Chemical Reactivity Theory Study of Advanced Glycation Endproduct Inhibitors


Abstract
:
1. Introduction
2. Theoretical Background
3. Settings and Computational Methods
4. Results and Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DFT | Density Functional Theory |
| AGE | Advanced Glycation Endproducts |
| SCF | Self Consistent Field |
| KID | Koopmans in DFT |
| HOMO | Higher Ocuppied Molecular Orbital |
| LUMO | Lower Unocuppied Molecular Orbital |
| IEF-PCM | Integral Equation Formalism - Polarized Continuum Model |
| SMD | Solvation Model Density |
References
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced Glycation End-Products: A Review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Ma, J.; Chen, F.; Wang, M. Naturally Occurring Inhibitors Against the Formation of Advanced Glycation End-Products. Food Funct. 2011, 2, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Chattaraj, P. (Ed.) Chemical Reactivity Theory—A Density Functional View; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2009.
- Lewars, E. Computational Chemistry—Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003. [Google Scholar]
- Young, D. Computational Chemistry—A Practical Guide for Applying Techniques to Real-World Problems; John Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
- Jensen, F. Introduction to Computational Chemistry, 2nd ed.; John Wiley & Sons: Chichester, UK, 2007. [Google Scholar]
- Cramer, C. Essentials of Computational Chemistry—Theories and Models, 2nd ed.; John Wiley & Sons: Chichester, UK, 2004. [Google Scholar]
- Frau, J.; Muñoz, F.; Glossman-Mitnik, D. A Molecular Electron Density Theory Study of the Chemical Reactivity of Cis- and Trans-Resveratrol. Molecules 2016, 21, 1650. [Google Scholar] [CrossRef] [PubMed]
- Peverati, R.; Truhlar, D.G. Quest for a Universal Density Functional: The Accuracy of Density Functionals Across a Broad Spectrum of Databases in Chemistry and Physics. Philos. Trans. Ser. A Math. Phys. Eng. Sci. 2014, 372. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, S.; Figarola, J.L. Novel Inhibitors of Advanced Glycation Endproducts. Arch. Biochem. Biophys. 2003, 419, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Peyroux, J.; Sternberg, M. Advanced Glycation Endproducts (AGEs): Pharmacological Inhibition in Diabetes. Pathol. Biol. 2006, 54, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Glossman-Mitnik, D. A Comparison of the Chemical Reactivity of Naringenin Calculated with M06 Family of Density Functionals. Chem. Cent. J. 2013, 7, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Araya, J.I.; Salgado-Morán, G.; Glossman-Mitnik, D. Computational Nanochemistry Report on the Oxicams—Conceptual DFT and Chemical Reactivity. J. Phys. Chem. B 2013, 117, 6639–6651. [Google Scholar] [CrossRef] [PubMed]
- Glossman-Mitnik, D. Computational Nanochemistry Study of the Chemical Reactivity Properties of the Rhodamine B Molecule. Proc. Comput. Sci. 2013, 18, 816–825. [Google Scholar] [CrossRef]
- Martínez-Araya, J.I.; Salgado-Morán, G.; Glossman-Mitnik, D. Computational Nutraceutics: Chemical Reactivity Properties of the Flavonoid Naringin by Means of Conceptual DFT. J. Chem. 2013, 2013, 850270. [Google Scholar] [CrossRef]
- Glossman-Mitnik, D. Chemical Reactivity Theory within DFT Applied to the Study of the Prunin Flavonoid. Eur. Int. J. Sci. Techno. 2014, 3, 195–207. [Google Scholar]
- Glossman-Mitnik, D. Computational Chemistry of Natural Products: A Comparison of the Chemical Reactivity of Isonaringin Calculated with the M06 Family of Density Functionals. J. Mol. Model. 2014, 20, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.; Yang, W. Density Functional Approach to the Frontier-Electron Theory of Chemical Reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Geerlings, P.; de Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.; Szentpaly, L.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Gázquez, J.; Cedillo, A.; Vela, A. Electrodonating and Electroaccepting Powers. J. Phys. Chem. A 2007, 111, 1966–1970. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, P.; Chakraborty, A.; Giri, S. Net Electrophilicity. J. Phys. Chem. A 2009, 113, 10068–10074. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting Basis Sets for H to R. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Peverati, R.; Truhlar, D.G. Improving the Accuracy of Hybrid Meta-GGA Density Functionals by Range Separation. J. Phys. Chem. Lett. 2011, 2, 2810–2817. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. M11-L: A Local Density Functional That Provides Improved Accuracy for Electronic Structure Calculations in Chemistry and Physics. J. Phys. Chem. Lett. 2012, 3, 117–124. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. An Improved and Broadly Accurate Local Approximation to the Exchange-Correlation Density Functional: the MN12-L Functional for Electronic Structure Calculations in Chemistry and Physics. Phys. Chem. Chem. Phys. 2012, 14, 13171–13174. [Google Scholar] [CrossRef] [PubMed]
- Peverati, R.; Truhlar, D.G. Screened-Exchange Density Functionals with Broad Accuracy for Chemistry and Solid-State Physics. Phys. Chem. Chem. Phys. 2012, 14, 16187–16191. [Google Scholar] [CrossRef] [PubMed]
- Peverati, R.; Truhlar, D.G. Exchange-Correlation Functional with Good Accuracy for Both Structural and Energetic Properties while Depending Only on the Density and Its Gradient. J. Chem. Theory Comput. 2012, 8, 2310–2319. [Google Scholar] [CrossRef] [PubMed]
- Peverati, R.; Zhao, Y.; Truhlar, D.G. Generalized Gradient Approximation That Recovers the Second-Order Density-Gradient Expansion with Optimized Across-the-Board Performance. J. Phys. Chem. Lett. 2011, 2, 1991–1997. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. Communication: A Global Hybrid Generalized Gradient Approximation to the Exchange-Correlation Functional That Satisfies the Second-Order Density-Gradient Constraint and Has Broad Applicability in Chemistry. J. Chem. Phys. 2011, 135, 191102. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.; Cramer, C.; Truhlar, D. Universal Solvation Model Based on Solute Electron Density and a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Avogadro: An Open-Source Molecular Builder and Visualization Tool—Version 1.2.0. 2016. Available online: http://avogadro.openmolecules.net (accessed on 6 October 2016).
- Hanweel, M.; Curtis, D.E.; Lonie, D.; Vandermeersch, T.; Zurek, E.; Hutchison, G. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General AMBER Force fFeld. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Kronik, L.; Stein, T.; Refaely-Abramson, S.; Baer, R. Excitation Gaps of Finite-Sized Systems from Optimally Tuned Range-Separated Hybrid Functionals. J. Chem. Theory Comput. 2012, 8, 1515–1531. [Google Scholar] [CrossRef] [PubMed]
- Lima, I.T.; Prado, A.d.S.; Martins, J.B.L.; de Oliveira Neto, P.H.; Ceschin, A.M.; da Cunha, W.F.; da Silva Filho, D.A. Improving the Description of the Optical Properties of Carotenoids by Tuning the Long-Range Corrected Functionals. J. Phys. Chem. A 2016, 120, 4944–4950. [Google Scholar] [CrossRef] [PubMed]
- Colzani, M.; de Maddis, D.; Casali, G.; Carini, M.; Vistoli, G.; Aldini, G. Reactivity, Selectivity, and Reaction Mechanisms of Aminoguanidine, Hydralazine, Pyridoxamine, and Carnosine as Sequestering Agents of Reactive Carbonyl Species: A Comparative Study. ChemMedChem 2016, 11, 1778–1789. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Perez, P. The Nucleophilicity N Index in Organic Chemistry. Org. Biomol. Chem. 2011, 9, 7168–7175. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Chamorro, E.; Perez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef] [PubMed]
- Pratihar, S.; Roy, S. Nucleophilicity and Site Selectivity of Commonly Used Arenes and Heteroarenes. J. Org. Chem. 2010, 75, 4957–4963. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not Available.
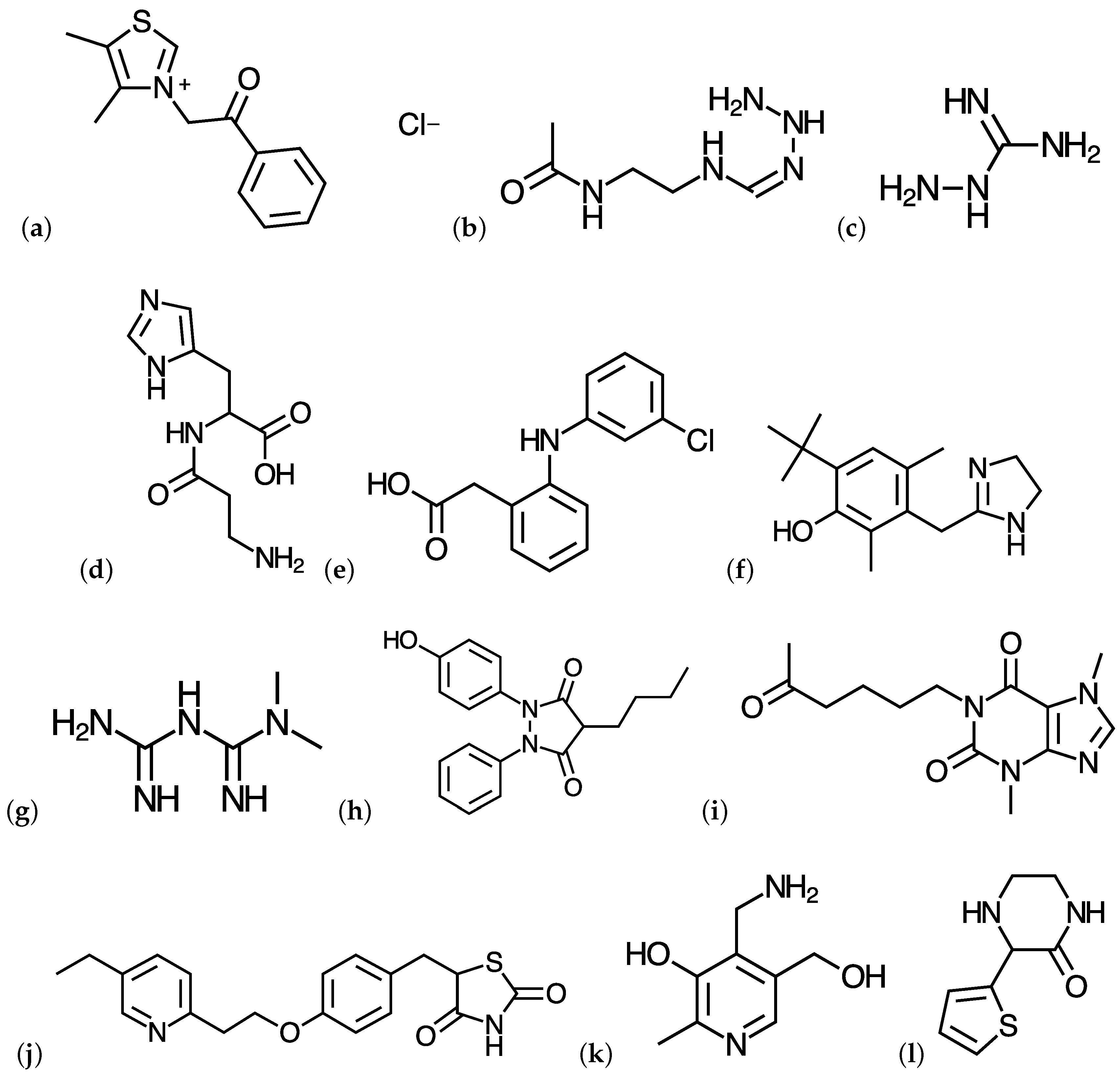

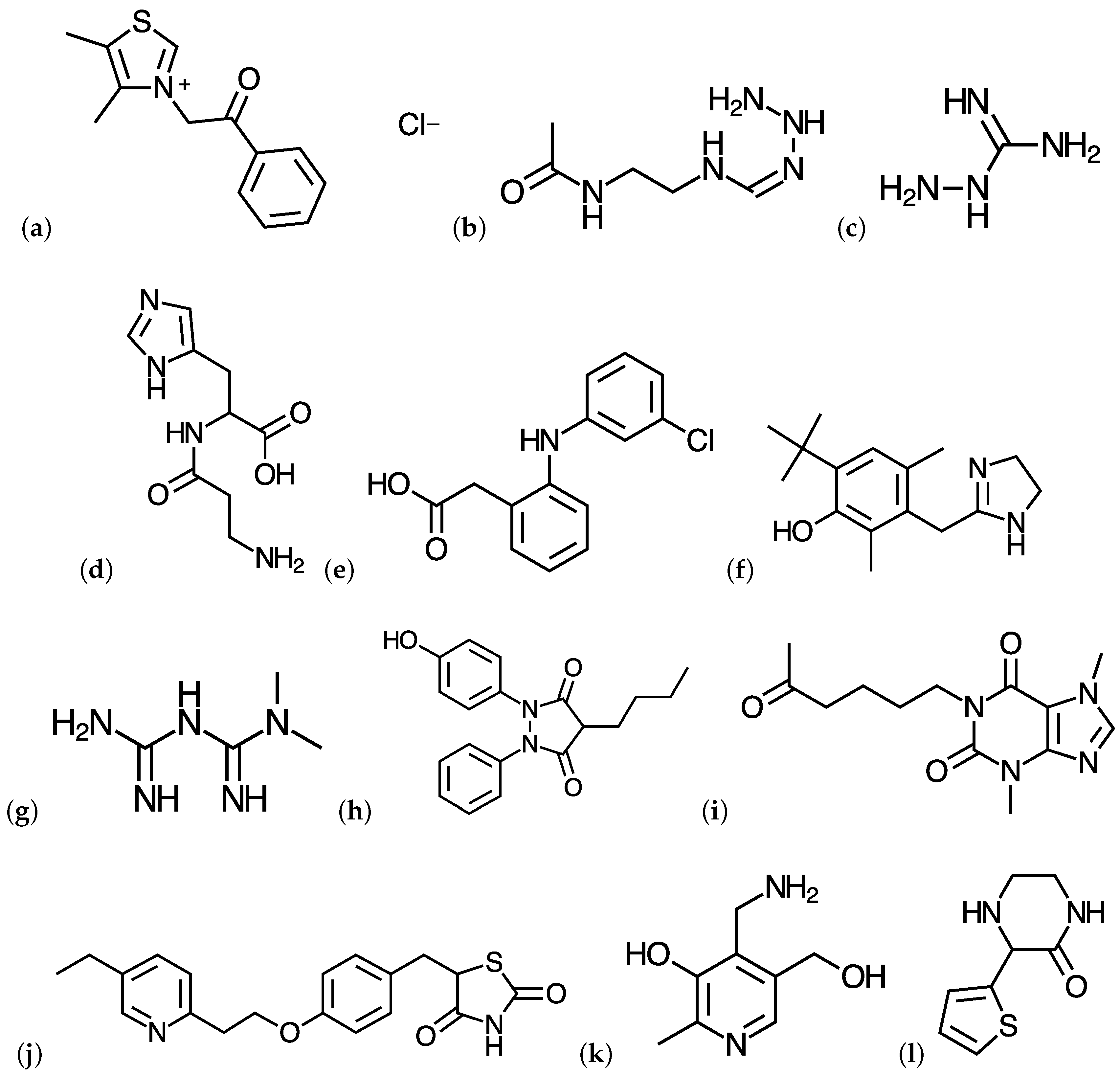
{kind=link}
{kind=link}
| J | J | J | J | J | J | J | J | J | J | J | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| M11 | 2.446 | 2.333 | 3.382 | 0.074 | 4.779 | 0.661 | 4.838 | 0.999 | 1.052 | 2.046 | 2.513 |
| M11L | 0.352 | 0.196 | 0.420 | 0.092 | 0.548 | 0.181 | 0.601 | 0.344 | 0.347 | 0.681 | 0.846 |
| MN12L | 0.311 | 0.198 | 0.377 | 0.072 | 0.491 | 0.133 | 0.528 | 0.242 | 0.249 | 0.487 | 0.604 |
| MN12SX | 0.069 | 0.108 | 0.143 | 0.067 | 0.125 | 0.054 | 0.164 | 0.137 | 0.070 | 0.207 | 0.260 |
| N12 | 0.459 | 0.355 | 0.601 | 0.088 | 0.811 | 0.306 | 0.896 | 0.568 | 0.567 | 1.135 | 1.396 |
| N12SX | 0.070 | 0.102 | 0.146 | 0.054 | 0.164 | 0.043 | 0.184 | 0.103 | 0.068 | 0.156 | 0.204 |
| SOGGA11 | 0.489 | 0.448 | 0.699 | 0.134 | 0.937 | 0.412 | 1.060 | 0.809 | 0.759 | 1.551 | 1.918 |
| SOGGA11X | 0.944 | 0.958 | 1.346 | 0.032 | 1.902 | 0.381 | 1.953 | 0.646 | 0.639 | 1.286 | 1.575 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frau, J.; Glossman-Mitnik, D. Chemical Reactivity Theory Study of Advanced Glycation Endproduct Inhibitors. Molecules 2017, 22, 226. https://doi.org/10.3390/molecules22020226
Frau J, Glossman-Mitnik D. Chemical Reactivity Theory Study of Advanced Glycation Endproduct Inhibitors. Molecules. 2017; 22(2):226. https://doi.org/10.3390/molecules22020226
Chicago/Turabian StyleFrau, Juan, and Daniel Glossman-Mitnik. 2017. "Chemical Reactivity Theory Study of Advanced Glycation Endproduct Inhibitors" Molecules 22, no. 2: 226. https://doi.org/10.3390/molecules22020226