Application of the Crystalline Sponge Method to Revise the Structure of the Phenalenone Fuliginone

 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
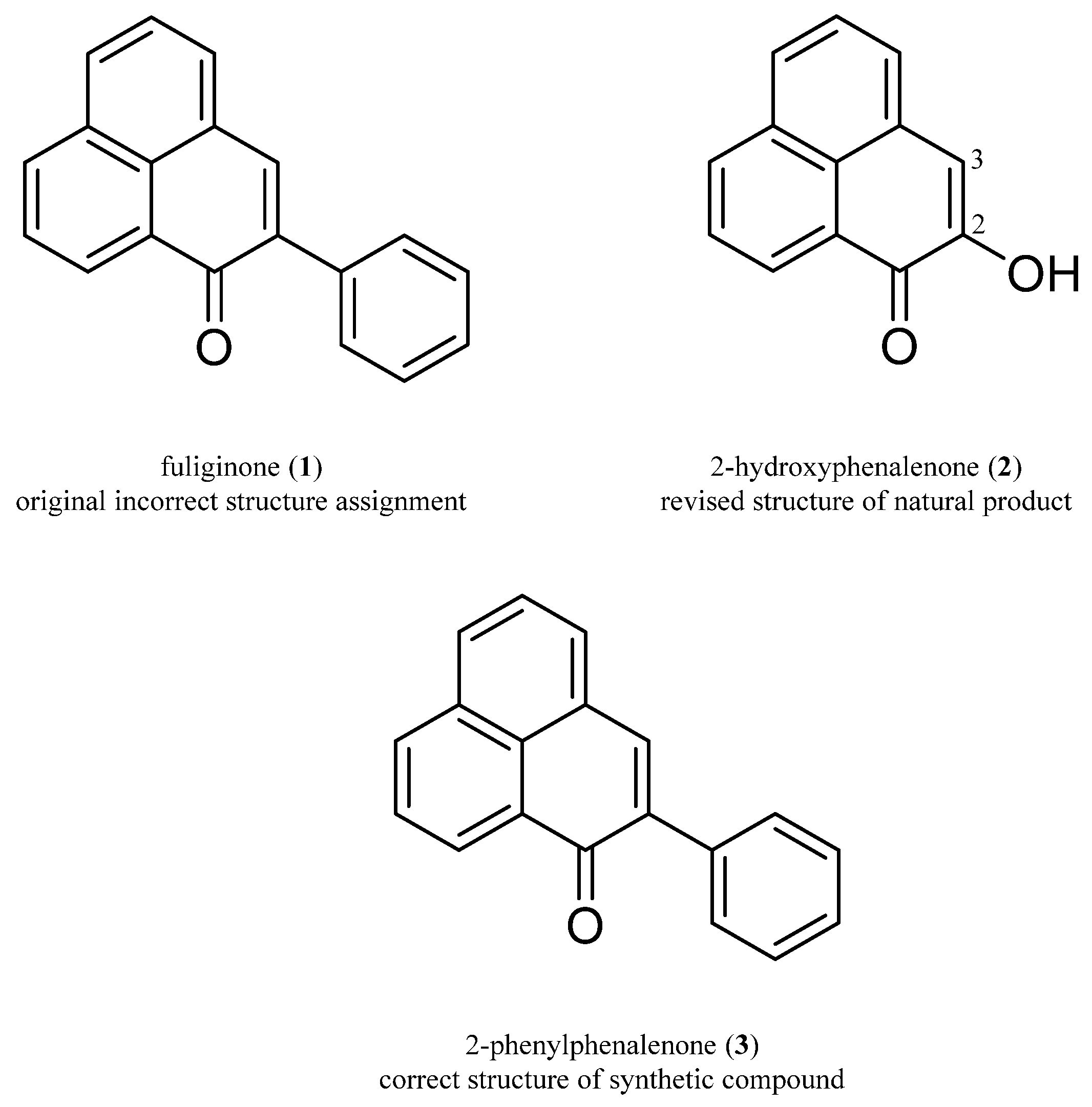
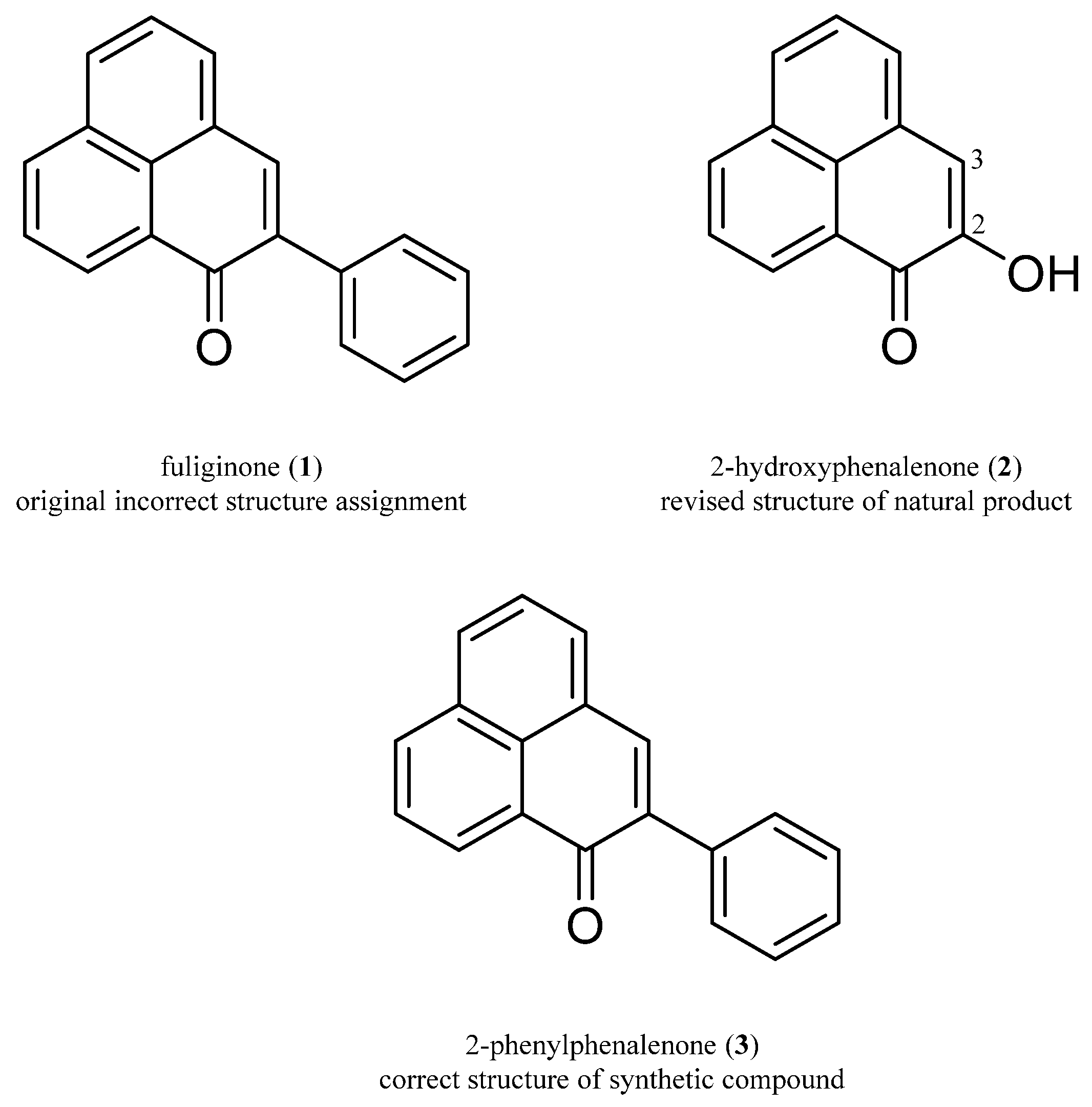
2.1. Comparison of NMR and MS Data of Isolated Natural Product and Synthetic Product
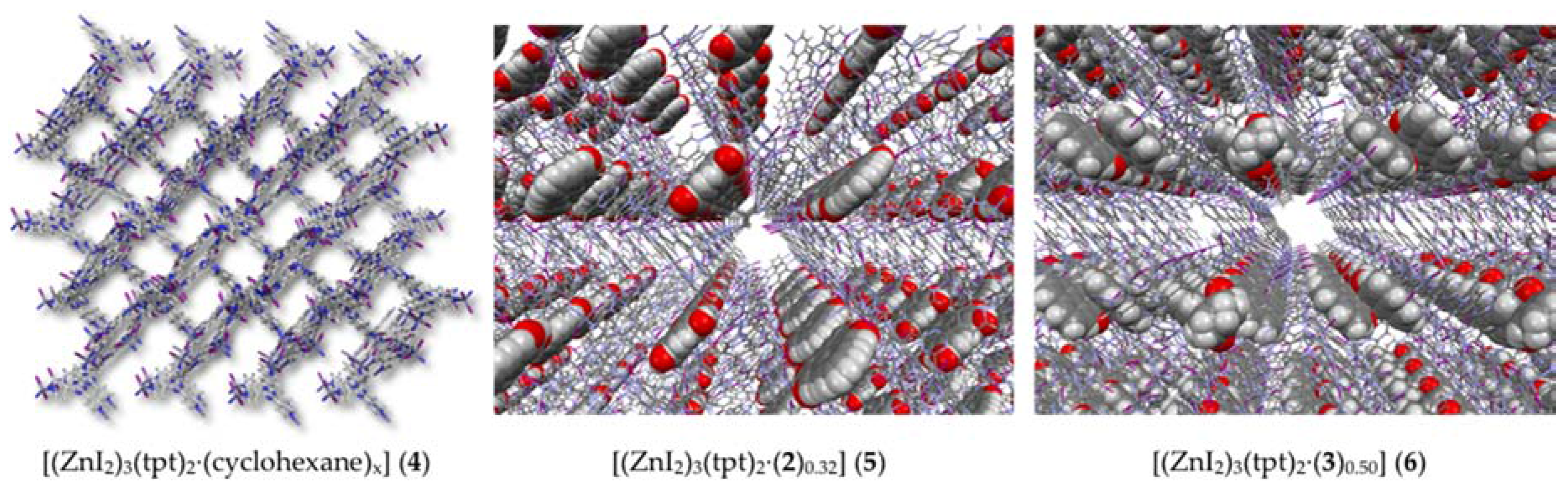
2.2. Crystalline Sponge Method
2.3. Chemical Profiling of Natural Products Present in the Bulbs of M. fuliginosa
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Natural Product Material
3.4. Re-purification of Natural Product Material
3.5. Crystalline Sponge Method
3.6. GC/MS Identification of 2-Hydroxyphenalen-1-one
3.7. Identification of 2-Hydroxyphenalen-1-one in the Bulbs of M. fuliginosa
3.8. Synthesis of 2-Phenylphenalen-1-one and 2-Hydroxyphenalen-1-one
3.9. On-Line (HPLC-NMR & HPLC-MS) Characterization of Compounds
3.10. Off-Line Characterization of Compounds
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brkljaca, R.; White, J.M.; Urban, S. Phytochemical investigation of the constituents derived from the Australian plant Macropidia fuliginosa. J. Nat. Prod. 2015, 78, 1600–1608. [Google Scholar] [CrossRef] [PubMed]
- Cooke, R.G.; Edwards, J.M. Naturally occurring phenalenones and related compounds. Prog. Chem. Org. Nat. Prod. 1980, 40, 153–190. [Google Scholar]
- Luis, J.G.; Echeverri, F.; Quinones, W.; Brito, I.; Lopez, M.; Torres, F.; Cardona, G.; Aguiar, Z.; Pelaez, C.; Rojas, M. Irenolone and emenolone: Two new types of phytoalexin from Musa paradisiaca. J. Org. Chem. 1993, 58, 4306–4308. [Google Scholar] [CrossRef]
- Hidalgo, W.; Chandran, J.N.; Menezes, R.C.; Otálvaro, F.; Schneider, B. Phenylphenalenones protect banana plants from infection by Mycosphaerella fijiensis and are deactivated by metabolic conversion. Plant Cell Environm. 2016, 39, 492–513. [Google Scholar] [CrossRef] [PubMed]
- Otálvaro, F.; Nanclares, J.; Vasquez, L.E.; Quinones, W.; Echeverri, F.; Arango, R.; Schneider, B. Phenalenone-type compounds from Musa acuminata var. "Yangambi km 5′′ (AAA) and their activity against Mycosphaerella fijiensis. J. Nat. Prod. 2007, 70, 887–890. [Google Scholar] [CrossRef] [PubMed]
- Hölscher, D.; Dhakshinamoorthy, S.; Alexandrov, T.; Becker, M.; Bretschneider, T.; Buerkert, A.; Crecelius, A.C.; de Waele, D.; Elsen, A.; Heckel, D.G.; et al. Phenalenone-type phytoalexins mediate resistance of banana plants (Musa spp.) to the burrowing nematode Radopholus similis. Proc. Natl. Acad. Sci. USA 2014, 111, 105–110. [Google Scholar]
- Rosquete, L.I.; Cabrera-Serra, M.G.; Pinero, J.E.; Martin-Rodriguez, P.; Fernandez-Perez, L.; Luis, J.G.; McNaughton-Smith, G.; Abad-Grillo, T. Synthesis and in vitro antiprotozoal evaluation of substituted phenalenone analogues. Bioorg. Med. Chem. 2010, 18, 4530–4534. [Google Scholar] [CrossRef] [PubMed]
- Brkljaca, R.; Urban, S. HPLC-NMR and HPLC-MS profiling and bioassay-guided identification of secondary metabolites from the Australian plant Haemodorum spicatum. J. Nat. Prod. 2015, 78, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Duque, L.; Zapata, C.; Rojano, B.; Schneider, B.; Otálvaro, F. Radical scavenging capacity of 2,4-dihydroxy-9-phenyl-1H-phenalen-1-one: A functional group exclusion approach. Org. Lett. 2013, 15, 3542–3545. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Paetz, C.; Menezes, R.C.; Schneider, B. Phenylbenzoisoquinolindione alkaloids accumulate in stamens of Xiphidium caeruleum aubl. Flowers. Phytochemistry 2016, 128, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, W.; Cano, M.; Arbelaez, M.; Zarrazola, E.; Gil, J.; Schneider, B.; Otálvaro, F. 4-Phenylphenalenones as a template for new photodynamic compounds against Mycosphaerella fijiensis. Pest Man. Sci. 2016, 72, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Ospina, F.; Hidalgo, W.; Cano, M.; Schneider, B.; Otálvaro, F. Synthesis of 8-phenylphenalenones: 2-hydroxy-8-(4-hydroxyphenyl)-1H-phenalen-1-one from Eichhornia crassipes. J. Org. Chem. 2016, 81, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Asscher, Y.; Agranat, I. Intramolecular friedel-crafts acylation of a lactone in polyphosphoric acid. Synthesis of 2-phenylphenalen-1-one. J. Org. Chem. 1980, 45, 3364–3366. [Google Scholar] [CrossRef]
- Hayes, L.M.; Knapp, C.E.; Nathoo, K.Y.; Press, N.J.; Tocher, D.A.; Carmalt, C.J. The crystalline sponge method: A systematic study of the reproducibility of simple aromatic molecule encapsulation and guest-host interactions. Cryst. Growth Des. 2016, 16, 3465–3472. [Google Scholar] [CrossRef]
- Hoshino, M.; Khutia, A.; Xing, H.; Inokuma, Y.; Fujita, M. The crystalline sponge method updated. IUCrJ 2016, 3, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Inokuma, Y.; Yoshioka, S.; Ariyoshi, J.; Arai, T.; Hitora, Y.; Takada, K.; Matsunaga, S.; Rissanen, K.; Fujita, M. X-ray analysis on the nanogram to microgram scale using porous complexes. Nature 2013, 495, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Inokuma, Y.; Yoshioka, S.; Ariyoshi, J.; Arai, T.; Hitora, Y.; Takada, K.; Matsunaga, S.; Rissanen, K.; Fujita, M. X-ray analysis on the nanogram to microgram scale using porous complexes. Nature 2013, 501, 262. [Google Scholar] [CrossRef]
- Lee, S.; Kapustin, E.A.; Yaghi, O.M. Coordinative alignment of molecules in chiral metal-organic frameworks. Science 2016, 353, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Ramadhar, T.R.; Zheng, S.-L.; Chen, Y.-S.; Clardy, J. Analysis of rapidly synthesized guest-filled porous complexes with synchrotron radiation: Practical guidelines for the crystalline sponge method. Acta Crystallogr. Sect. A 2015, 71, 46–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Hoshino, M.; Fujita, M.; Urban, S. Cycloelatanene A and B: Absolute configuration determination and structural revision by the crystalline sponge method. Chem. Sci. 2017. [Google Scholar] [CrossRef]
- Matsuda, Y.; Mitsuhashi, T.; Lee, S.; Hoshino, M.; Mori, T.; Okada, M.; Zhang, H.; Hayashi, F.; Fujita, M.; Abe, I. Astellifadiene: Structure determination by NMR spectroscopy and crystalline sponge method, and elucidation of its biosynthesis. Angew. Chem., Int. Ed. 2016, 55, 5785–5788. [Google Scholar] [CrossRef] [PubMed]
- Urban, S.; Brkljaca, R.; Hoshino, M.; Lee, S.; Fujita, M. Determination of the absolute configuration of the pseudo-symmetric natural product elatenyne by the crystalline sponge method. Angew. Chem. Int. Ed. 2016, 55, 2678–2682. [Google Scholar] [CrossRef] [PubMed]
- Biradha, K.; Fujita, M. A spring-like 3D-coordination network that shrinks or swells in crystal-to-crystal manner upon guest removal or readsorption. Angew. Chem., Int. Ed. 2002, 41, 3392–3395. [Google Scholar] [CrossRef]
- Smallcombe, S.H.; Patt, S.L.; Keifer, P.A. Wet solvent suppression and its applications to lC-NMR and high-resolution nmr spectroscopy. J. Magn. Reson. Ser. A 1995, 117, 295–303. [Google Scholar] [CrossRef]
- Otálvaro, F.; Ramirez, A.; Cano, M.; Upegui, J.; Hidalgo, W.; Schneider, B.; Robledo, S.; Otalvaro, F. Synthesis of positional isometric phenylphenalenones and their application in Plasmodium falciparum pharmacophore recognition. (manuscript in preparation).
- Sample Availability: Samples are not available.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brkljača, R.; Schneider, B.; Hidalgo, W.; Otálvaro, F.; Ospina, F.; Lee, S.; Hoshino, M.; Fujita, M.; Urban, S. Application of the Crystalline Sponge Method to Revise the Structure of the Phenalenone Fuliginone. Molecules 2017, 22, 211. https://doi.org/10.3390/molecules22020211
Brkljača R, Schneider B, Hidalgo W, Otálvaro F, Ospina F, Lee S, Hoshino M, Fujita M, Urban S. Application of the Crystalline Sponge Method to Revise the Structure of the Phenalenone Fuliginone. Molecules. 2017; 22(2):211. https://doi.org/10.3390/molecules22020211
Chicago/Turabian StyleBrkljača, Robert, Bernd Schneider, William Hidalgo, Felipe Otálvaro, Felipe Ospina, Shoukou Lee, Manabu Hoshino, Makoto Fujita, and Sylvia Urban. 2017. "Application of the Crystalline Sponge Method to Revise the Structure of the Phenalenone Fuliginone" Molecules 22, no. 2: 211. https://doi.org/10.3390/molecules22020211