
Synthesis, Anti-Breast Cancer Activity, and Molecular Docking Study of a New Group of Acetylenic Quinolinesulfonamide Derivatives

 and
and
Abstract
:
1. Introduction
2. Results and Discussion
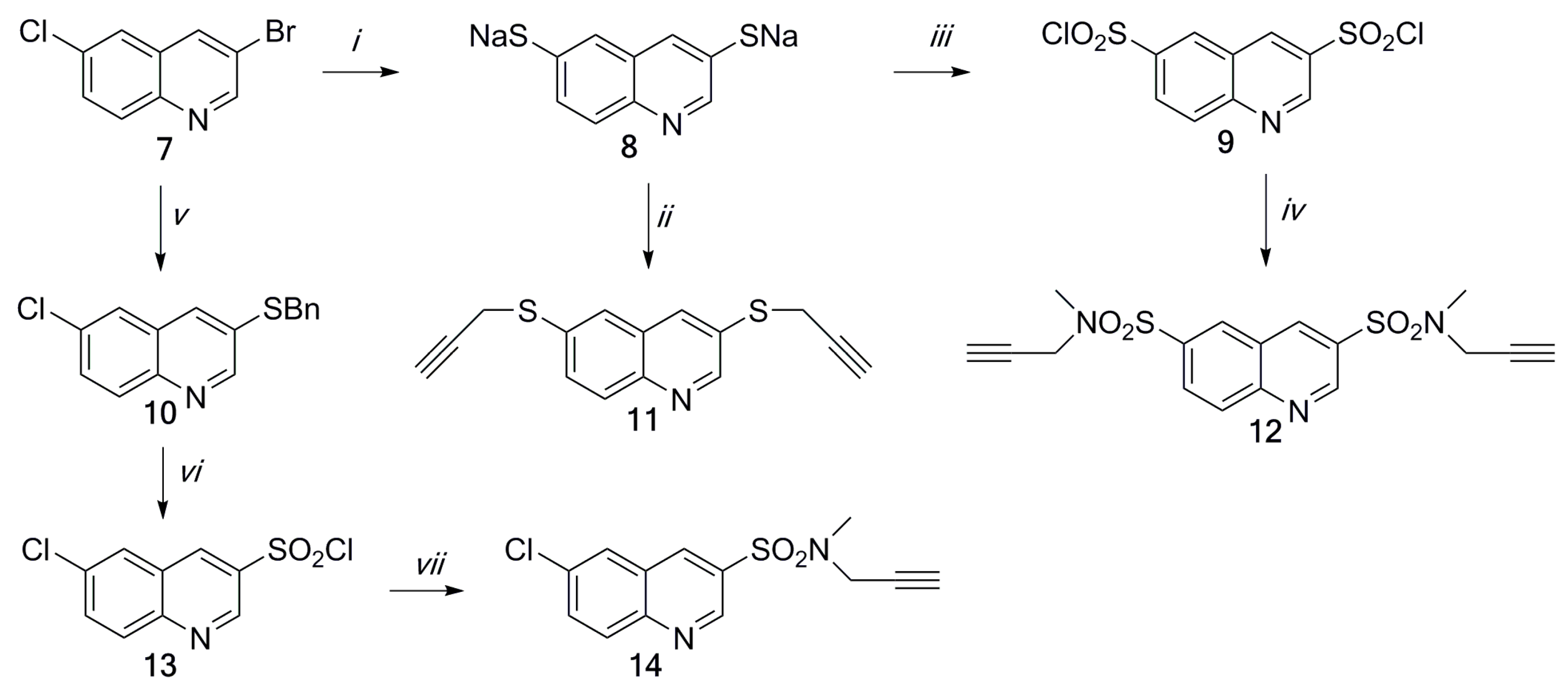
2.1. Chemistry
2.2. Biological Evaluation and SAR Analysis
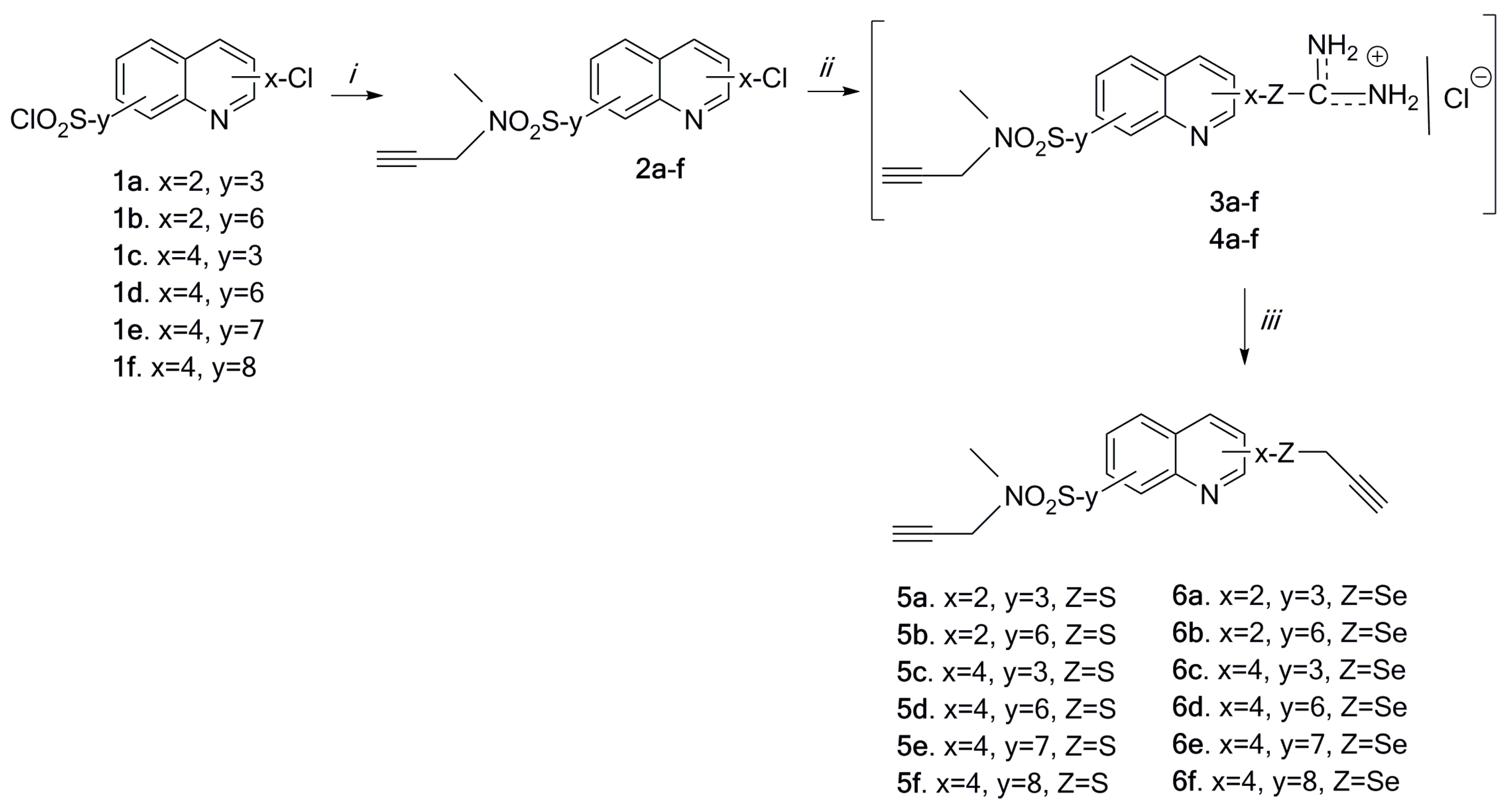
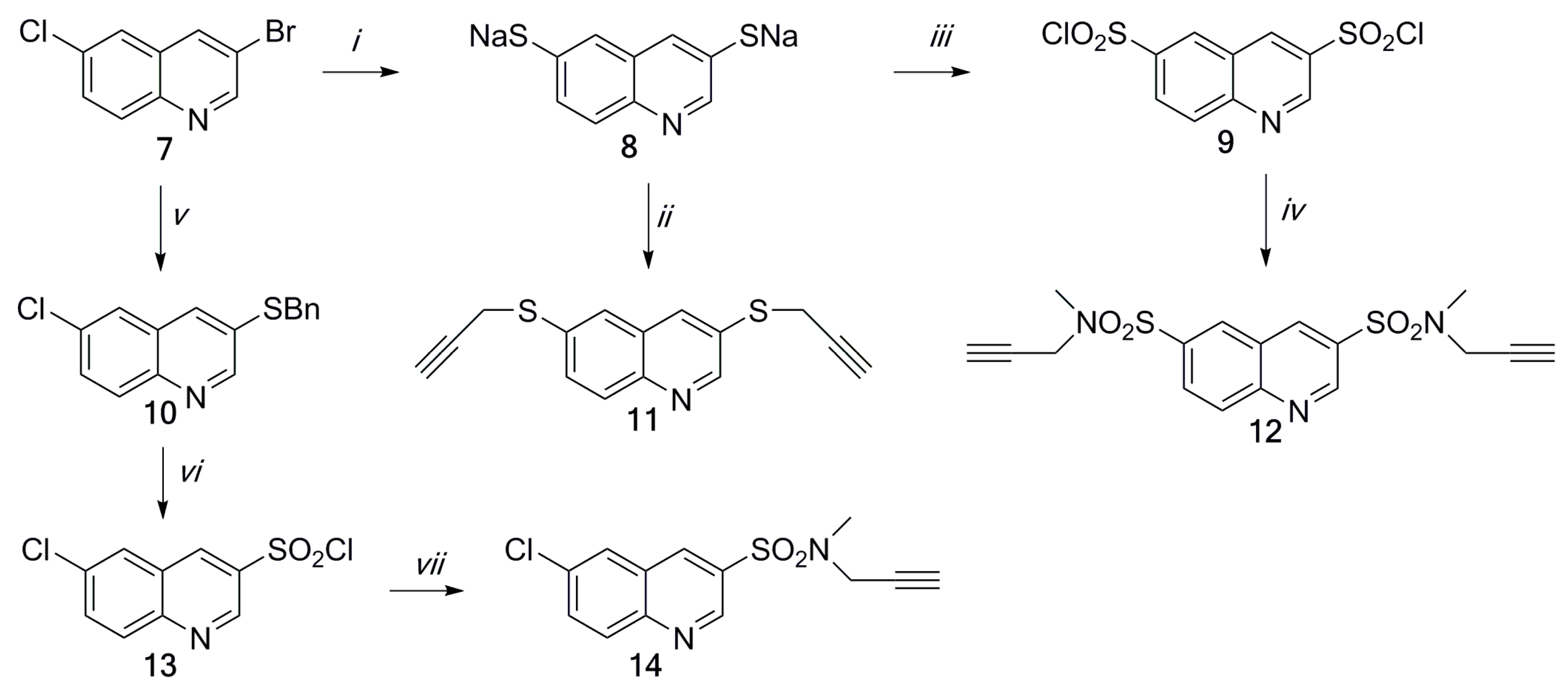
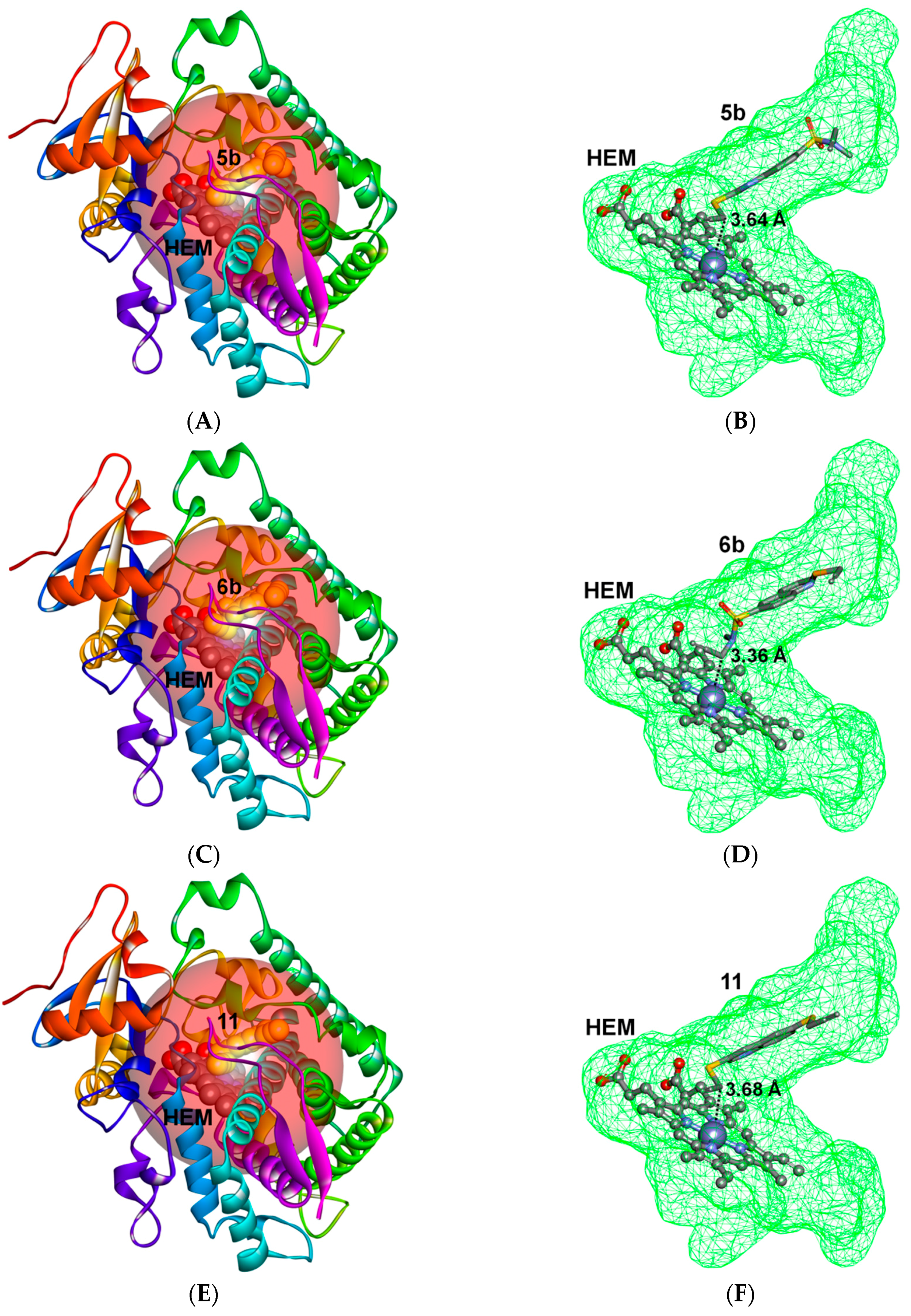
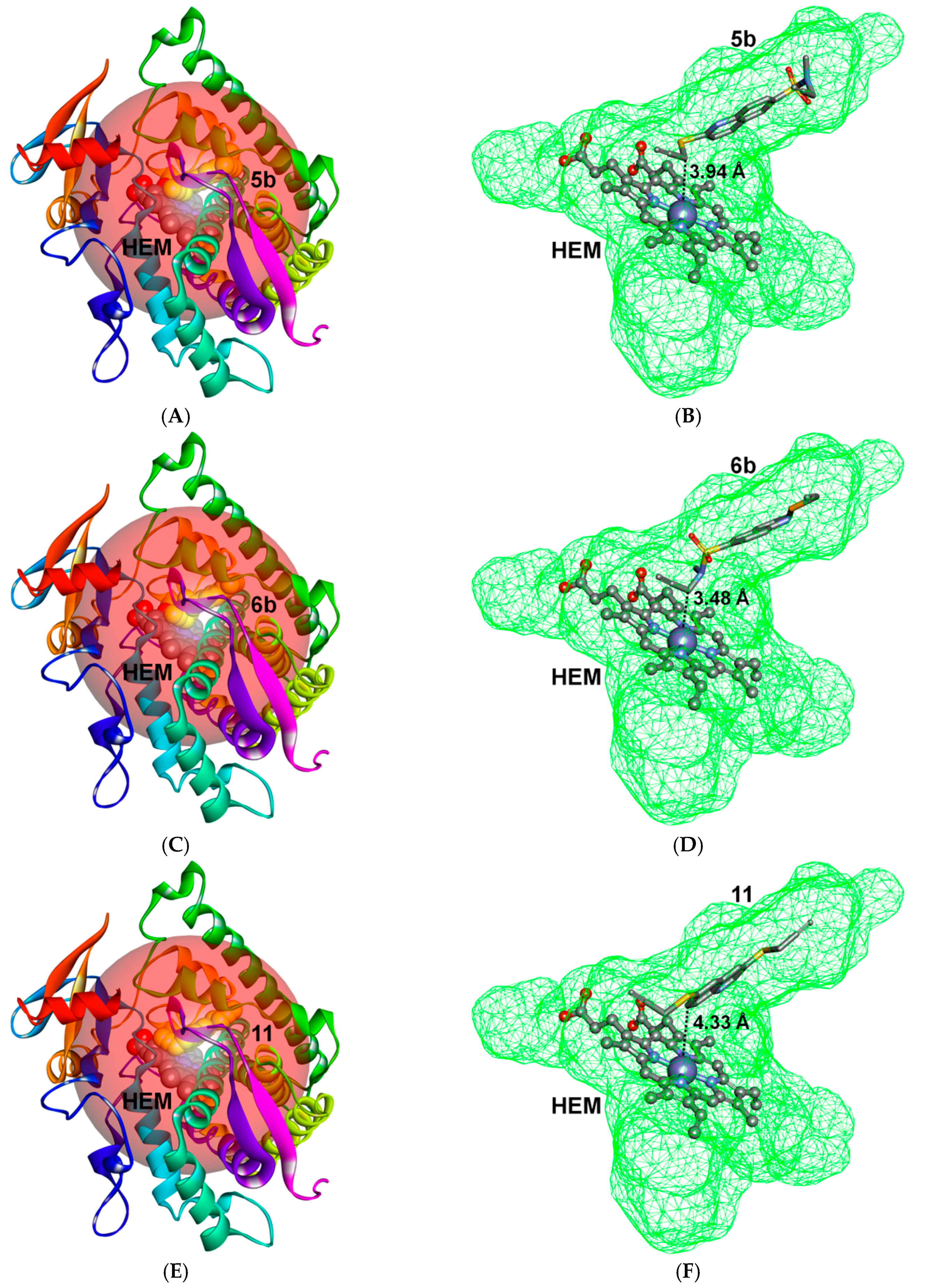
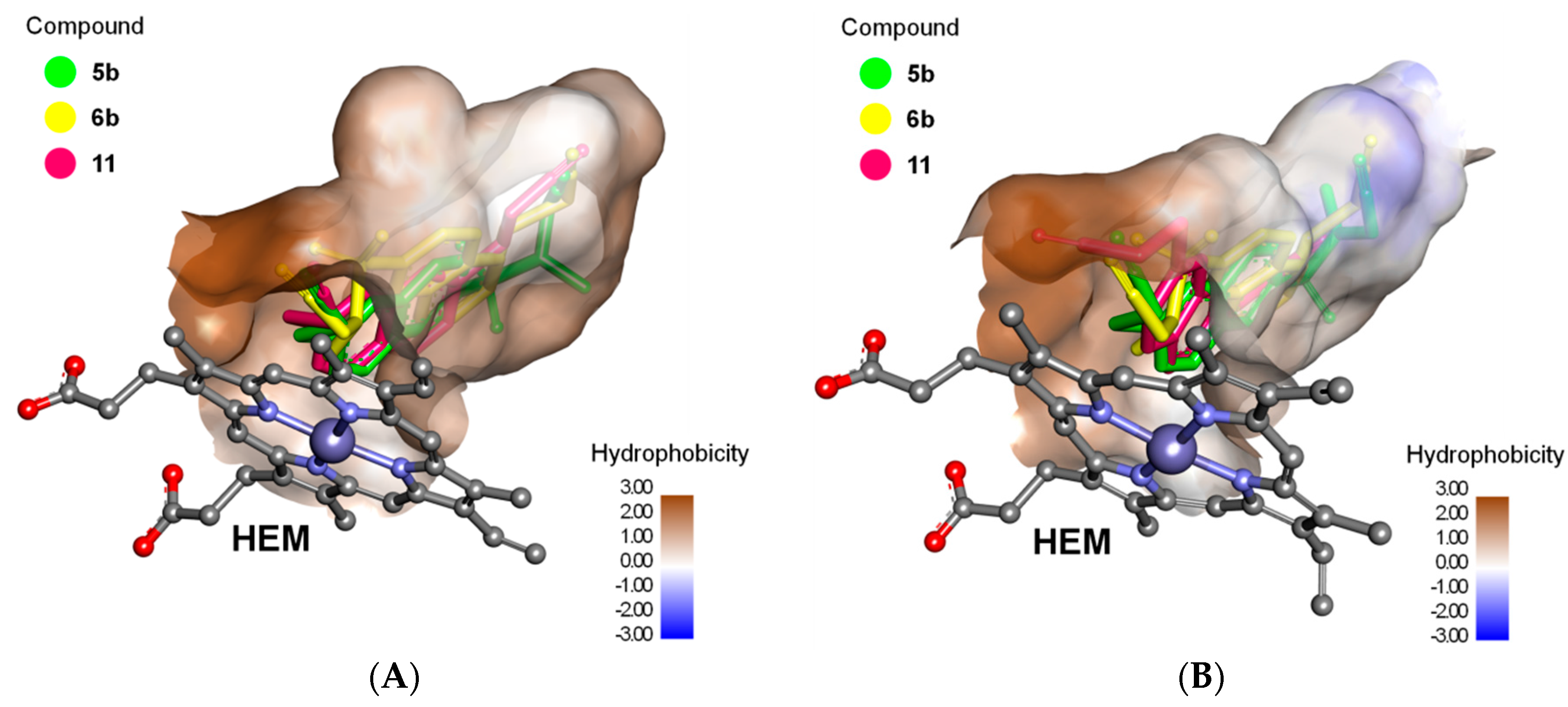
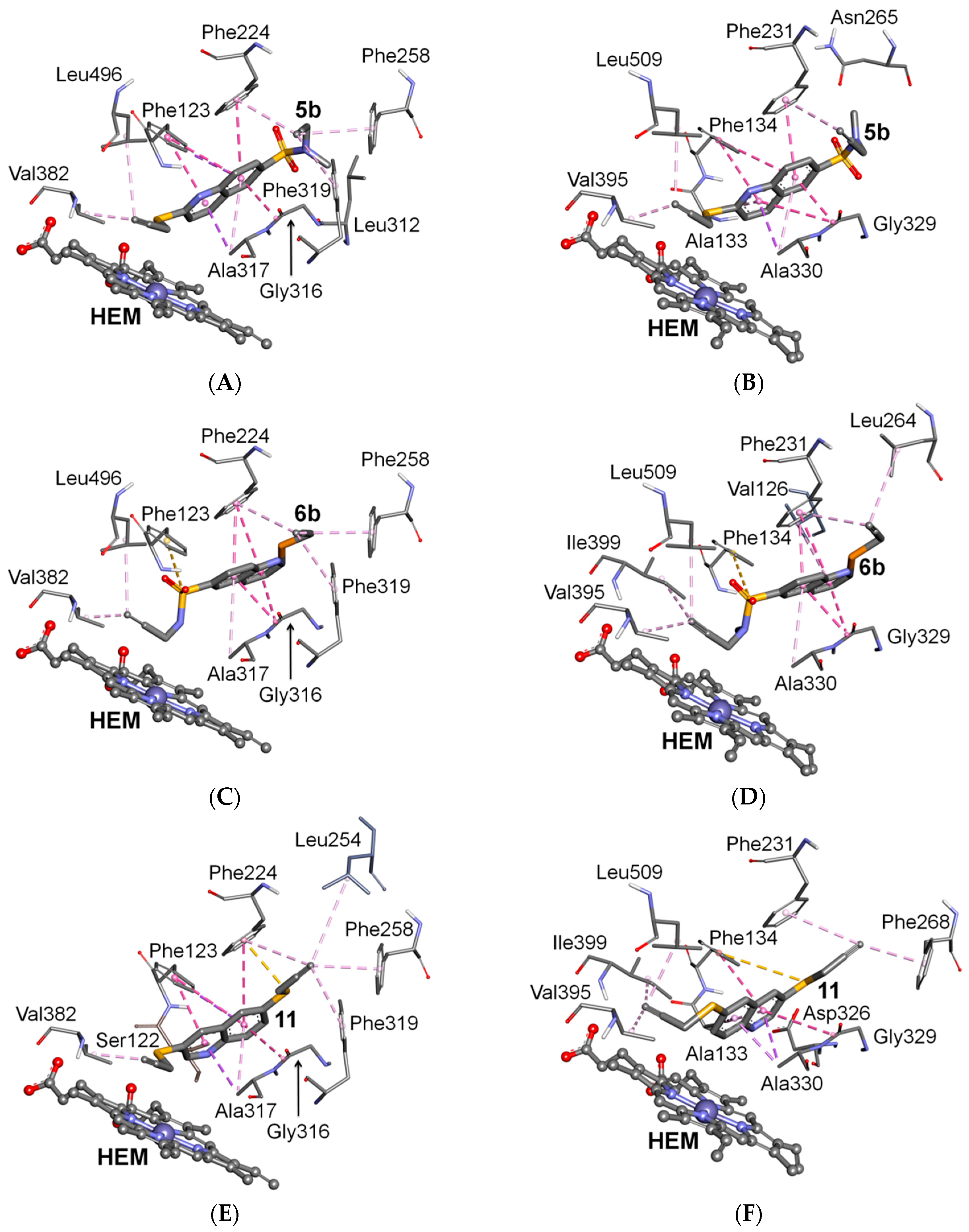
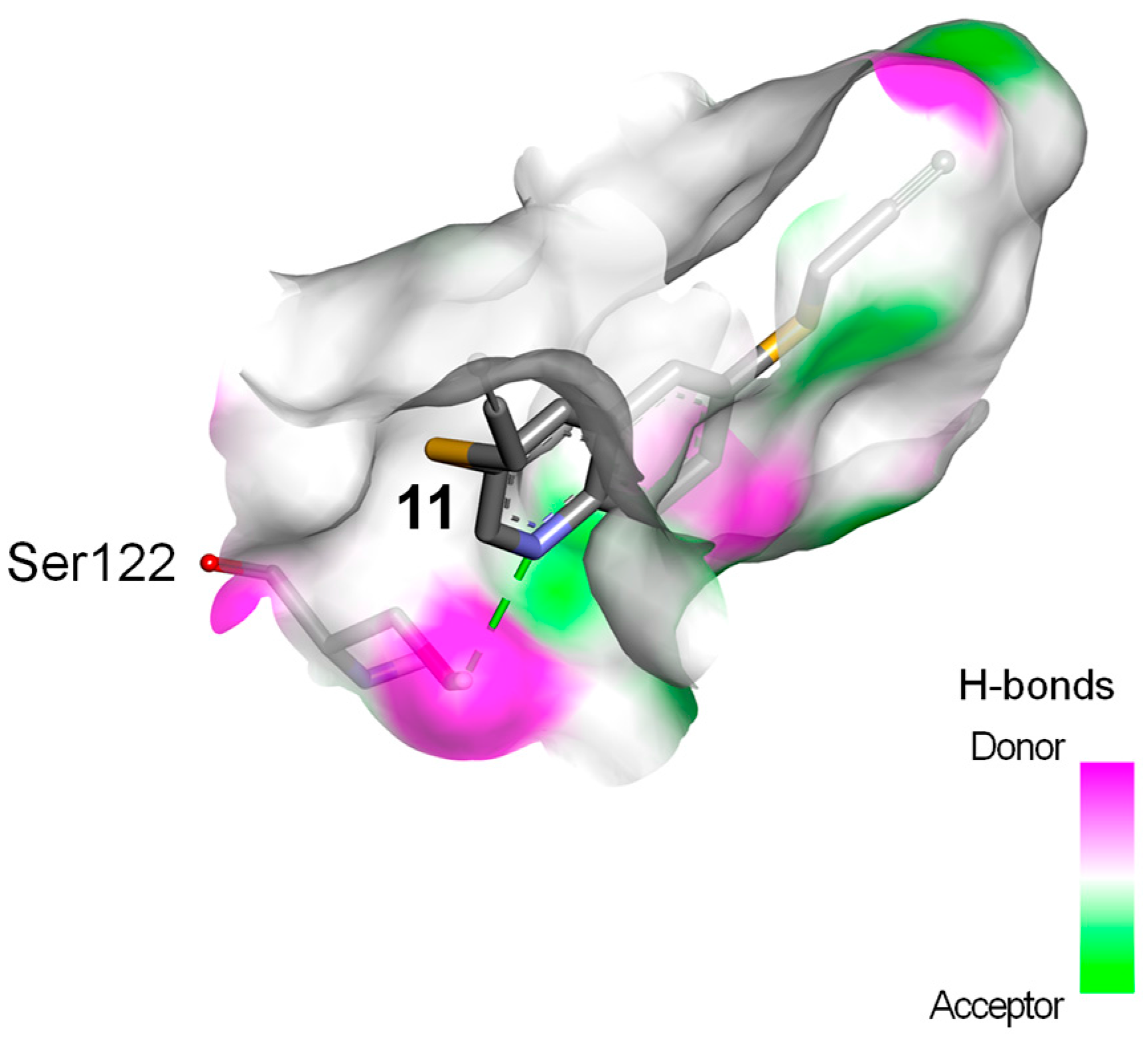
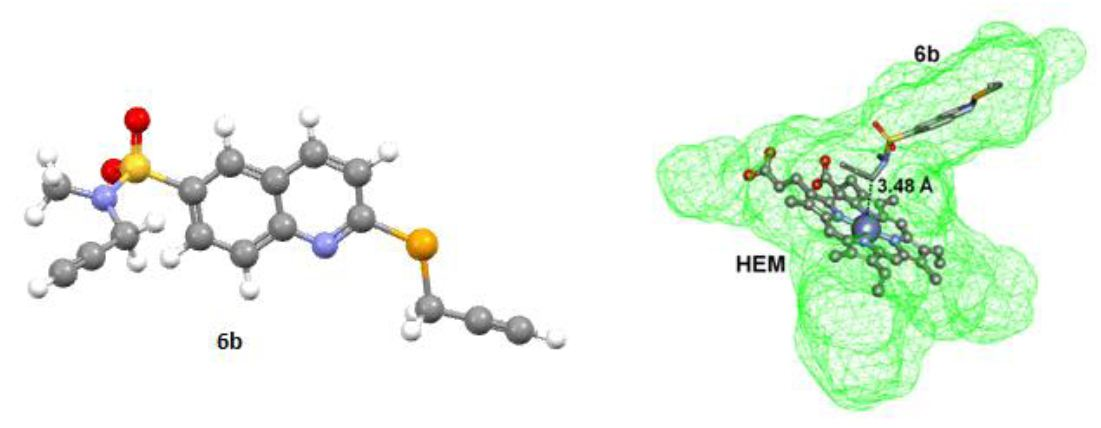
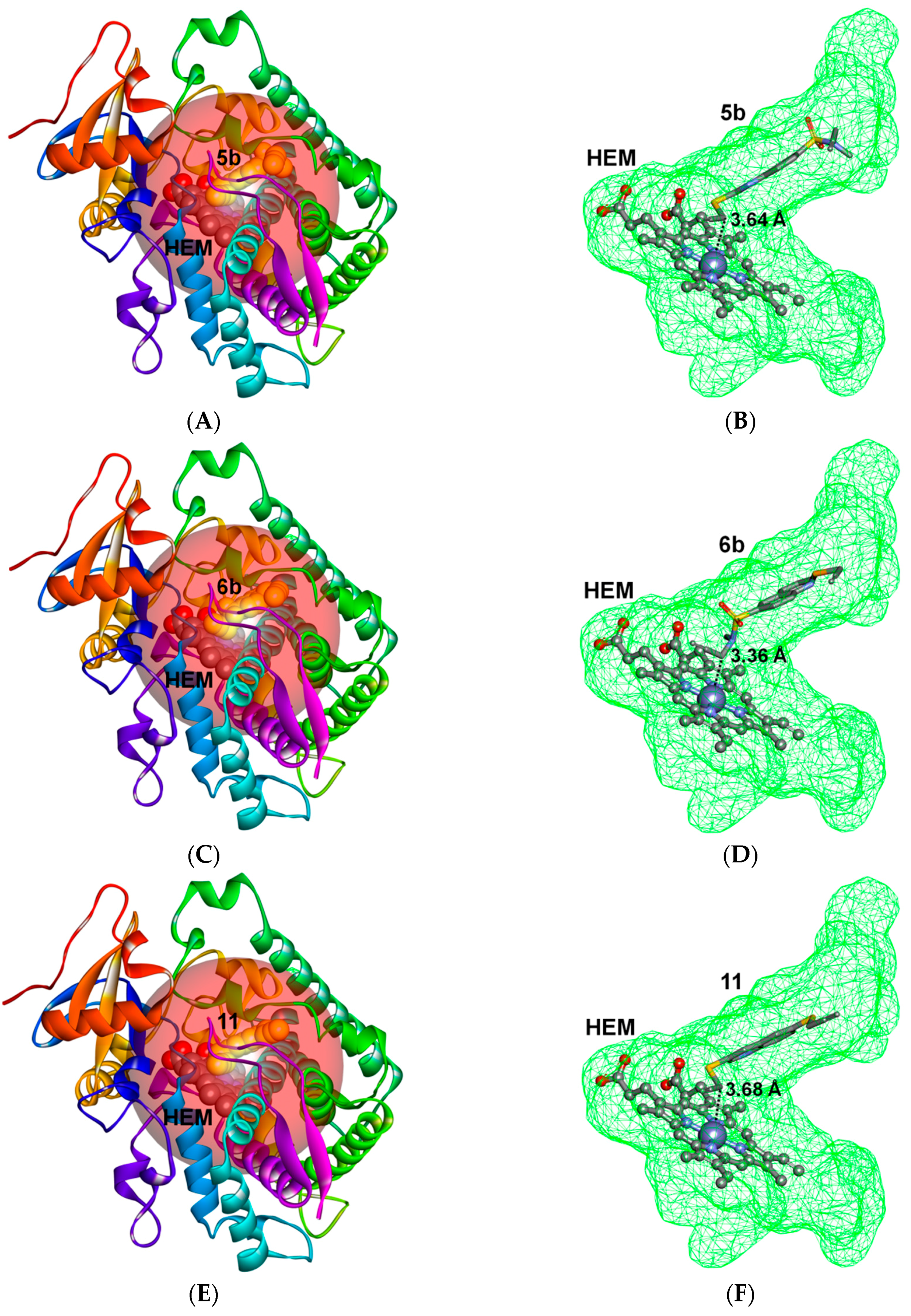
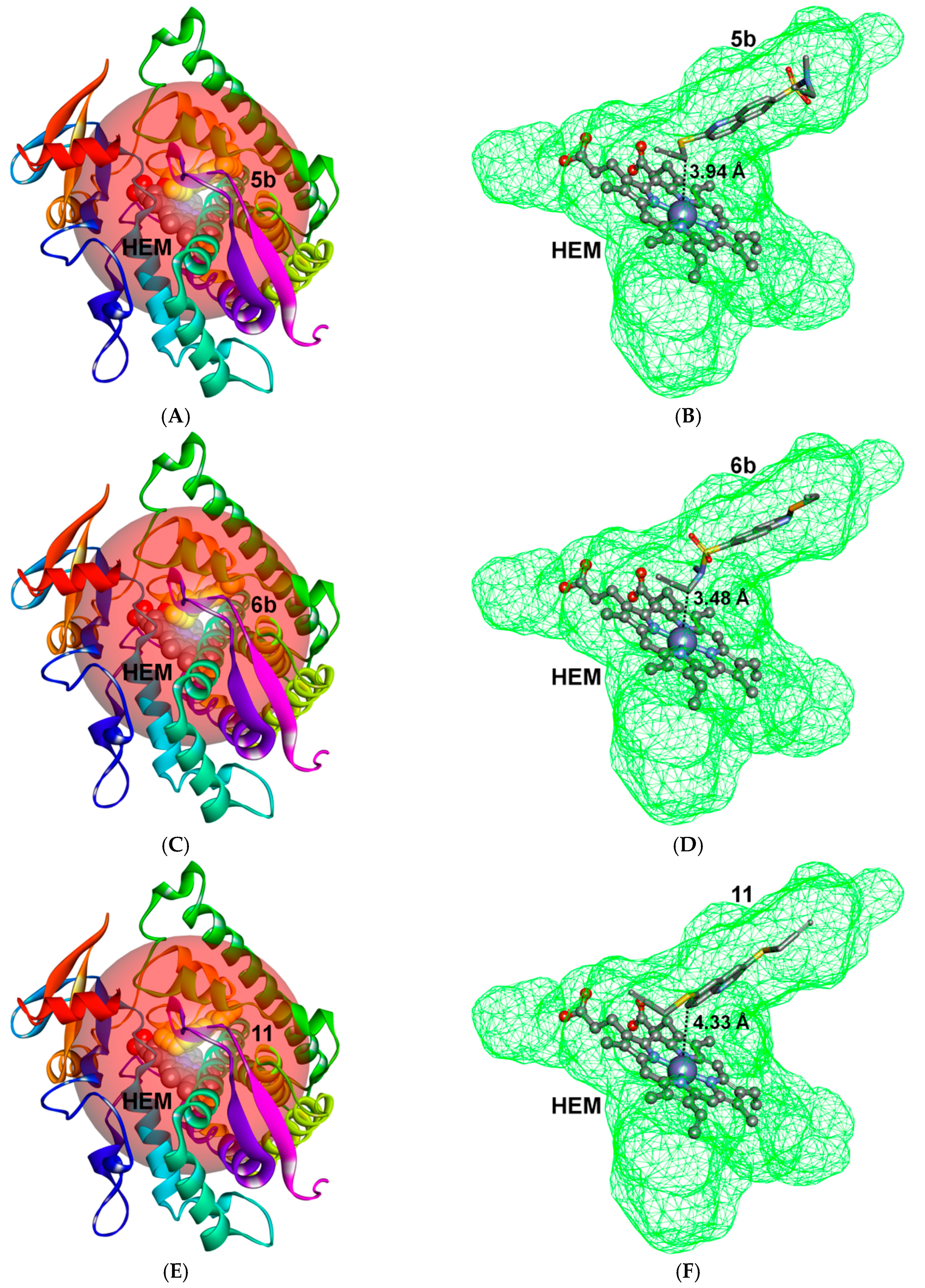
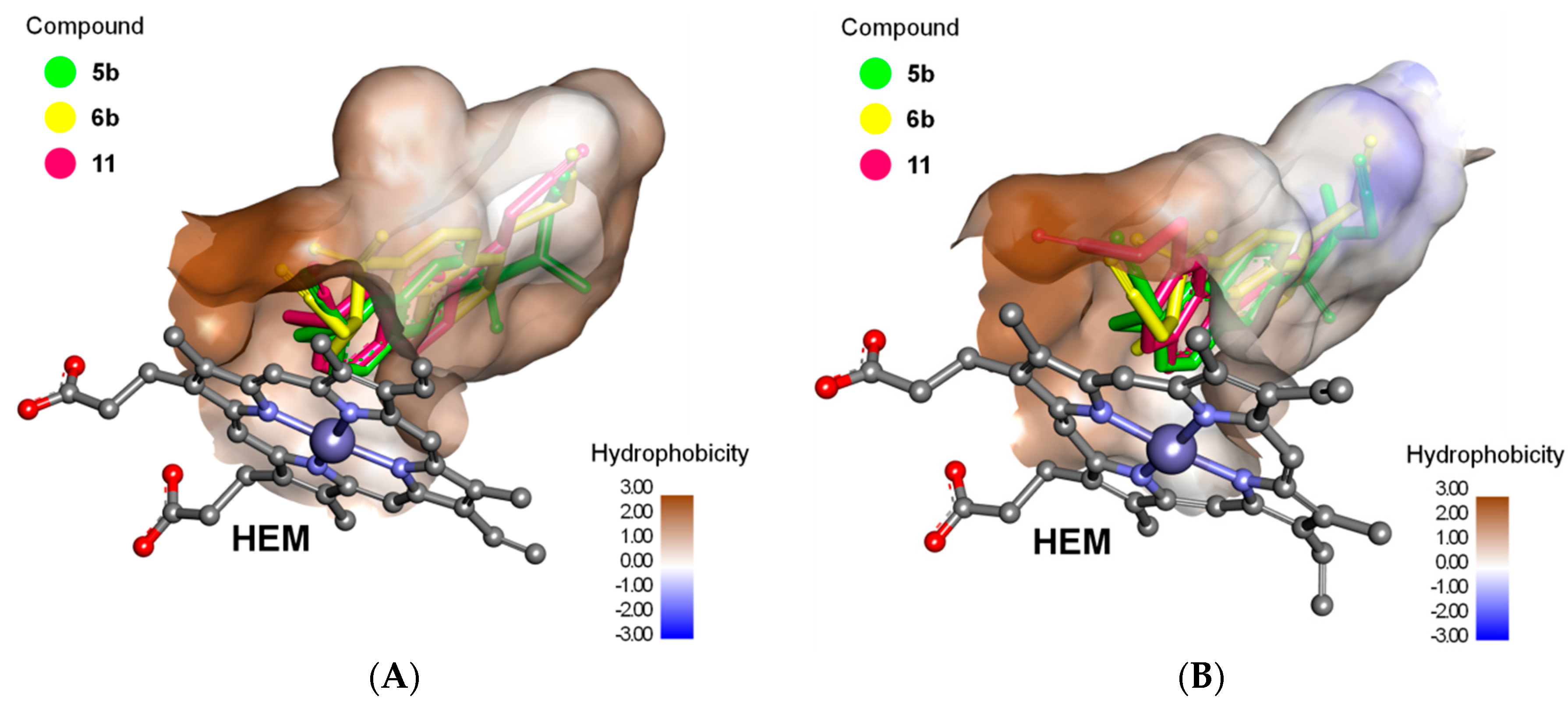
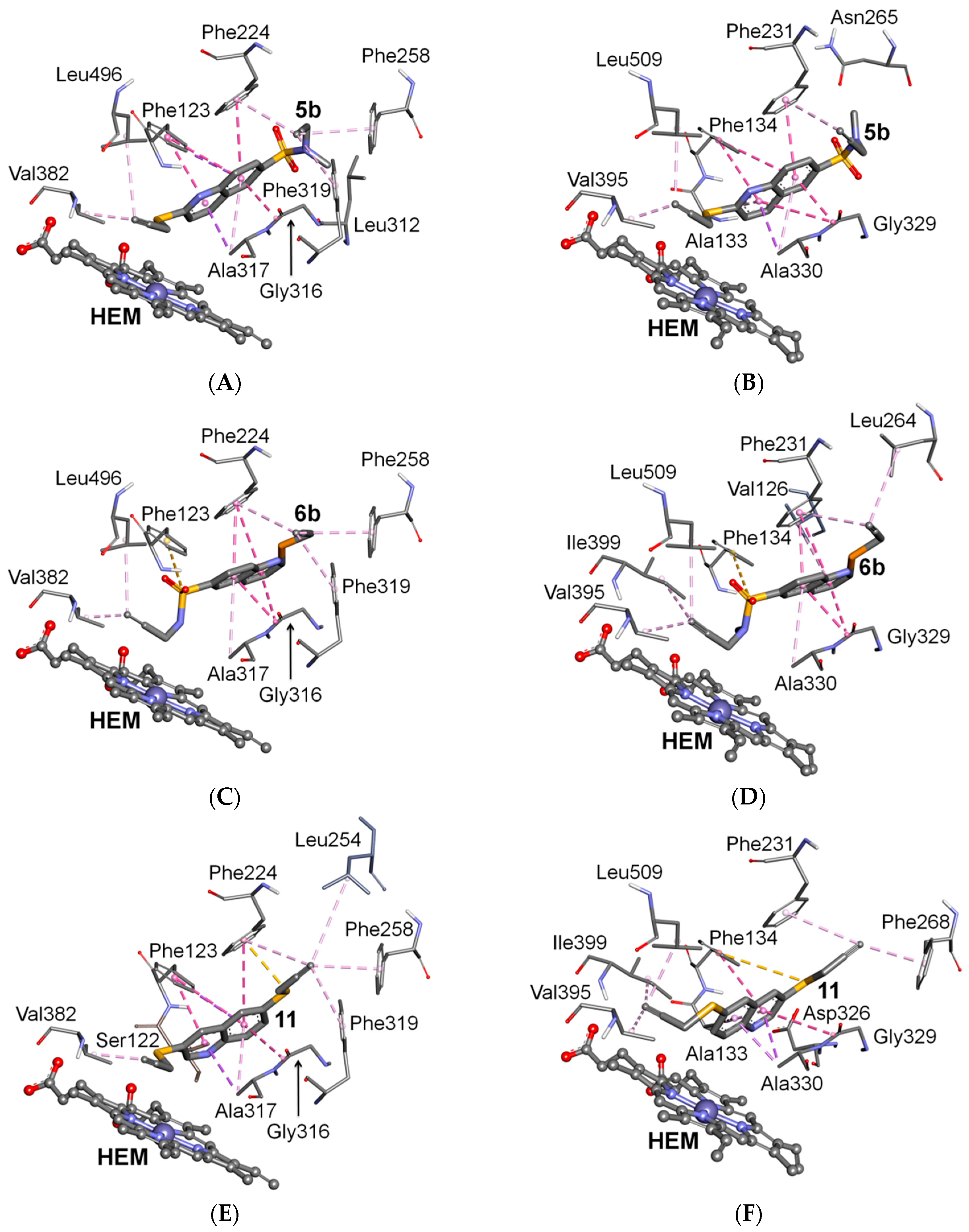
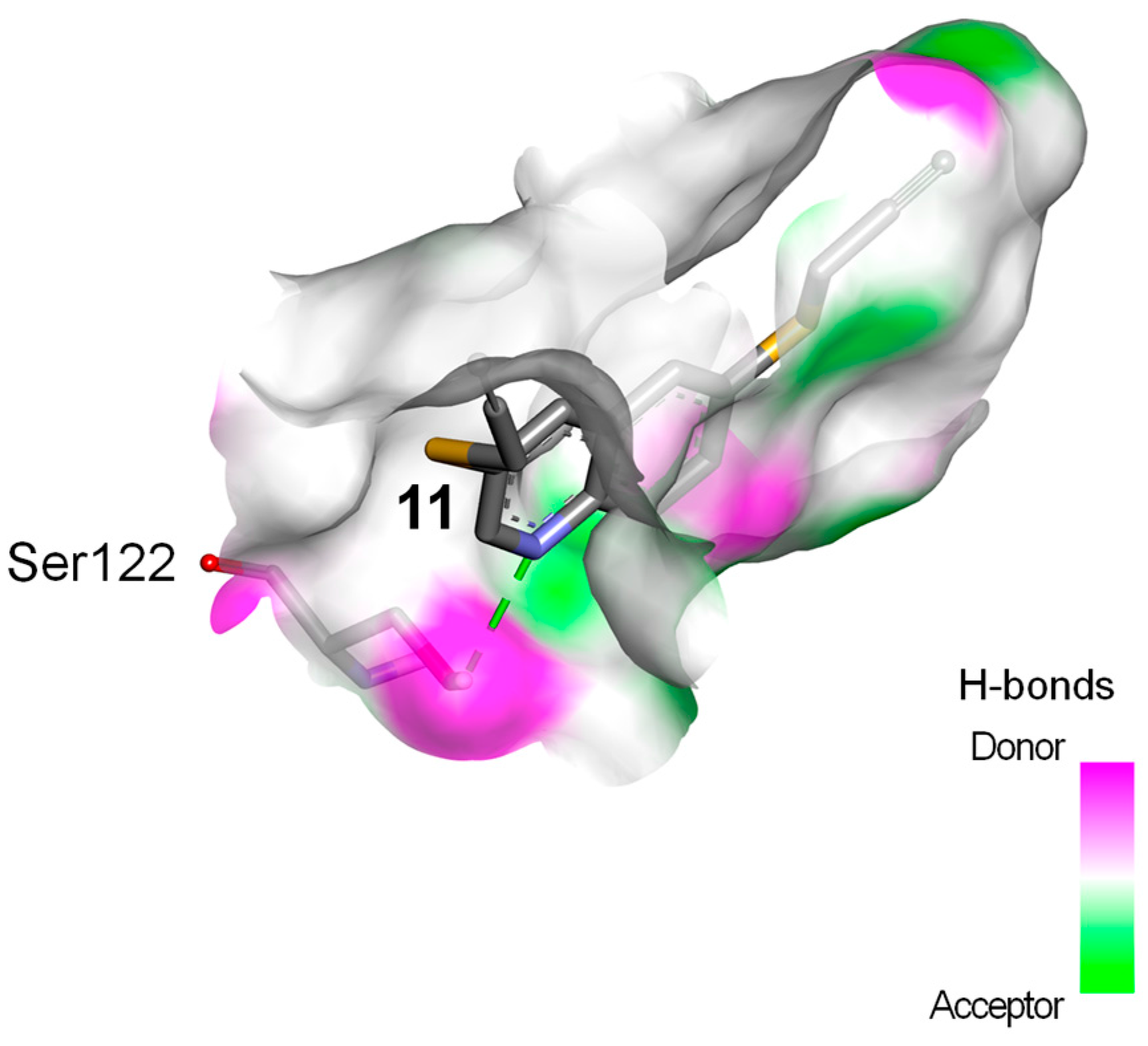
2.3. Docking Study
3. Materials and Methods
3.1. General Techniques
3.2. General Procedure for the Synthesis of Chloro-[N-methyl-N-(3-propynyl)sulfamoyl]quinolines 2a–f and 14
3.3. General Procedure for the Synthesis of Propynylthio- or Propynylseleno-[(N-methyl-N-(3-propynyl)sulfamoyl]quinolines 5a–f and 6a–f
3.4. Procedure for the Synthesis of 3,6-Di(3-propynylsulfanyl)quinoline (11)
3.5. Procedure for the Synthesis of 3,6-Di[N-methyl-N-(3-propynyl)sulfamoyl]quinoline (12)
3.6. Procedure for the Synthesis of 6-Chloro-3-quinolinesulfochloride (13)
3.6.1. Procedure for the Synthesis of 6-Chloro-3-phenylmethylsulfanylquinoline (10)
3.6.2. Chlorination of 6-Chloro-3-phenylmethylsulfanylquinoline (10)
3.7. Biological Study
3.8. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Clegg, L.X.; Feuer, E.J.; Midthune, D.N.; Fay, M.P.; Hankey, B.F. Impact of reporting delay and reporting error on cancer incidence rates and trends. J. Natl. Cancer Inst. 2002, 94, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- McGuire, A.; Brown, J.A.L.; Malone, C.; McLaughlin, R.; Kerin, M.J. Effects of age on the detection and management of breast cancer. Cancers 2015, 7, 908–929. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, M.; Bary, P.G.; Hawley, S.R.; Ward, S.A.; Park, B.K. 4-Aminoquinolines-past, present, and future; A chemical perspective. Pharmacol. Ther. 1998, 77, 29–58. [Google Scholar] [CrossRef]
- Afzal, O.; Kumar, S.; Haider, M.R.; Ali, M.R.; Kumar, R.; Jaggi, M.; Bawa, S. A review on anticancer ptential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 2015, 97, 871–910. [Google Scholar] [CrossRef] [PubMed]
- Chung, P.Y.; Bian, Z.X.; Pun, H.Y.; Chan, D.; Chan, A.S.; Chui, C.H.; Tang, J.C.; Lam, K.H. Recent avances in research of natural and synthetic bioactive quinolines. Future Med. Chem. 2015, 7, 947–967. [Google Scholar] [CrossRef] [PubMed]
- Marciniec, K.; Latocha, M.; Boryczka, S.; Kurczab, R. Synthesis, molecular docking study, and evalution of the antiproliferative action of a new group of propargylthio- and propargylselenoquinolines. Med. Chem. Res. 2014, 23, 3468–3477. [Google Scholar] [CrossRef]
- Scozzafava, A.; Owa, T.; Mastrolorenzo, A.; Supuran, C.T. Anticancer and Antiviral Sulfonamide. Curr. Med. Chem. 2003, 10, 925–953. [Google Scholar] [CrossRef] [PubMed]
- Dorn, C.P.; Finke, P.E.; Oates, B.; Budhu, R.J.; Mills, S.G.; MacCoss, M.; Malkowitz, L.; Springer, M.L.; Daugherty, B.L.; Gould, S.L.; et al. Antagonists of the human CCR5 receptor as anti-HIV-1 agents. Part 1: Discovery and initial structure-activity relationships for 1-amino-2-phenyl-4-(piperidin-1-yl)butanes. Bioorg. Med. Chem. Lett. 2001, 11, 259–264. [Google Scholar] [CrossRef]
- Zajdel, P.; Partyka, A.; Marciniec, K.; Bojarski, A.; Pawłowski, M.; Wesołowska, A. Quinoline- and isquinoline-sulfonamide analogs of aripiprazole: Novel antipsychotic agents? Future Med. Chem. 2014, 6, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Borras, J.; Scozzafava, A.; Menabuoni, L.; Mincione, F.; Briganti, F.; Mincione, G.; Supuran, C.T. Carbonic anhydrase inhibitors: Synthesis of water-soluble, topically effective intraocular pressure lowering aromatic/heterocyclic sulfonamides containing 8-quinoline-sulfonyl moieties: Is the tail more important than the ring? Bioorg. Med. Chem. 1999, 7, 2397–2406. [Google Scholar] [CrossRef]
- Galm, U.; Hager, M.H.; van Lanen, S.G.; Ju, J.; Thorson, J.S.; Shen, B. Antitumor antibiotics: Bleomycin, enediynes, and mitomycin. Chem. Rev. 2005, 105, 739–758. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Joshi, M.C.; Kumar, V.; Malhotra, S.V.; Rawat, D.S. Synthesis and anticancer activity of 13-membered cyclic enediynes. Arch. Pharm. 2011, 344, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.C. Synthesis and characterization of novel acyclic asymmetrical and symmetrical endiyne-triazole conjugates. ARKIVOC 2011, 10, 139–147. [Google Scholar]
- Amit, B.; Kumar, R.S.; Basab, R.; Ajoy, B. Synthesis of highly strained enediynes and dienediynes. Curr. Top. Med. Chem. 2008, 8, 487–504. [Google Scholar]
- Gleiter, R.; Merger, R. Synthesis and properties of skipped cyclic CH dienediynes. Tetrahedron Lett. 1990, 31, 1845–1848. [Google Scholar] [CrossRef]
- Boryczka, S.; Wietrzyk, J.; Opolski, A. Synthesis and antiproliferative activity in vitro of new propargyl thioquinolines. Pharmazie 2002, 57, 151–154. [Google Scholar] [PubMed]
- Boryczka, S.; Wietrzyk, J.; Nasulewicz, A.; Pelczynska, M.; Opolski, A. New propargyl thioquinlines-synthesis, antiproliferative activity in vitro and structure–activity relationships. Pharmazie 2002, 57, 733–739. [Google Scholar] [PubMed]
- Boryczka, S.; Jastrzebska, M.; Nowak, M.; Kusz, J.; Wrzalik, R.; Wietrzyk, J.; Matyja, M. Synthesis, X-ray structure and antiproliferative activity of 3-benzylthio-4-propargylselenoquinoline. Med. Chem. Res. 2010, 19, 551–564. [Google Scholar] [CrossRef]
- Boryczka, S.; Mól, W.; Milczarek, M.; Wietrzyk, J.; Bębenek, E. Synthesis and in vitro antiproliferative activity of novel (4-chloro- and 4-acyloxy-2-butynyl)thioquinolines. Med. Chem. Res. 2011, 20, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Boryczka, S.; Kulig, K.; Malawska, B. RP TLC determination of the lipophilicity of anticancer-active propargyl thioquinolines. J. Planar Chromatogr. 2003, 16, 117–120. [Google Scholar] [CrossRef]
- Bajda, M.; Boryczka, S.; Wietrzyk, J.; Malawska, B. Investigation of lipophilicity of anticancer-active thiquinoline derivatives. Biomed. Chromatogr. 2007, 21, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.C.; Angus, W.G.; Brake, P.B.; Eltom, S.E.; Sukow, K.A.; Jefcoate, C.R. Charakterization of CYP1B1 and CYP1A1 expression in human mammary epithelial cells: Role of the aryl hydrocarbon recetor in polycylic aromatic hydrocarbon metabolism. Cancer Res. 1998, 58, 2366–2374. [Google Scholar] [PubMed]
- Shimada, T.; Tanaka, N.K.; Takenaka, S.; Imai, Y.; Hopkins, N.E.; Foroozesh, M.K.; Alworth, W.L.; Yamazaki, H.; Guengerich, F.P.; Komori, M. Interaction of polycyclic aromatic hydrocarbons with human cytchrome P450 1B1 in inhibiting catalytic activity. Chem. Res. Toxicol. 2008, 21, 2313–2323. [Google Scholar] [CrossRef] [PubMed]
- Gajjar, K.; Martin-Hirsch, P.L.; Martin, F.L. CYP1B1 and hormone–induced cancer. Cancer Lett. 2012, 324, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Cai, M.J.; Thomas, P.E.; Conney, A.H.; Zhu, B.T. Characterization of the oxidative matabolites of 15b-estradiol and estrone formed by 15 selectively expressed human cytochrome P450 isoforms. Endocrnology 2003, 144, 3382–3398. [Google Scholar] [CrossRef] [PubMed]
- Bhat, H.K.; Calaf, G.; Hei, T.K.; Loya, T.; Vadgama, J.V. Critical role of oxidative stress in estrogen-induced carcinogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 3913–3918. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, E.; Chakravarti, D.; Guttenplan, J.; Hart, E.; Ingle, J.; Jankowiak, R.; Muti, P.; Rogan, E.; Russo, J.; Santen, R.; et al. Catechol estrogen quinones as initiators of breast and other human cancers: Implictions for biomarkers of susceptibility and cancer prevention. Biochim. Biophys. Acta. 2006, 1766, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Yager, J.D.; Davidson, N.E. Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 2006, 354, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Mense, S.M.; Hei, T.K.; Ganju, R.K.; Bhat, H.K. Phytoestrogens and Breast Cancer Prevention. Environ. Health Perspect. 2008, 116, 426–433. [Google Scholar] [PubMed]
- Nelson, R. Steroidal oestrogens added to list of known human carcinogens. Lancet 2002, 360, 2053. [Google Scholar] [CrossRef]
- Shimada, T.; Yamazaki, H.; Foroozesh, M.; Hopkins, N.E.; Alworth, W.L.; Guengerich, F.P. Selectivity of polycyclic inhibitors for human cytochrome P450s 1A1, 1A2, and 1B1. Chem. Res. Toxicol. 1998, 11, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Marciniec, K.; Maślankiewicz, A. Synthesis of 4- and 7-quinolinesulfonamides from 4,7-dichloroquinoline. Heterocycles 2009, 7, 93–101. [Google Scholar]
- Marciniec, K.; Maślankiewicz, A. From 2,3-, 2,6-, 3,4-, and 4,6-dichloquinolines to isomeric chloroquinlinesulfonyl chlorides. Heterocycles 2010, 81, 305–316. [Google Scholar] [CrossRef]
- Marciniec, K.; Maślankiewicz, A.; Maślankiewicz, M.J. Synthesis of 6- and 8-halogenosubstituted 3-quinoline-sulfonic acid derivatives. J. Heterocycl. Chem. 2015, 52, 1019–1025. [Google Scholar] [CrossRef]
- Walsh, A.A.; Szklarz, G.D.; Scott, E.E. Human cytochrome P450 1A1 structure and utility in understanding drug and xenobiotic metabolism. J. Biol. Chem. 2013, 288, 12932–12943. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Savas, U.; Stout, C.D.; Johnson, E.F. Structural characterization of the complex between apha-naphthoflavone and human cytochrome P450 1B1. J. Biol. Chem. 2011, 286, 5736–5743. [Google Scholar] [CrossRef] [PubMed]
- RCSB Protein Data Bank. Available online: http://www.rcsb.org (accessed on 22 December 2016).
- Vainio, M.J.; Johnson, M.S. Generating conformer ensembles using a multiobjective genetic algorithm. J. Chem. Inf. Model. 2007, 47, 2462–2474. [Google Scholar] [CrossRef] [PubMed]
- Balloon Version 1.6.4.1258. Available online: http://users.abo.fi/mivainio/balloon/download.php (accessed on 22 December 2016).
- CLC Drug Discovery Workbench Version 2.5; CLC Bio, a QIAGEN Company: Aarhus, Denmark, 2015.
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced Protein-Ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Dessault Systemes BIOVIA. Discovery Studio Modeling Environment; Release 2017; Dessault Systemes: San Diego, CA, USA, 2016. [Google Scholar]
- Molegro Molecular Viewer Version 2012.2.5.0; CLC Bio Company: Aarhus, Denmark, 2012.
- Sample Availability: Samples of the compounds 2, 5, 6, 11, 12 and 14 and screening results (in CLC or Mol Files) are available from the authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Human Cell Line/IC50 ± SD [µM] | |||
|---|---|---|---|---|
| MCF-7 | T47D | MDA-MB-231 | HFF-1 | |
| 2a | 351.36 ± 6.32 | neg 1 | 139.82 ± 1.98 | neg |
| 2b | 22.89 ± 1.45 | 0.30 ± 0.03 | 0.24 ± 0.01 | neg |
| 2c | 0.17 ± 0.02 | 2.24 ± 0.11 | 0.27 ± 0.02 | 85.37 ± 1.60 |
| 2d | 233.84 ± 1.56 | 292.04 ± 2.34 | 180.27 ± 2.35 | 425.17 ± 1.46 |
| 2e | 1.29 ± 0.09 | 26.80 ± 0.67 | 59.31 ± 1.56 | neg |
| 2f | 61.22 ± 1.65 | 295.64 ± 2.56 | 204.08 ± 3.10 | 153.40 ± 3.0 |
| 5a | 287.87 ± 3.45 | neg | 11.75 ± 0.87 | neg |
| 5b | 18.36 ± 0.98 | 181.81 ± 3.23 | 0.27 ± 0.03 | neg |
| 5c | neg | 205.36 ± 6.02 | 116.15 ± 2.13 | neg |
| 5d | 236.81 ± 2.34 | neg | 202.00 ± 5.46 | neg |
| 5e | 0.27 ± 0.03 | 0.21 ± 0.01 | 7.12 ± 0.78 | 276.06 ± 2.60 |
| 5f | 13.09 ± 0.11 | 0.27 ± 0.03 | 21.21 ± 0.96 | 366.06 ± 1.46 |
| 6a | 237.93 ± 2.11 | 146.26 ± 1.98 | 0.42 ± 0.01 | neg |
| 6b | 22.72 ± 0.12 | 10.58 ± 0.15 | 1.32 ± 0.12 | 92.85 ± 2.0 |
| 6c | 112.67 | 313.48 ± 7.98 | 17.56 ± 0.87 | neg |
| 6d | 259.25 ± 5.45 | neg | 0.68 ± 0.09 | 330.95 ± 4.0 |
| 6e | 108.75 ± 2.11 | 159.86 ± 3.23 | 23.46 ± 1.02 | neg |
| 6f | 0.26 ± 0.01 | 55.63 ± 1.78 | 64.47 ± 1.98 | 317.46 ± 5.46 |
| 11 | 7.36 ± 0.45 | 126.84 ± 2.01 | 8.73 ± 0.78 | 175.83 ± 3.0 |
| 12 | 3.14 ± 0.34 | 2.50 ± 0.09 | 2.86 ± 0.34 | 375.47 ± 3.0 |
| 14 | 118.29 ± 2.04 | 11.73 ± 1.11 | 26.83 ± 1.45 | 71.76 ± 1.60 |
| Cisplatin | 77.76 ± 0.76 | 30.26 ± 0.34 | 17.63 ± 0.31 | 7.2 ± 0.60 |
| Compound | Interaction Energy (Score) between the Ligand and CYPs (Arbitrary Unit) * | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Active Site of CYP1A1 (Volume 250.37 Å3) | Active Site of CYP1B1 (Volume 214.53 Å3) | |||||||||
| No. | Weight (Da) | Flexible Bonds | Total | H-Bond | Steric | Ligand Penalty | Total | H-Bond | Steric | Ligand Penalty |
| 2a | 294.75 | 4 | −69.57 | 0.00 | −71.29 | 1.72 | −70.02 | 0.00 | −72.00 | 1.97 |
| 2b | 294.75 | 4 | −73.39 | 0.00 | −74.40 | 1.00 | −71.11 | 0.00 | −72.12 | 1.01 |
| 2c | 294.75 | 4 | −68.07 | 0.00 | −69.29 | 1.22 | −71.88 | 0.00 | −73.41 | 1.53 |
| 2d | 294.75 | 4 | −70.40 | 0.00 | −71.62 | 1.22 | −74.76 | 0.00 | −76.36 | 1.61 |
| 2e | 294.75 | 4 | −70.12 | 0.00 | −71.13 | 1.02 | −72.70 | 0.00 | −73.80 | 1.10 |
| 2f | 294.75 | 4 | −72.18 | 0.00 | −73.89 | 1.72 | −74.12 | 0.00 | −75.80 | 1.68 |
| 5a | 330.42 | 7 | −73.22 | 0.00 | −76.14 | 2.92 | −70.11 | 0.00 | −72.24 | 2.14 |
| 5b | 330.42 | 7 | −81.05 | 0.00 | −84.89 | 3.83 | −76.45 | 0.00 | −80.29 | 3.84 |
| 5c | 330.42 | 7 | −77.64 | 0.00 | −80.96 | 3.32 | −73.07 | 0.00 | −76.84 | 3.77 |
| 5d | 330.42 | 7 | −76.83 | 0.00 | −80.24 | 3.40 | −71.88 | 0.00 | −75.08 | 3.20 |
| 5e | 330.42 | 7 | −76.49 | −1.59 | −78.25 | 3.36 | −74.32 | 0.00 | −77.37 | 3.05 |
| 5f | 330.42 | 7 | −75.33 | 0.00 | −78.49 | 3.16 | −76.95 | 0.00 | −79.79 | 2.84 |
| 6a | 377.32 | 7 | −72.57 | 0.00 | −80.45 | 7.87 | −65.20 | 0.00 | −73.18 | 7.98 |
| 6b | 377.32 | 7 | −79.23 | 0.00 | −81.22 | 1.99 | −81.55 | 0.00 | −83.49 | 1.93 |
| 6c | 377.32 | 7 | −80.72 | 0.00 | −83.19 | 2.47 | −70.84 | 0.00 | −72.35 | 1.50 |
| 6d | 377.32 | 7 | −79.98 | 0.00 | −82.45 | 2.47 | −83.69 | 0.00 | −85.93 | 2.25 |
| 6e | 377.32 | 7 | −72.09 | −2.00 | −71.53 | 1.44 | −73.53 | 0.00 | −76.02 | 2.49 |
| 6f | 377.32 | 7 | −79.11 | 0.00 | −81.49 | 2.38 | −80.21 | 0.00 | −82.29 | 2.08 |
| 11 | 269.38 | 6 | −80.80 | −1.88 | −81.63 | 2.71 | −76.50 | 0.00 | −78.79 | 2.29 |
| 12 | 391.46 | 8 | −77.86 | 0.00 | −81.48 | 3.61 | −78.01 | 0.00 | −80.19 | 2.18 |
| 13 | 294.75 | 4 | −68.83 | 0.00 | −70.68 | 1.86 | −73.50 | 0.00 | −74.31 | 0.81 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marciniec, K.; Pawełczak, B.; Latocha, M.; Skrzypek, L.; Maciążek-Jurczyk, M.; Boryczka, S. Synthesis, Anti-Breast Cancer Activity, and Molecular Docking Study of a New Group of Acetylenic Quinolinesulfonamide Derivatives. Molecules 2017, 22, 300. https://doi.org/10.3390/molecules22020300
Marciniec K, Pawełczak B, Latocha M, Skrzypek L, Maciążek-Jurczyk M, Boryczka S. Synthesis, Anti-Breast Cancer Activity, and Molecular Docking Study of a New Group of Acetylenic Quinolinesulfonamide Derivatives. Molecules. 2017; 22(2):300. https://doi.org/10.3390/molecules22020300
Chicago/Turabian StyleMarciniec, Krzysztof, Bartosz Pawełczak, Małgorzata Latocha, Leszek Skrzypek, Małgorzata Maciążek-Jurczyk, and Stanisław Boryczka. 2017. "Synthesis, Anti-Breast Cancer Activity, and Molecular Docking Study of a New Group of Acetylenic Quinolinesulfonamide Derivatives" Molecules 22, no. 2: 300. https://doi.org/10.3390/molecules22020300