Theoretical Study of Intramolecular Interactions in Peri-Substituted Naphthalenes: Chalcogen and Hydrogen Bonds



Abstract
:1. Introduction
2. Results
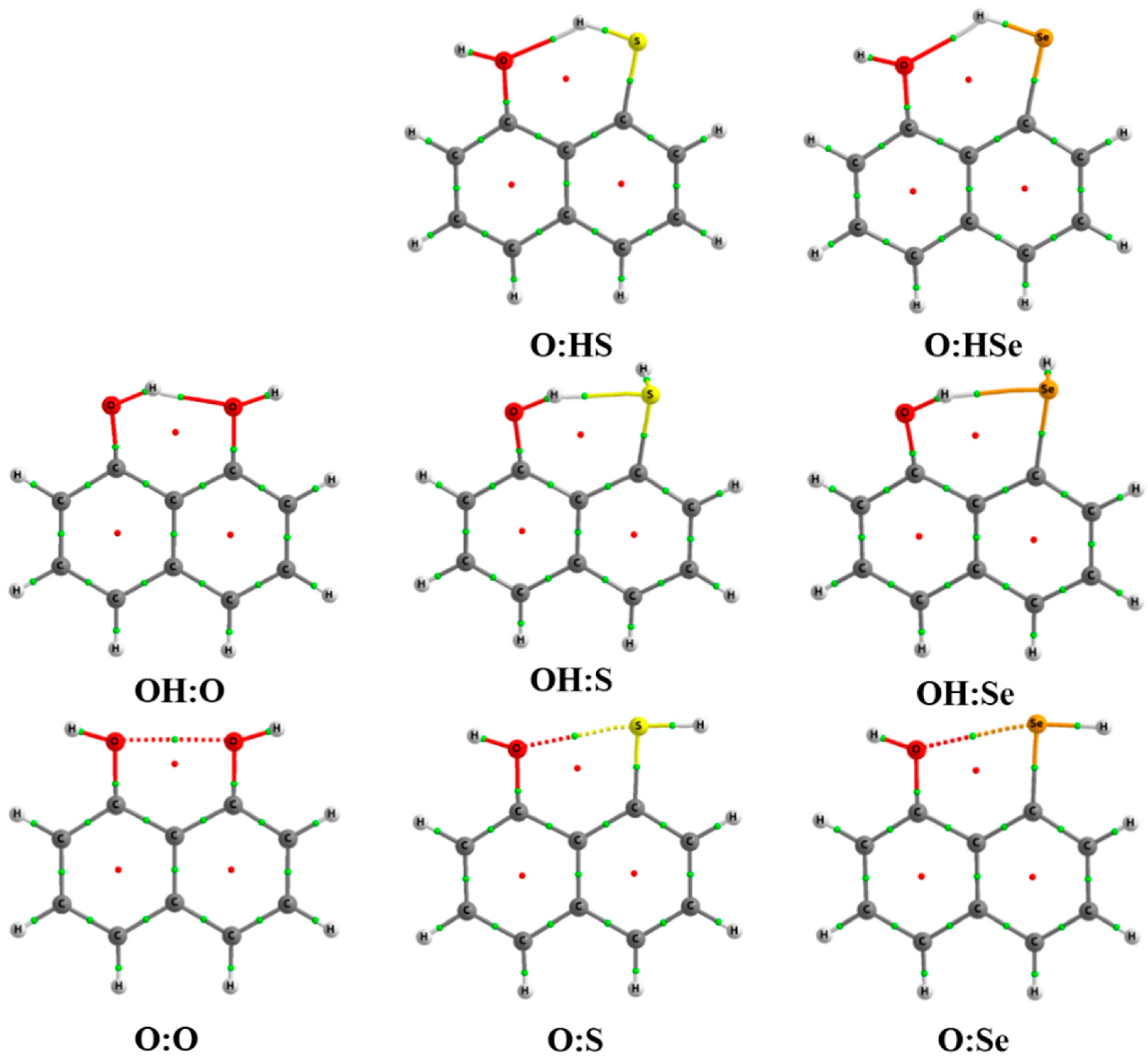
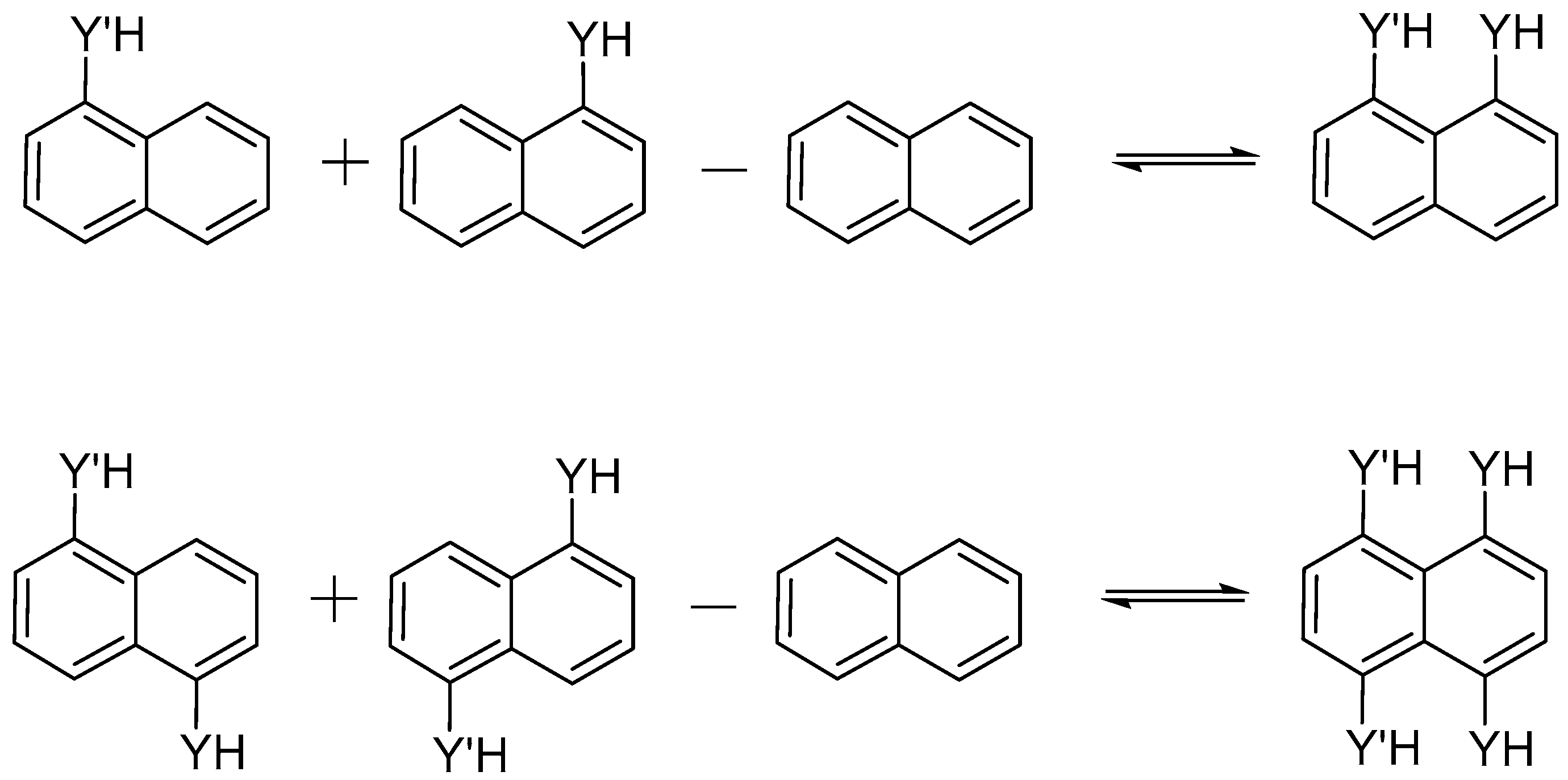
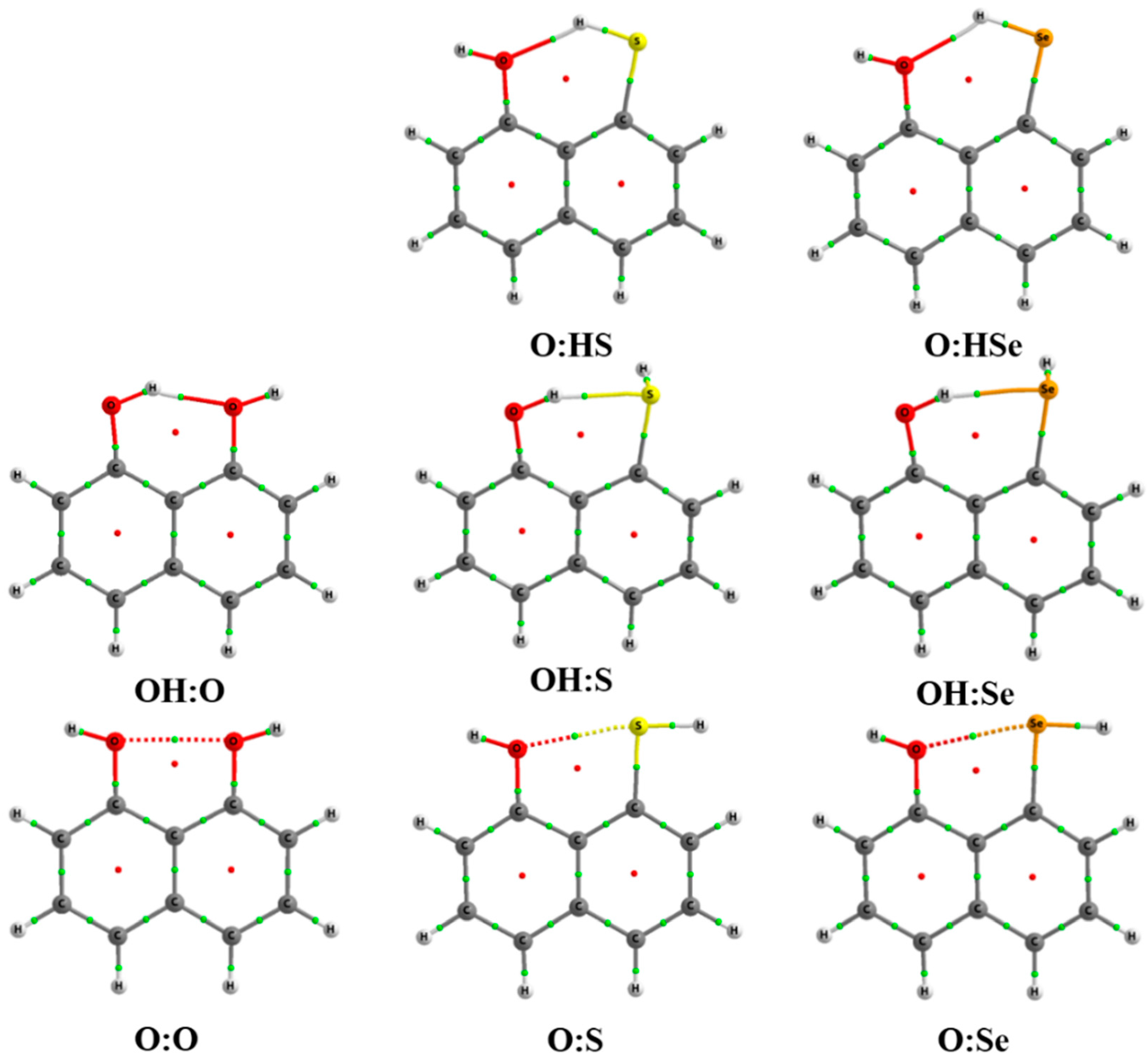
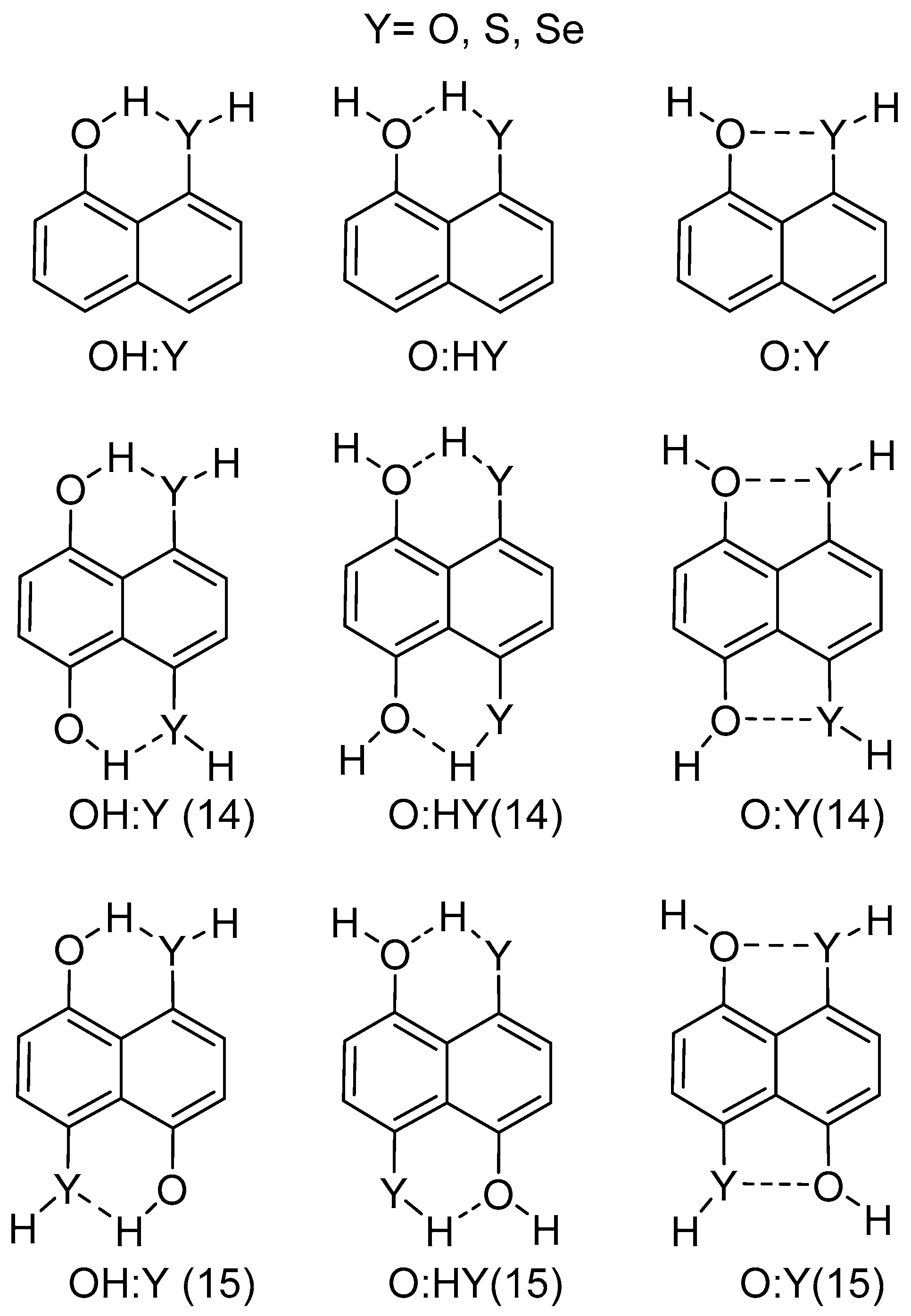
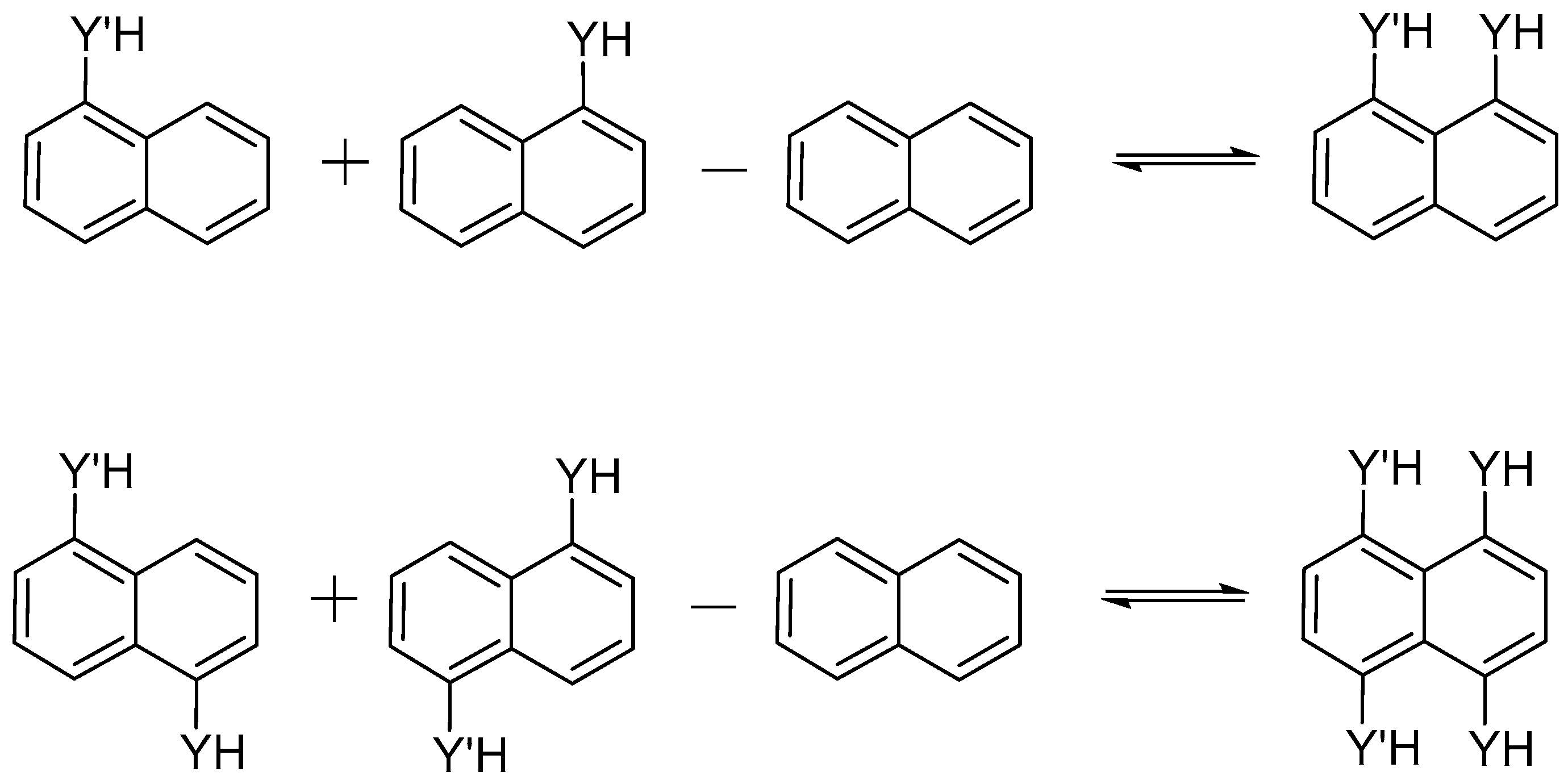
2.1. Structure and Energy of IMHB1 and IMYB1 Compounds
2.2. Structure and Energy of IMHB2 and IMYB2 Compounds
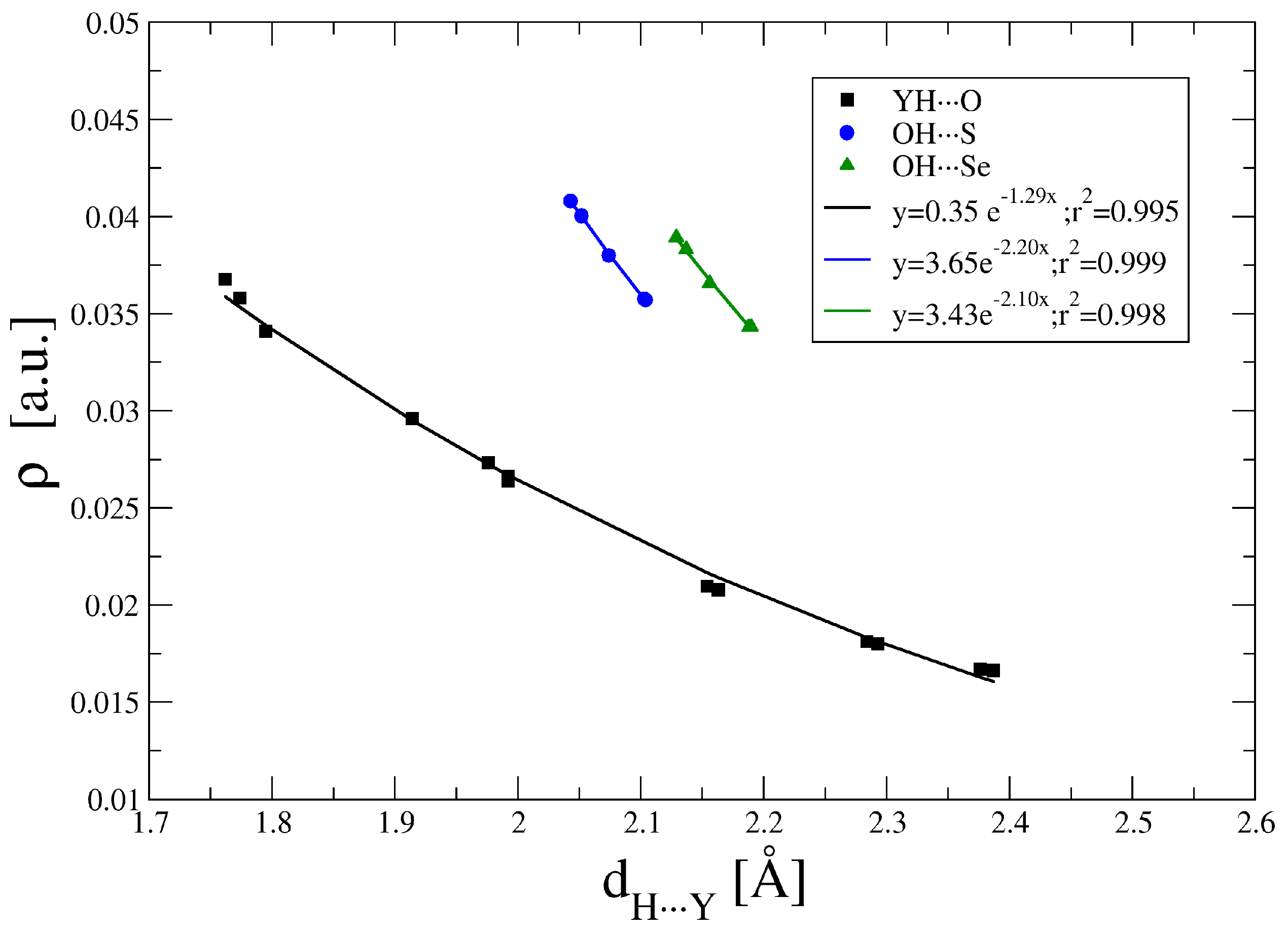
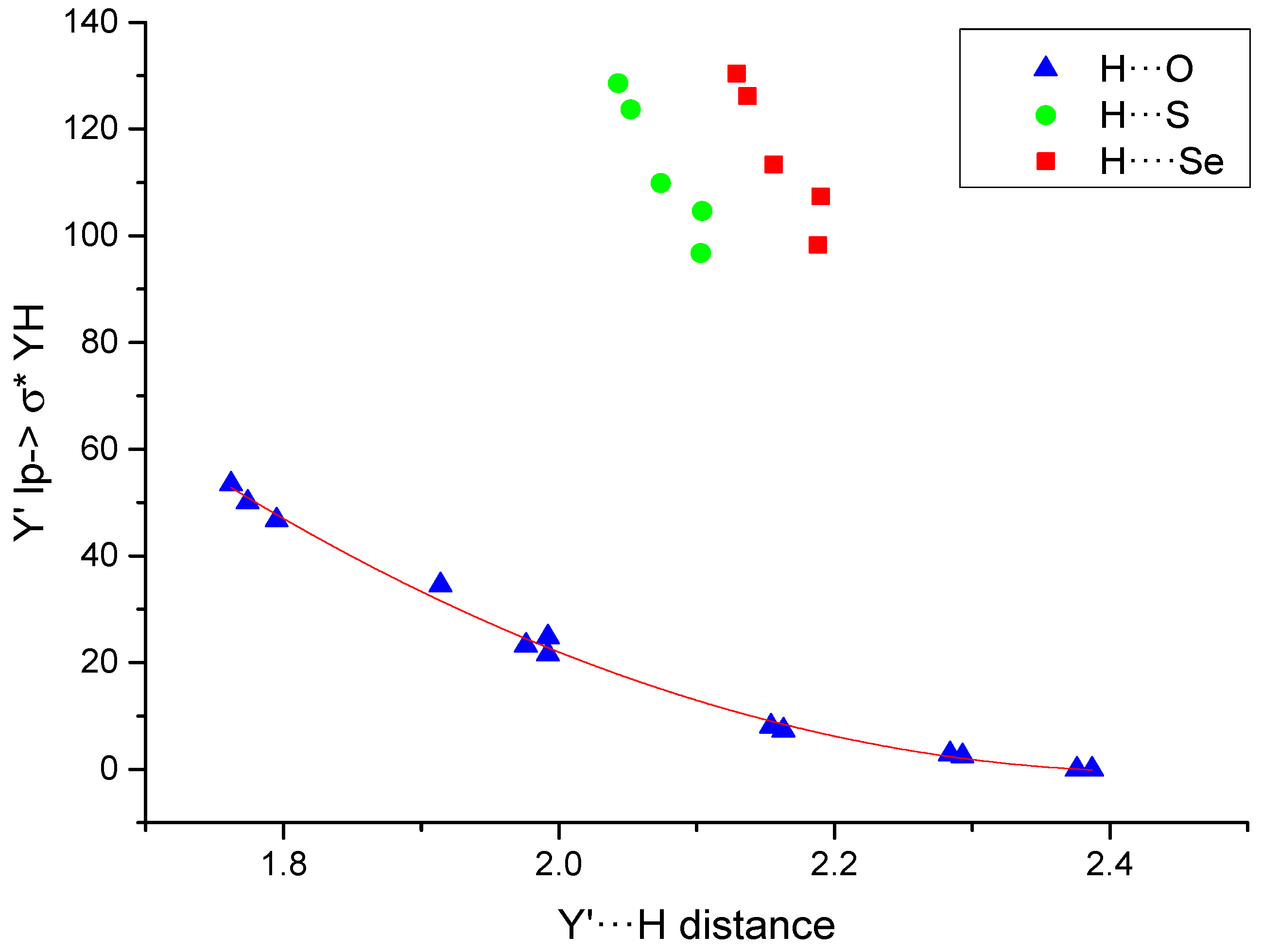
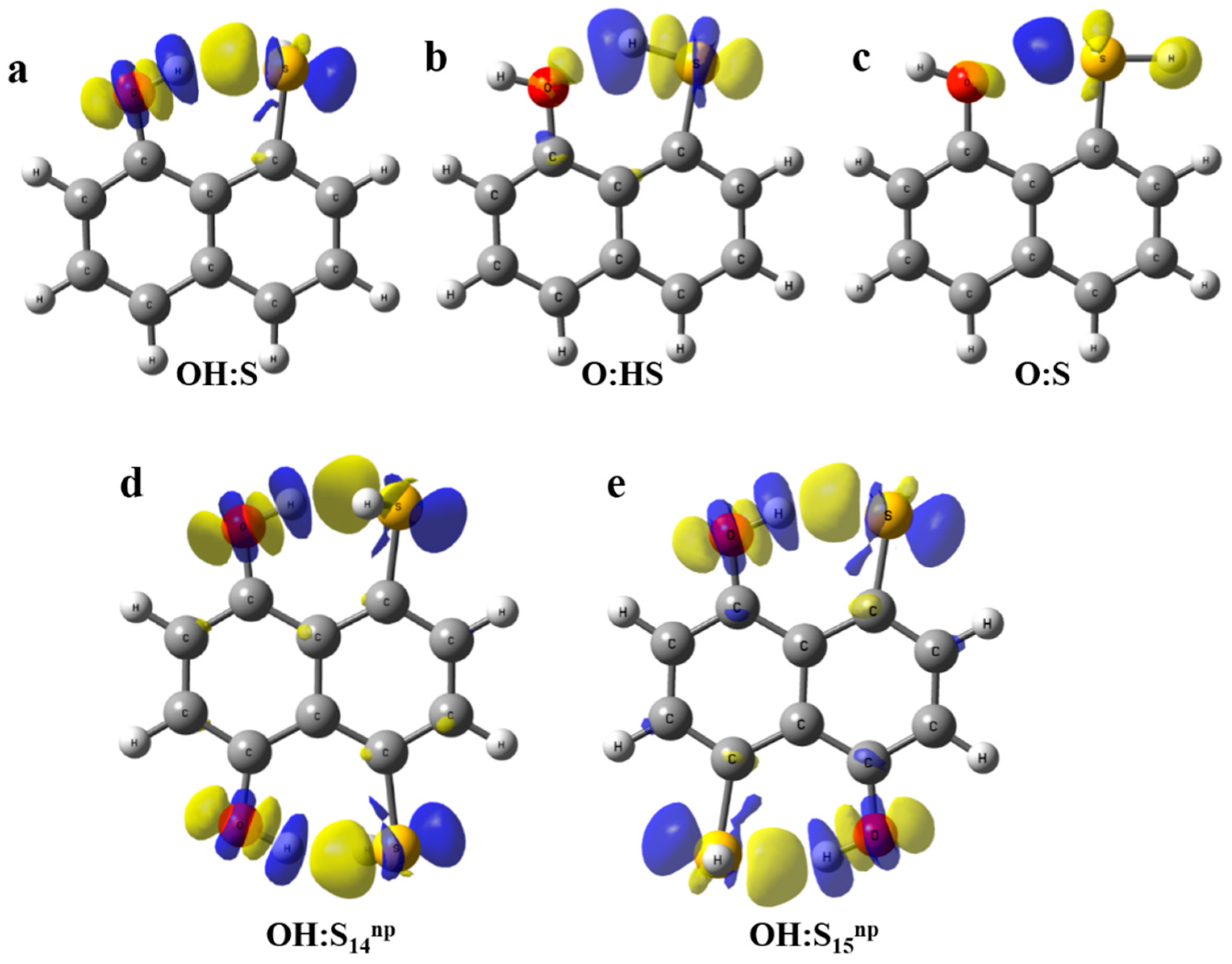
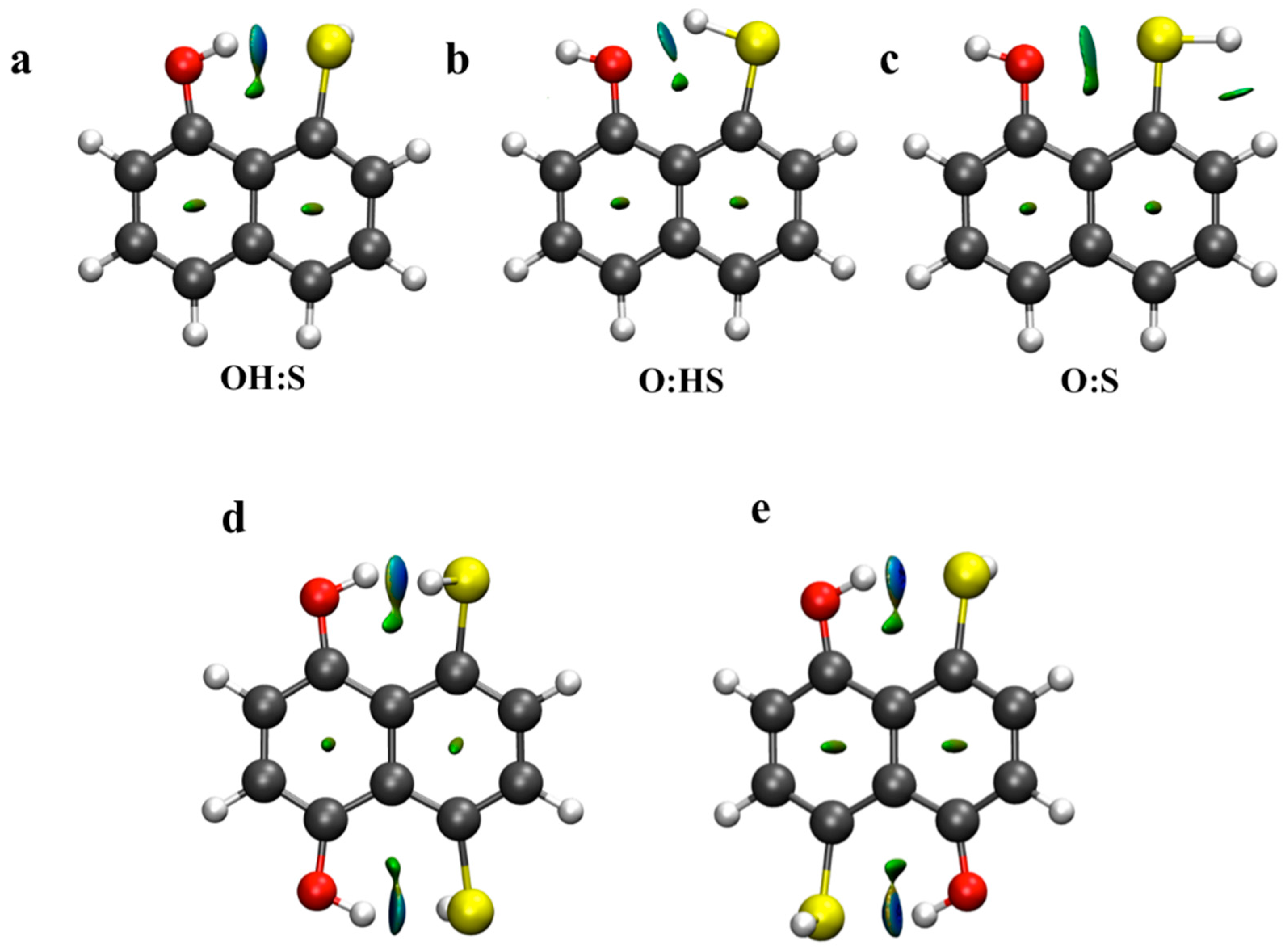
2.3. Atoms in Molecules (AIM) and Natural Bond Orbital (NBO) Analysis
3. Materials and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Mulliken, R.S. Structures of Complexes Formed by Halogen Molecules with Aromatic and with Oxygenated Solvents1. J. Am. Chem. Soc. 1950, 72, 600–608. [Google Scholar]
- Politzer, P.; Lane, P.; Concha, M.; Ma, Y.; Murray, J. An overview of halogen bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Meyer, F.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen Bonding in Supramolecular Chemistry. Angew. Chem. Int. Ed. 2008, 47, 6114–6127. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sanz, G.; Alkorta, I.; Trujillo, C.; Elguero, J. Intramolecular Pnicogen Interactions in PHF(CH2)nPHF (n =2–6) Systems. ChemPhysChem 2013, 14, 1656–1665. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Modulating intramolecular P···N pnictogen interactions. Phys. Chem. Chem. Phys. 2016, 18, 9148–9160. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Rozas, I.; Elguero, J. Molecular Complexes between Silicon Derivatives and Electron-Rich Groups. J. Phys. Chem. A 2001, 105, 743–749. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-Bonding Interaction: Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef] [PubMed]
- Sovago, I.; Gutmann, M.J.; Hill, J.G.; Senn, H.M.; Thomas, L.H.; Wilson, C.C.; Farrugia, L.J. Experimental Electron Density and Neutron Diffraction Studies on the Polymorphs of Sulfathiazole. Cryst. Growth Des. 2014, 14, 1227–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.P.; Jayatilaka, D.; Guru Row, T.N. S[three dots, centered]O chalcogen bonding in sulfa drugs: insights from multipole charge density and X-ray wavefunction of acetazolamide. Phys. Chem. Chem. Phys. 2015, 17, 25411–25420. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.P.; Veccham, S.P.K.P.; Farrugia, L.J.; Guru Row, T.N. “Conformational Simulation” of Sulfamethizole by Molecular Complexation and Insights from Charge Density Analysis: Role of Intramolecular S···O Chalcogen Bonding. Cryst. Growth Des. 2015, 15, 2110–2118. [Google Scholar] [CrossRef]
- Kilian, P.; Knight, F.R.; Woollins, J.D. Naphthalene and Related Systems peri-Substituted by Group 15 and 16 Elements. Chem. Eur. J. 2011, 17, 2302–2328. [Google Scholar] [CrossRef] [PubMed]
- Brea, O.; Corral, I.; Mó, O.; Yáñez, M.; Alkorta, I.; Elguero, J. Beryllium-Based Anion Sponges: Close Relatives of Proton Sponges. Chem. Eur. J. 2016, 22, 18322–18325. [Google Scholar] [CrossRef] [PubMed]
- Cormanich, R.A.; Rittner, R.; O’Hagan, D.; Bühl, M. Analysis of CF···FC Interactions on Cyclohexane and Naphthalene Frameworks. J. Phys. Chem. A 2014, 118, 7901–7910. [Google Scholar] [CrossRef] [PubMed]
- Llamas-Saiz, A.L.; Foces-Foces, C.; Elguero, J. Proton sponges. J. Mol. Struct. 1994, 328, 297–323. [Google Scholar] [CrossRef]
- Matta, C.F.; Castillo, N.; Boyd, R.J. Characterization of a Closed-Shell Fluorine−Fluorine Bonding Interaction in Aromatic Compounds on the Basis of the Electron Density. J. Phys. Chem. A 2005, 109, 3669–3681. [Google Scholar] [CrossRef] [PubMed]
- Minyaev, R.M.; Minkin, V.I. Theoretical study of O - > X (S, Se, Te) coordination in organic compounds. Can. J. Chem. 1998, 76, 776–788. [Google Scholar] [CrossRef]
- Esseffar, M.H.; Herrero, R.; Quintanilla, E.; Dávalos, J.Z.; Jiménez, P.; Abboud, J.-L.M.; Yáñez, M.; Mó, O. Activation of the Disulfide Bond and Chalcogen–Chalcogen Interactions: An Experimental (FTICR) and Computational Study. Chem. Eur. J. 2007, 13, 1796–1803. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Sanz, G.; Alkorta, I.; Elguero, J. Theoretical study of the HXYH dimers (X,Y=O, S, Se). Hydrogen bonding and chalcogen–chalcogen interactions. Mol. Phys. 2011, 109, 2543–2552. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Intermolecular Weak Interactions in HTeXH Dimers (X=O, S, Se, Te): Hydrogen Bonds, Chalcogen–Chalcogen Contacts and Chiral Discrimination. ChemPhysChem 2012, 13, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, U.; Scheiner, S. Effects of Charge and Substituent on the S···N Chalcogen Bond. J. Phys. Chem. A 2014, 118, 3183–3192. [Google Scholar] [CrossRef] [PubMed]
- Alikhani, E.; Fuster, F.; Madebene, B.; Grabowski, S.J. Topological reaction sites—very strong chalcogen bonds. Phys. Chem. Chem. Phys. 2014, 16, 2430–2442. [Google Scholar] [CrossRef] [PubMed]
- Sanz, P.; Yáñez, M.; Mó, O. Competition between X···H···Y Intramolecular Hydrogen Bonds and X····Y (X = O, S, and Y = Se, Te) Chalcogen−Chalcogen Interactions. J. Phys. Chem. A 2002, 106, 4661–4668. [Google Scholar] [CrossRef]
- Sanz, P.; Mó, O.; Yañez, M. Characterization of intramolecular hydrogen bonds and competitive chalcogen-chalcogen interactions on the basis of the topology of the charge density. Phys. Chem. Chem. Phys. 2003, 5, 2942–2947. [Google Scholar] [CrossRef]
- Sanz, P.; Yáñez, M.; Mó, O. Resonance-Assisted Intramolecular Chalcogen–Chalcogen Interactions? Chem. Eur. J. 2003, 9, 4548–4555. [Google Scholar] [CrossRef] [PubMed]
- Iwaoka, M.; Komatsu, H.; Katsuda, T.; Tomoda, S. Nature of Nonbonded Se···O Interactions Characterized by 17O NMR Spectroscopy and NBO and AIM Analyses. J. Am. Chem. Soc. 2004, 126, 5309–5317. [Google Scholar] [CrossRef] [PubMed]
- Nziko, V.d.P.N.; Scheiner, S. Intramolecular S···O Chalcogen Bond as Stabilizing Factor in Geometry of Substituted Phenyl-SF3 Molecules. J. Org. Chem. 2015, 80, 2356–2363. [Google Scholar] [CrossRef] [PubMed]
- Shishkin, O.V.; Omelchenko, I.V.; Kalyuzhny, A.L.; Paponov, B.V. Intramolecular S···O chalcogen bond in thioindirubin. Struct. Chem. 2010, 21, 1005–1011. [Google Scholar] [CrossRef]
- Mikherdov, A.S.; Kinzhalov, M.A.; Novikov, A.S.; Boyarskiy, V.P.; Boyarskaya, I.A.; Dar’in, D.V.; Starova, G.L.; Kukushkin, V.Y. Difference in Energy between Two Distinct Types of Chalcogen Bonds Drives Regioisomerization of Binuclear (Diaminocarbene)PdII Complexes. J. Am. Chem. Soc. 2016, 138, 14129–14137. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.P.; Sathishkumar, R.; Guru Row, T.N. Organic alloys of room temperature liquids thiophenol and selenophenol. Chem. Commun. 2015, 51, 14255–14258. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Sanz, G.; Alkorta, I.; Elguero, J. A theoretical study of the conformation of 2,2′-bifuran, 2,2′-bithiophene, 2,2′-bitellurophene and mixed derivatives: Chalcogen–chalcogen interactions or dipole–dipole effects? Comput. Theor. Chem. 2011, 974, 37–42. [Google Scholar] [CrossRef]
- Mukherjee, A.J.; Zade, S.S.; Singh, H.B.; Sunoj, R.B. Organoselenium Chemistry: Role of Intramolecular Interactions. Chem. Rev. 2010, 110, 4357–4416. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Lane, P.; Politzer, P. A predicted new type of directional noncovalent interaction. Int. J. Quantum Chem. 2007, 107, 2286–2292. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.; Concha, M. σ-hole bonding between like atoms; a fallacy of atomic charges. J. Mol. Model. 2008, 14, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Mohajeri, A.; Pakiari, A.H.; Bagheri, N. Theoretical studies on the nature of bonding in σ-hole complexes. Chem. Phys. Lett. 2009, 467, 393–397. [Google Scholar] [CrossRef]
- Buckingham, A.D.; Fowler, P.W. A model for the geometries of Van der Waals complexes. Can. J. Chem. 1985, 63, 2018–2025. [Google Scholar] [CrossRef]
- Legon, A.C.; Millen, D.J. Angular geometries and other properties of hydrogen-bonded dimers: A simple electrostatic interpretation of the success of the electron-pair model. Chem. Soc. Rev. 1987, 16, 467–498. [Google Scholar] [CrossRef]
- Stone, A.J.; Price, S.L. Some new ideas in the theory of intermolecular forces: Anisotropic atom-atom potentials. J. Phys. Chem. 1988, 92, 3325–3335. [Google Scholar] [CrossRef]
- Brinck, T.; Murray, J.S.; Politzer, P. Surface electrostatic potentials of halogenated methanes as indicators of directional intermolecular interactions. Int. J. Quantum Chem. 1992, 44, 57–64. [Google Scholar] [CrossRef]
- Burling, F.T.; Goldstein, B.M. Computational studies of nonbonded sulfur-oxygen and selenium-oxygen interactions in the thiazole and selenazole nucleosides. J. Am. Chem. Soc. 1992, 114, 2313–2320. [Google Scholar] [CrossRef]
- Price, S.L. Applications of realistic electrostatic modelling to molecules in complexes, solids and proteins. J. Chem. Soc. Faraday Trans. 1996, 92, 2997–3008. [Google Scholar] [CrossRef]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef] [PubMed]
- Awwadi, F.F.; Willett, R.D.; Peterson, K.A.; Twamley, B. The Nature of Halogen⋅⋅⋅Halogen Synthons: Crystallographic and Theoretical Studies. Chem. Eur. J. 2006, 12, 8952–8960. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Riley, K.E.; Bulat, F.A.; Murray, J.S. Perspectives on halogen bonding and other σ-hole interactions: Lex parsimoniae (Occam’s Razor). Comput. Theor. Chem. 2012, 998, 2–8. [Google Scholar] [CrossRef]
- Hennemann, M.; Murray, J.; Politzer, P.; Riley, K.; Clark, T. Polarization-induced σ-holes and hydrogen bonding. J. Mol. Model. 2012, 18, 2461–2469. [Google Scholar] [CrossRef] [PubMed]
- Clark, T. σ-Holes. WIREs Comput. Mol. Sci. 2013, 3, 13–20. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Halogen Bonding: An Interim Discussion. ChemPhysChem 2013, 14, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Sanchez-Sanz, G.; Elguero, J. Linear free energy relationships in halogen bonds. CrystEngComm 2013, 15, 3178–3186. [Google Scholar] [CrossRef]
- Nagels, N.; Geboes, Y.; Pinter, B.; De Proft, F.; Herrebout, W.A. Tuning the Halogen/Hydrogen Bond Competition: A Spectroscopic and Conceptual DFT Study of Some Model Complexes Involving CHF2I. Chem. Eur. J. 2014, 20, 8433–8443. [Google Scholar] [CrossRef] [PubMed]
- Lo, R.; Fanfrlik, J.; Lepsik, M.; Hobza, P. The properties of substituted 3D-aromatic neutral carboranes: the potential for [sigma]-hole bonding. Phys. Chem. Chem. Phys. 2015, 17, 20814–20821. [Google Scholar] [CrossRef] [PubMed]
- Fanfrlík, J.; Holub, J.; Růžičková, Z.; Řezáč, J.; Lane, P.D.; Wann, D.A.; Hnyk, D.; Růžička, A.; Hobza, P. Competition between Halogen, Hydrogen and Dihydrogen Bonding in Brominated Carboranes. ChemPhysChem 2016, 17, 3373–3376. [Google Scholar] [CrossRef] [PubMed]
- Bauza, A.; Quinonero, D.; Frontera, A.; Deya, P.M. Substituent effects in halogen bonding complexes between aromatic donors and acceptors: A comprehensive ab initio study. Phys. Chem. Chem. Phys. 2011, 13, 20371–20379. [Google Scholar] [CrossRef]
- Kolář, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef] [PubMed]
- Hobza, P.; Řezáč, J. Introduction: Noncovalent Interactions. Chem. Rev. 2016, 116, 4911–4912. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H[centered ellipsis]F–Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Mata, I.; Alkorta, I.; Molins, E.; Espinosa, E. Universal Features of the Electron Density Distribution in Hydrogen-Bonding Regions: A Comprehensive Study Involving H⋅⋅⋅X (X=H, C, N, O, F, S, Cl, π) Interactions. Chem. Eur. J. 2010, 16, 2442–2452. [Google Scholar] [CrossRef] [PubMed]
- Knop, O.; Boyd, R.J.; Choi, S.C. Sulfur-sulfur bond lengths, or can a bond length be estimated from a single parameter? J. Am. Chem. Soc. 1988, 110, 7299–7301. [Google Scholar] [CrossRef]
- Knop, O.; Rankin, K.N.; Boyd, R.J. Coming to Grips with N−H···N Bonds. 1. Distance Relationships and Electron Density at the Bond Critical Point. J. Phys. Chem. A 2001, 105, 6552–6566. [Google Scholar] [CrossRef]
- Knop, O.; Rankin, K.N.; Boyd, R.J. Coming to Grips with N−H···N Bonds. 2. Homocorrelations between Parameters Deriving from the Electron Density at the Bond Critical Point1. J. Phys. Chem. A 2002, 107, 272–284. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Del Bene, J.E. Pnicogen Bonded Complexes of PO2X (X = F, Cl) with Nitrogen Bases. J. Phys. Chem. A 2013, 117, 10497–10503. [Google Scholar] [CrossRef] [PubMed]
- Rozas, I.; Alkorta, I.; Elguero, J. Behavior of ylides containing N, O, and C atoms as hydrogen bond accepters. J. Am. Chem. Soc. 2000, 122, 11154–11161. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. B4H4 and B4(CH3)4 as Unique Electron Donors in Hydrogen-Bonded and Halogen-Bonded Complexes. J. Phys. Chem. A 2016, 120, 5745–5751. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Substituent Effects on the Properties of Pnicogen-Bonded Complexes H2XP:PYH2, for X, Y = F, Cl, OH, NC, CCH, CH3, CN, and H. J. Phys. Chem. A 2015, 119, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. V. Core-valence basis sets for boron through neon. J. Chem. Phys. 1995, 103, 4572–4585. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian-Basis Sets for Use in Correlated Molecular Calculations. 1. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision d1, Inc.: Wallingford, CT, USA, 2009.
- Halkier, A.; Helgaker, T.; Jørgensen, P.; Klopper, W.; Olsen, J. Basis-set convergence of the energy in molecular Hartree–Fock calculations. Chem. Phys. Lett. 1999, 302, 437–446. [Google Scholar] [CrossRef]
- Halkier, A.; Klopper, W.; Helgaker, T.; Jørgensen, P.; Taylor, P.R. Basis set convergence of the interaction energy of hydrogen-bonded complexes. J. Chem. Phys. 1999, 111, 9157–9167. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Popelier, P.L.A. Atoms in Molecules. An introduction; Prentice Hall: Harlow, UK, 2000. [Google Scholar]
- Keith, T.A. TK Gristmill Software. Available online: aim.tkgristmill.com (accessed on 14 December 2016).
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Electron density shift description of non-bonding intramolecular interactions. Comput. Theor. Chem. 2012, 991, 124–133. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IMHB1 | H···Y′ | Y–H···Y′ | C–Y···Y′–C |
|---|---|---|---|
| OH:O | 1.795 | 144.2 | 0.0 |
| OH:S | 2.104 | 152.8 | 0.0 |
| OH:Se | 2.190 | 154.6 | 0.0 |
| O:HS | 1.914 | 134.6 | 0.0 |
| O:HSe a | 1.992 | 128.0 | 0.0 |
| IMYB1 | O···Y | O···Y′–H | C–O···Y′–C |
| O:O | 2.574 | 163.4 | 0.0 |
| O:S | 2.702 | 170.2 | 0.0 |
| O:Se | 2.734 | 165.2 | 0.0 |
| Compound | Eint | Eiso | Edef | Erel OO | Erel SO | Erel SeO |
|---|---|---|---|---|---|---|
| IMHB1 | ||||||
| OH:O | –12.6 | –3.6 | 9.0 | 0.0 | ||
| OH:S | –10.0 | 4.1 | 14.1 | 0.0 | ||
| OH:Se | –12.3 | 3.7 | 16.0 | 0.0 | ||
| O:HS | 4.1 | 18.9 | 14.9 | 14.9 | ||
| O:HSe | 6.1 | 25.5 | 19.5 | 21.9 | ||
| IMYB1 | ||||||
| O:O | 14.6 | 23.7 | 9.2 | 27.8 | ||
| O:S | 9.1 | 14.6 | 5.4 | 10.5 | ||
| O:Se | 5.2 | 12.5 | 7.2 | 8.8 |
| H···Y′ | ΔH···Y′ b | Y–H···Y′ | C–Y···Y′–C | |
|---|---|---|---|---|
| IMHB2 a | ||||
| OH:O14 | 1.774 | –0.021 | 143.8 | 0.0 |
| OH:O15 | 1.762 | –0.033 | 144.5 | 0.0 |
| OH:S14p | 2.074 | –0.030 | 151.2 | 5.9 |
| OH:S14np | 2.103 | –0.001 | 148.0 | 12.0 |
| OH:S15p | 2.052 | –0.053 | 152.9 | 7.9 |
| OH:S15np | 2.043 | –0.061 | 153.7 | 5.0 |
| OH:Se14p | 2.156 | –0.033 | 152.9 | 5.7 |
| OH:Se14 np | 2.188 | –0.002 | 149.0 | 12.1 |
| OH:Se15p | 2.137 | –0.053 | 154.5 | 8.0 |
| OH:Se15np | 2.129 | –0.061 | 155.3 | 5.0 |
| O:HS14p | 1.992 | 0.078 | 122.6 | 14.1 |
| O:HS14np | 1.976 | 0.062 | 123.7 | 11.9 |
| O:HS15p | 2.163 | 0.249 | 110.0 | 13.6 |
| O:HS15np | 2.154 | 0.240 | 110.4 | 15.1 |
| O:HSe14p | 2.284 | 0.292 | 102.3 | 13.4 |
| O:HSe14np | 2.293 | 0.300 | 101.6 | 11.7 |
| O:HSe15p | 2.387 | 0.395 | 97.2 | 16.0 |
| O:HSe15np | 2.376 | 0.383 | 97.7 | 15.6 |
| IMYB2 a | ||||
| HO···SH | O···Y' | ΔO··Y′ b | O···Y′–H | C–O···Y′–C |
| O:O | 2.525 | –0.049 | 163.2 | 0.0 |
| O:S14 | 2.654 | –0.049 | 169.6 | 0.0 |
| O:S15 | 2.657 | –0.046 | 169.7 | 0.0 |
| O:Se14 | 2.690 | –0.044 | 164.8 | 0.0 |
| O:Se15 | 2.694 | –0.040 | 164.9 | 0.0 |
| IMHB2 | Eint | Eiso | Edef | Erel SO | Erel SeO |
|---|---|---|---|---|---|
| OH:O14 | –11.8 | 9.1 | 20.9 | 4.3 a | |
| OH:O15 | –13.8 | 4.8 | 18.5 | 0.0 a | |
| OH:S14p | –7.5 | 27.3 | 34.9 | 19.4 | |
| OH:S14np | –8.8 | 24.5 | 33.4 | 16.6 | |
| OH:S15p | –19.4 | 7.1 | 26.5 | 0.0 | |
| OH:S15np | –20.7 | 7.5 | 28.2 | 0.4 | |
| OH:Se14p | –12.0 | 26.1 | 38.2 | 20.1 | |
| OH:Se14np | –10.3 | 23.8 | 34.1 | 17.8 | |
| OH:Se15p | –25.5 | 5.2 | 30.7 | 0.0 | |
| OH:Se15np | –26.7 | 5.9 | 32.6 | 0.7 | |
| O:HS14p | 19.4 | 54.5 | 35.1 | 46.6 | |
| O:HS14np | 20.1 | 55.0 | 34.8 | 47.0 | |
| O:HS15p | 24.9 | 53.1 | 28.2 | 46.1 | |
| O:HS15np | 23.8 | 52.1 | 28.3 | 45.1 | |
| O:HSe14p | 30.8 | 62.5 | 31.6 | 56.4 | |
| O:HSe14np | 32.2 | 62.9 | 30.6 | 56.8 | |
| O:HSe15p | 27.4 | 55.9 | 28.5 | 50.7 | |
| O:HSe15np | 27.5 | 55.6 | 28.0 | 50.4 | |
| IMYB2 | |||||
| HO···SH | |||||
| O:O | 52.9 | 65.9 | 13.0 | 61.2 a | |
| O:S14 | 27.5 | 40.0 | 12.5 | 32.1 | |
| O:S15 | 27.4 | 38.1 | 10.8 | 31.0 | |
| O:Se14 | 16.8 | 32.3 | 15.5 | 26.2 | |
| O:Se15 | 17.3 | 31.0 | 13.7 | 25.9 |
| HB | YB | ||
|---|---|---|---|
| Y′lp→σ*HY | Olp→σ*YH | ||
| OH:O | 46.69 | O:O | - |
| OH:S | 104.63 | O:S | 10.58 |
| OH:Se | 107.38 | O:Se | 15.26 |
| O:HS | 34.53 | ||
| O:HSe | 24.79 | ||
| Y′lp→σ*HY | Olp→σ*YH | ||
| OH:O14 | 50.03 | O:O | - |
| OH:O15 | 53.42 | O:S14 | 12.71 |
| OH:S14p | 109.85 | O:S15 | 12.46 |
| OH:S14np | 96.73 | O:Se14 | 17.68 |
| OH:S15p | 123.69 | O:Se15 | 17.35 |
| OH:S15np | 128.58 | ||
| OH:Se14p | 113.36 | ||
| OH:Se14np | 98.27 | ||
| OH:Se15p | 126.19 | ||
| OH:Se15np | 130.37 | ||
| O:HS14p | 21.57 | ||
| O:HS14np | 23.24 | ||
| O:HS15p | 7.32 | ||
| O:HS15np | 8.03 | ||
| O:HSe14p | 2.84 | ||
| O:HSe14np | 2.47 | ||
| O:HSe15p | 0.00 | ||
| O:HSe15np | 0.00 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez–Sanz, G.; Alkorta, I.; Elguero, J. Theoretical Study of Intramolecular Interactions in Peri-Substituted Naphthalenes: Chalcogen and Hydrogen Bonds. Molecules 2017, 22, 227. https://doi.org/10.3390/molecules22020227
Sánchez–Sanz G, Alkorta I, Elguero J. Theoretical Study of Intramolecular Interactions in Peri-Substituted Naphthalenes: Chalcogen and Hydrogen Bonds. Molecules. 2017; 22(2):227. https://doi.org/10.3390/molecules22020227
Chicago/Turabian StyleSánchez–Sanz, Goar, Ibon Alkorta, and José Elguero. 2017. "Theoretical Study of Intramolecular Interactions in Peri-Substituted Naphthalenes: Chalcogen and Hydrogen Bonds" Molecules 22, no. 2: 227. https://doi.org/10.3390/molecules22020227