits. J. Agric. Food. Chem. 2015, 63, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Dolezal, M.; Tumova, L.; Kesetovicova, D.; Tuma, J.; Kral‘ova, K. Substituted N-phenylpyrazine-2-carboxamides, their synthesis and evaluation as herbicides and abiotic elicitors. Molecules 2007, 12, 2589–2598. [Google Scholar] [CrossRef] [PubMed]
- Tumova, L.; Tuma, J.; Dolezal, M. Pyrazinecarboxamides as Potential Elicitors of Flavonolignan and Flavonoid Production in Silybum marianum and Ononis arvensis Cultures In Vitro. Molecules 2011, 16, 9142–9152. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.N.; Barry, A.L. Optimal dilution susceptibility testing conditions, recommendations for MIC interpretation, and quality control guidelines for the ampicillin-sulbactam combination. J. Clin. Microbiol. 1987, 25, 1920–1925. [Google Scholar] [PubMed]
- Murashige, T.; Skoog, F. A Revised Medium for Rapid Growth and Bio Assays with Tobacco Tissue Cultures. Physiol. Plant. 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Kreft, I.; Fabjan, N.; Yasumoto, K. Rutin content in buckwheat (Fagopyrum esculentum Moench) food materials and products. Food Chem. 2006, 98, 508–512. [Google Scholar] [CrossRef]
Sample Availability: Samples of the title compounds are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | B | C & D | ||||
|---|---|---|---|---|---|---|
 |  |  | ||||
| No. | R | log P | Antimycobacterial Activity MIC (μg/mL) | |||
| Mtb | M. kansasii | M. avium | ||||
| A | 1 | H | −0.75 | >100 | >100 | >100 |
| 2 | propyl | 0.34 | >100 | >100 | >100 | |
| B | 3 | benzyl | 1.69 | >100 | >100 | >100 |
| 4 | phenyl | 1.62 | 1.56 | >100 | >100 | |
| 5 a | phenyl | 0.54 | >100 | >100 | >100 | |
| 6 | 4-methoxyphenyl | 1.50 | 6.25 | 25 | >100 | |
| 7 | 2-chlorophenyl | 2.18 | >100 | >100 | >100 | |
| 8 | 4-chlorophenyl | 2.18 | 25 | >100 | >100 | |
| 9 | 3,4-dichlorophenyl | 2.74 | >100 | >100 | >100 | |
| C | 10 | propyl | −0.28 | >100 | >100 | >100 |
| 11 | butyl | 0.13 | >100 | >100 | >100 | |
| 12 | pentyl | 0.55 | 100 | >100 | >100 | |
| 13 | octyl | 1.8 | 25 | 25 | >100 | |
| 14 | decyl | 2.64 | >100 | >100 | >100 | |
| D | 15 | benzyl | 0.62 | >100 | >100 | >100 |
| 16 | 4-methoxyphenyl | 0.43 | >100 | >100 | >100 | |
| 17 | 2-chlorophenyl | 1.11 | >100 | >100 | >100 | |
| 18 | 4-chlorophenyl | 1.11 | 12.5 | >100 | >100 | |
| 19 | 3,4-dichlorophenyl | 1.67 | >100 | >100 | >100 | |
| 20 | 2-chlorobenzyl | 0.14 | >100 | >100 | >100 | |
| PZA | −1.31 | >100 b | >100 | >100 | ||
| INH | −0.64 | 0.2–0.4 | 6.25–12.5 | 6.25–12.5 | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouz, G.; Juhás, M.; Niklová, P.; Janďourek, O.; Paterová, P.; Janoušek, J.; Tůmová, L.; Kovalíková, Z.; Kastner, P.; Doležal, M.; et al. Ureidopyrazine Derivatives: Synthesis and Biological Evaluation as Anti-Infectives and Abiotic Elicitors. Molecules 2017, 22, 1797. https://doi.org/10.3390/molecules22101797
Bouz G, Juhás M, Niklová P, Janďourek O, Paterová P, Janoušek J, Tůmová L, Kovalíková Z, Kastner P, Doležal M, et al. Ureidopyrazine Derivatives: Synthesis and Biological Evaluation as Anti-Infectives and Abiotic Elicitors. Molecules. 2017; 22(10):1797. https://doi.org/10.3390/molecules22101797
Chicago/Turabian StyleBouz, Ghada, Martin Juhás, Pavlína Niklová, Ondřej Janďourek, Pavla Paterová, Jiří Janoušek, Lenka Tůmová, Zuzana Kovalíková, Petr Kastner, Martin Doležal, and et al. 2017. "Ureidopyrazine Derivatives: Synthesis and Biological Evaluation as Anti-Infectives and Abiotic Elicitors" Molecules 22, no. 10: 1797. https://doi.org/10.3390/molecules22101797