γPNA FRET Pair Miniprobes for Quantitative Fluorescent In Situ Hybridization to Telomeric DNA in Cells and Tissue

Abstract
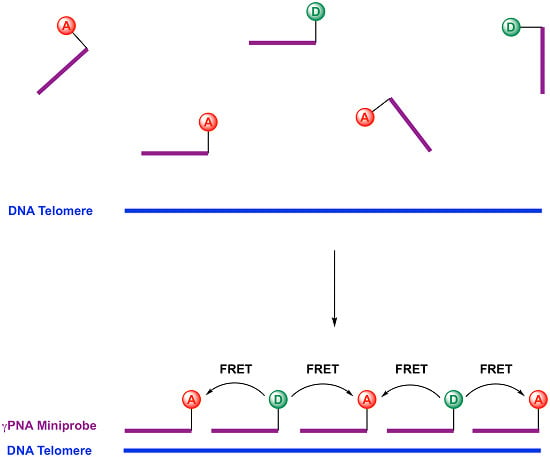
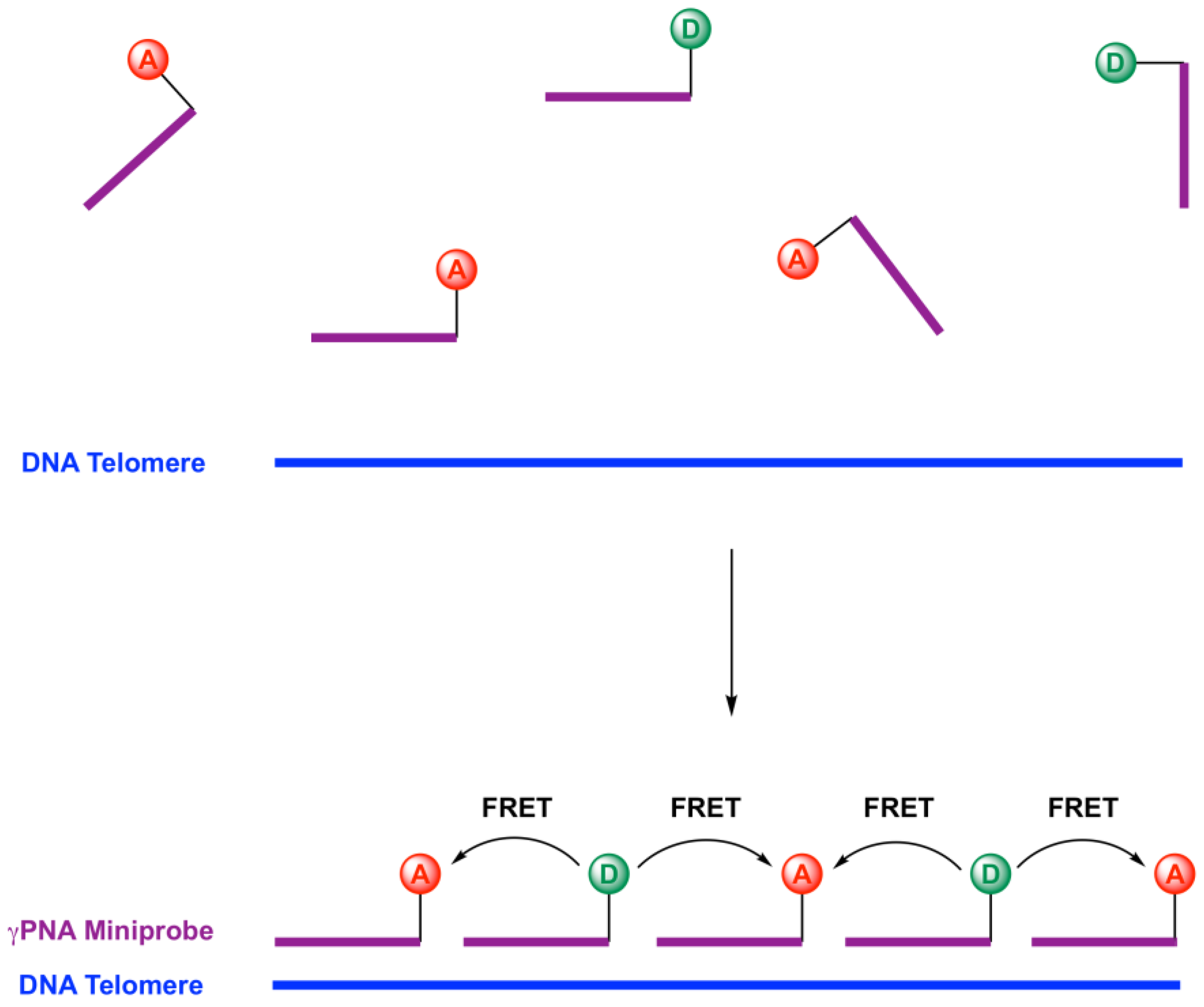
:
1. Introduction
2. Results
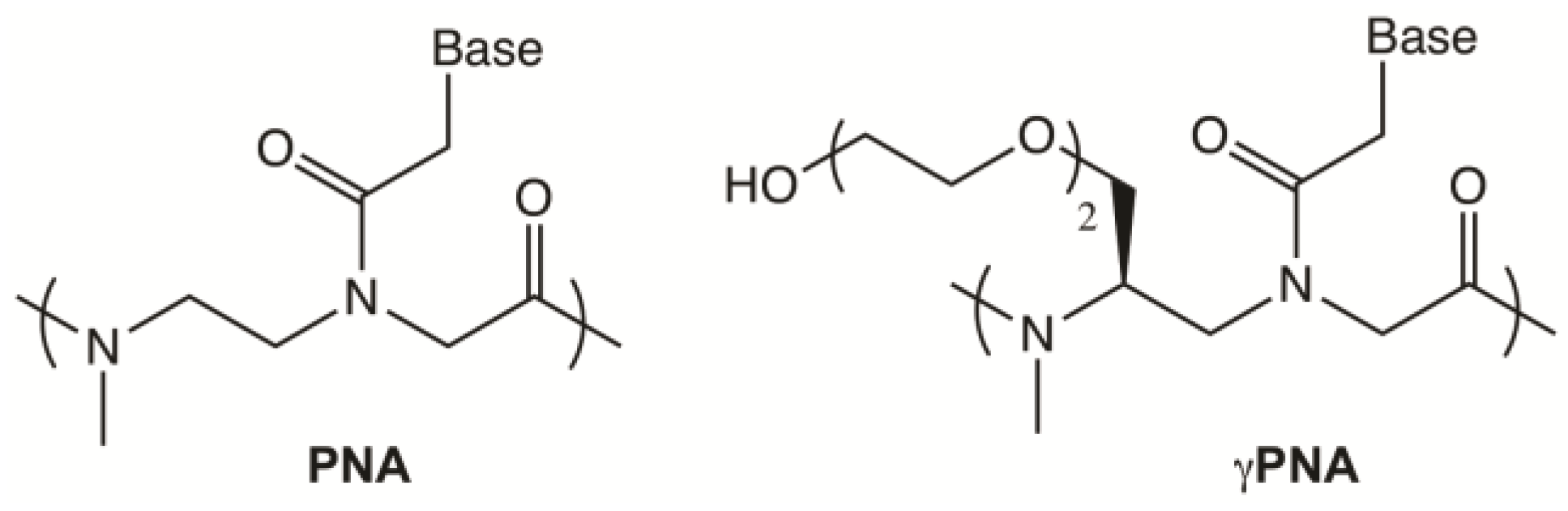
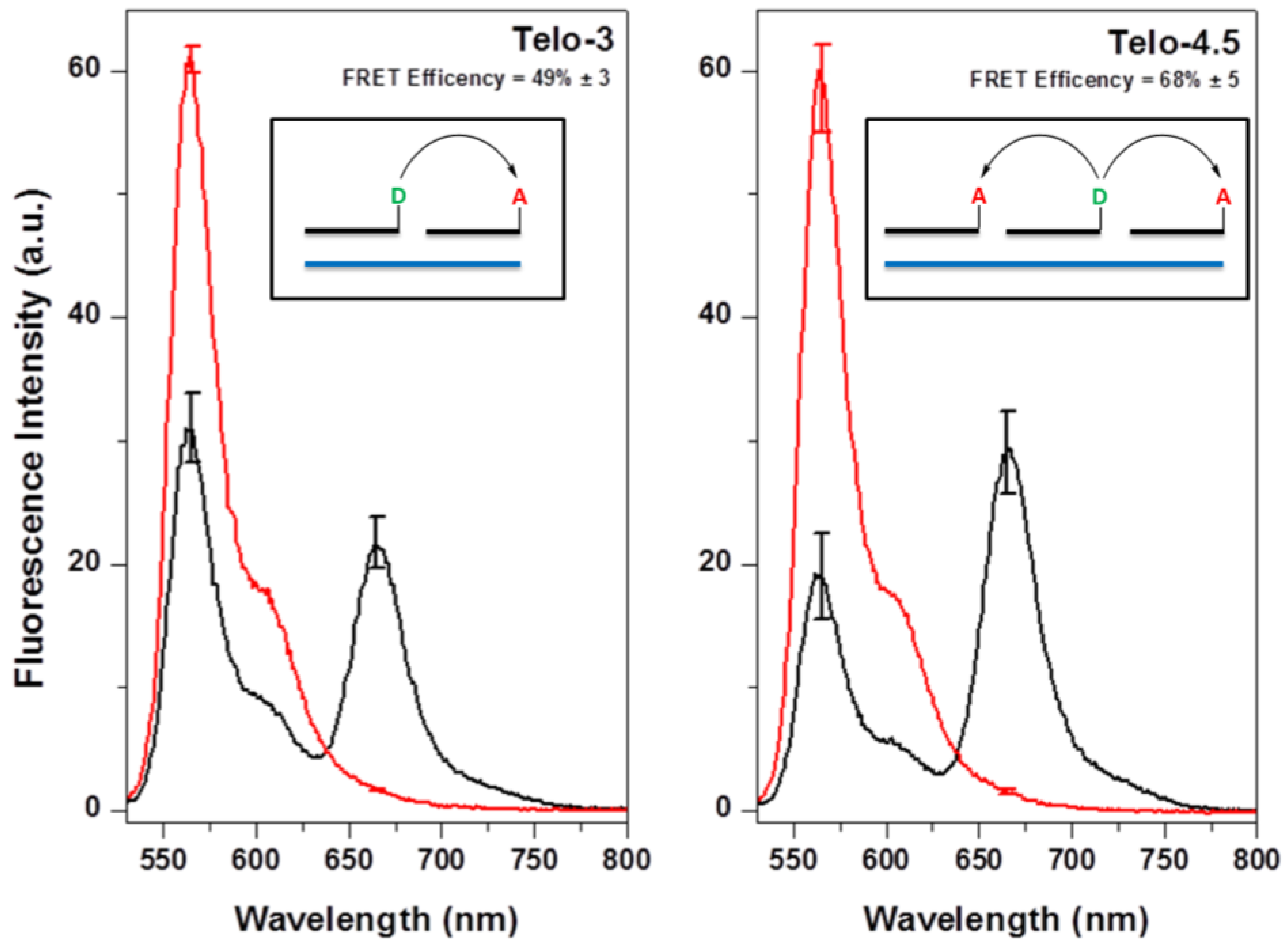
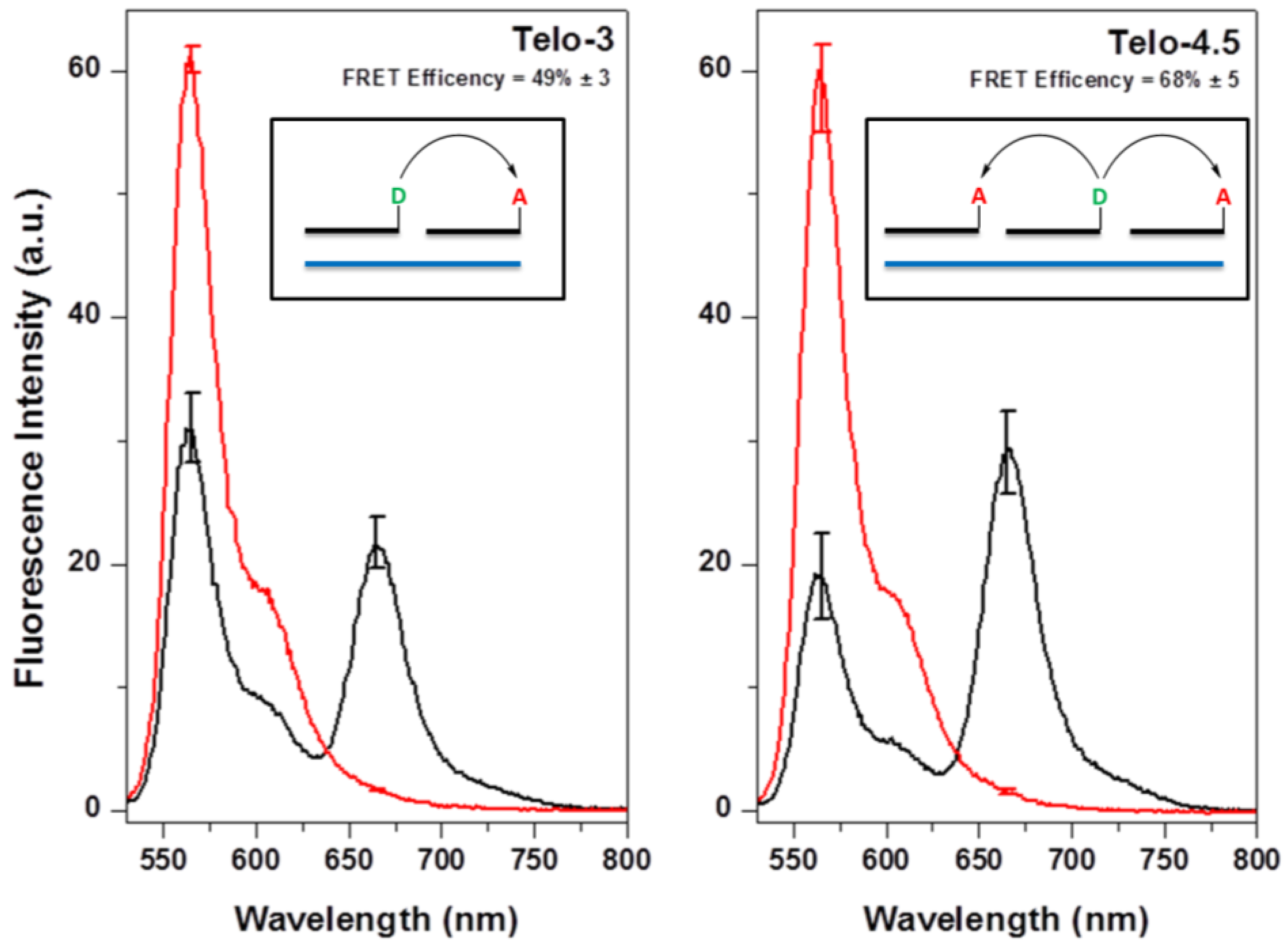
2.1. Design of Telomeric γPNA FRET Pair Miniprobes
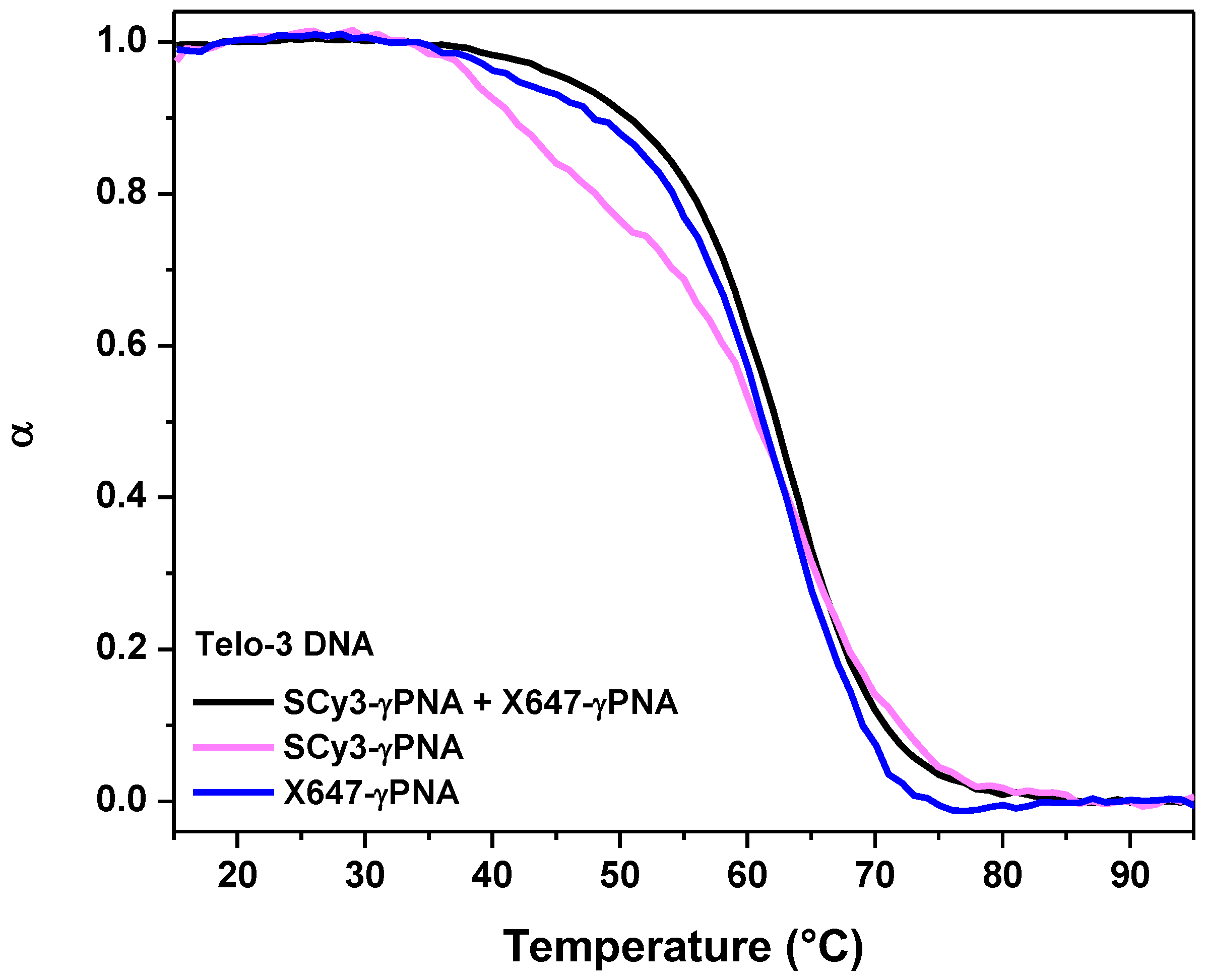
2.2. Hybridization of γPNA Miniprobes to DNA Oligonucleotides
2.3. Telomere Staining by FRET γPNA Miniprobes in Human Cells
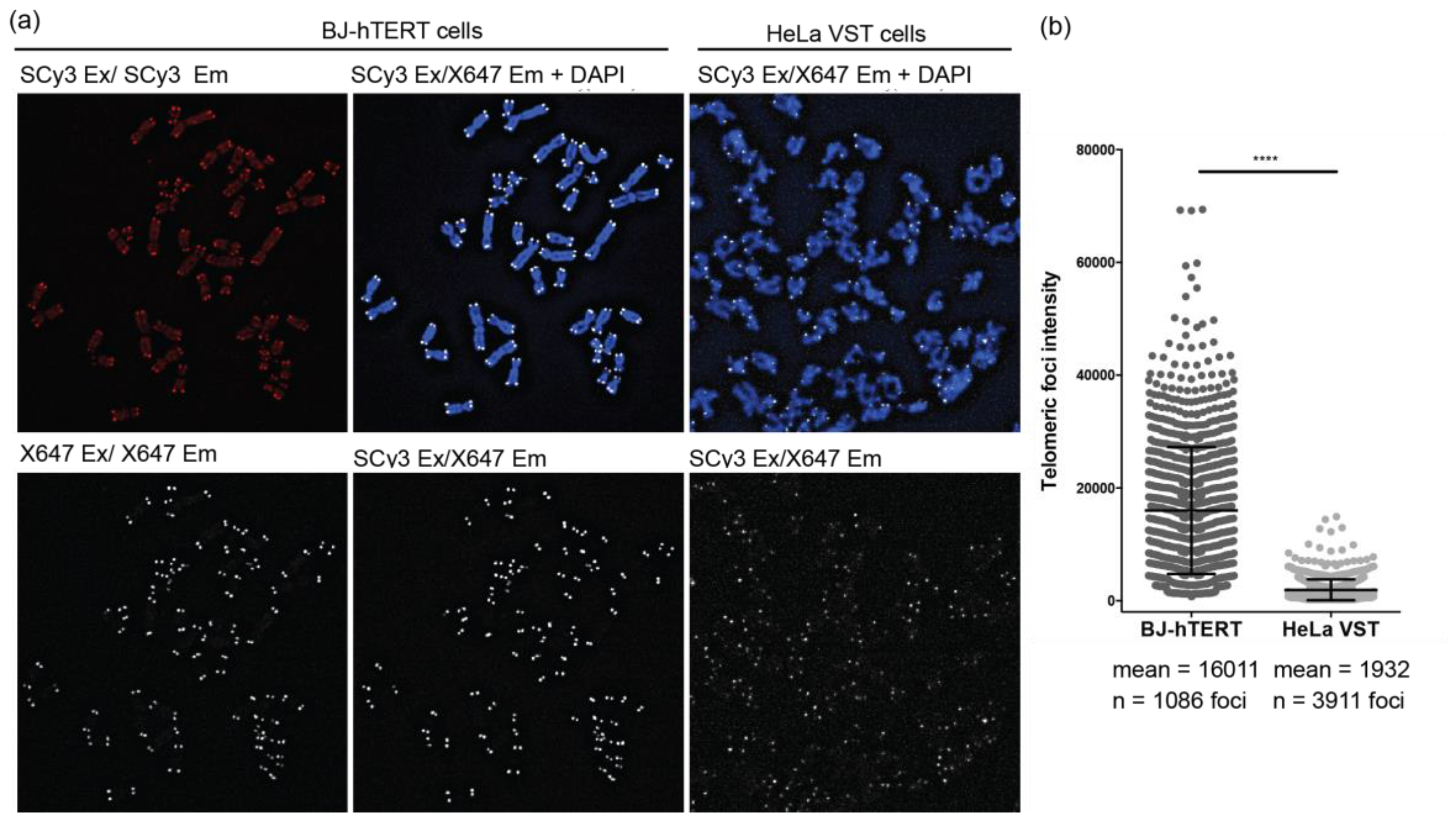
2.4. FRET Miniprobes Exhibit Reduced Background Staining on Chromosome Spreads
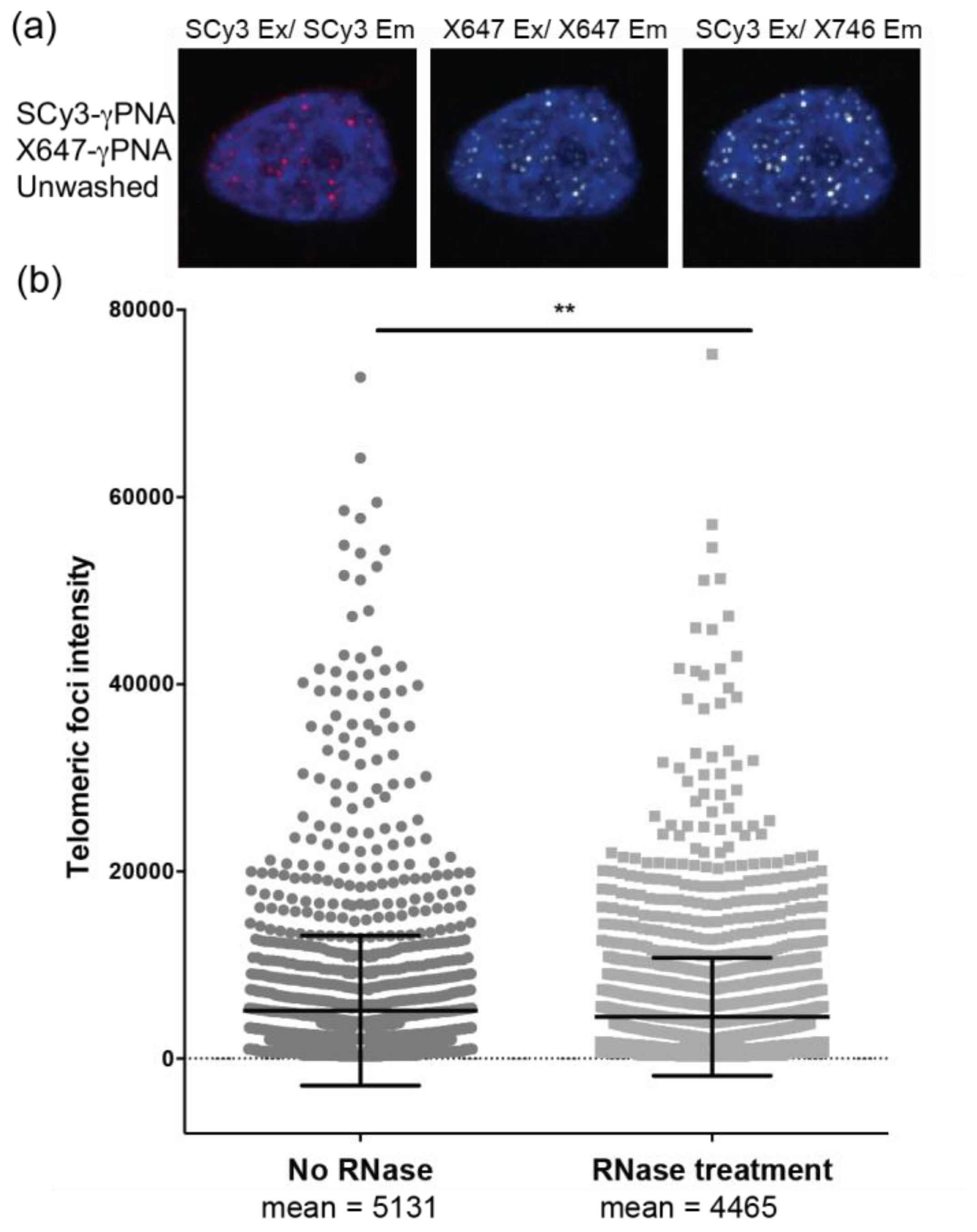
2.5. Elimination of Wash Steps Improves qFISH Detection of Telomeres in Tissue Sections
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Thermal Melting Analysis
4.3. Circular Dichroism (CD) Spectroscopy
4.4. Fluorescence Spectroscopy
4.5. Cell Culture
4.6. Fluorescent In Situ Hybridization of Interphase Cells
4.7. Fluorescent In Situ Hybridization of Metaphase Chromosomes
4.8. Tissue Fluorescent In Situ Hybridization
4.9. Imaging
4.10. γPNA Fluorescence Intensity Measurements
4.11. Statistical Analyses
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008, 42, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Calado, R.T.; Young, N.S. Telomere diseases. N. Engl. J. Med. 2009, 361, 2353–2365. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.L.; Cauley, J.A.; Boudreau, R.M.; Zmuda, J.M.; Strotmeyer, E.S.; Opresko, P.L.; Hsueh, W.C.; Cawthon, R.M.; Li, R.; Harris, T.B.; et al. Leukocyte telomere length is not associated with bmd, osteoporosis, or fracture in older adults: Results from the health, aging and body composition study. J. Bone Miner. Res. 2009, 24, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- Jaskelioff, M.; Muller, F.L.; Paik, J.H.; Thomas, E.; Jiang, S.; Adams, A.C.; Sahin, E.; Kost-Alimova, M.; Protopopov, A.; Cadinanos, J.; et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 2011, 469, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Zou, Y.; Hiyama, E.; Wright, W.E. Telomerase and cancer. Hum. Mol. Genet. 2001, 10, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Vera, E.; Blasco, M.A. Beyond average: Potential for measurement of short telomeres. Aging (Albany NY) 2012, 4, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Calado, R.; Young, N. Telomeres in disease. F1000 Med. Rep. 2012, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Stone, R.C.; Hunt, S.C.; Skurnick, J.; Lu, X.; Cao, X.; Harley, C.B.; Aviv, A. Measurement of telomere length by the southern blot analysis of terminal restriction fragment lengths. Nat. Protoc. 2010, 5, 1596–1607. [Google Scholar] [CrossRef] [PubMed]
- Cawthon, R.M. Telomere measurement by quantitative pcr. Nucleic Acids Res. 2002, 30, e47. [Google Scholar] [CrossRef] [PubMed]
- Lansdorp, P.M.; Verwoerd, N.P.; van de Rijke, F.M.; Dragowska, V.; Little, M.T.; Dirks, R.W.; Raap, A.K.; Tanke, H.J. Heterogeneity in telomere length of human chromosomes. Hum. Mol. Genet. 1996, 5, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Takai, H.; Smogorzewska, A.; de Lange, T. DNA damage foci at dysfunctional telomeres. Curr. Biol. 2003, 13, 1549–1556. [Google Scholar] [CrossRef]
- Kaul, Z.; Cesare, A.J.; Huschtscha, L.I.; Neumann, A.A.; Reddel, R.R. Five dysfunctional telomeres predict onset of senescence in human cells. EMBO Rep. 2012, 13, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.-J.; Barchowsky, A.; Opresko, P.L. The werner syndrome protein suppresses telomeric instability caused by chromium (vi) induced DNA replication stress. PLoS ONE 2010, 5, e11152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zijlmans, J.M.; Martens, U.M.; Poon, S.S.; Raap, A.K.; Tanke, H.J.; Ward, R.K.; Lansdorp, P.M. Telomeres in the mouse have large inter-chromosomal variations in the number of t2ag3 repeats. Proc. Natl. Acad. Sci. USA 1997, 94, 7423–7428. [Google Scholar] [CrossRef] [PubMed]
- Canela, A.; Vera, E.; Klatt, P.; Blasco, M.A. High-throughput telomere length quantification by fish and its application to human population studies. Proc. Natl. Acad. Sci. USA 2007, 104, 5300–5305. [Google Scholar] [CrossRef] [PubMed]
- Rufer, N.; Dragowska, W.; Thornbury, G.; Roosnek, E.; Lansdorp, P.M. Telomere length dynamics in human lymphocyte subpopulations measured by flow cytometry. Nat. Biotechnol. 1998, 16, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Flores, I.; Canela, A.; Vera, E.; Tejera, A.; Cotsarelis, G.; Blasco, M.A. The longest telomeres: A general signature of adult stem cell compartments. Genes Dev. 2008, 22, 654–667. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.N.; Finley, J.C.; Risques, R.A.; Shen, W.T.; Gollahon, K.A.; Rabinovitch, P.S. Quantitative fluorescence in situ hybridization (qfish) of telomere lengths in tissue and cells. Curr. Protoc. Cytom. 2005. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Meeker, A.K.; Makambi, K.H.; Kosti, O.; Kallakury, B.V.; Sidawy, M.K.; Loffredo, C.A.; Zheng, Y.L. Telomere length variation in normal epithelial cells adjacent to tumor: Potential biomarker for breast cancer local recurrence. Carcinogenesis 2012, 33, 113–118. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.N.; Finley, J.C.; Risques, R.A.; Shen, W.T.; Gollahon, K.A.; Moskovitz, A.H.; Gryaznov, S.; Harley, C.B.; Rabinovitch, P.S. Telomere length assessment in tissue sections by quantitative fish: Image analysis algorithms. Cytom. A 2004, 58, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Schnell, S.A.; Staines, W.A.; Wessendorf, M.W. Reduction of lipofuscin-like autofluorescence in fluorescently labeled tissue. J. Hist. Cytochem. 1999, 47, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Buscone, S.; Argentieri, M.C.; Pilla, D.; Cattoretti, G. Whole-slide, quadruple immunofluorescence labeling of routinely processed paraffin sections. Appl. Immunohistochem. Mol. Morphol. 2014, 22, e1–e7. [Google Scholar] [CrossRef] [PubMed]
- Sahu, B.; Sacui, I.; Rapireddy, S.; Zanotti, K.J.; Bahal, R.; Armitage, B.A.; Ly, D.H. Synthesis and characterization of conformationally preorganized, (r)-diethylene glycol-containing gamma-peptide nucleic acids with superior hybridization properties and water solubility. J. Org. Chem. 2011, 76, 5614–5627. [Google Scholar] [CrossRef] [PubMed]
- Pham, H.H.; Murphy, C.T.; Sureshkumar, G.; Ly, D.H.; Opresko, P.L.; Armitage, B.A. Cooperative hybridization of gammapna miniprobes to a repeating sequence motif and application to telomere analysis. Org. Biomol. Chem. 2014, 12, 7345–7354. [Google Scholar] [CrossRef] [PubMed]
- Cardullo, R.A.; Parpura, V. Fluorescence resonance energy transfer microscopy: Theory and instrumentation. Methods Cell Biol. 2003, 72, 415–430. [Google Scholar] [PubMed]
- O’Sullivan, R.J.; Arnoult, N.; Lackner, D.H.; Oganesian, L.; Haggblom, C.; Corpet, A.; Almouzni, G.; Karlseder, J. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone asf1. Nat. Struct. Mol. Biol. 2014, 21, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Fouquerel, E.; Lormand, J.; Bose, A.; Lee, H.T.; Kim, G.S.; Li, J.; Sobol, R.W.; Freudenthal, B.D.; Myong, S.; Opresko, P.L. Oxidative guanine base damage regulates human telomerase activity. Nat. Struct. Mol. Biol. 2016, 23, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Pickett, H.A.; Reddel, R.R. Molecular mechanisms of activity and derepression of alternative lengthening of telomeres. Nat. Struct. Mol. Biol. 2015, 22, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Marras, S.A.; Kramer, F.R.; Tyagi, S. Efficiencies of fluorescence resonance energy transfer and contact-mediated quenching in oligonucleotide probes. Nucleic Acids Res. 2002, 30, e122. [Google Scholar] [CrossRef] [PubMed]
- Artandi, S.E.; DePinho, R.A. A critical role for telomeres in suppressing and facilitating carcinogenesis. Curr. Opin. Genet. Dev. 2000, 10, 39–46. [Google Scholar] [CrossRef]
- O’Sullivan, J.N.; Bronner, M.P.; Brentnall, T.A.; Finley, J.C.; Shen, W.T.; Emerson, S.; Emond, M.J.; Gollahon, K.A.; Moskovitz, A.H.; Crispin, D.A.; et al. Chromosomal instability in ulcerative colitis is related to telomere shortening. Nat. Genet. 2002, 32, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Bisoffi, M.; Heaphy, C.M.; Griffith, J.K. Telomeres: Prognostic markers for solid tumors. Int. J. Cancer 2006, 119, 2255–2260. [Google Scholar] [CrossRef] [PubMed]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. Brafe600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Meeker, A.K.; Hicks, J.L.; Iacobuzio-Donahue, C.A.; Montgomery, E.A.; Westra, W.H.; Chan, T.Y.; Ronnett, B.M.; De Marzo, A.M. Telomere length abnormalities occur early in the initiation of epithelial carcinogenesis. Clin. Cancer Res. 2004, 10, 3317–3326. [Google Scholar] [CrossRef] [PubMed]
- Marky, L.A.; Breslauer, K.J. Calculating thermodynamic data for transitions of any molecularity from equilibrium melting curves. Biopolymers 1987, 26, 1601–1620. [Google Scholar] [CrossRef] [PubMed]
- Pope-Varsalona, H.; Liu, F.J.; Guzik, L.; Opresko, P.L. Polymerase eta suppresses telomere defects induced by DNA damaging agents. Nucleic Acids Res. 2014, 42, 13096–13109. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, B.; Qiu, W.; Buchanan, M.E.; Zou, F.; Vernon, P.; Moyer, M.P.; Yin, X.M.; Schoen, R.E.; Yu, J.; Zhang, L. Bid mediates selective killing of apc-deficient cells in intestinal tumor suppression by nonsteroidal antiinflammatory drugs. Proc. Natl. Acad. Sci. USA 2014, 111, 16520–16525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the ©PNA FRET miniprobes are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (C-to-N for γPNA) |
|---|---|
| SCy3-γPNA | H2N–TCC CAA TCC–SCy3 |
| X647-γPNA | H2N–CAA TCC CAA–X647 |
| Telo-3 DNA | 5′–(AGG GTT)3–3′ |
| Telo-4.5 DNA | 5′–(GTT AGG)4 GTT–3′ |
| 9-mer A DNA | 5′–AGG GTT AGG–3′ |
| 9-mer B DNA | 5′–GTT AGG GTT–3′ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orenstein, A.; Berlyoung, A.S.; Rastede, E.E.; Pham, H.H.; Fouquerel, E.; Murphy, C.T.; Leibowitz, B.J.; Yu, J.; Srivastava, T.; Armitage, B.A.; et al. γPNA FRET Pair Miniprobes for Quantitative Fluorescent In Situ Hybridization to Telomeric DNA in Cells and Tissue. Molecules 2017, 22, 2117. https://doi.org/10.3390/molecules22122117
Orenstein A, Berlyoung AS, Rastede EE, Pham HH, Fouquerel E, Murphy CT, Leibowitz BJ, Yu J, Srivastava T, Armitage BA, et al. γPNA FRET Pair Miniprobes for Quantitative Fluorescent In Situ Hybridization to Telomeric DNA in Cells and Tissue. Molecules. 2017; 22(12):2117. https://doi.org/10.3390/molecules22122117
Chicago/Turabian StyleOrenstein, Alexander, April S. Berlyoung, Elizabeth E. Rastede, Ha H. Pham, Elise Fouquerel, Connor T. Murphy, Brian J. Leibowitz, Jian Yu, Tumul Srivastava, Bruce A. Armitage, and et al. 2017. "γPNA FRET Pair Miniprobes for Quantitative Fluorescent In Situ Hybridization to Telomeric DNA in Cells and Tissue" Molecules 22, no. 12: 2117. https://doi.org/10.3390/molecules22122117