
A Computational Investigation of the Substituent Effects on Geometric, Electronic, and Optical Properties of Siloles and 1,4-Disilacyclohexa-2,5-dienes


Abstract
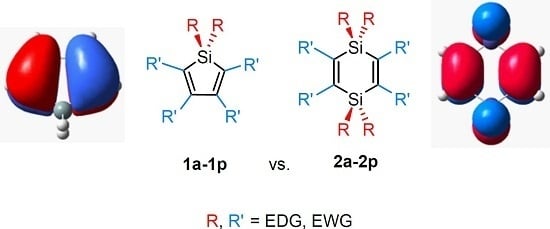
:
1. Introduction
2. Results and Discussion
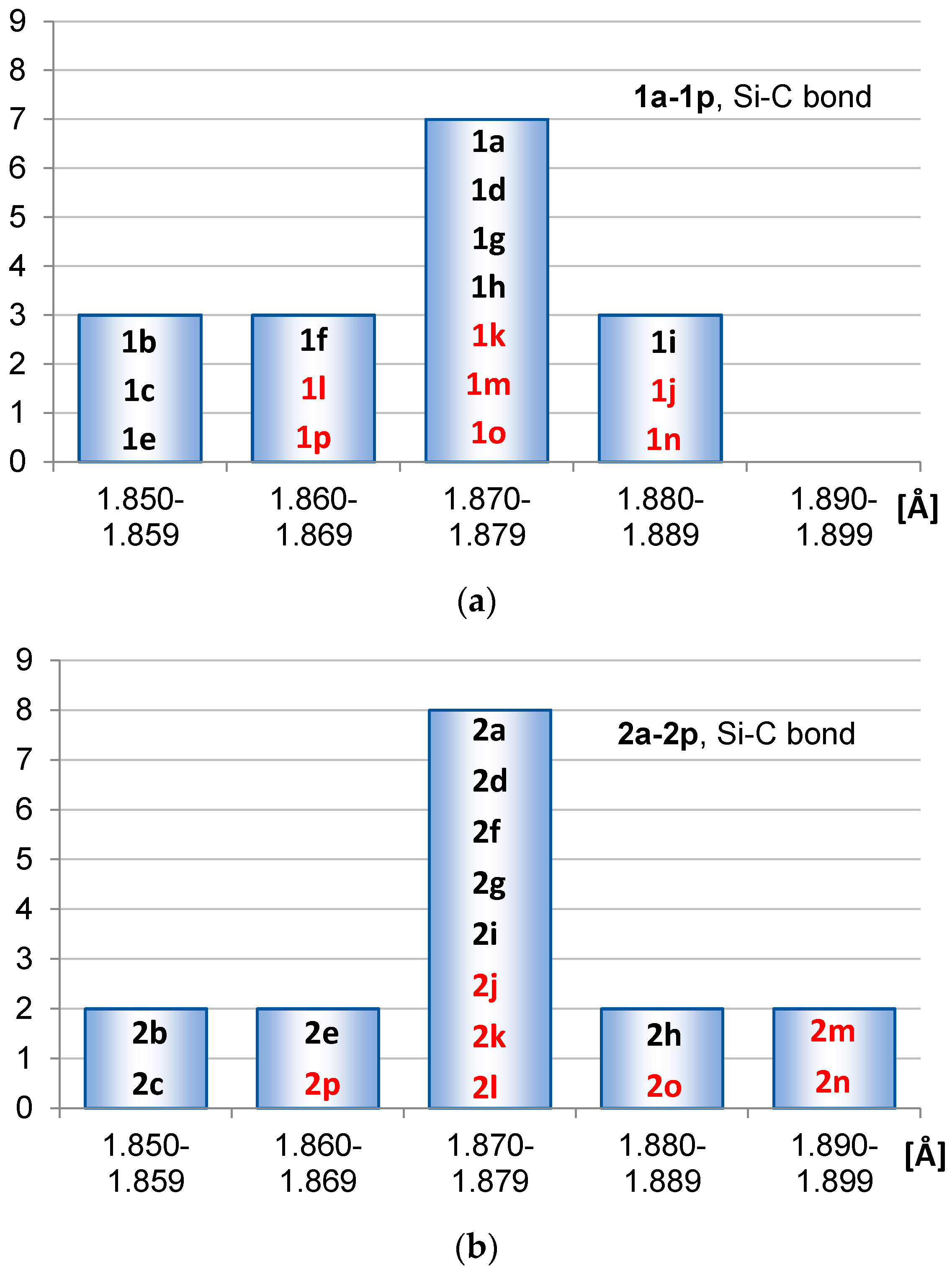
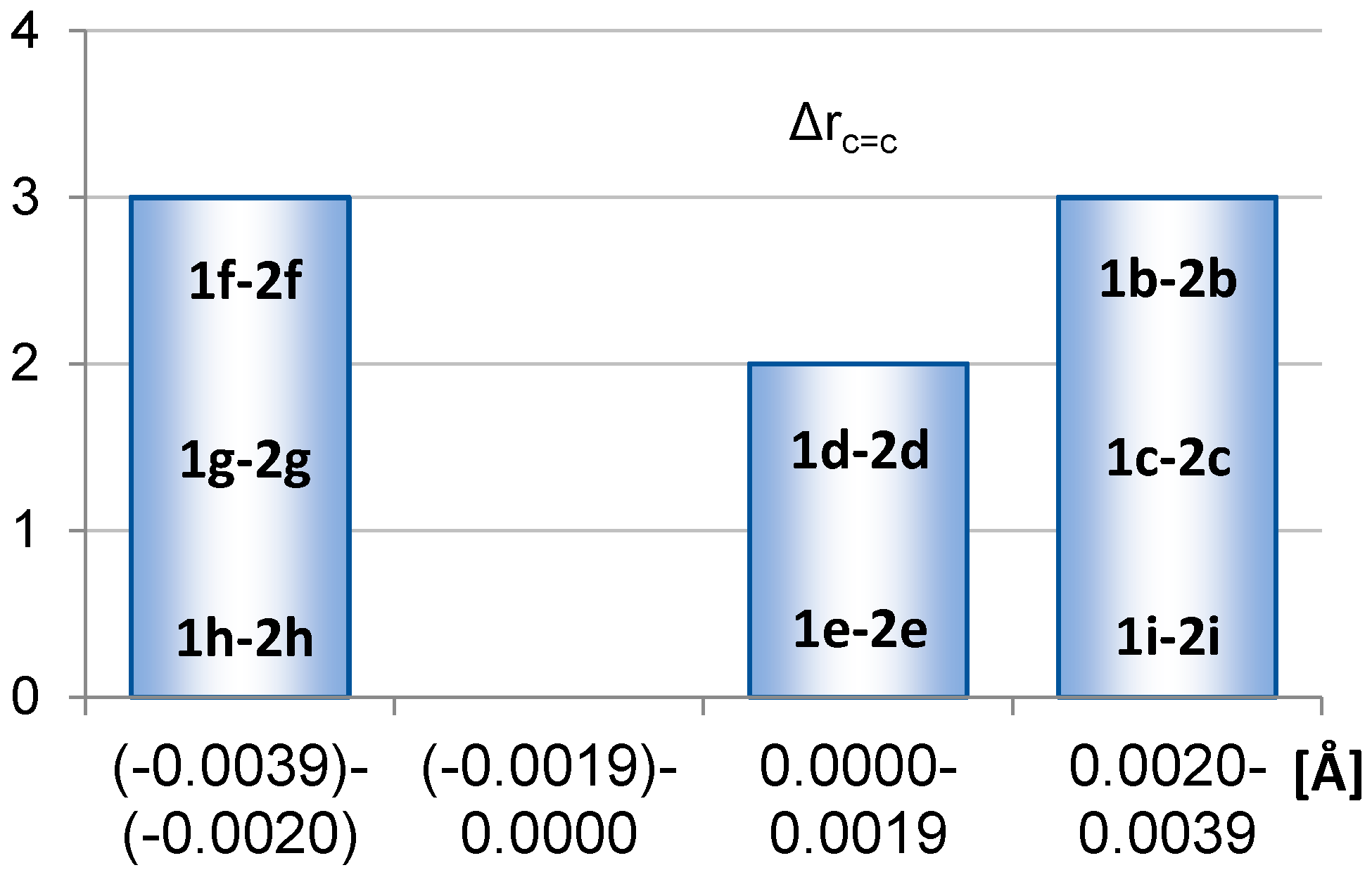
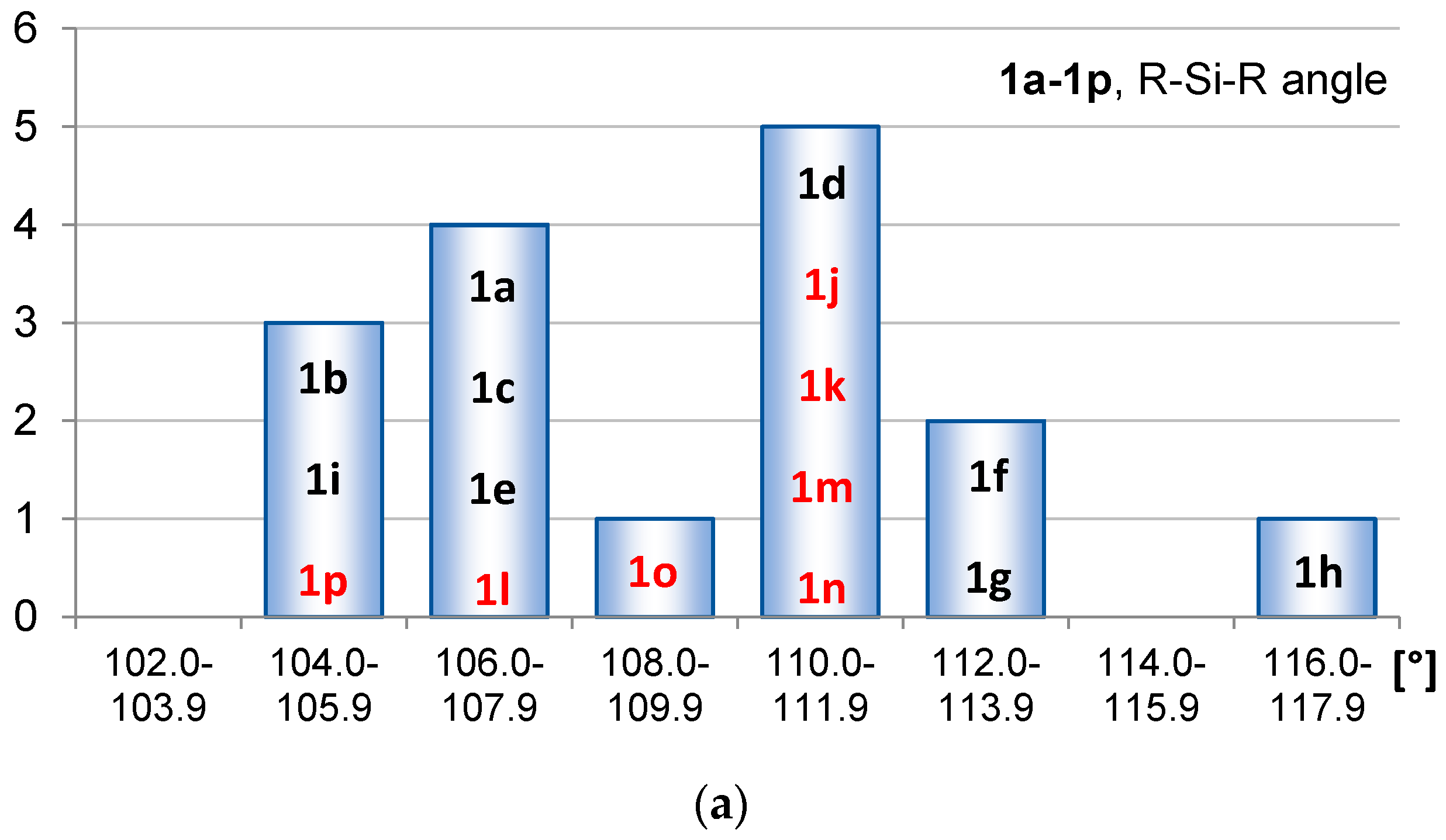
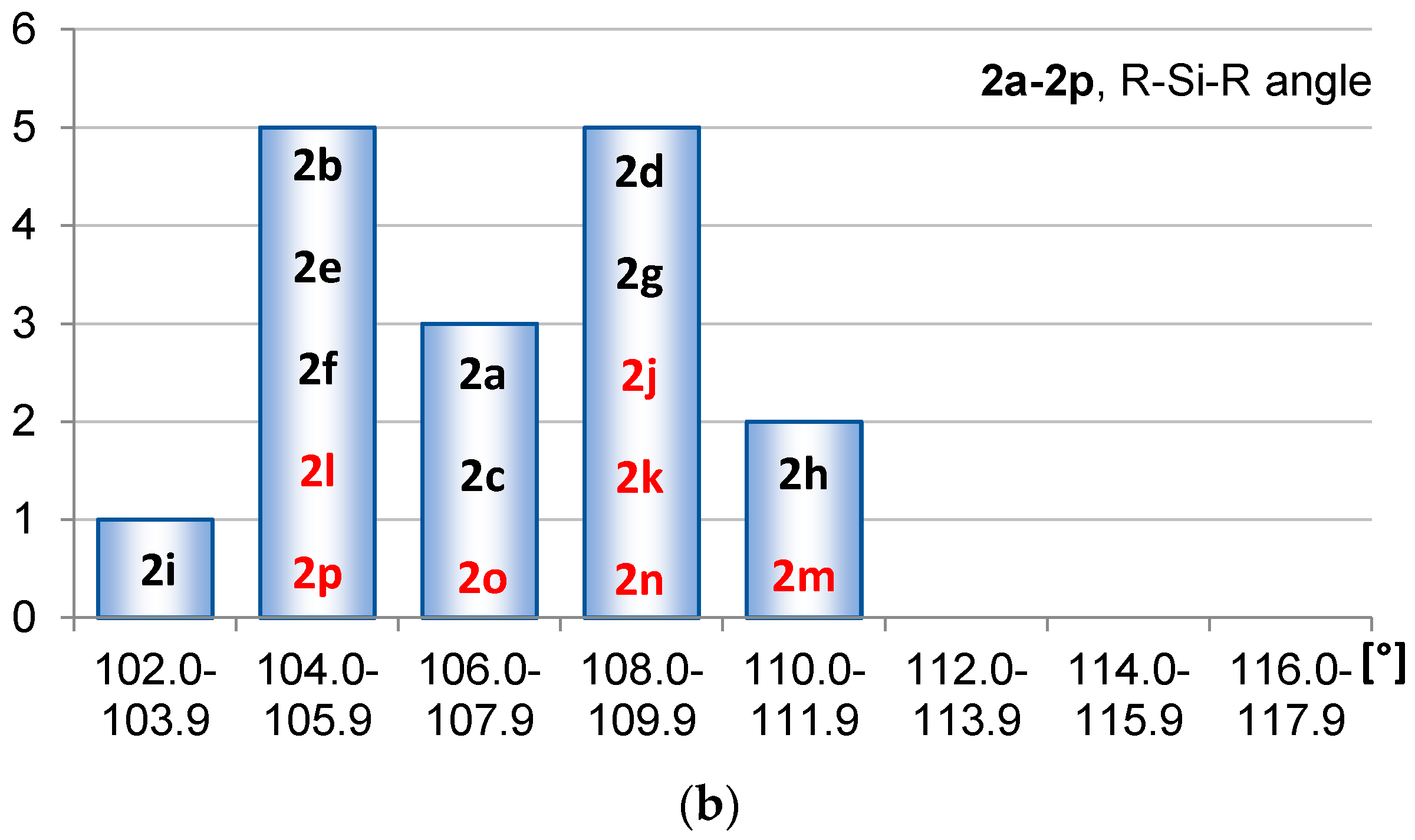
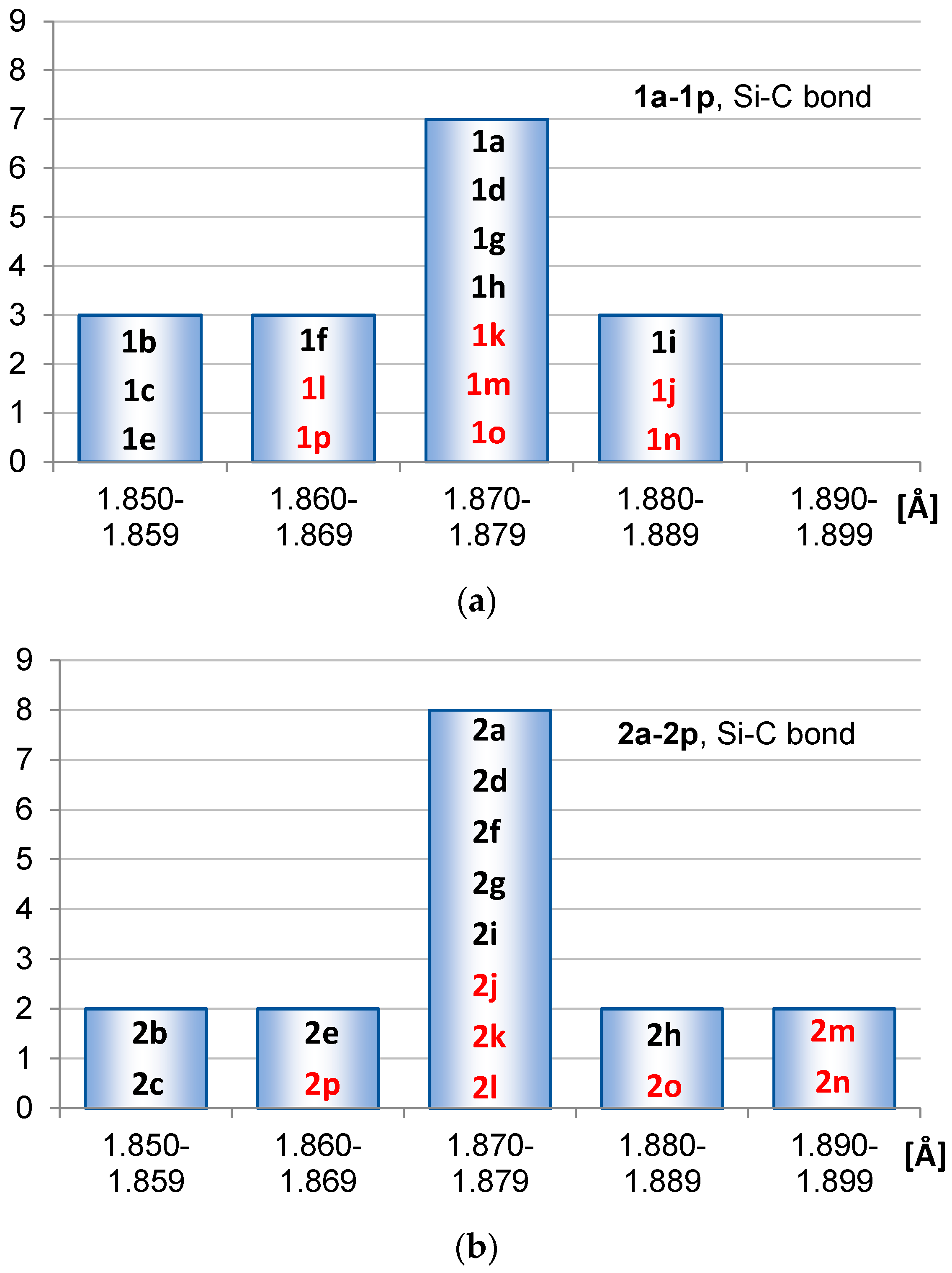
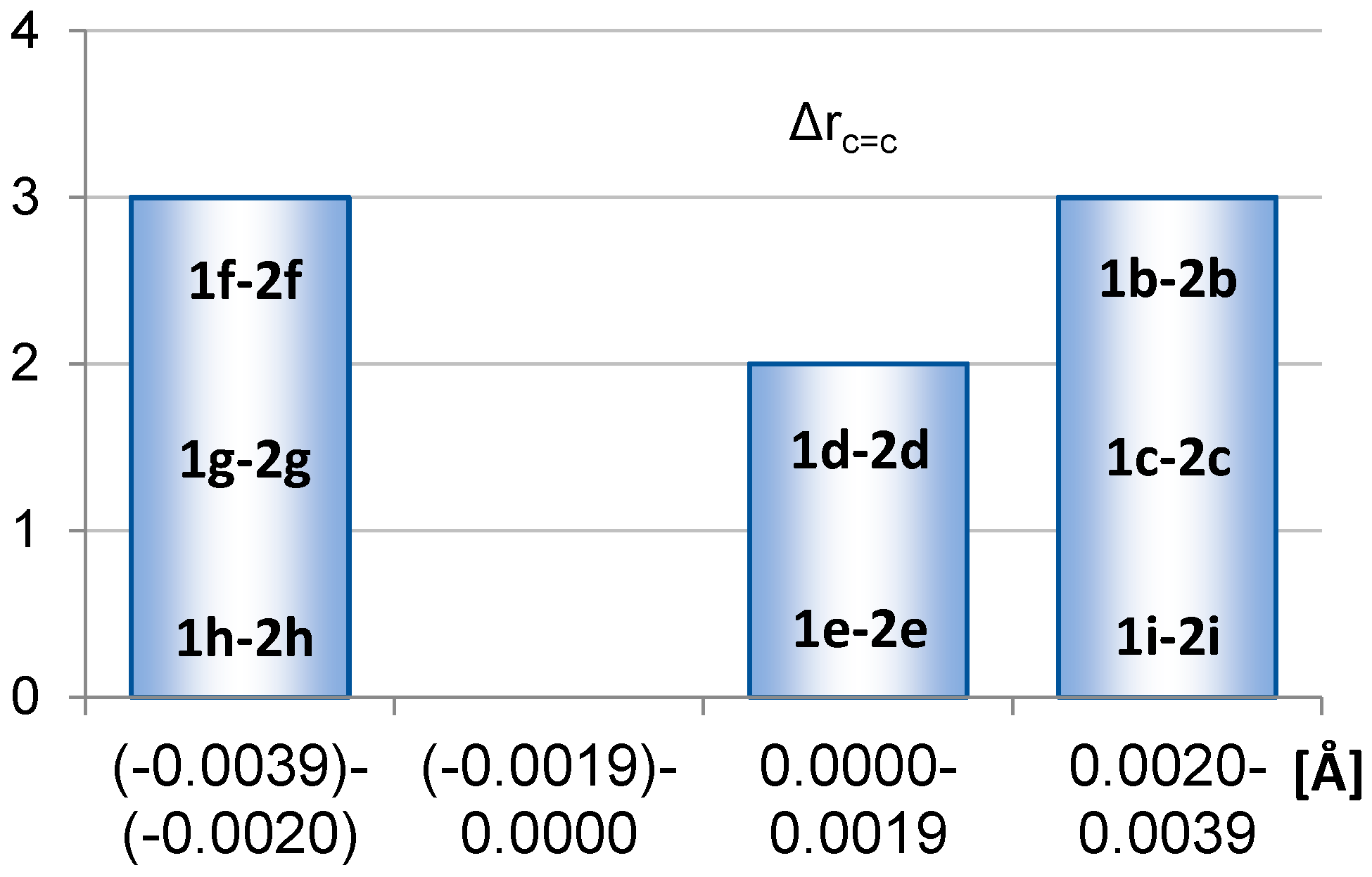
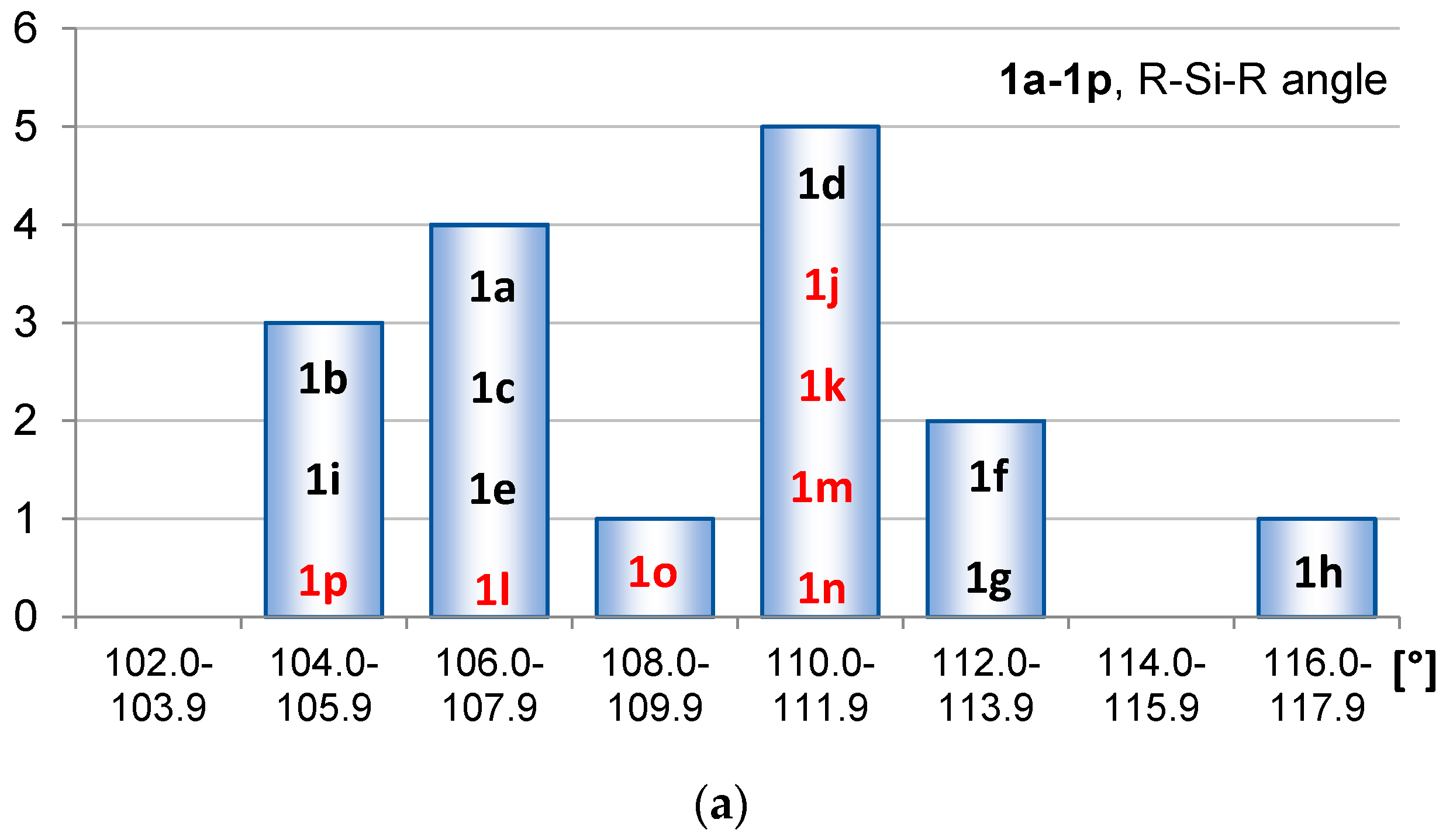
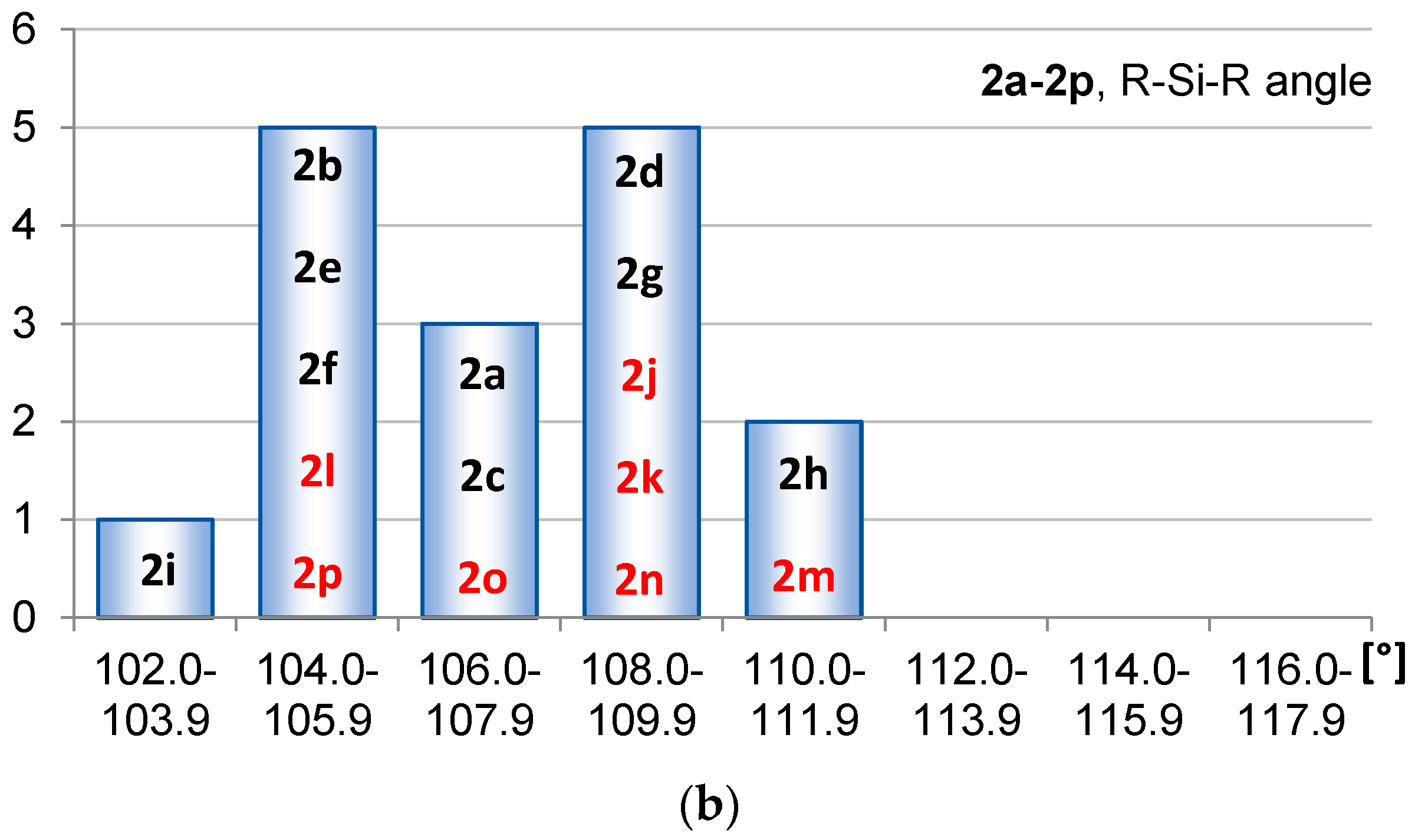
2.1. Geometric Structure
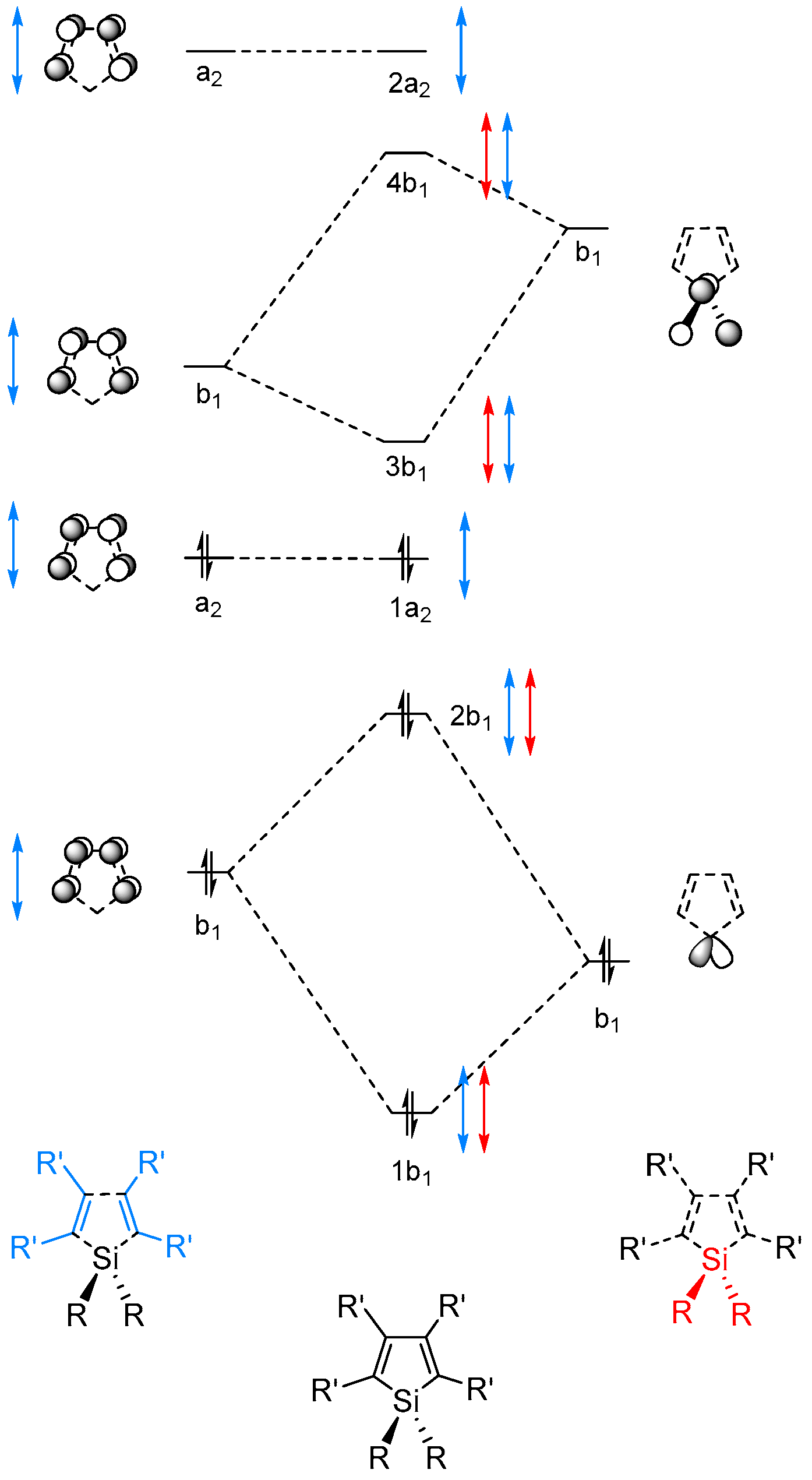
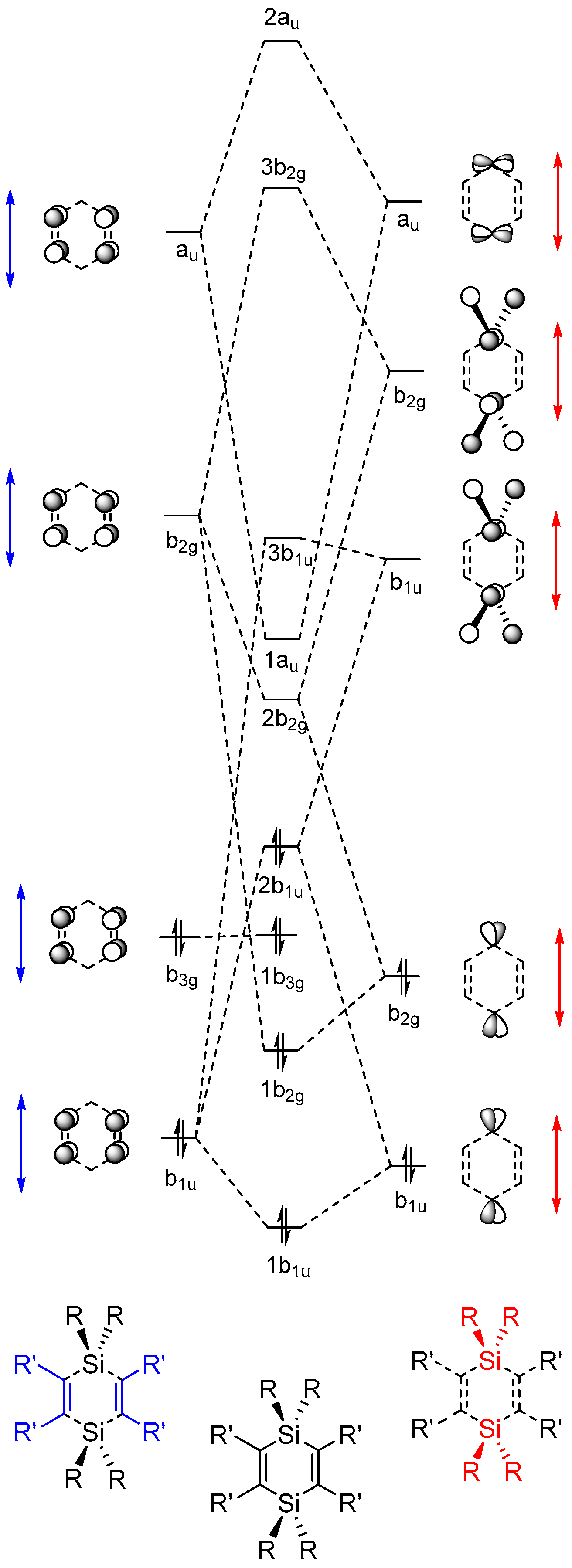
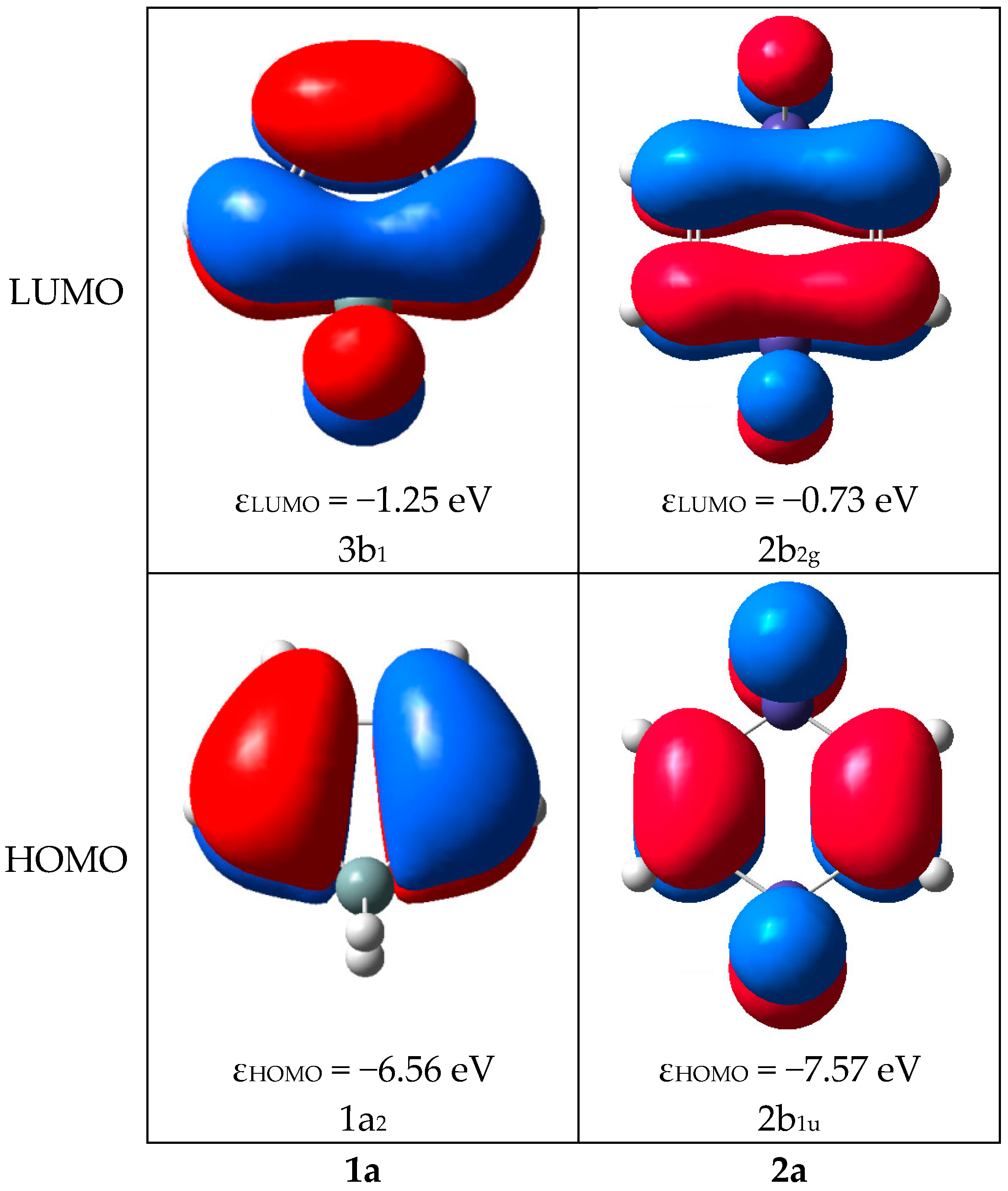
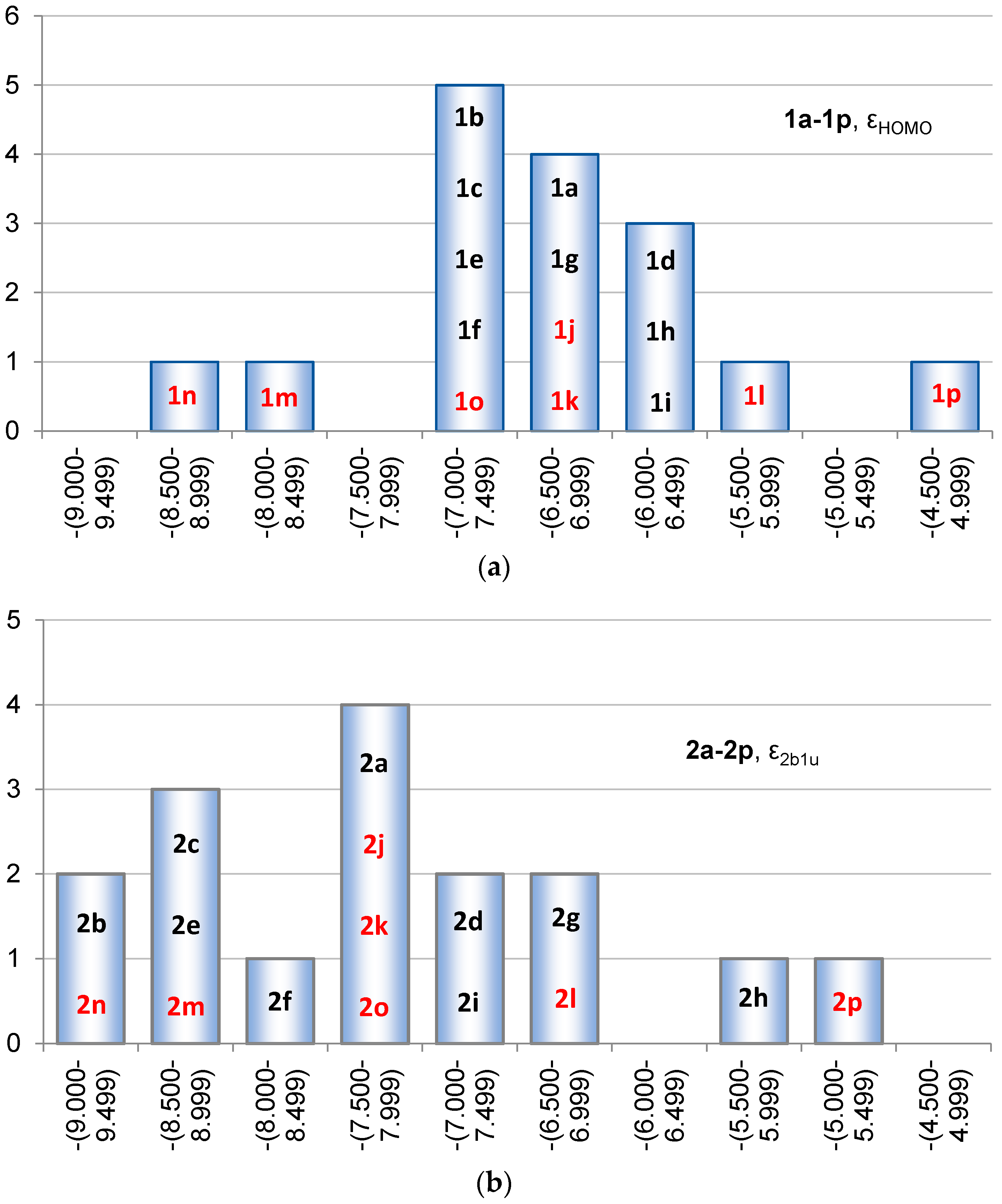
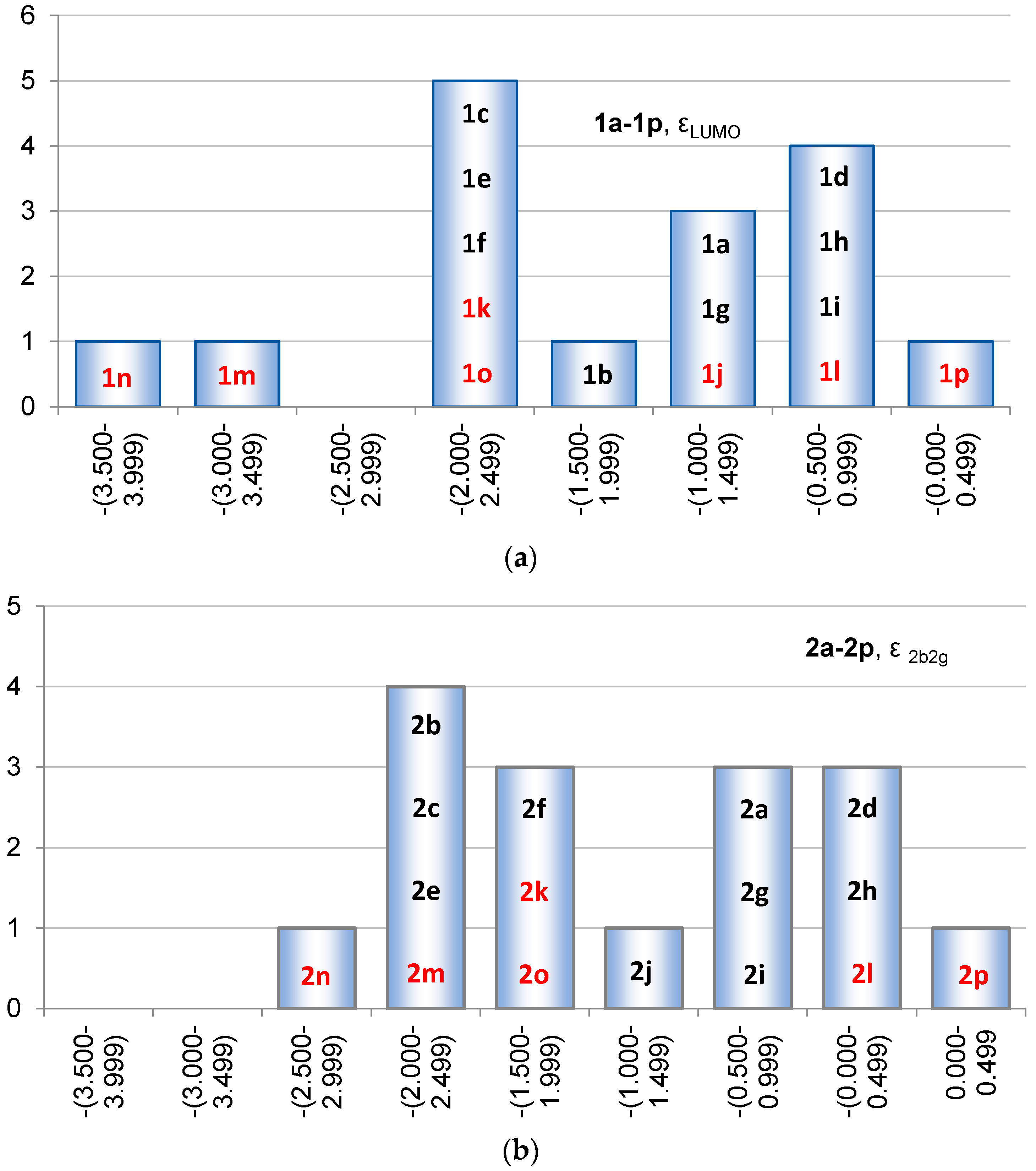
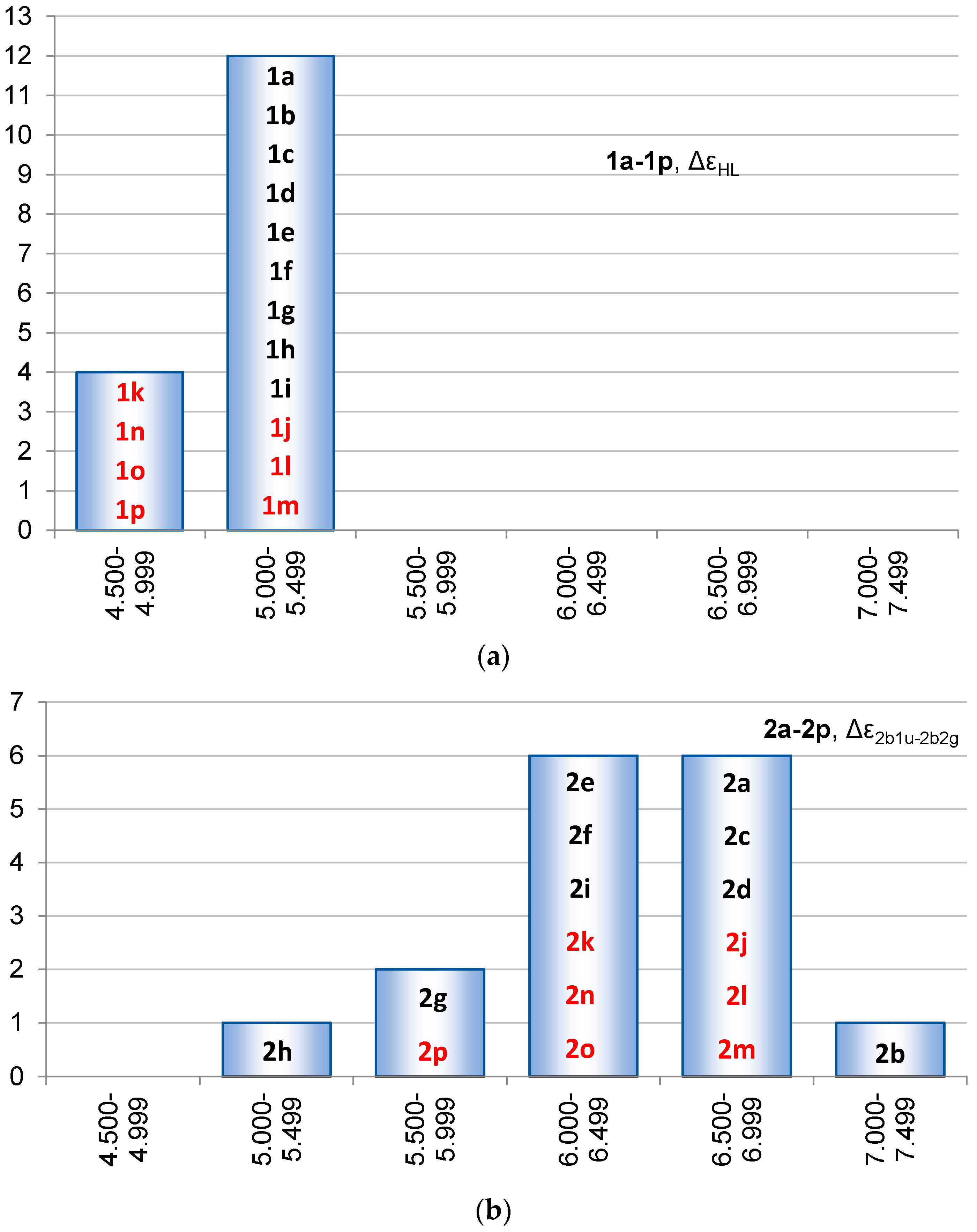
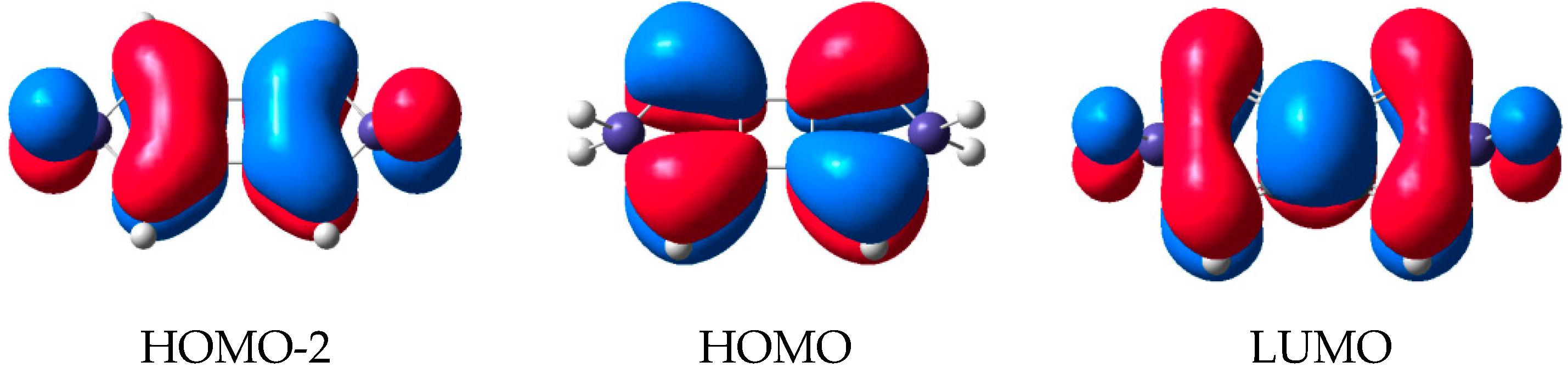
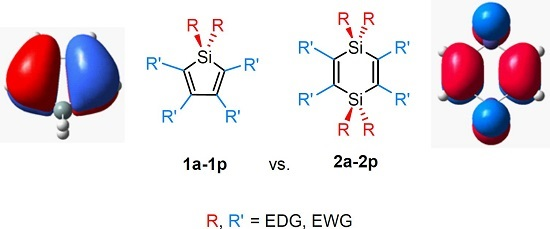
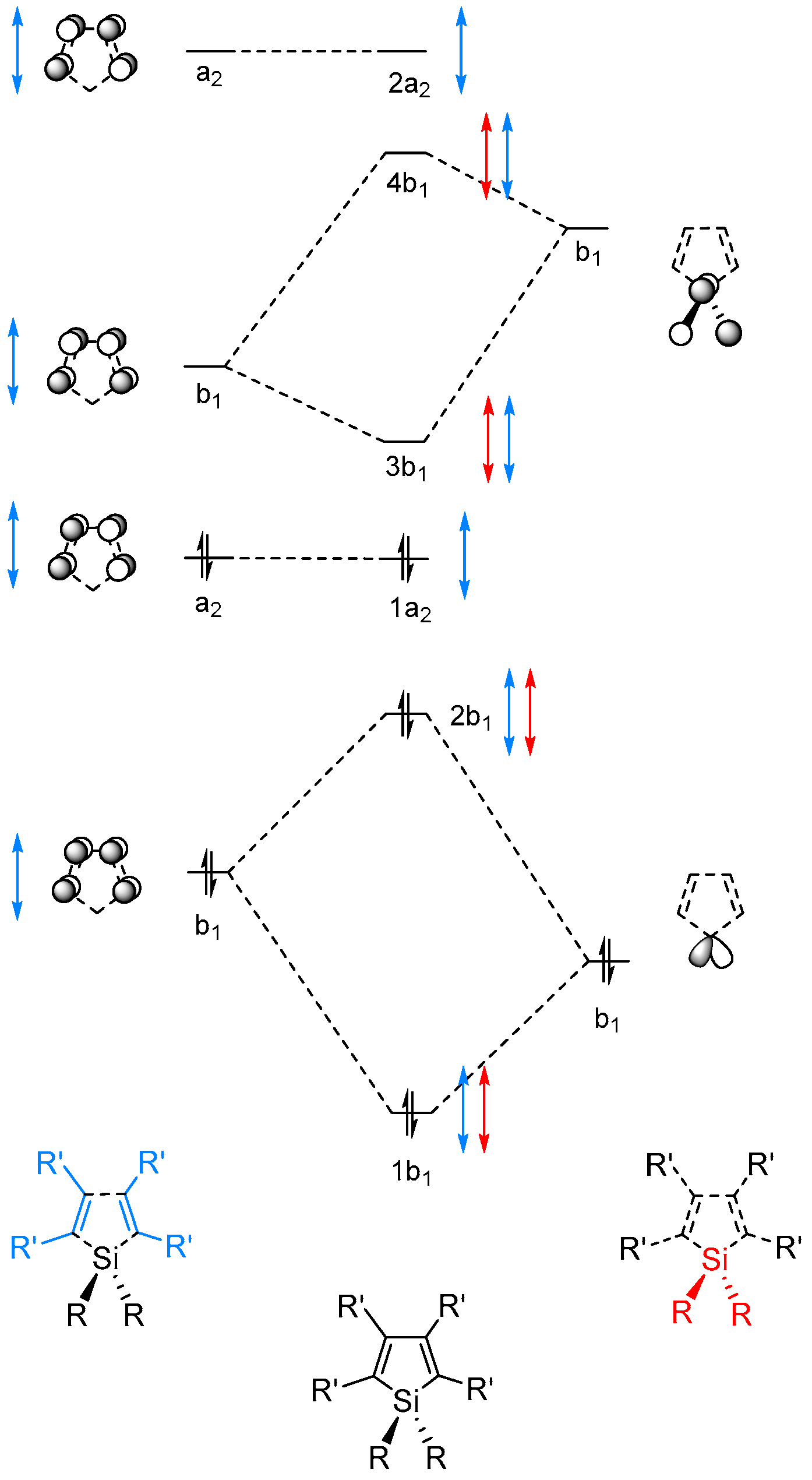
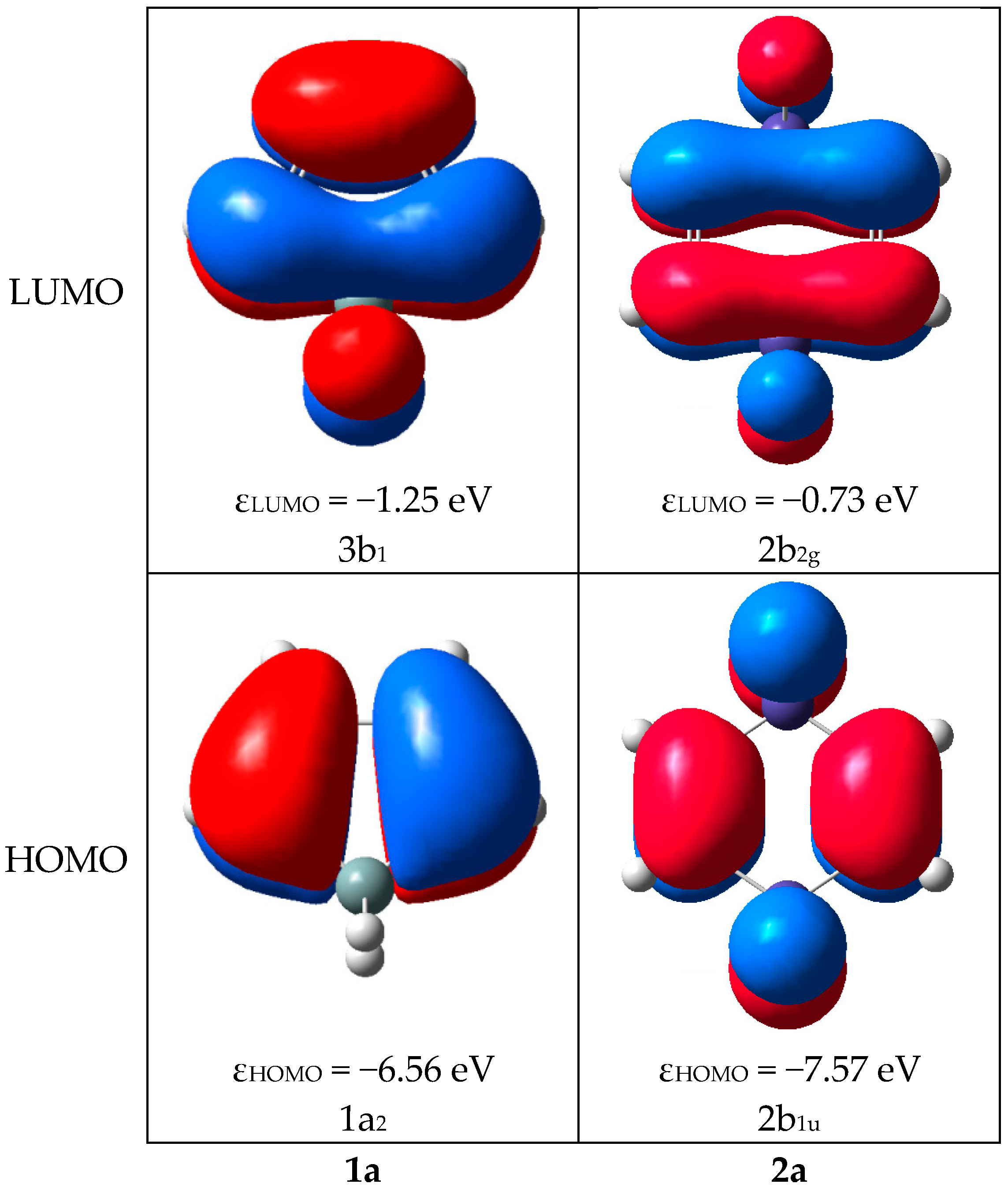
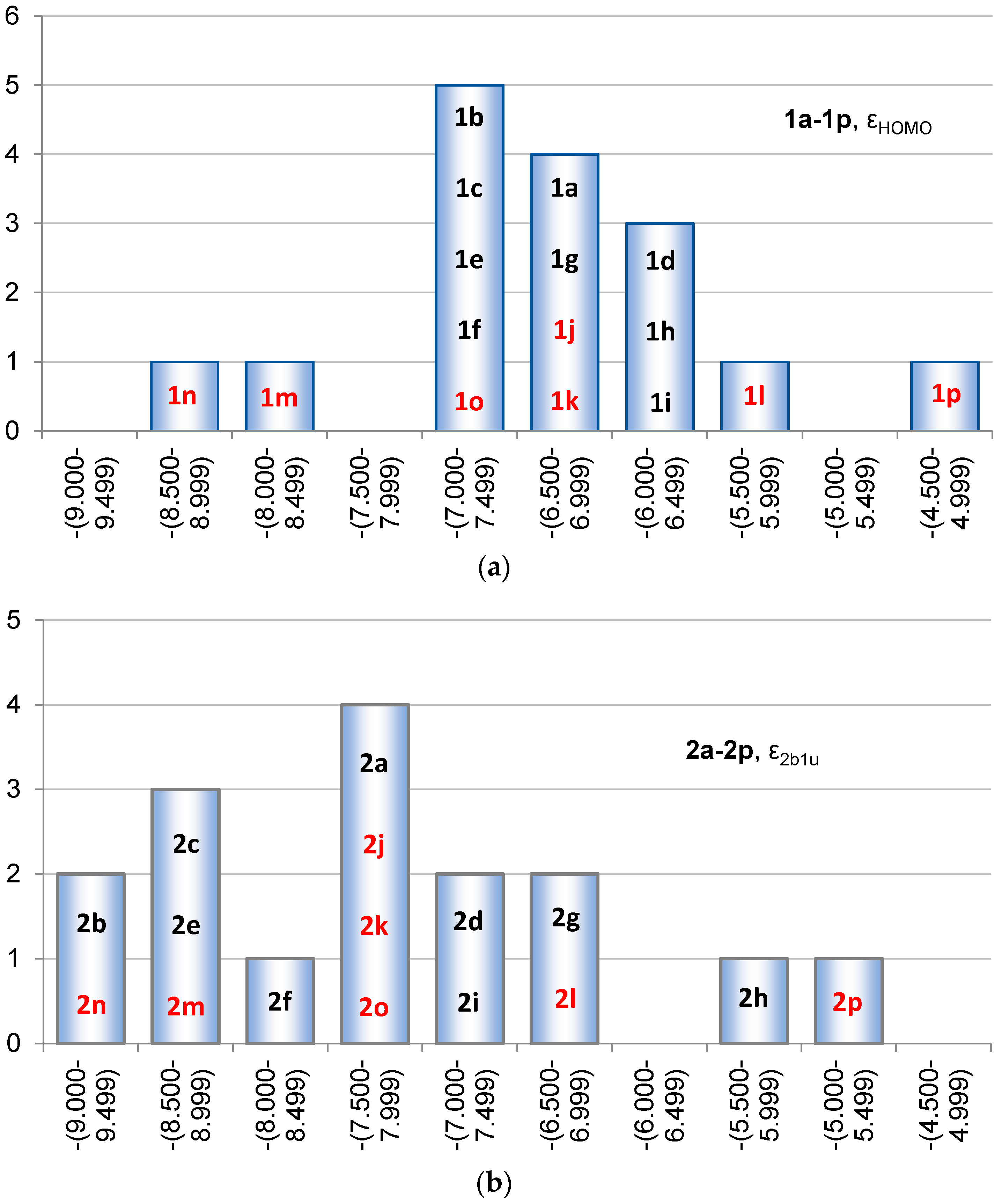
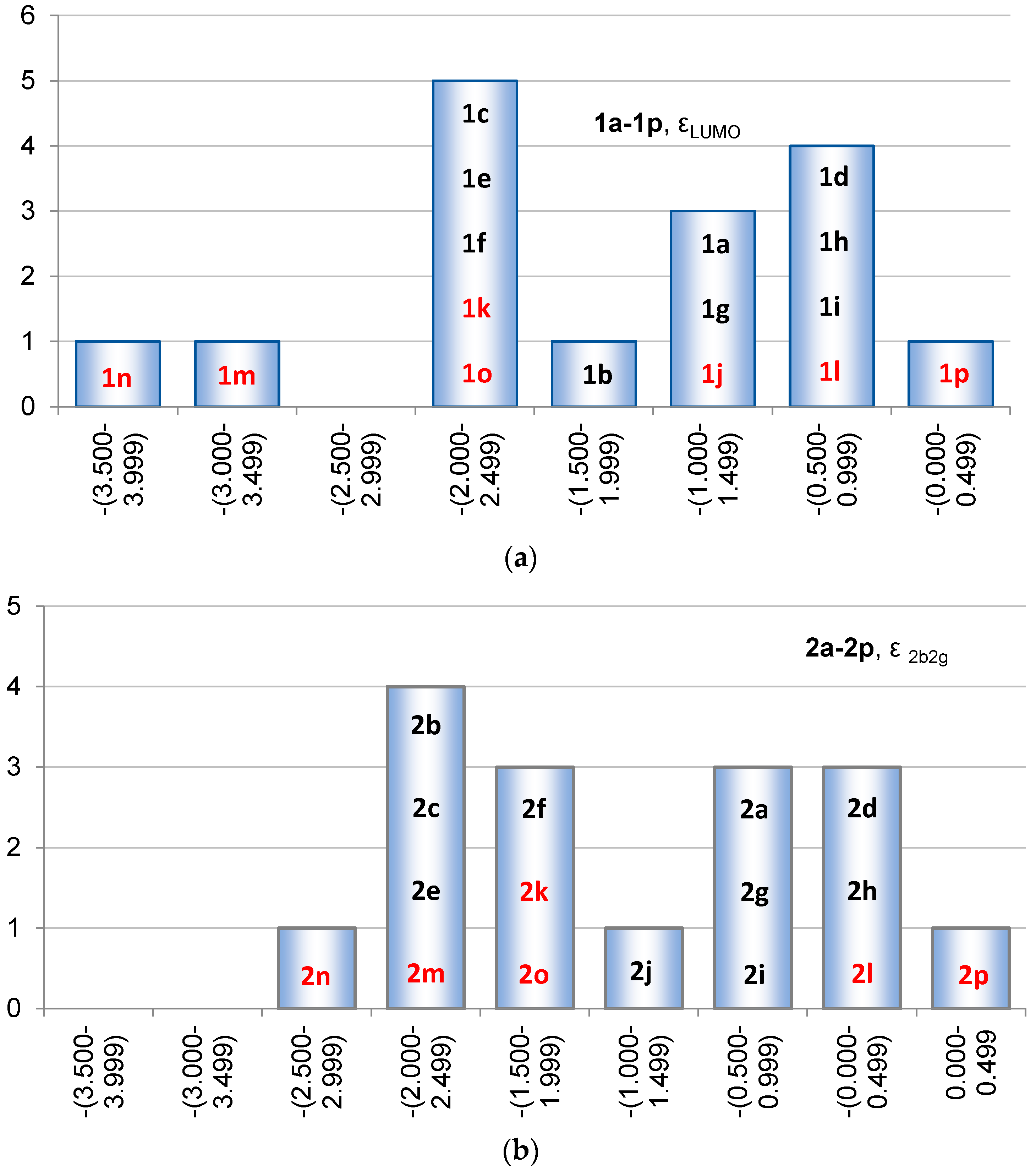
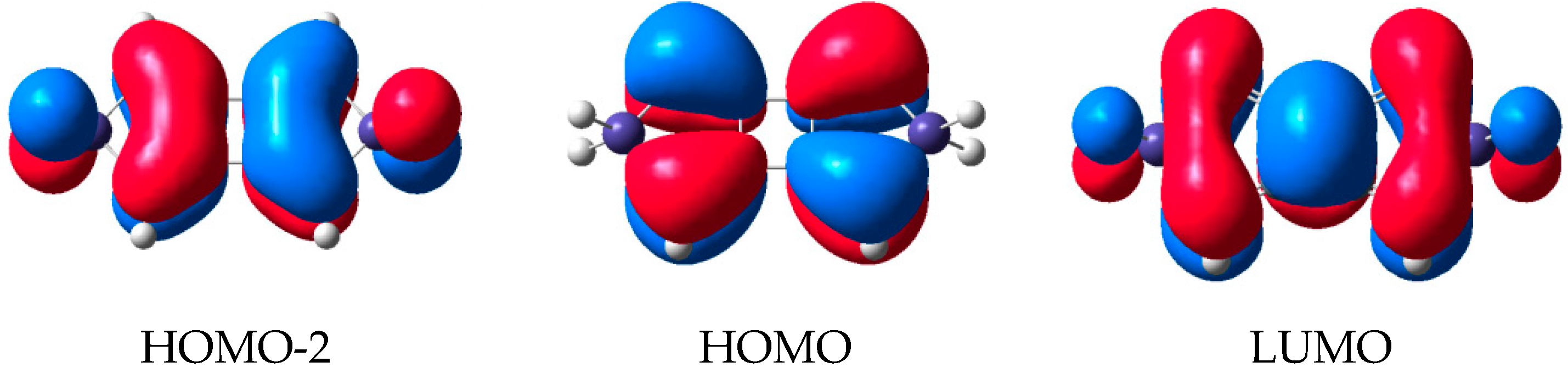
2.2. Molecular Orbitals
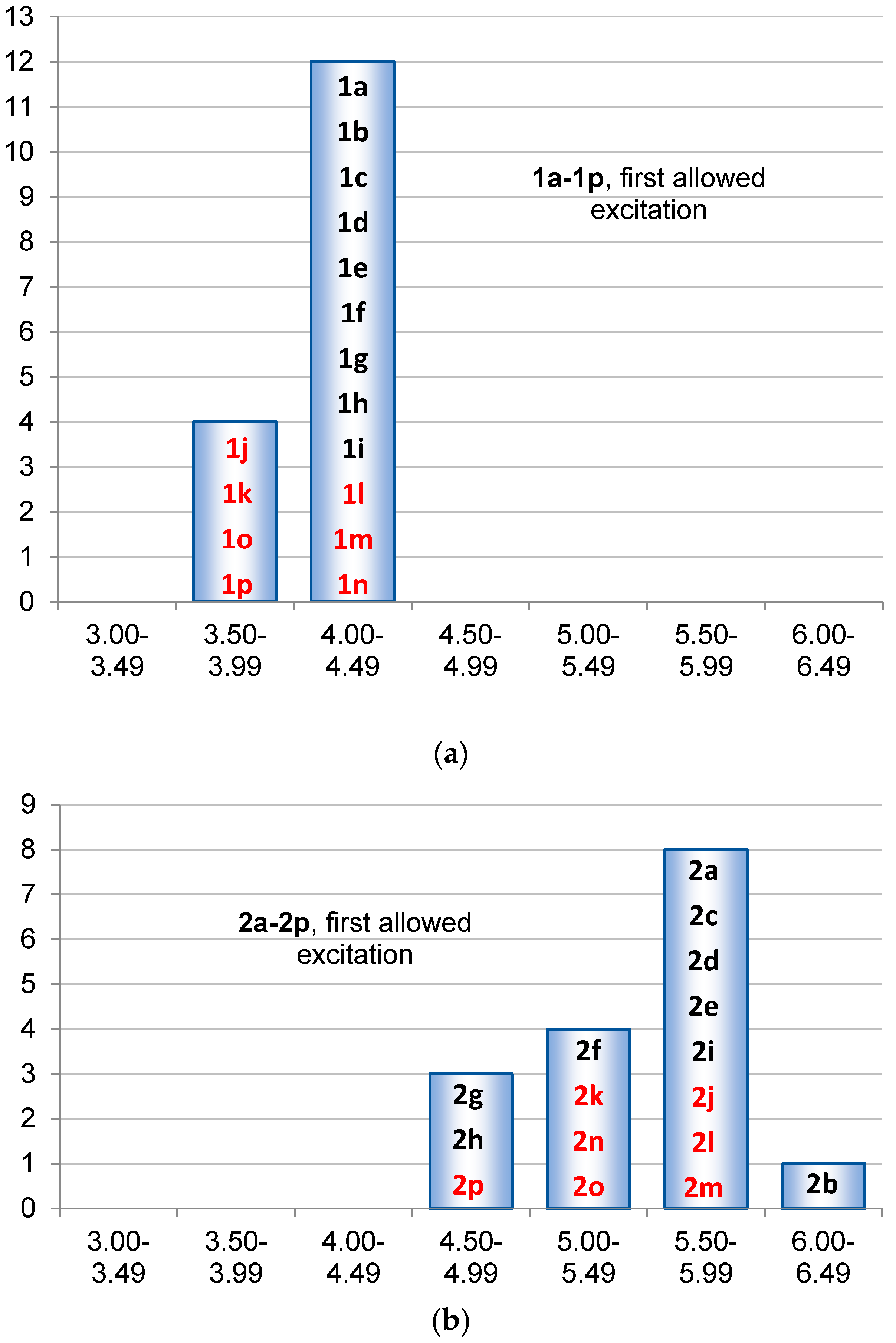
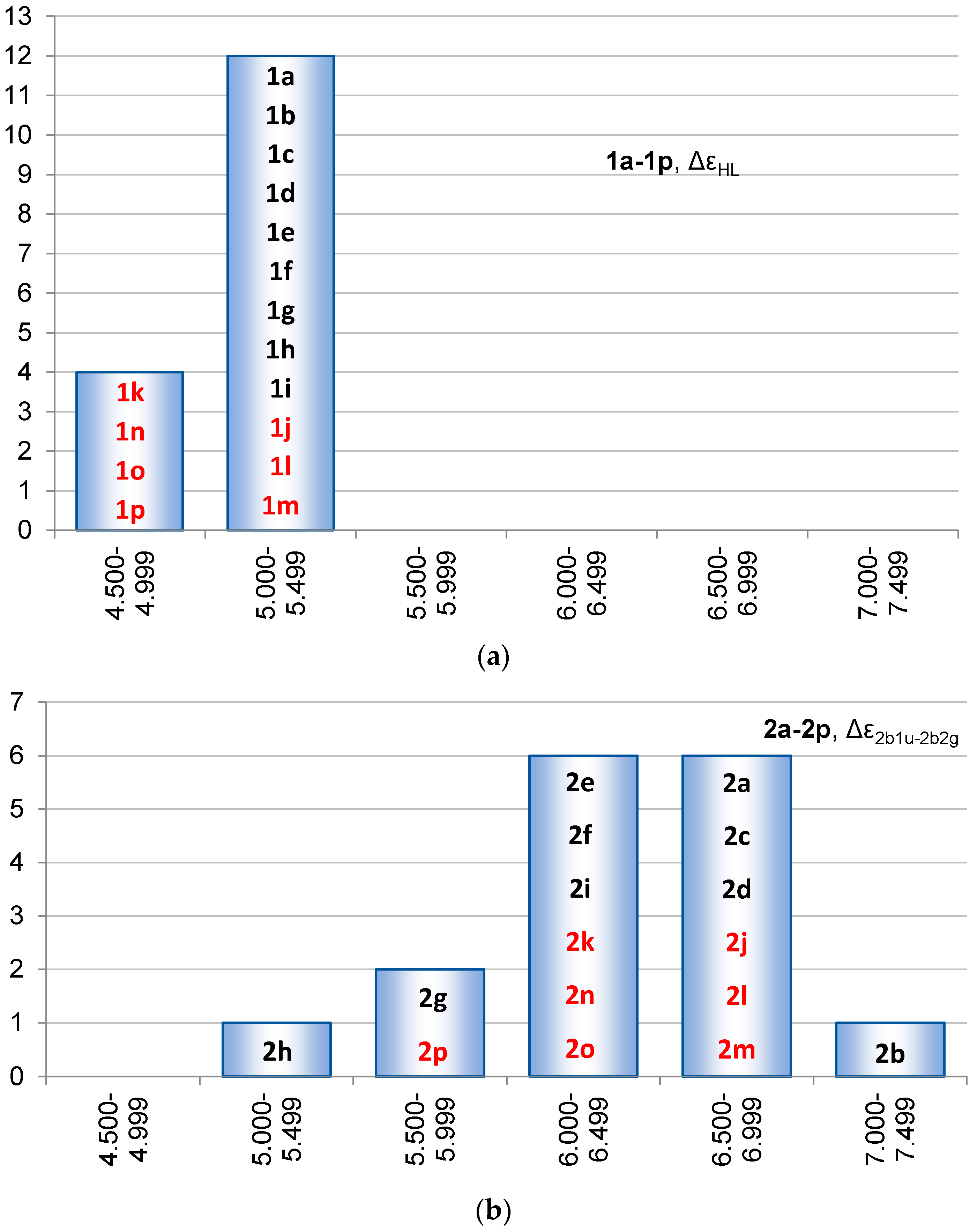
2.3. Singlet State Excitation Energies
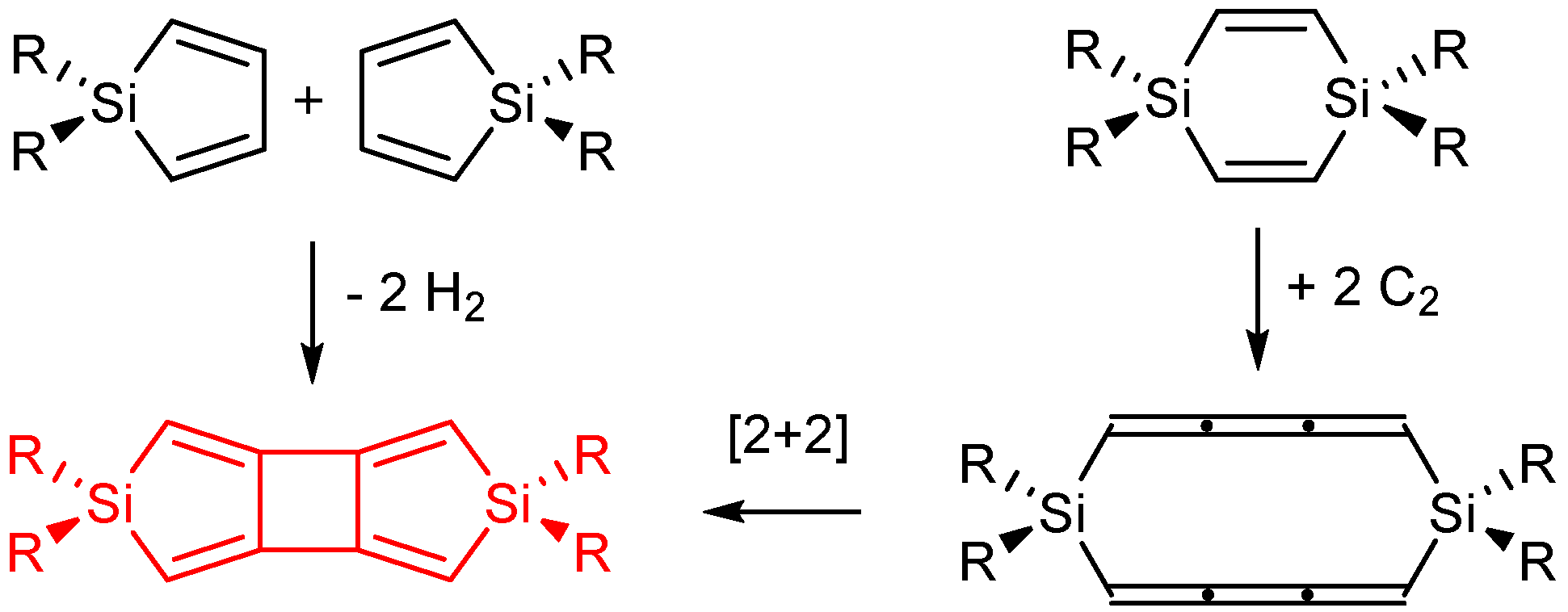
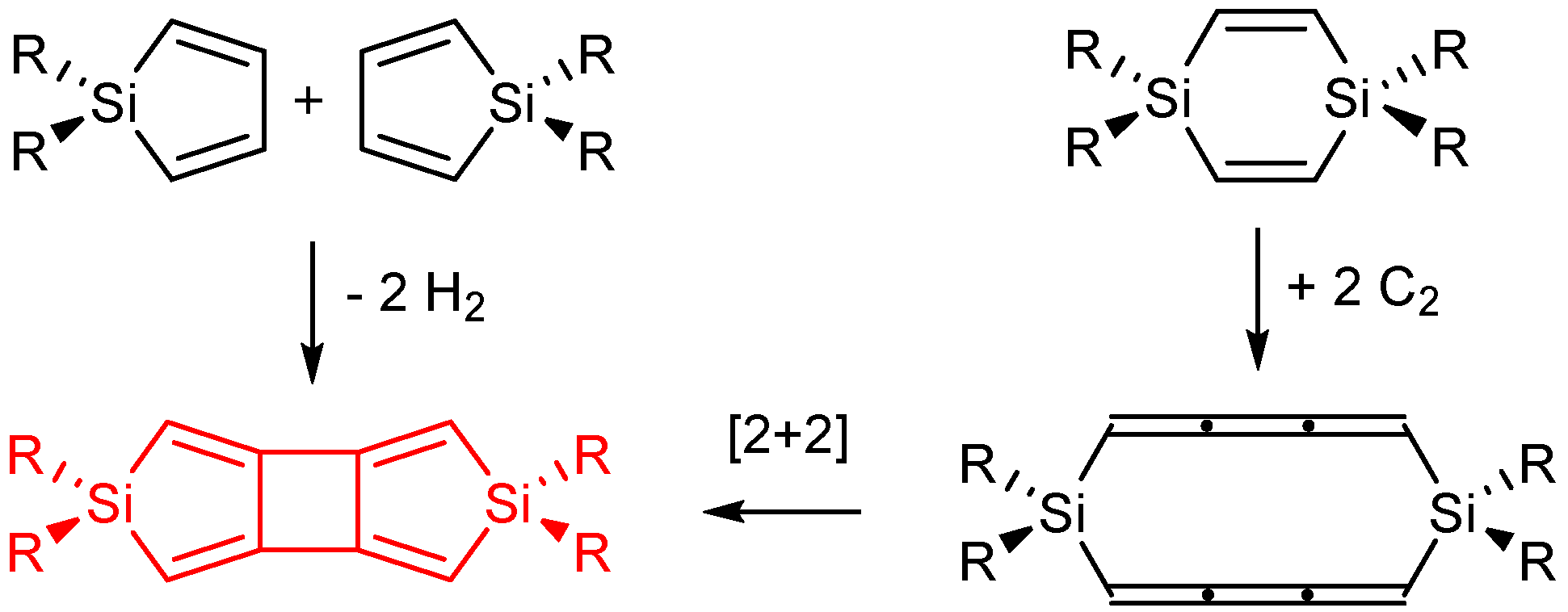
2.4. On Dimerization Reactions
2.5. Cyclobutadisiloles
3. Computational Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Murata, H.; Kafafi, Z.H.; Uchida, M. Efficient organic light-emitting diodes with undoped active layers based on silole derivatives. Appl. Phys. Lett. 2002, 80, 189–191. [Google Scholar] [CrossRef]
- Murata, H.; Malliaras, G.G.; Uchida, M.; Shen, Y.; Kafafi, Z.H. Non-dispersive and air-stable electron transport in an amorphous organic semiconductor. Chem. Phys. Lett. 2001, 339, 161–166. [Google Scholar] [CrossRef]
- Makinen, A.J.; Malliaras, G.G.; Uchida, M.; Kafafi, Z.H. Electronic structure of a silole derivative-magnesium thin film interface. J. Appl. Phys. 2004, 95, 2832–2838. [Google Scholar] [CrossRef]
- Chen, L.; Nie, H.; Chen, B.; Lin, G.; Luo, W.; Hu, R.; Huang, F.; Qin, A.; Zhao, Z.; Tang, B.Z. 2,5-Dicarbazole-functioned siloles with aggregation-enhanced emission for application in organic light-emitting diodes. J. Photonic Energy 2015, 5, 053598. [Google Scholar] [CrossRef]
- Nie, H.; Chen, B.; Quan, C.; Zhou, J.; Qiu, H.; Hu, R.; Su, S.-J.; Qin, A.; Zhao, Z.; Tang, B.Z. Modulation of Aggregation-Induced Emission and Electroluminescence of Silole Derivatives by a Covalent Bonding Pattern. Chem. Eur. J. 2015, 21, 8137–8147. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Sun, N.; Huang, J.; Li, Q.; Peng, Q.; Tang, X.; Dong, Y.; Ma, D.; Li, Z. New AIEgens containing tetraphenylethene and silole moieties: tunable intramolecular conjugation, aggregation-induced emission characteristics and good device performance. J. Mater. Chem. C 2015, 3, 2624–2631. [Google Scholar] [CrossRef]
- Chen, B.; Nie, H.; Hu, R.; Qin, A.; Zhao, Z.; Tang, B.Z. Red fluorescent siloles with aggregation-enhanced emission characteristics. Sci. China Chem. 2016, 59, 699–706. [Google Scholar] [CrossRef]
- Zhang, F.B.; Adachi, Y.; Ooyama, Y.; Ohshita, J. Synthesis and Properties of Benzofuran-Fused Silole and Germole Derivatives: Reversible Dimerization and Crystal Structures of Monomers and Dimers. Organometallics 2016, 35, 2327–2332. [Google Scholar] [CrossRef]
- Zhan, X.; Barlow, S.; Marder, S.R. Substituent effects on the electronic structure of siloles. Chem. Commun. 2009, 1948–1955. [Google Scholar] [CrossRef] [PubMed]
- Ohshita, J. Conjugated Oligomers and Polymers Containing Dithienosilole Units. Macromol. Chem. Phys. 2009, 210, 1360–1370. [Google Scholar]
- Chen, J.; Cao, Y. Silole-Containing Polymers: Chemistry and Optoelectronic Properties. Macromol. Rapid Commun. 2007, 28, 1714–1742. [Google Scholar] [CrossRef]
- Ohshita, J.; Kurushima, Y.; Lee, K.-H.; Kunai, A.; Ooyama, Y.; Harima, Y. Synthesis of Bis(diarylphosphino)dithienosilole Derivatives as Novel Photo- and Electroluminescence Materials. Organometallics 2007, 26, 6591–6595. [Google Scholar] [CrossRef]
- Mulliken, R.S. Intensities of Electronic Transitions in Molecular Spectra IV. Cyclic Dienes and Hyperconjugation. J. Chem. Phys. 1939, 7, 339–352. [Google Scholar] [CrossRef]
- Mulliken, R.S.; Rieke, C.A.; Brown, W.G. Hyperconjugation. J. Am. Chem. Soc. 1941, 63, 41–56. [Google Scholar] [CrossRef]
- Emanuelsson, R.; Wallner, A.; Ng, E.A.M.; Smith, J.R.; Nauroozi, D.; Ott, S.; Ottosson, H. Cross-Hyperconjugation: An Unexplored Orbital Interaction between π-Conjugated and Saturated Molecular Segments. Angew. Chem. Int. Ed. 2013, 52, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Göransson, E.; Emanuelsson, R.; Jorner, K.; Markle, T.F.; Hammarström, L.; Ottosson, H. Charge transfer through cross-hyperconjugated versus cross-π-conjugated bridges: An intervalence charge transfer study. Chem. Sci. 2013, 4, 3522–3532. [Google Scholar] [CrossRef]
- Emanuelsson, R.; Löfås, H.; Wallner, A.; Nauroozi, D.; Baumgartner, J.; Marschner, C.; Ahuja, R.; Ott, S.; Grigoriev, A.; Ottosson, H. Configuration- and Conformation-Dependent Electronic Structure Variations in 1,4-Disubstituted Cyclohexanes Enabled by a Carbon-to-Silicon Exchange. Chem. Eur. J. 2014, 20, 9304–9311. [Google Scholar] [CrossRef] [PubMed]
- Möllerstedt, H.; Piqueras, M.C.; Crespo, R.; Ottosson, H. Fulvenes, Fulvalenes, and Azulene: Are They Aromatic Chameleons? J. Am. Chem. Soc. 2004, 126, 13938–13939. [Google Scholar] [CrossRef] [PubMed]
- Jorner, K.; Emanuelsson, R.; Dahlstrand, C.; Tong, H.; Denisova, A.V.; Ottosson, H. Impact of Ground- and Excited-State Aromaticity on Cyclopentadiene and Silole Excitation Energies and Excited-State Polarities. Chem. Eur. J. 2014, 20, 9295–9303. [Google Scholar] [CrossRef] [PubMed]
- Baird, N.C. Quantum organic photochemistry. II. Resonance and aromaticity in the lowest 3ππ* state of cyclic hydrocarbons. J. Am. Chem. Soc. 1972, 94, 4941–4948. [Google Scholar] [CrossRef]
- Ottosson, H. Exciting Excited-State Aromaticity. Nat. Chem. 2012, 4, 969–971. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.; Dahlstrand, C.; Kilså, K.; Ottosson, H. Excited State Aromaticity and Antiaromaticity: Opportunities for Photophysical and Photochemical Rationalizations. Chem. Rev. 2014, 114, 5379–5425. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, R.; Ottosson, H. The excited state antiaromatic benzene ring: a molecular Mr Hyde? Chem. Soc. Rev. 2015, 44, 6472–6493. [Google Scholar] [CrossRef] [PubMed]
- Tibbelin, J.; Wallner, A.; Emanuelsson, R.; Heijkenskjöld, F.; Rosenberg, M.; Yamazaki, K.; Nauroozi, D.; Karlsson, L.; Feifel, R.; Pettersson, R.; et al. 1,4-Disilacyclohexa-2,5-diene: A molecular building block that allows for remarkably strong neutral cyclic cross-hyperconjugation. Chem. Sci. 2014, 5, 360–371. [Google Scholar] [CrossRef]
- Emanuelsson, R.; Löfås, H.; Zhu, J.; Ahuja, R.; Grigoriev, A.; Ottosson, H. In Search of Flexible Molecular Wires with Near Conformer-Independent Conjugation and Conductance: A Computational Study. J. Phys. Chem. C 2014, 118, 5637–5649. [Google Scholar] [CrossRef]
- Emanuelsson, R.; Denisova, A.V.; Baumgartner, J.; Ottosson, H. Optimization of the Cyclic Cross-Hyperconjugation in 1,4-Ditetrelcyclohexa-2,5-dienes. Organometallics 2014, 33, 2997–3004. [Google Scholar] [CrossRef]
- Denisova, A.; Emanuelsson, R.; Ottosson, H. Expanding the (Cross-)Hyperconjugation of 1,4-Disilacyclohexa-2,5-dienes to Larger Monomers and Oligomers: A Computational Investigation. RSC Adv. 2016, 6, 36961–36970. [Google Scholar] [CrossRef]
- Titilas, I.; Kidonakis, M.; Gryparis, C.; Stratakis, M. Tandem Si–Si and Si–H Activation of 1,1,2,2-Tetramethyldisilane by Gold Nanoparticles in Its Reaction with Alkynes: Synthesis of Substituted 1,4-Disila-2,5-cyclohexadienes. Organometallics 2015, 34, 1597–1600. [Google Scholar] [CrossRef]
- Zhan, X.; Risko, C.; Korlyukov, A.; Sena, F.; Timofeeva, T.V.; Antipin, M.Y.; Barlow, S.; Brédas, J.L.; Marder, S.R. Comparative studies of the geometric and electronic properties of 1,1-disubstituted-2,3,4,5-tetraphenylsiloles and 1,1,2,2-tetramethyl-3,4,5,6-tetraphenyl-1,2-disila-3,5-cyclo-hexadiene. J. Mater. Chem. 2006, 16, 3814–3822. [Google Scholar] [CrossRef]
- Párkányi, L. The crystal structure of 1,1-dimethyl-2,3,4,5-tetraphenyl-1-silacyclopentadiene. J. Organomet. Chem. 1981, 216, 9–16. [Google Scholar] [CrossRef]
- Zhan, X.; Haldi, A.; Risko, C.; Chan, C.K.; Zhao, W.; Timofeeva, T.V.; Korlyukov, A.; Antipin, M.Y.; Montgomery, S.; Thompson, E.; et al. Fluorenyl-substituted silole molecules: geometric, electronic, optical, and device properties. J. Mater. Chem. 2008, 18, 3157–3166. [Google Scholar] [CrossRef]
- Yin, S.; Liu, Y.; Chen, J.; Xu, X.; Sun, X.; Ma, D.; Zhan, X.; Peng, Q.; Shuai, Z.; Tang, B.; et al. Structures, Electronic States, Photoluminescence, and Carrier Transport Properties of 1,1-Disubstituted 2,3,4,5-Tetraphenylsiloles. J. Am. Chem. Soc. 2005, 127, 6335–6346. [Google Scholar]
- West, R.; Sohn, H.; Powell, D.R.; Müller, T.; Apeloig, Y. The Dianion of Tetraphenylgermole is Aromatic. Angew. Chem. Int. Ed. Engl. 1996, 35, 1002–1004. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Jin, R.Z.; Tamao, K. Modification of the electronic structure of silole by the substituents on the ring silicon. J. Organomet. Chem. 1998, 559, 73–80. [Google Scholar] [CrossRef]
- Viehe, H.G.; Janousek, Z.; Merényi, R. Substituent Effects in Radical Chemistry; Springer: Dordrecht, The Netherlands, 2012. [Google Scholar]
- Dougherty, D.A.; Anslyn, E.V. Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, USA, 2006. [Google Scholar]
- Burns, G.T.; Barton, T.J. 1,1-Dimethylsilole. J. Organomet. Chem. 1981, 209, C25–C27. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. (Erratum). Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, J. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef]
- Poirier, R.; Kari, R.; Csizmadia, I.G. Handbook of Gaussian Basis Sets; Elsevier: Amsterdam, The Netherlands, 1985. [Google Scholar]
- Sample Availability: The paper reports on a purely computational study. Detailed data from the computations are available upon request from the authors.

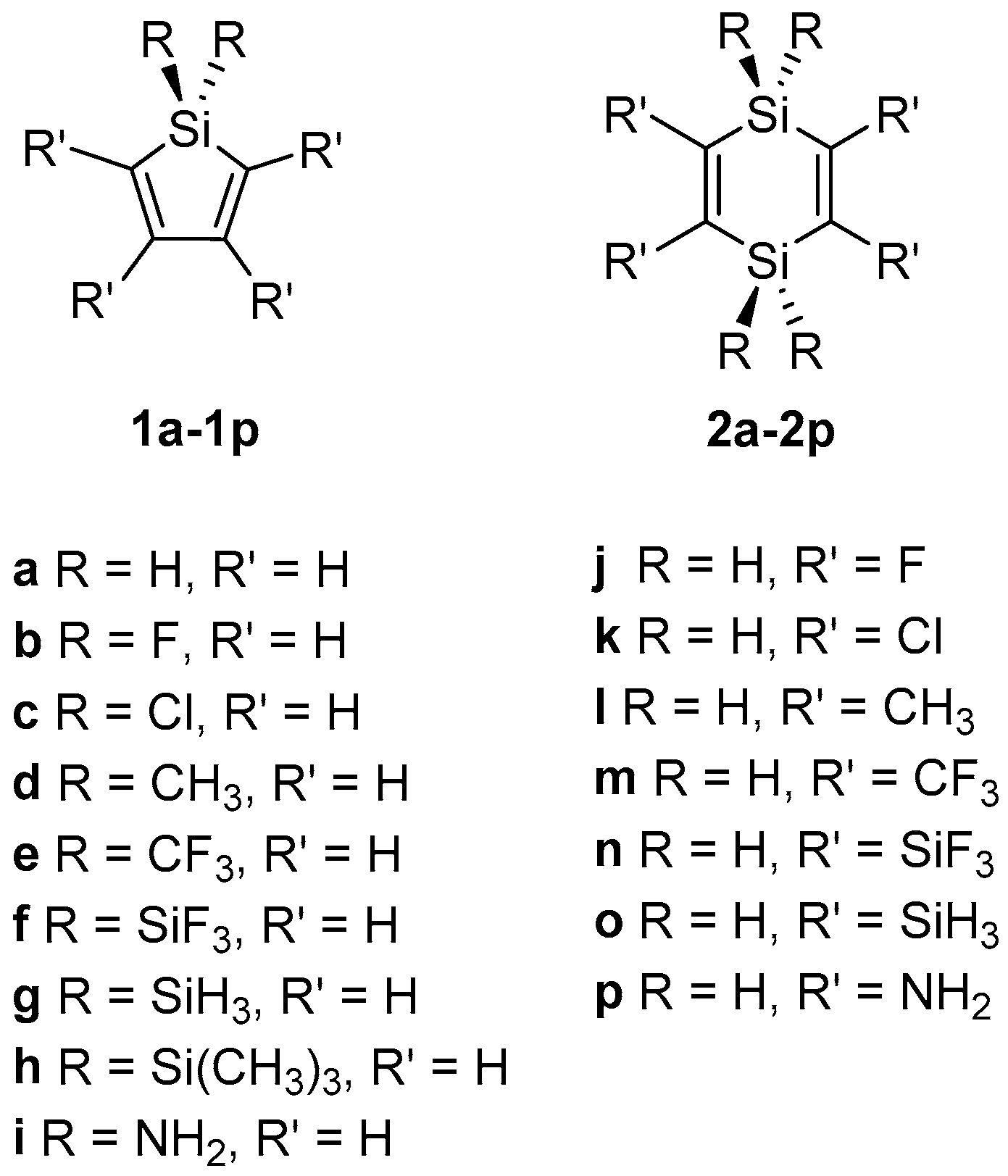

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R = | H (1a) | F (1b) | SiH3 (1g) |
|---|---|---|---|
| ∆G‡ | 21.5 | 18.6 | 29.2 |
| ∆G | −23.8 | −30.4 | −15.2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Denisova, A.V.; Tibbelin, J.; Emanuelsson, R.; Ottosson, H. A Computational Investigation of the Substituent Effects on Geometric, Electronic, and Optical Properties of Siloles and 1,4-Disilacyclohexa-2,5-dienes. Molecules 2017, 22, 370. https://doi.org/10.3390/molecules22030370
Denisova AV, Tibbelin J, Emanuelsson R, Ottosson H. A Computational Investigation of the Substituent Effects on Geometric, Electronic, and Optical Properties of Siloles and 1,4-Disilacyclohexa-2,5-dienes. Molecules. 2017; 22(3):370. https://doi.org/10.3390/molecules22030370
Chicago/Turabian StyleDenisova, Aleksandra V., Julius Tibbelin, Rikard Emanuelsson, and Henrik Ottosson. 2017. "A Computational Investigation of the Substituent Effects on Geometric, Electronic, and Optical Properties of Siloles and 1,4-Disilacyclohexa-2,5-dienes" Molecules 22, no. 3: 370. https://doi.org/10.3390/molecules22030370