
Complete Chloroplast Genome of Medicinal Plant Lonicera japonica: Genome Rearrangement, Intron Gain and Loss, and Implications for Phylogenetic Studies

Abstract
:1. Introduction
2. Results and Discussion
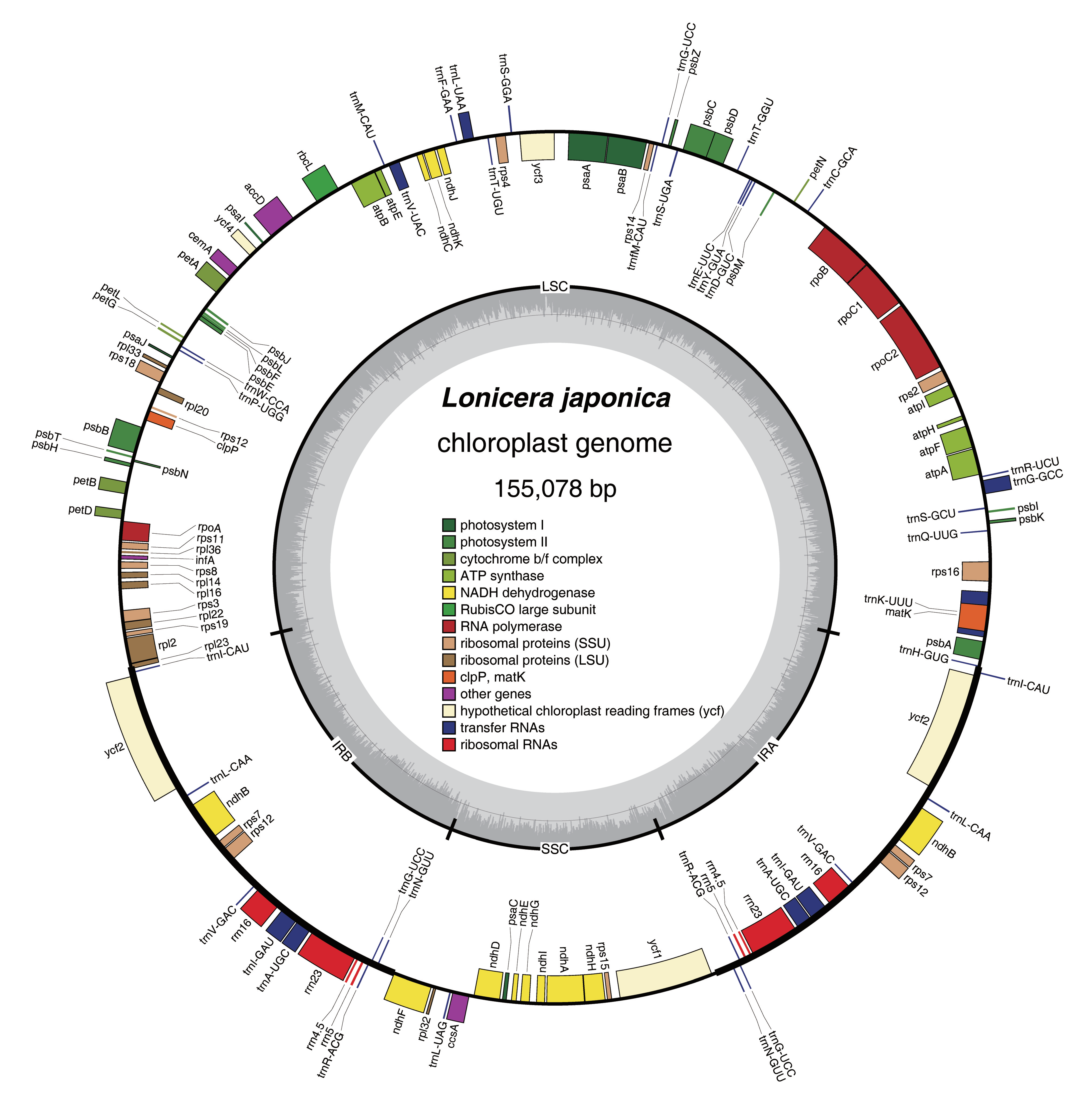
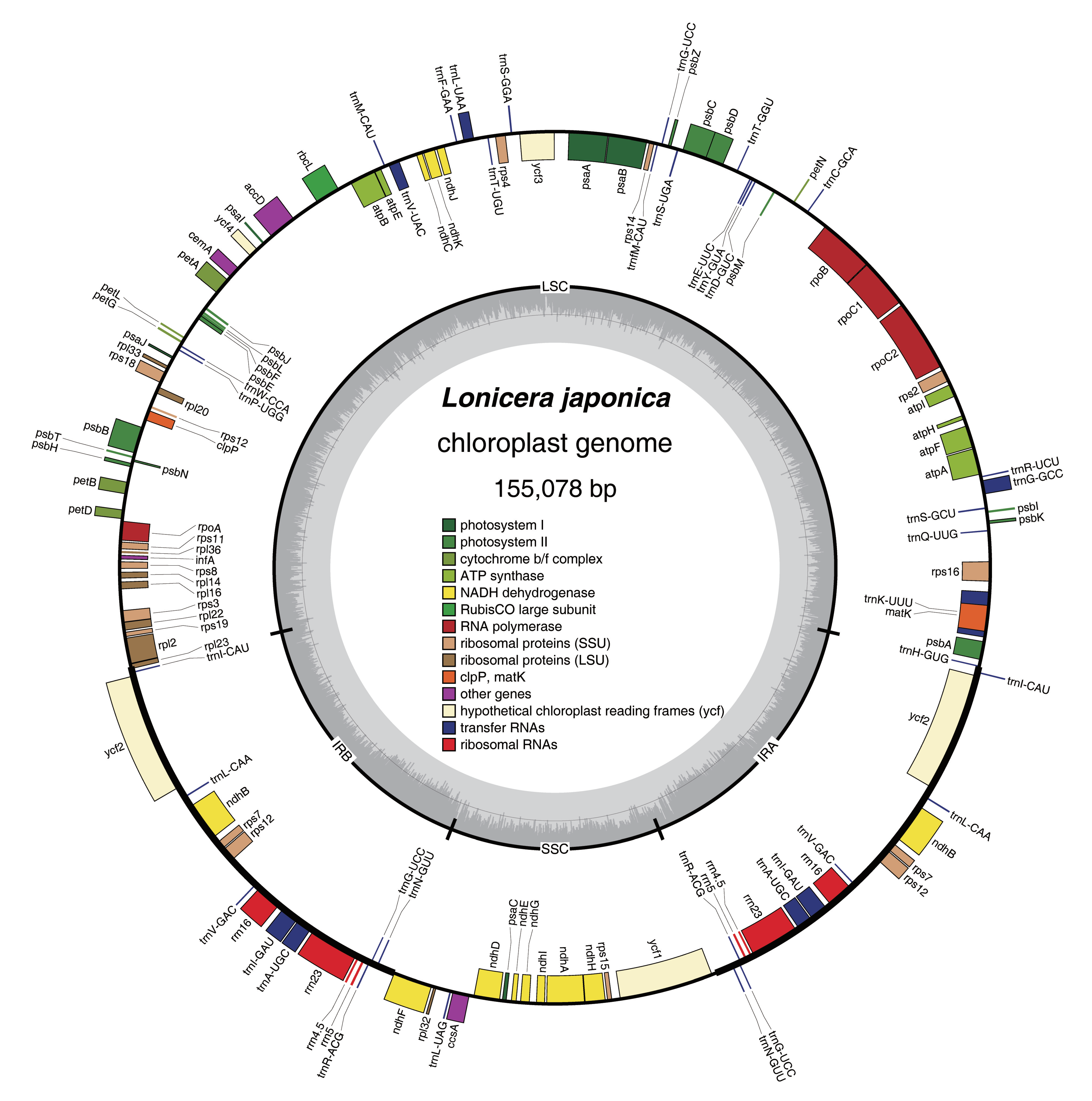
2.1. Characteristics of L. japonica cpDNA
2.2. Intron Gain and Loss
2.3. Comparison with Other cp Genomes in the Order Apiales
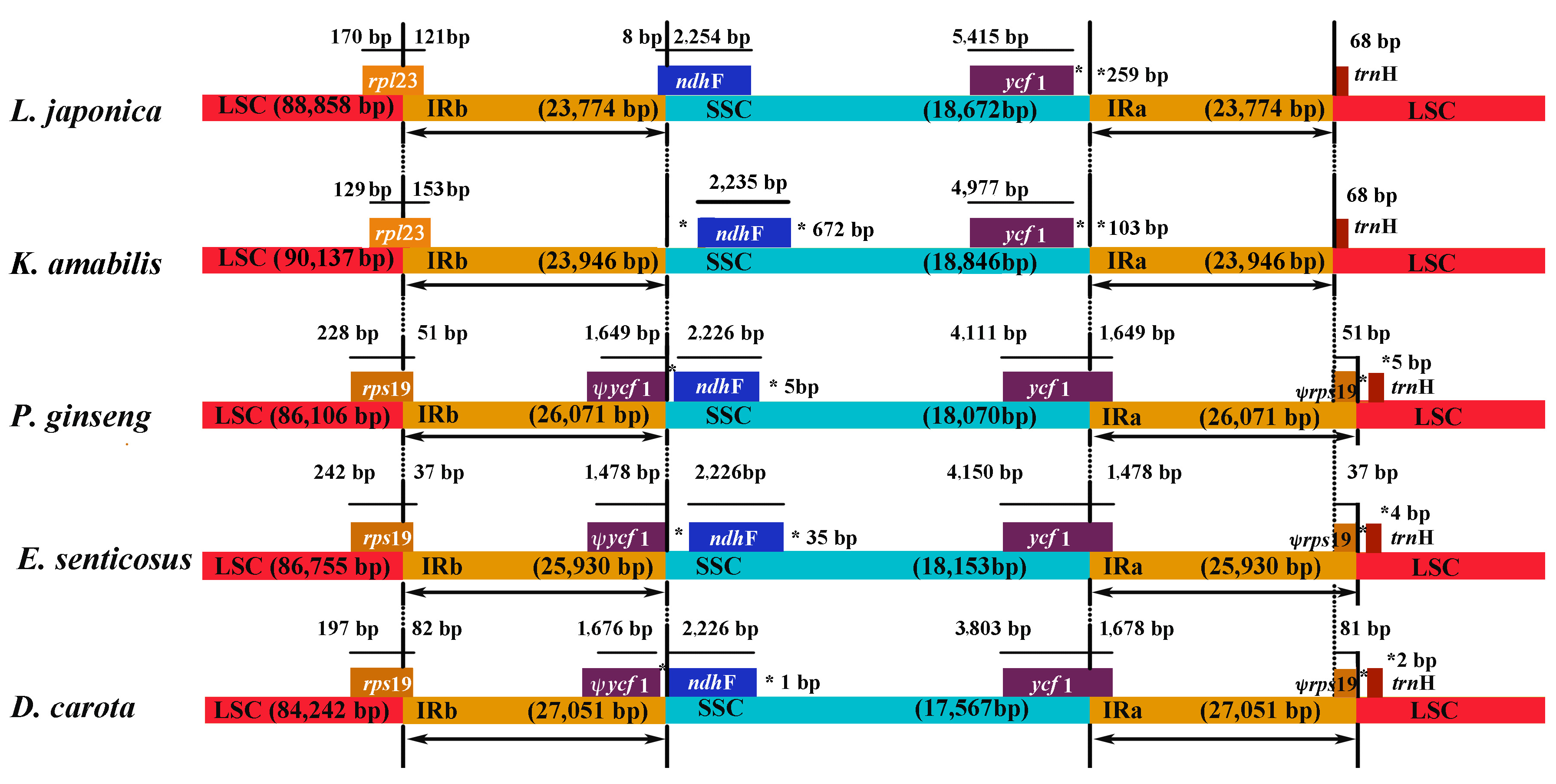
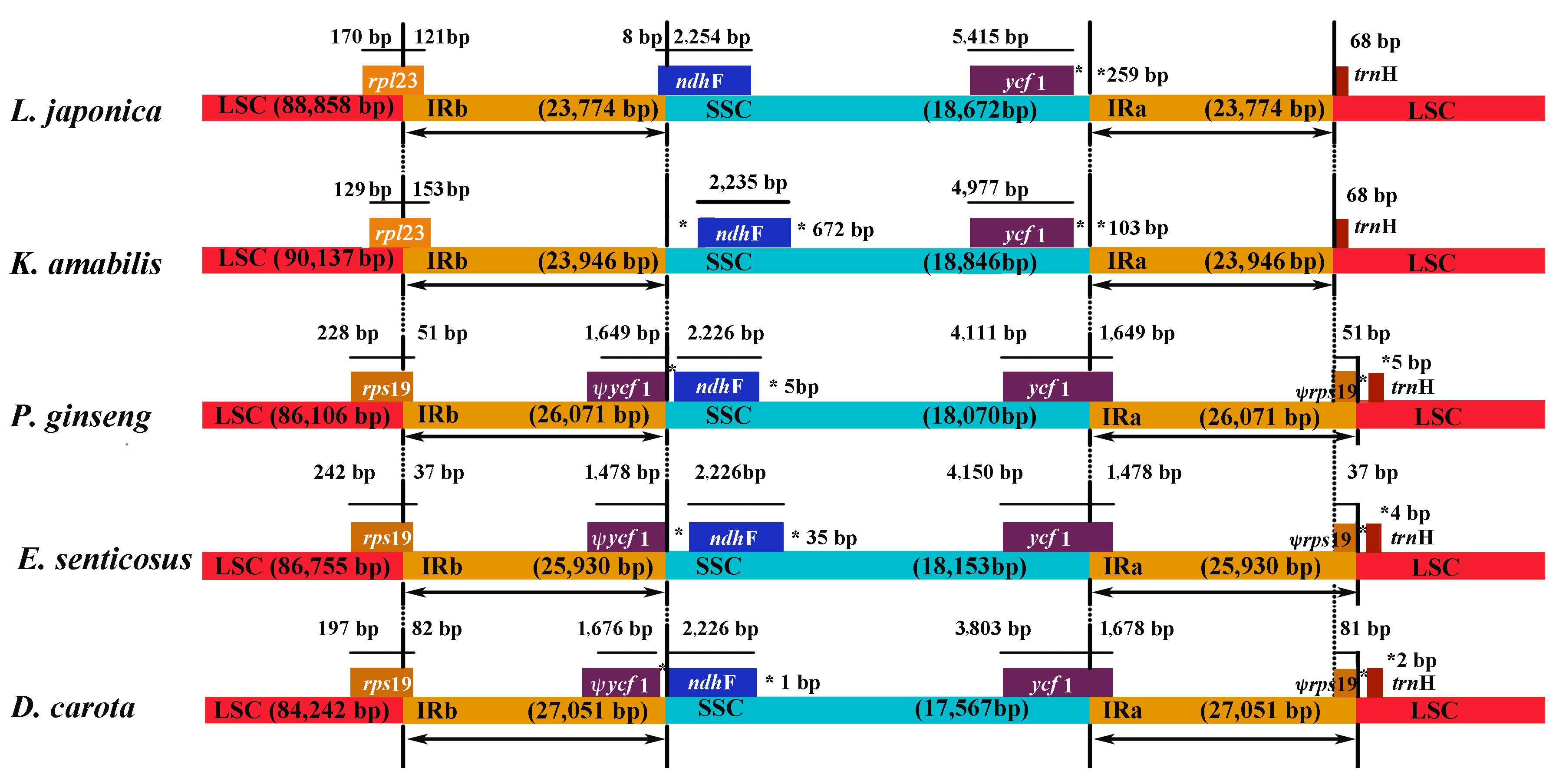
2.4. IR Contraction in the L. japonica cp Genome
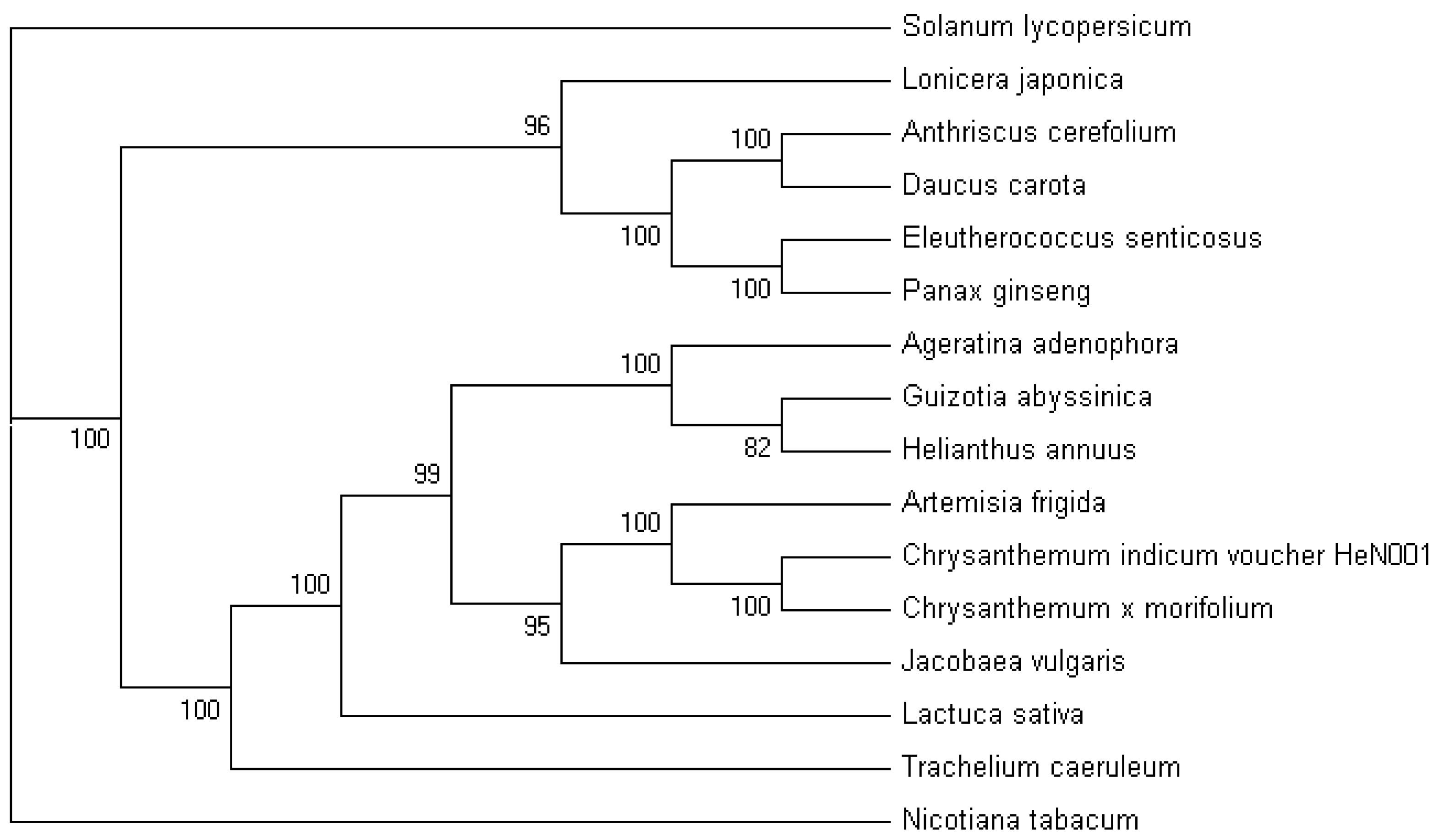
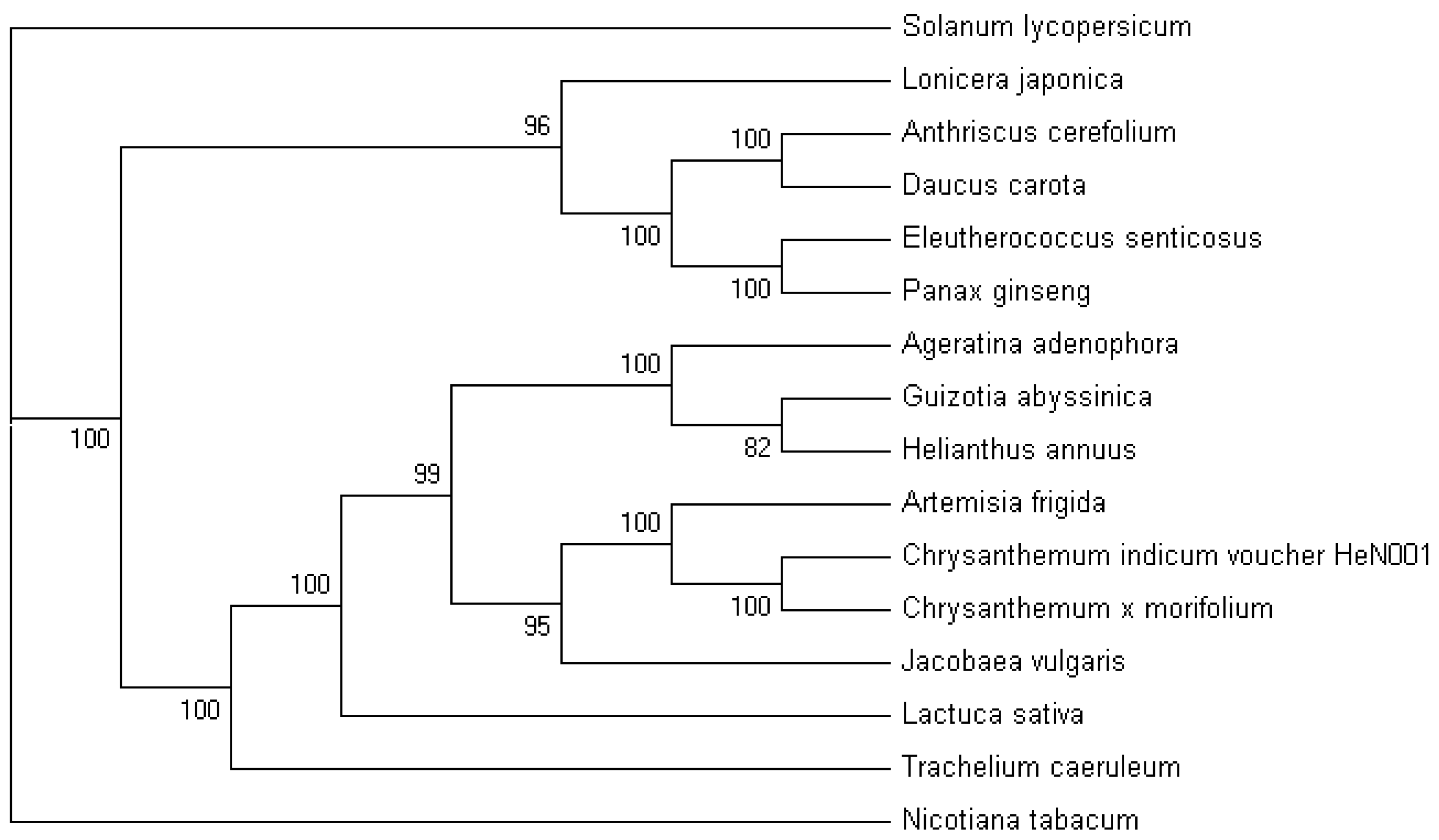
2.5. Phylogenetic Analysis
3. Materials and Methods
3.1. DNA Sequencing, Genome Assembly, and Validation
3.2. Gene Annotation and Sequence Analyses
3.3. Genome Comparison
3.4. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, J.; Zhao, X.Z.; Qi, Q.; Tao, L.; Zhao, Q.; Mu, R.; Gu, H.Y.; Wang, M.; Feng, X.; Guo, Q.L. Macranthoside B, a hederagenin saponin extracted from Lonicera macranthoides and its anti-tumor activities in vitro and in vivo. Food Chem. Toxicol. 2009, 47, 1716–1721. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.M.; Wang, T.Z.; Wang, Z.X. Studies on chemical constituents in dried buds of Lonicera similis Hemsl. Zhongguo Zhong Yao Za Zhi 2001, 26, 45–47. [Google Scholar] [PubMed]
- Tang, D.; Li, H.J.; Li, P.; Wen, X.D.; Qian, Z.M. Interaction of bioactive components caffeoylquinic Acid derivatives in Chinese medicines with bovine serum albumin. Chem. Pharm. Bull. 2008, 56, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Tseng, C.P.; Tsai, K.C.; Lin, C.F.; Wen, C.Y.; Tsay, H.S.; Sakamoto, N.; Tseng, C.H.; Cheng, J.C. Bioactivity-guided screening identifies pheophytin a as a potent anti-hepatitis C virus compound from Lonicera hypoglauca Miq. Biochem. Biophys. Res. Commun. 2009, 385, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.F.; Pan, H.; Li, M.X.; Miao, X.L.; Ding, H. Lonicera japonica Thunb.: Ethnopharmacology, phytochemistry and pharmacology of an important traditional Chinese medicine. J. Ethnopharmacol. 2011, 138, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Muluye, R.A.; Bian, Y.H.; Alemu, P.N. Anti-inflammatory and Antimicrobial effects of Heat-Clearing Chinese herbs: A Current Review. J. Tradit. Complement. Med. 2014, 4, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Schierenbeck, K.A. Japanese Honeysuckle (Lonicera japonica) as an invasive species; history, ecology, and context. Crit. Rev. Plant Sci. 2004, 23, 391–400. [Google Scholar] [CrossRef]
- Yao, H.; Song, J.Y.; Liu, C.; Luo, K.; Han, J.P.; Li, Y.; Pang, X.H.; Xu, H.X.; Zhu, Y.J.; Xiao, P.G.; et al. Use of ITS2 region as the universal DNA barcode for plants and animals. PLoS ONE 2010, 5, e13102. [Google Scholar] [CrossRef] [PubMed]
- Li, X.W.; Yang, Y.; Henry, R.J.; Rossetto, M.; Wang, Y.T.; Chen, S.L. Plant DNA barcoding: From gene to genome. Biol. Rev. 2015, 90, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Schierenbeck, K.A.; Marshall, J.D. Seasonal and diurnal patterns of photosynthetic gas exchange for Lonicera sempervirens and L. japonica (Caprifoliaceae). Am. J. Bot. 1993, 80, 1292–1299. [Google Scholar] [CrossRef]
- Chen, S.L.; Song, J.Y. Herbgenomics. China J. Chin. Mater. Med. 2016, 41, 3881–3889. [Google Scholar]
- Chen, S.L.; Song, J.Y.; Sun, C.; Xu, J.; Zhu, Y.J.; Verpoorte, R.; Fan, T.P. Herbal genomics: Examining the biology of traditional medicines. Science 2015, 347, S27–S29. [Google Scholar]
- He, L.; Xu, X.L.; Li, Y.; Li, C.F.; Zhu, Y.J.; Yan, H.X.; Sun, Z.Y.; Sun, C.; Song, J.Y.; Bi, Y.A.; et al. Transcriptome analysis of buds and leaves using 454 pyrosequencing to discover genes associated with the biosynthesis of active ingredients in Lonicera japonica Thunb. PLoS ONE 2013, 8, e62922. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Lv, S.; Zhang, Y.; Du, X.; Wang, L.; Biradar, S.S.; Tan, X.; Wan, F.; Weining, S. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 2012, 7, e36869. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.K.; Kim, K.J. Complete chloroplast genome sequences of important oilseed crop Sesamum indicum L. PLoS ONE 2012, 7, e35872. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.K.; Lee, H.L.; Sun, B.Y.; Chung, M.Y.; Kim, K.J. The complete chloroplast DNA sequence of Eleutherococcus senticosus (Araliaceae); comparative evolutionary analyses with other three asterids. Mol. Cells 2012, 33, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Ruhlman, T.; Lee, S.B.; Jansen, R.K.; Hostetler, J.B.; Tallon, L.J.; Town, C.D.; Daniell, H. Complete plastid genome sequence of Daucus carota: Implications for biotechnology and phylogeny of angiosperms. BMC Genom. 2006, 7, 222. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Clegg, M.T.; Gaut, B.S.; Learn, G.H.; Morton, B.R. Rates and patterns of chloroplast DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6795–6801. [Google Scholar] [CrossRef] [PubMed]
- Yue, F.; Cui, L.; de Pamphilis, C.W.; Moret, B.M.; Tang, J. Gene rearrangement analysis and ancestral order inference from chloroplast genomes with inverted repeat. BMC Genom. 2008, 9, S25. [Google Scholar] [CrossRef] [PubMed]
- Haberle, R.C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Extensive rearrangements in the chloroplast genome of Trachelium caeruleum are associated with repeats and tRNA genes. J. Mol. Evol. 2008, 66, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Cai, Z.Q.; Raubeson, L.A.; Daniell, H.; dePamphilis, C.W.; Leebeans-Mack, J.; Müller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Fujimoto, M.; Arimura, S.; Murata, J.; Tsutsumi, N.; Kadowaki, K. Loss of the rpl32 gene from the chloroplast genome and subsequent acquisition of a preexisting transit peptide within the nuclear gene in Populus. Gene 2007, 402, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Downie, S.R.; Llanas, E.; KatzDownie, D.S. Multiple independent losses of the rpoC1 intron in angiosperm chloroplast DNA’s. Syst. Bot. 1996, 21, 135–151. [Google Scholar] [CrossRef]
- Downie, S.R.; Olmstead, R.G.; Zurawski, G.; Soltis, D.E.; Soltis, P.S. Six independent losses of the chloroplast DNA rpl2 intron in dicotyledons: Molecular and phylogenetic implications. Evolution 1991, 45, 1245–1259. [Google Scholar] [CrossRef]
- Graveley, B.R. Alternative splicing: Increasing diversity in the proteomic world. Trends Genet. 2001, 17, 100–107. [Google Scholar] [CrossRef]
- Guisinger, M.M.; Chumley, T.W.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Implications of the plastid genome sequence of Typha (Typhaceae, Poales) for understanding genome evolution in Poaceae. J. Mol. Evol. 2010, 70, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Wurdack, K.J.; Kanagaraj, A.; Lee, S.B.; Saski, C.; Jansen, R.K. The complete nucleotide sequence of the cassava (Manihot esculenta) chloroplast genome and the evolution of atpF in Malpighiales: RNA editing and multiple losses of a group II intron. Theor. Appl. Genet. 2008, 116, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Rogalski, M.; Ruf, S.; Bock, R. Tobacco plastid ribosomal protein S18 is essential for cell survival. Nucleic Acids Res. 2006, 34, 4537–4545. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, E.; Takahashi, Y.; Lemieux, C.; Turmel, M.; Rochaix, J.D. The chloroplast ycf3 and ycf4 open reading frames of Chlamydomonas reinhardtii are required for the accumulation of the photosystem I complex. EMBO J. 1997, 16, 6095–6104. [Google Scholar] [CrossRef] [PubMed]
- Chorev, M.; Carmel, L. The function of introns. Front. Genet. 2012, 3, 55. [Google Scholar] [CrossRef] [PubMed]
- Drescher, A.; Ruf, S.; Calsa, T.; Carrer, H.; Bock, R. The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 2000, 22, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.L.; Jansen, R.K.; Chumley, T.W.; Kim, K.J. Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Mol. Biol. Evol. 2007, 24, 1161–1180. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S.M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Ohme, M.; Tanaka, M.; Wakasugi, T.; Hayashida, N.; Matsubayashi, T.; Zaita, N.; Chunwongse, J.; Obokata, J.; Yamaguchi-Shinozaki, K.; et al. The complete nucleotide sequence of the tobacco chloroplast genome: Its gene organization and expression. EMBO J. 1986, 5, 2043–2049. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.P.; Xu, C.; Cheng, T.; Zhou, S.L. Complete chloroplast genome of Sedum sarmentosum and chloroplast genome evolution in Saxifragales. PLoS ONE 2013, 8, e77965. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Manhart, J.R. The chloroplast rpl23 gene cluster of Spirogyra maxima (Charophyceae) shares many similarities with the Angiosperm rpl23 Operon. Algae 2002, 17, 59–68. [Google Scholar] [CrossRef]
- Lee, J.; Kang, Y.; Shin, S.C.; Park, H.; Lee, H. Combined analysis of the chloroplast genome and transcriptome of the Antarctic vascular plant Deschampsia antarctica Desv. PLoS ONE 2014, 9, e92501. [Google Scholar] [CrossRef] [PubMed]
- Li, X.W.; Hu, Z.G.; Lin, X.H.; Li, Q.; Gao, H.H.; Luo, G.A.; Chen, S.L. High-throughput pyrosequencing of the complete chloroplast genome of Magnolia officinalis and its application in species identification. Acta Pharm. Sin. 2012, 47, 124–130. [Google Scholar]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. Organellar Genome DRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000, 16, 1046–1047. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids. Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods); Version 4.0b10; Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar]
- Sample Availability: Not available.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T(U) (%) | C (%) | A (%) | G (%) | Length (bp) | ||
|---|---|---|---|---|---|---|
| LSC | 32.1 | 19.0 | 30.8 | 18.1 | 88,858 | |
| SSC | 34.3 | 16.9 | 32.3 | 16.5 | 18,672 | |
| IRa | 28.6 | 23.1 | 27.9 | 20.4 | 23,774 | |
| IRb | 27.9 | 20.4 | 28.6 | 23.1 | 23,774 | |
| Total | 31.2 | 19.6 | 30.2 | 19.0 | 155,078 | |
| CDS | 31.3 | 18.1 | 30.0 | 20.6 | 74,724 | |
| 1st position | 23.7 | 19.2 | 30.0 | 27.1 | 24,908 | |
| 2nd position | 32.8 | 20.5 | 28.8 | 17.9 | 24,908 | |
| 3rd position | 37.5 | 14.4 | 31.3 | 16.8 | 24,908 |
| Amino Acid | Codon | No. | RSCU | tRNA | Amino Acid | Codon | No. | RSCU | tRNA |
|---|---|---|---|---|---|---|---|---|---|
| Phe | UUU | 911 | 1.27 | Tyr | UAU | 720 | 1.6 | ||
| Phe | UUC | 527 | 0.73 | trnF-GAA | Tyr | UAC | 182 | 0.4 | trnY-GUA |
| Leu | UUA | 785 | 1.75 | trnL-UAA | Stop | UAA | 44 | 1.63 | |
| Leu | UUG | 570 | 1.27 | trnL-CAA | Stop | UAG | 21 | 0.78 | |
| Leu | CUU | 589 | 1.31 | His | CAU | 448 | 1.54 | ||
| Leu | CUC | 202 | 0.45 | His | CAC | 135 | 0.46 | trnH-GUG | |
| Leu | CUA | 377 | 0.84 | trnL-UAG | Gln | CAA | 666 | 1.5 | trnQ-UUG |
| Leu | CUG | 169 | 0.38 | Gln | CAG | 220 | 0.5 | ||
| Ile | AUU | 972 | 1.42 | Asn | AAU | 857 | 1.5 | ||
| Ile | AUC | 418 | 0.61 | trnI-GAU | Asn | AAC | 284 | 0.5 | trnN-GUU |
| Ile | AUA | 660 | 0.97 | trnI-CAU | Lys | AAA | 942 | 1.43 | trnK-UUU |
| Met | AUG | 582 | 1 | trn(f)M-CAU | Lys | AAG | 371 | 0.57 | |
| Val | GUU | 521 | 1.5 | Asp | GAU | 827 | 1.6 | ||
| Val | GUC | 172 | 0.5 | trnV-GAC | Asp | GAC | 205 | 0.4 | trnD-GUC |
| Val | GUA | 490 | 1.41 | trnV-UAC | Glu | GAA | 902 | 1.44 | trnE-UUC |
| Val | GUG | 204 | 0.59 | Glu | GAG | 354 | 0.56 | ||
| Ser | UCU | 554 | 1.75 | Cys | UGU | 191 | 1.4 | ||
| Ser | UCC | 312 | 0.99 | trnS-GGA | Cys | UGC | 82 | 0.6 | trnC-GCA |
| Ser | UCA | 366 | 1.16 | trnS-UGA | Stop | UGA | 16 | 0.59 | |
| Ser | UCG | 171 | 0.54 | Trp | UGG | 454 | 1 | trnW-CCA | |
| Pro | CCU | 402 | 1.47 | Arg | CGU | 318 | 1.26 | trnR-ACG | |
| Pro | CCC | 209 | 0.77 | Arg | CGC | 95 | 0.38 | ||
| Pro | CCA | 330 | 1.21 | trnP-UGG | Arg | CGA | 367 | 1.45 | |
| Pro | CCG | 150 | 0.55 | Arg | CGG | 113 | 0.45 | ||
| Thr | ACU | 515 | 1.65 | Arg | AGA | 377 | 1.19 | trnR-UCU | |
| Thr | ACC | 226 | 0.72 | trnT-GGU | Arg | AGG | 117 | 0.37 | |
| Thr | ACA | 369 | 1.18 | trnT-UGU | Ser | AGU | 456 | 1.8 | |
| Thr | ACG | 140 | 0.45 | Ser | AGC | 168 | 0.66 | trnS-GCU | |
| Ala | GCU | 603 | 1.76 | Gly | GGU | 544 | 1.27 | ||
| Ala | GCC | 227 | 0.66 | Gly | GGC | 189 | 0.44 | trnG-GCC | |
| Ala | GCA | 388 | 1.13 | trnA-UGC | Gly | GGA | 646 | 1.51 | trnG-UCC |
| Ala | GCG | 155 | 0.45 | Gly | GGG | 331 | 0.77 |
| Gene | Location | Exon I (bp) | Intron I (bp) | Exon II (bp) | Intron II (bp) | Exon III (bp) |
|---|---|---|---|---|---|---|
| atpF | LSC | 411 | 735 | 144 | ||
| ndhA | SSC | 539 | 1093 | 553 | ||
| ndhB | IR | 756 | 679 | 777 | ||
| rpl2 | IR | 434 | 661 | 394 | ||
| ycf2 | IR | 498 | 249 | 6105 | ||
| rpoC1 | LSC | 430 | 775 | 1619 | ||
| rps12 * | LSC | 114 | - | 232 | 536 | 26 |
| rps16 | LSC | 230 | 869 | 40 | ||
| rps18 | LSC | 39 | 63 | 202 | 309 | 47 |
| trnA-UGC | IR | 35 | 809 | 38 | ||
| trnG-UCC | LSC | 47 | 717 | 23 | ||
| trnI-GAU | IR | 35 | 947 | 37 | ||
| trnK-UUU | LSC | 35 | 2535 | 37 | ||
| trnL-UAA | LSC | 49 | 515 | 36 | ||
| trnV-UAC | LSC | 36 | 565 | 40 | ||
| ycf3 | LSC | 155 | 758 | 226 | 732 | 126 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, L.; Qian, J.; Li, X.; Sun, Z.; Xu, X.; Chen, S. Complete Chloroplast Genome of Medicinal Plant Lonicera japonica: Genome Rearrangement, Intron Gain and Loss, and Implications for Phylogenetic Studies. Molecules 2017, 22, 249. https://doi.org/10.3390/molecules22020249
He L, Qian J, Li X, Sun Z, Xu X, Chen S. Complete Chloroplast Genome of Medicinal Plant Lonicera japonica: Genome Rearrangement, Intron Gain and Loss, and Implications for Phylogenetic Studies. Molecules. 2017; 22(2):249. https://doi.org/10.3390/molecules22020249
Chicago/Turabian StyleHe, Liu, Jun Qian, Xiwen Li, Zhiying Sun, Xiaolan Xu, and Shilin Chen. 2017. "Complete Chloroplast Genome of Medicinal Plant Lonicera japonica: Genome Rearrangement, Intron Gain and Loss, and Implications for Phylogenetic Studies" Molecules 22, no. 2: 249. https://doi.org/10.3390/molecules22020249