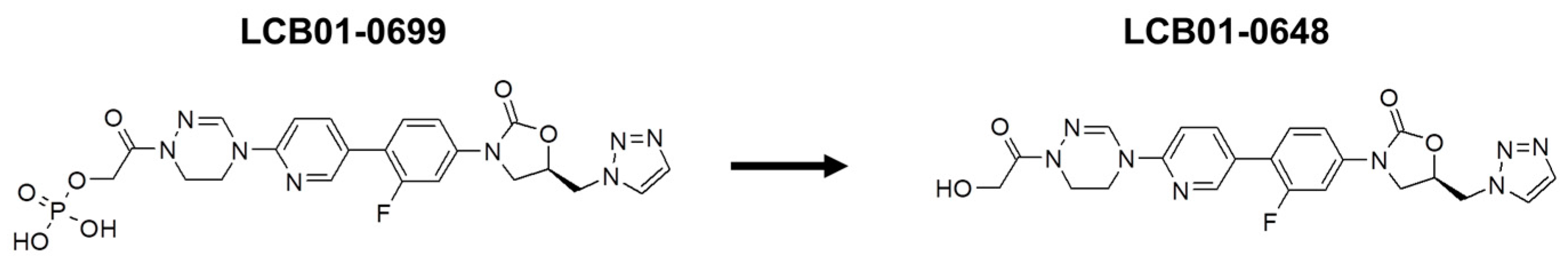
In Vivo Activity of LCB 01-0699, a Prodrug of LCB 01-0648, against Staphylococcus aureus

 and
and
Abstract
:1. Introduction
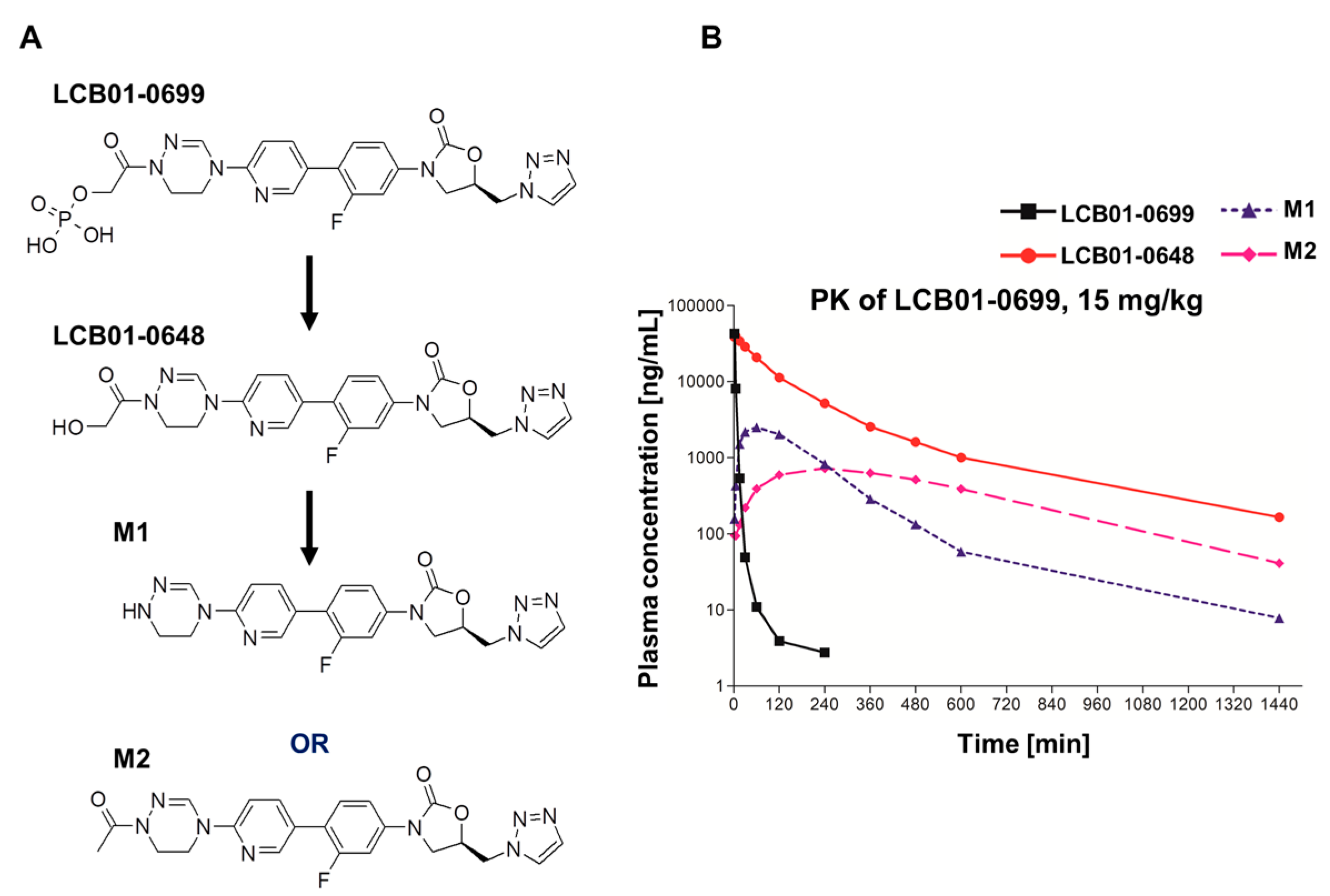
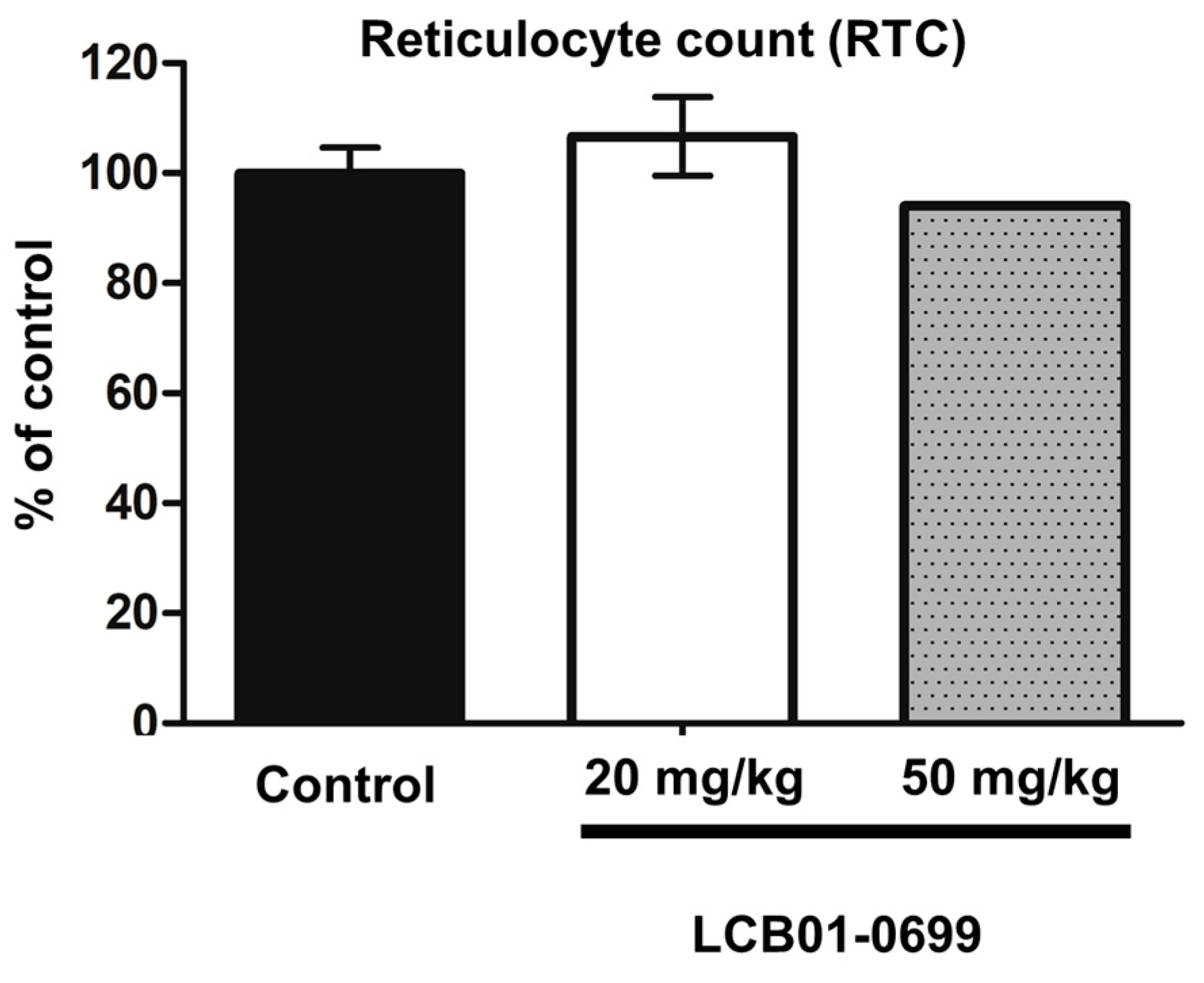
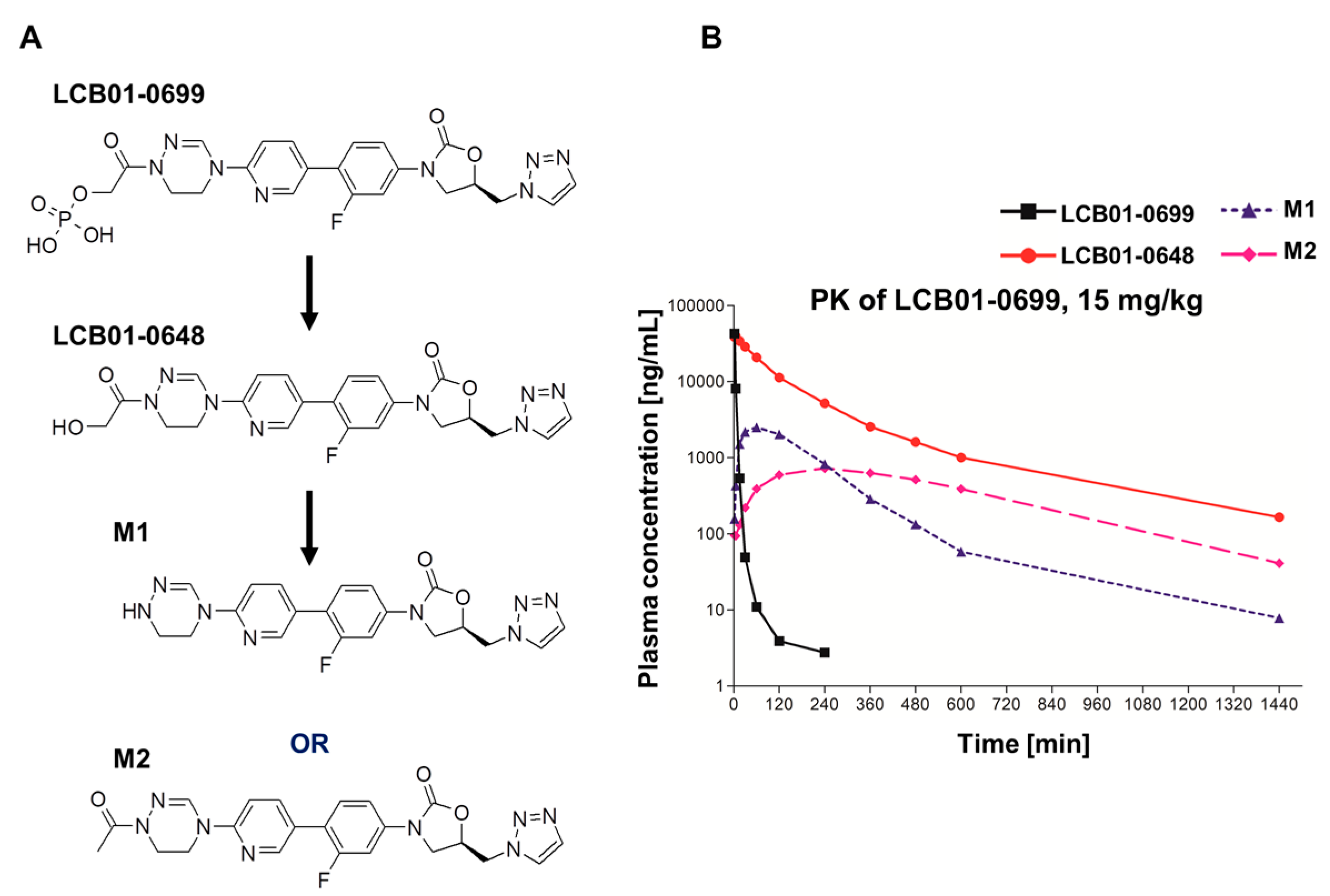
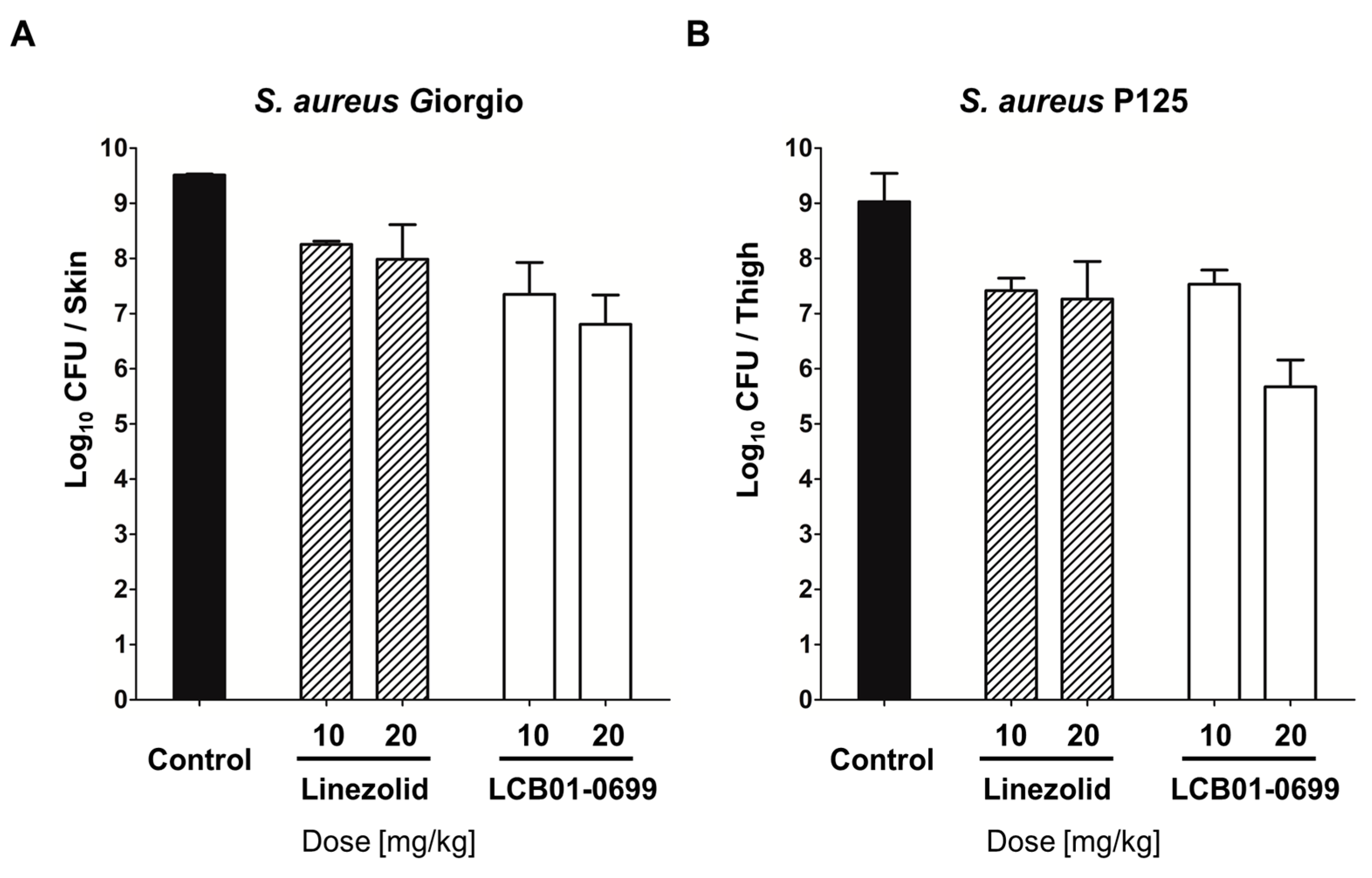
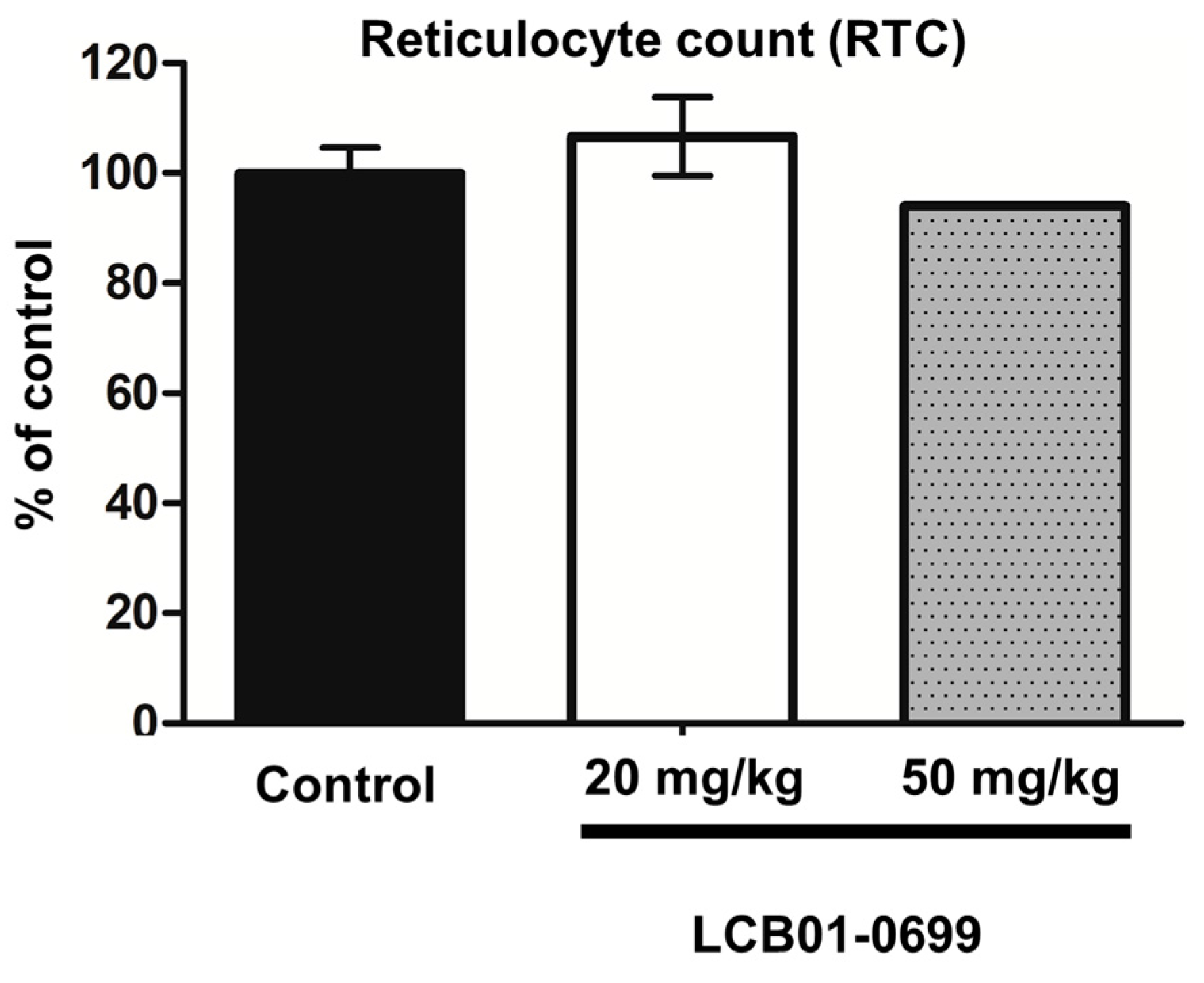
2. Results
3. Discussion
4. Materials and Methods
4.1. Antimicrobial Agents and Bacterial Strains
4.2. Systemic Infection Model
4.3. Soft Tissue Infection Model
4.4. Mice and Ethics Statement
4.5. Pharmacokinetic Analysis
4.6. Myelosuppression Assay
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Levy, S.B.; Marshall, B. Antibacterial resistance worldwide: Causes, challenges and responses. Nat. Med. 2004, 10, S122–S129. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Cynamon, M.H.; Klemens, S.P.; Sharpe, C.A.; Chase, S. Activities of several novel oxazolidinones against mycobacterium tuberculosis in a murine model. Antimicrob. Agents Chemother. 1999, 43, 1189–1191. [Google Scholar] [PubMed]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Draenert, R.; Seybold, U.; Grutzner, E.; Bogner, J.R. Novel antibiotics: Are we still in the pre-post-antibiotic era? Infection 2015, 43, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Bozdogan, B.; Appelbaum, P.C. Oxazolidinones: Activity, mode of action, and mechanism of resistance. Int. J. Antimicrob. Agents 2004, 23, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Barbachyn, M.R.; Ford, C.W. Oxazolidinone structure-activity relationships leading to linezolid. Angew. Chem. Int. Ed. Engl. 2003, 42, 2010–2023. [Google Scholar] [CrossRef] [PubMed]
- Diekema, D.J.; Jones, R.N. Oxazolidinone antibiotics. Lancet 2001, 358, 1975–1982. [Google Scholar] [CrossRef]
- Pandit, N.; Singla, R.K.; Shrivastava, B. Current updates on oxazolidinone and its significance. Int. J. Med. Chem. 2012, 2012, 159285. [Google Scholar] [CrossRef] [PubMed]
- Moellering, R.C. Linezolid: The first oxazolidinone antimicrobial. Ann. Intern. Med. 2003, 138, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Kelesidis, T.; Tsiodras, S.; Hindler, J.; Humphries, R.M. The emerging problem of linezolid-resistant staphylococcus. J. Antimicrob. Chemother. 2013, 68, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Rab, S. Tedizolid phosphate (sivextro): A second-generation oxazolidinone to treat acute bacterial skin and skin structure infections. Pharm. Ther. 2014, 39, 555–579. [Google Scholar]
- Oh, S.H.; Kim, J.; Baek, S.Y.; Chae, S.E.; Park, H.S.; Cho, Y.L.; Kwak, J.H. In vitro activities of LCB 01–0648, a novel oxazolidinone, against gram-positive bacteria. Molecules 2017, 22, 394. [Google Scholar] [CrossRef] [PubMed]
- Green, S.L.; Maddox, J.C.; Huttenbach, E.D. Linezolid and reversible myelosuppression. JAMA 2001, 285, 1291. [Google Scholar] [CrossRef] [PubMed]
- Locke, J.B.; Hilgers, M.; Shaw, K.J. Mutations in ribosomal protein L3 are associated with oxazolidinone resistance in staphylococci of clinical origin. Antimicrob. Agents Chemother. 2009, 53, 5275–5278. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Kim, H.J.; Seol, M.J.; Choi, D.R.; Choi, E.C.; Kwak, J.H. In vitro and in vivo antibacterial activities of DW-224a, a new fluoronaphthyridone. Antimicrob. Agents Chemother. 2006, 50, 2261–2264. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Parameters | LCB01-0699 (15 mg/kg) |
|---|---|---|
| LCB01-0699 | Tmax (h) a | 0.03 ± 0.00 |
| Cmax (mg/L) b | 42.9 ± 4.21 | |
| AUClast (mg·h/L) c | 5.08 ± 0.68 | |
| LCB01-0648 | Tmax (h) | 0.07 ± 0.03 |
| Cmax (mg/L) | 41.1 ± 8.09 | |
| AUClast (mg·h/L) | 85.0 ± 46.4 | |
| M1 | Tmax (h) | 1.00 ± 0.00 |
| Cmax (mg/L) | 2.51 ± 0.98 | |
| AUClast (mg·h/L) | 9.13 ± 4.23 | |
| M2 | Tmax (h) | 4.00 ± 0.00 |
| Cmax (mg/L) | 0.72 ± 0.27 | |
| AUClast (mg·h/L) | 8.45 ± 2.42 | |
| Sum | Cmax (mg/L) | 87.2 |
| AUClast (mg·h/L) | 107.7 |
| Microorganism Inoculum a (CFU/Mouse) b | Antimicrobial Agent c | MIC d (mg/L) | ED50 (mg/kg) e (95% Confidence Limits) | |
|---|---|---|---|---|
| p.o. f | s.c. g | |||
| S. aureus Giorgio (methicillin-susceptible S. aureus) (5 × 107) | ||||
| LCB01-0699 | 0.5 | 6.20 (3.58~10.65) | 5.51 (3.02~16.91) | |
| Linezolid | 2 | 7.07 (4.07~12.29) | 6.26 (3.55~13.89)) | |
| S. aureus P125 (methicillin-resistant S. aureus) (5 × 108) | ||||
| LCB01-0699 | 0.5 | 2.23 (0.94~5.28) | 2.73 (0.90~8.25) | |
| Linezolid | 2 | 7.07 (4.07~12.29) | 5.51 (3.02~16.9) | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, S.-H.; Park, H.-S.; Lee, J.-H.; Baek, S.-Y.; Chae, S.-E.; Oh, K.; Cho, Y.L.; Kwak, J.-H. In Vivo Activity of LCB 01-0699, a Prodrug of LCB 01-0648, against Staphylococcus aureus. Molecules 2017, 22, 2096. https://doi.org/10.3390/molecules22122096
Oh S-H, Park H-S, Lee J-H, Baek S-Y, Chae S-E, Oh K, Cho YL, Kwak J-H. In Vivo Activity of LCB 01-0699, a Prodrug of LCB 01-0648, against Staphylococcus aureus. Molecules. 2017; 22(12):2096. https://doi.org/10.3390/molecules22122096
Chicago/Turabian StyleOh, Sang-Hun, Hee-Soo Park, Jun-Hyung Lee, Sung-Yun Baek, Sang-Eun Chae, Kyuman Oh, Young Lag Cho, and Jin-Hwan Kwak. 2017. "In Vivo Activity of LCB 01-0699, a Prodrug of LCB 01-0648, against Staphylococcus aureus" Molecules 22, no. 12: 2096. https://doi.org/10.3390/molecules22122096