
Hydrogen Atomic Positions of O–H···O Hydrogen Bonds in Solution and in the Solid State: The Synergy of Quantum Chemical Calculations with 1H-NMR Chemical Shifts and X-ray Diffraction Methods

Abstract
:
1. Introduction
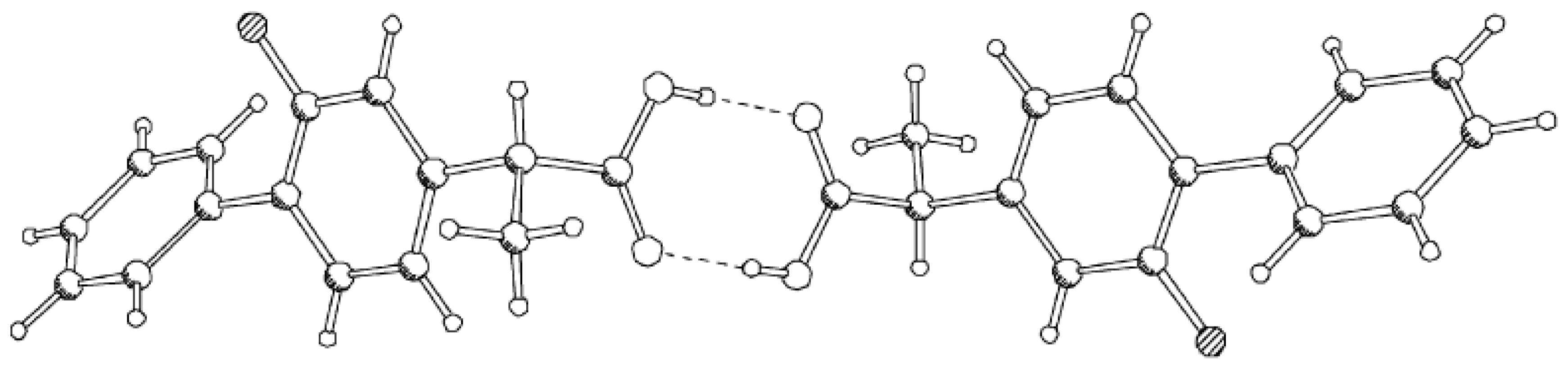
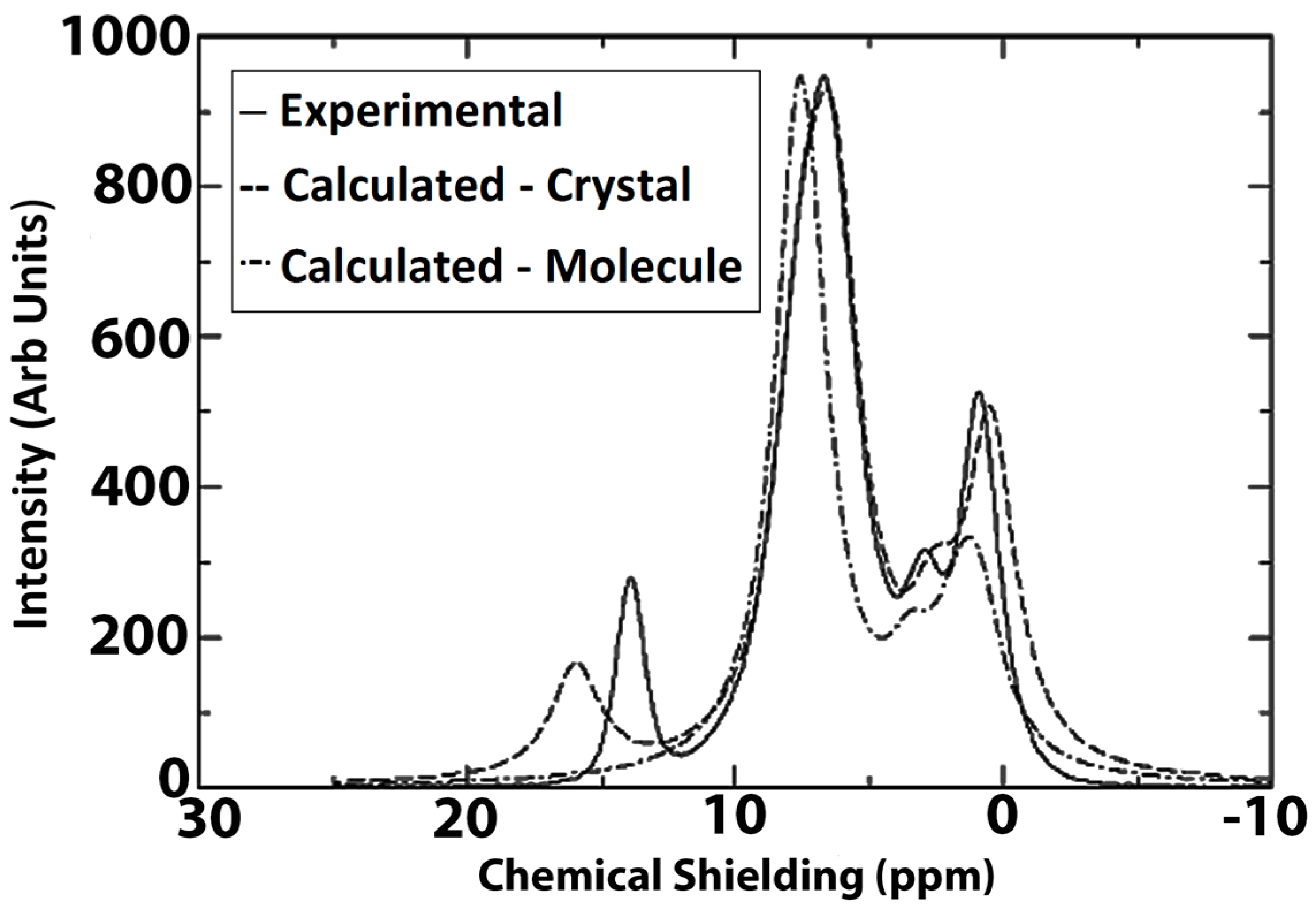
2. Accurate Hydrogen Atom Positions with the Combined Use of X-ray and Quantum Chemical Methods
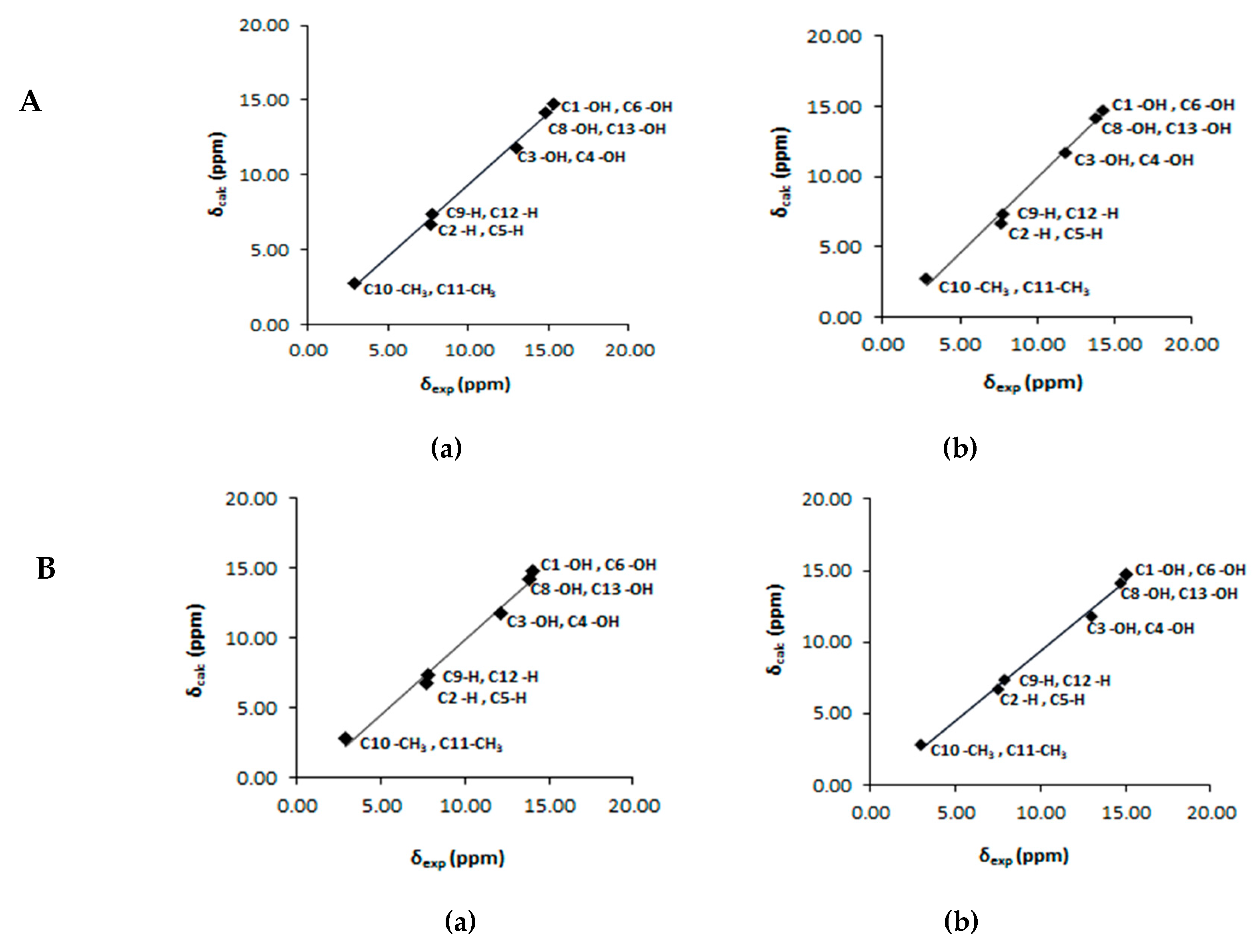
3. Hydrogen Atomic Positions of Intramolecular O–H···O Hydrogen Bonds with the Combined use of 1H-NMR Chemical Shifts and Quantum Chemical Methods
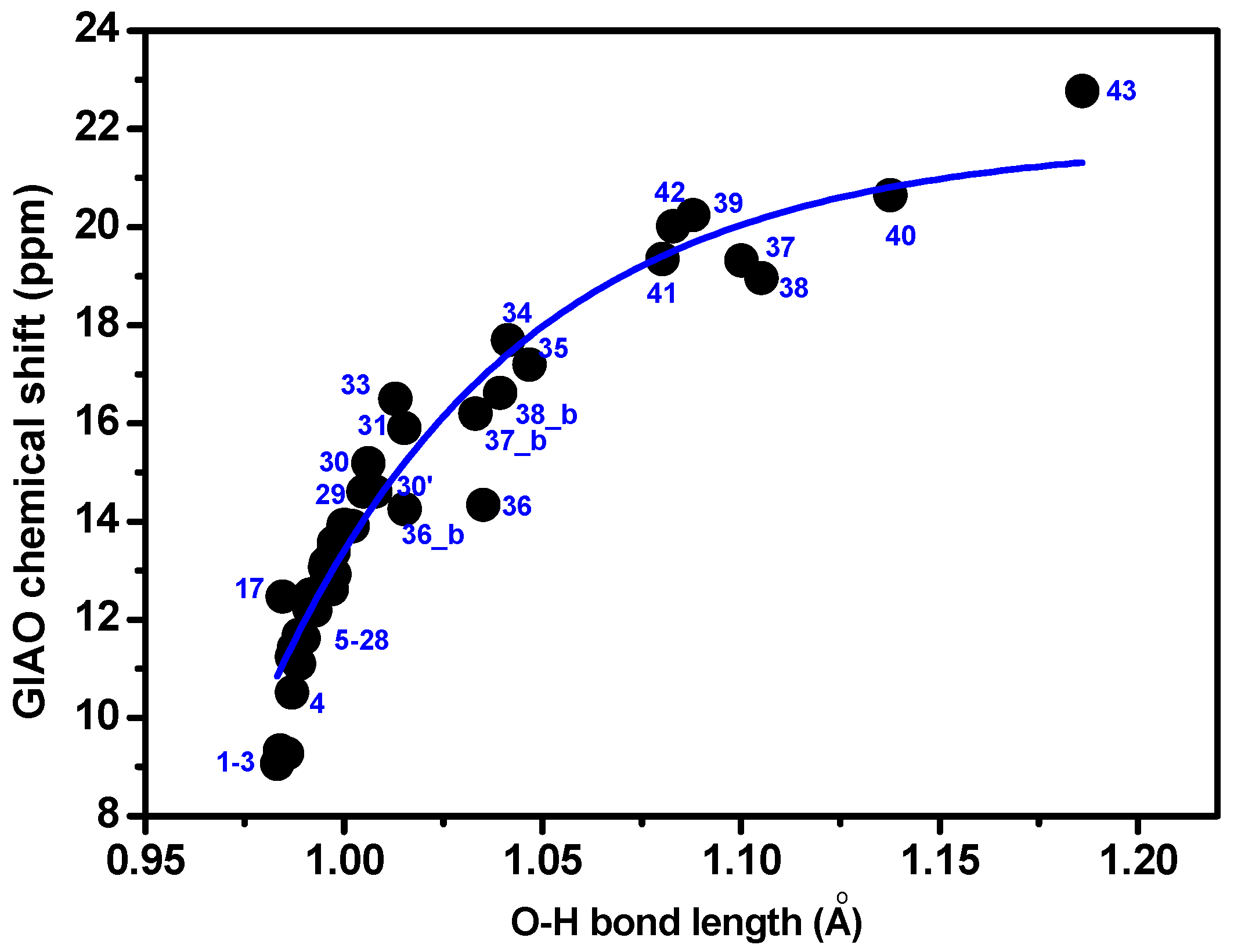
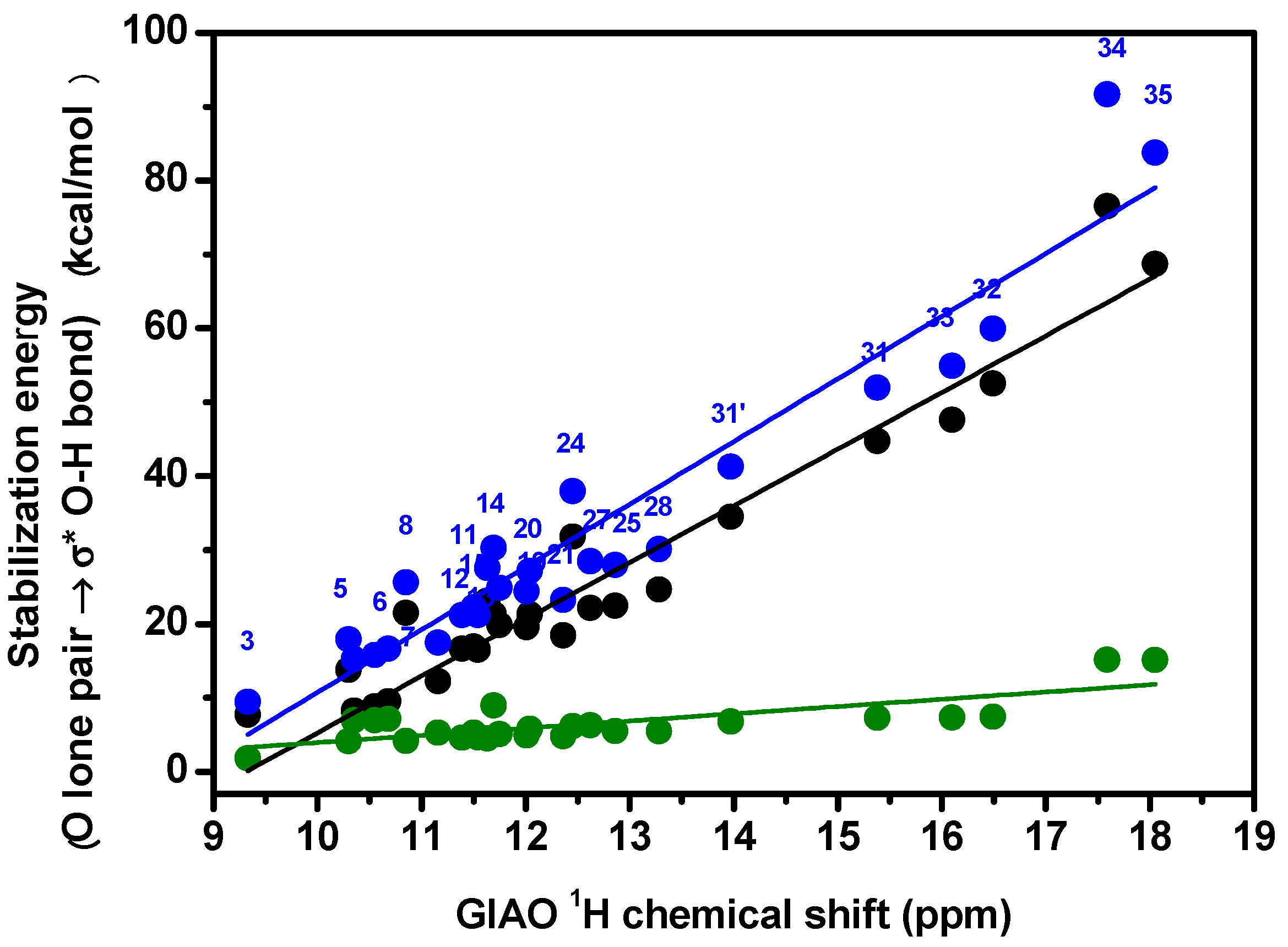
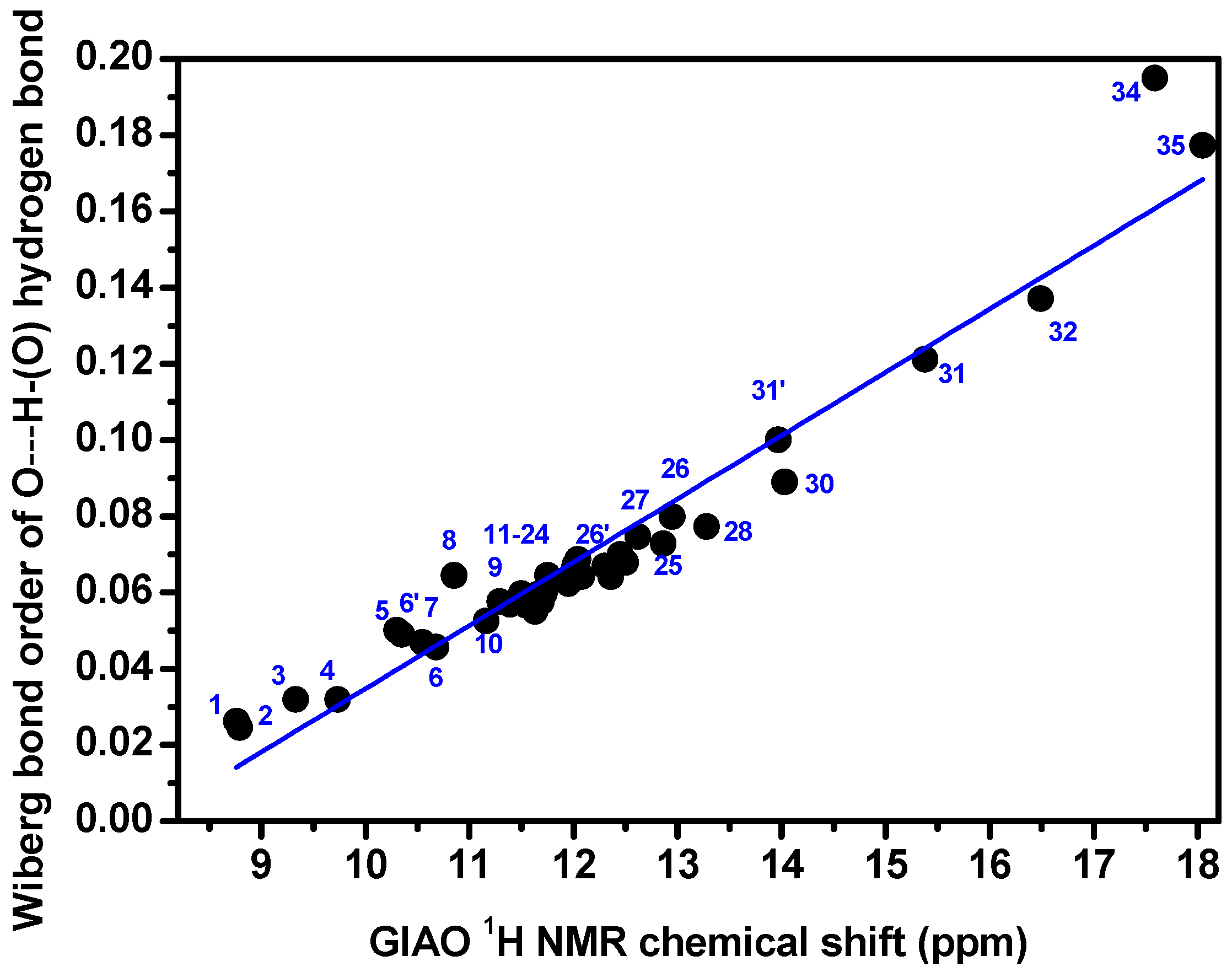
3.1. Factors Influencing OH 1H-NMR Chemical Shifts
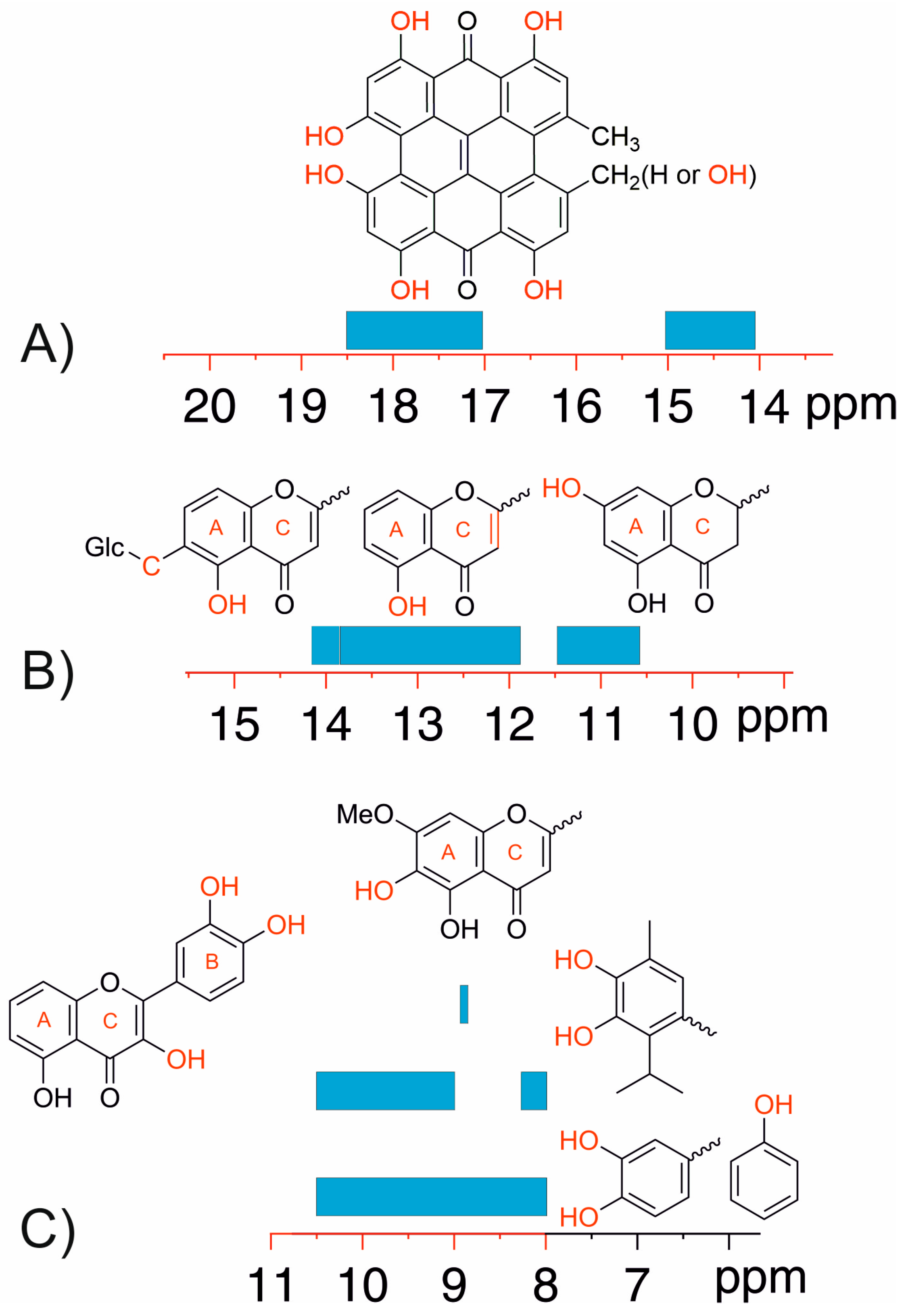
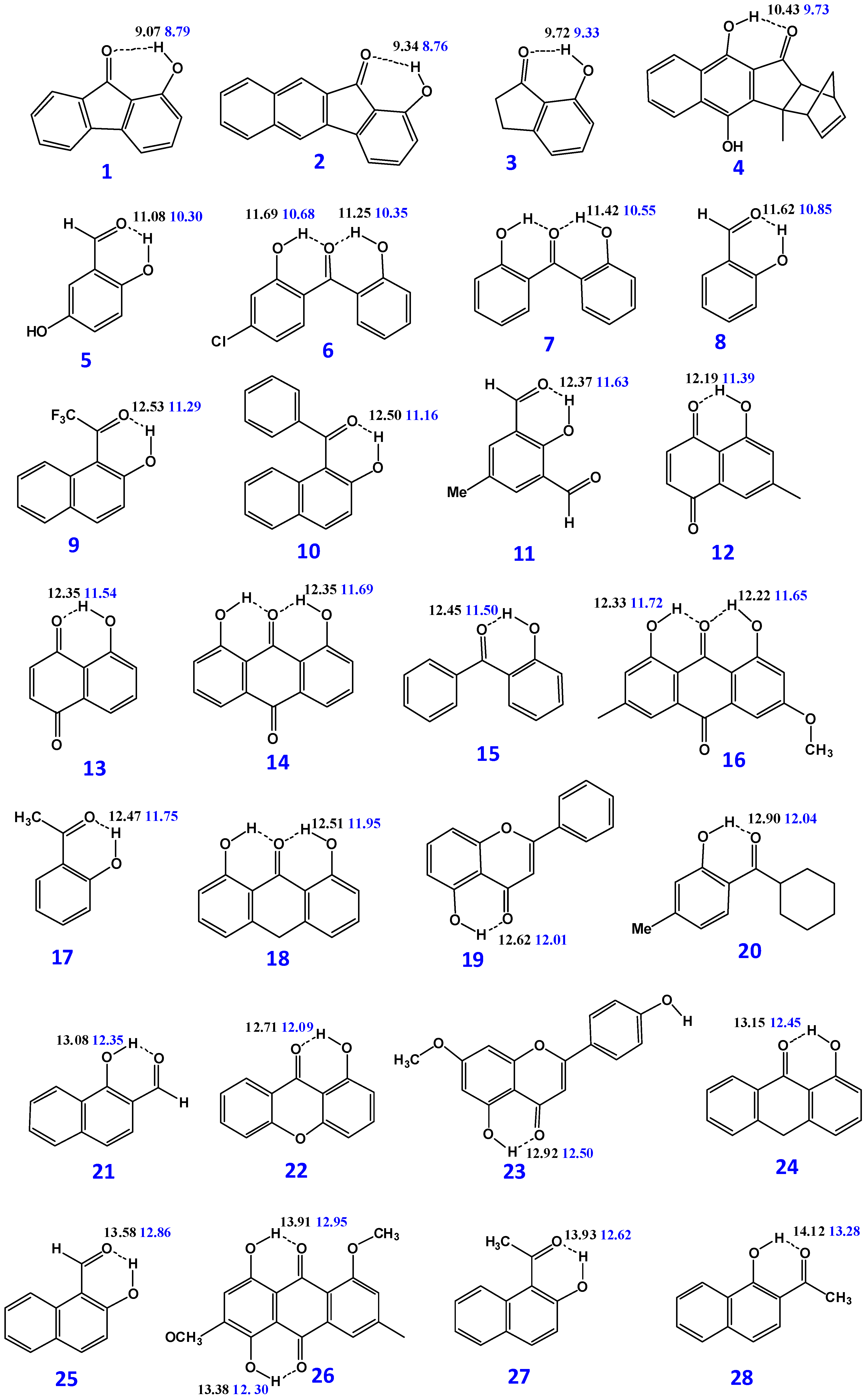
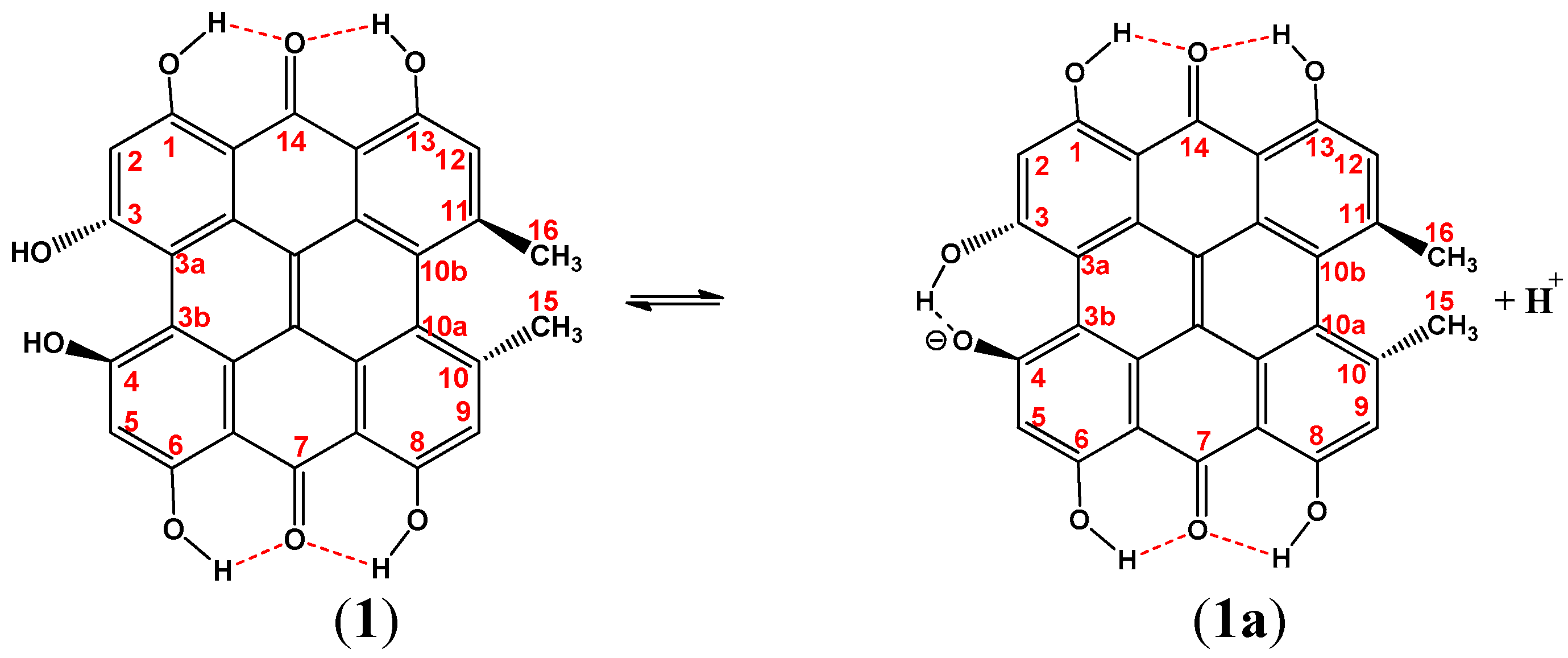
3.2. Resolving Conflicting Literature OH Resonance Assignments
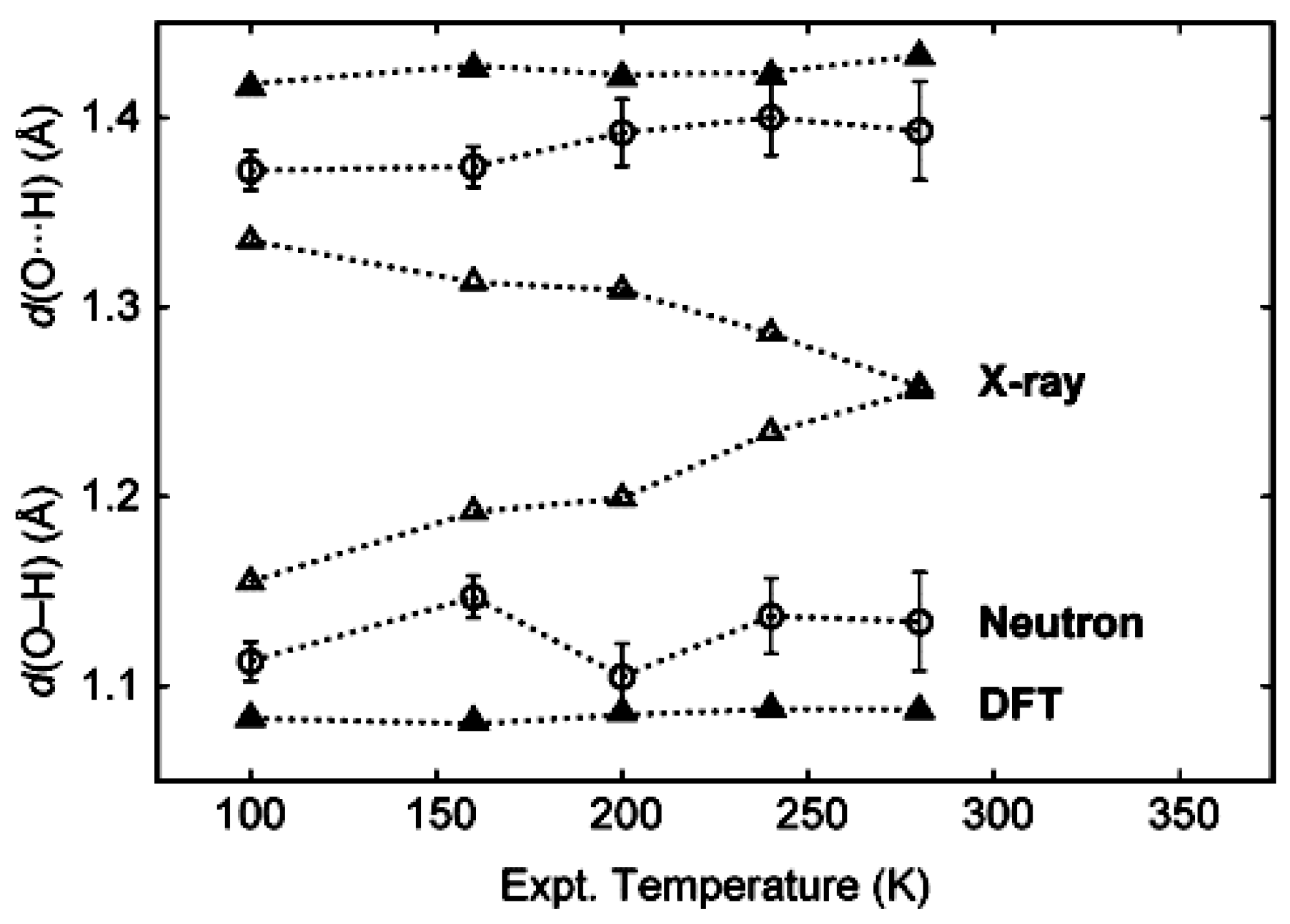
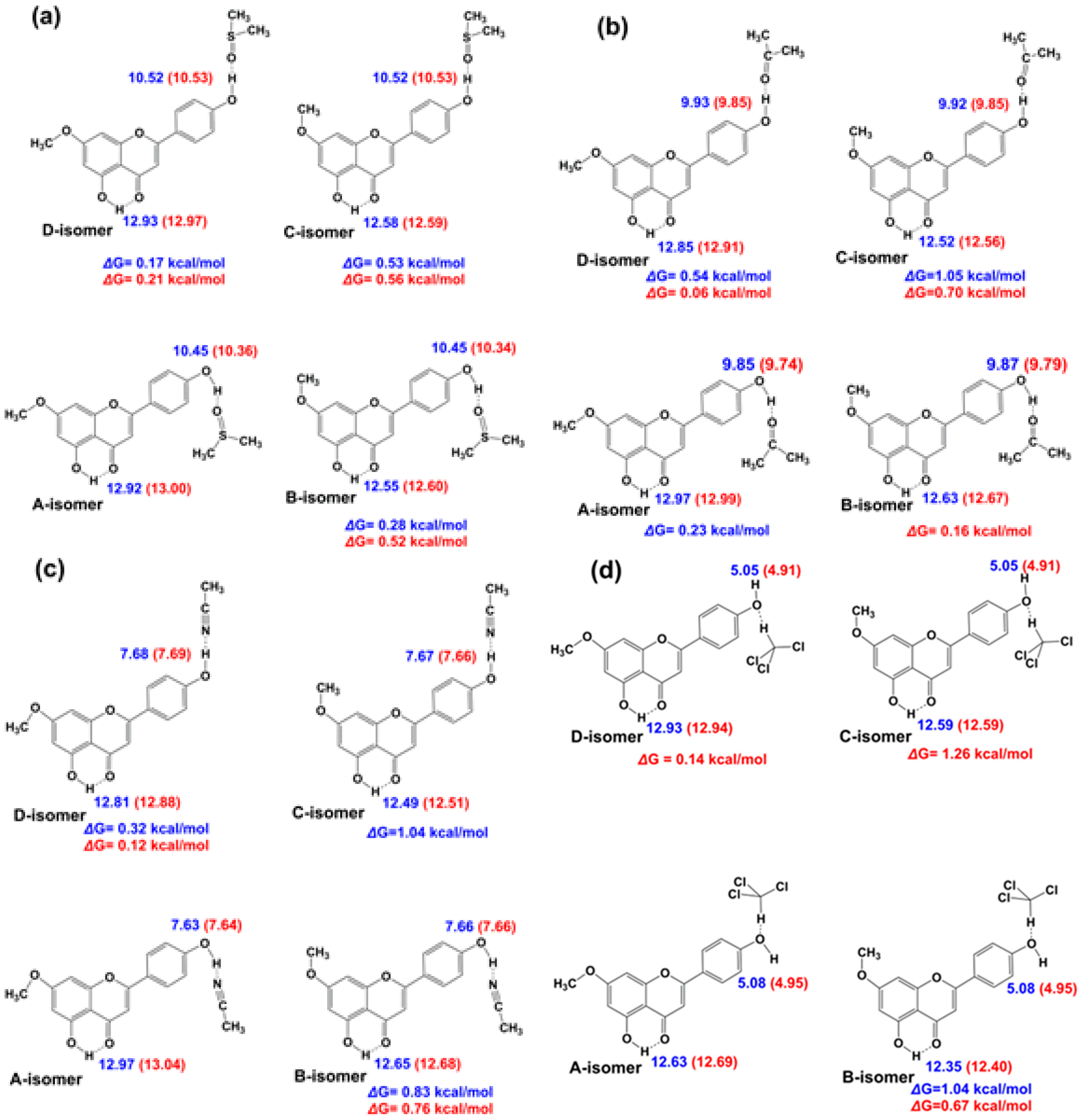
3.3. Comparison of X-ray with NMR Crystallography in the Solid State—The Location of Labile Hydrogens
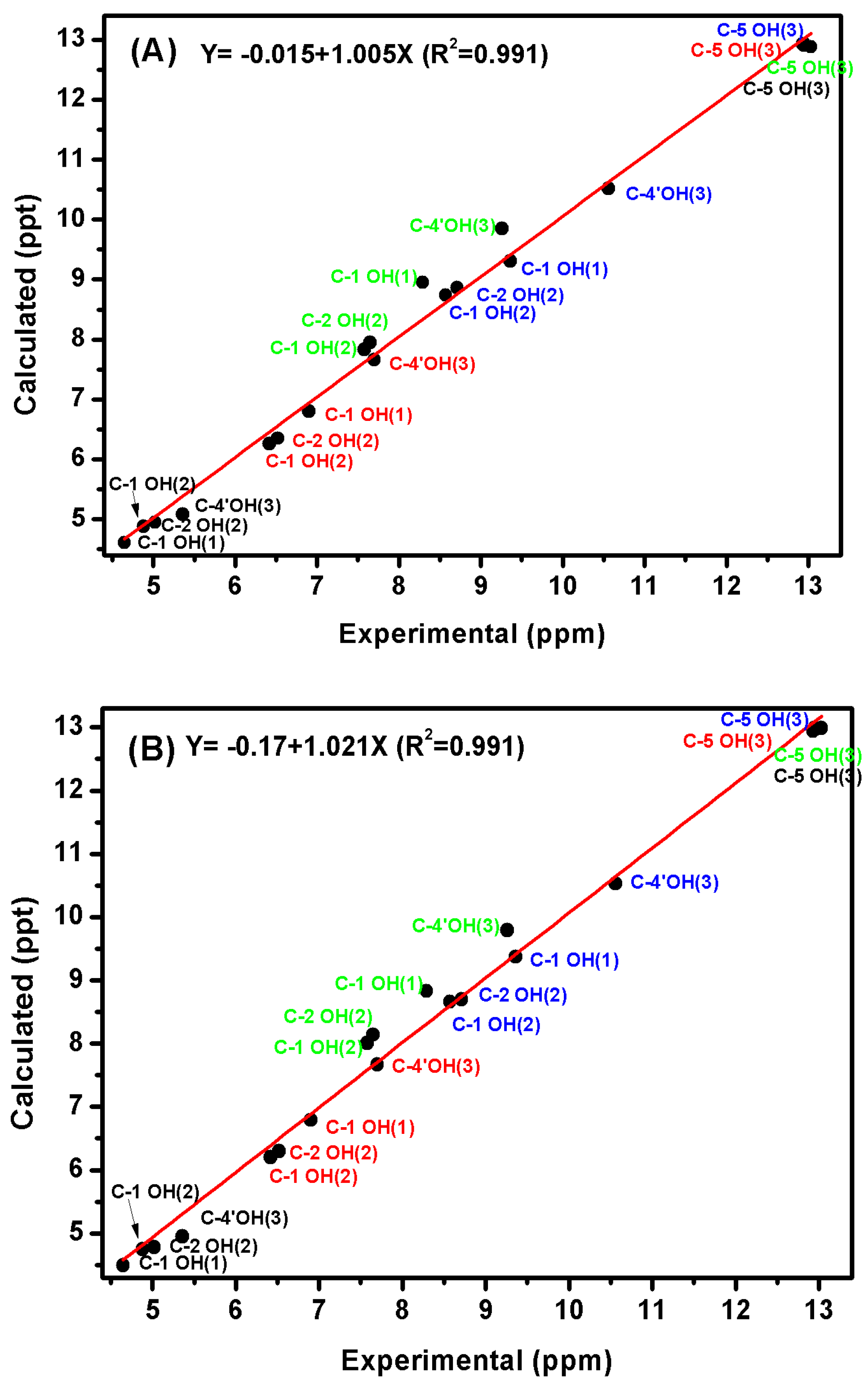
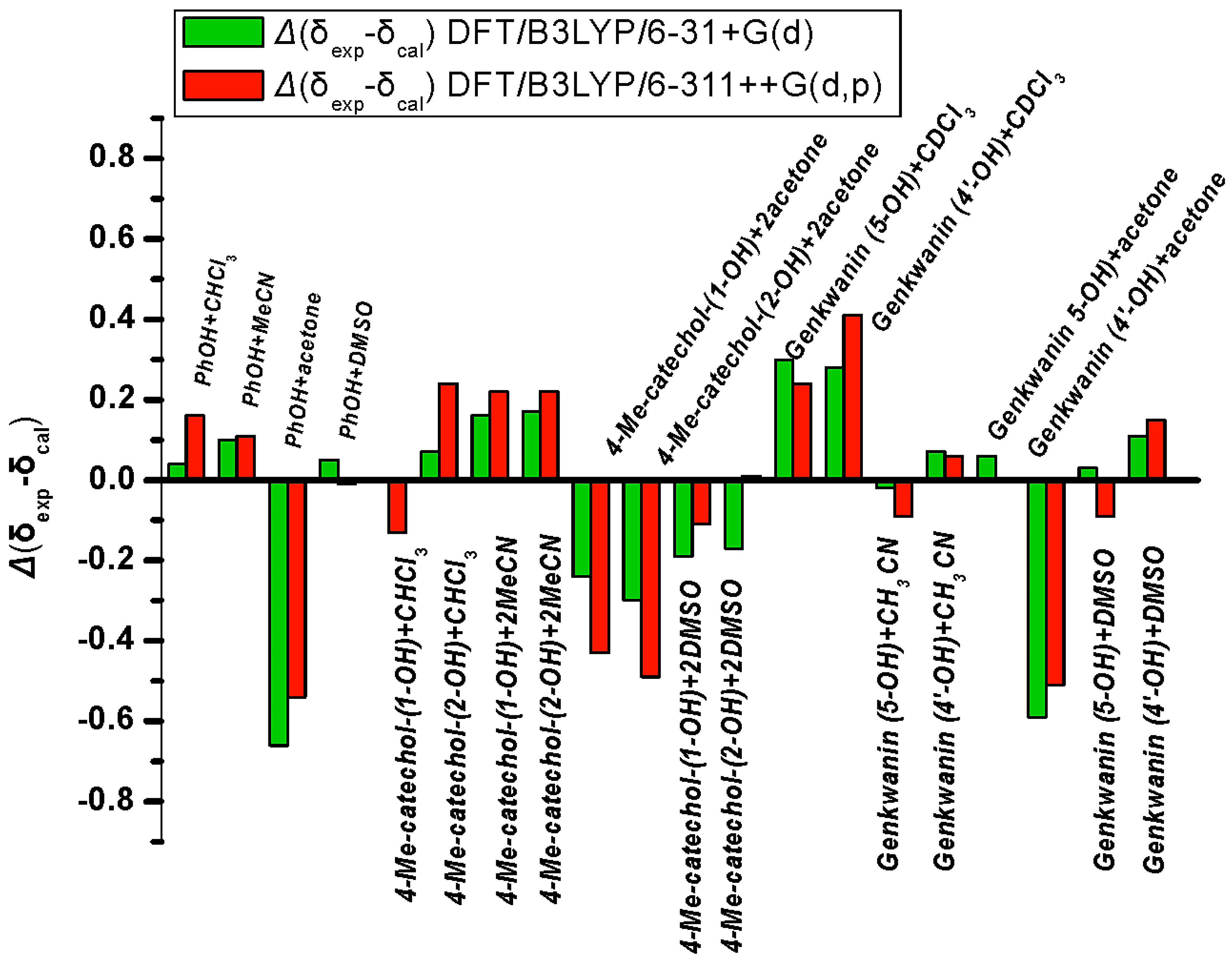
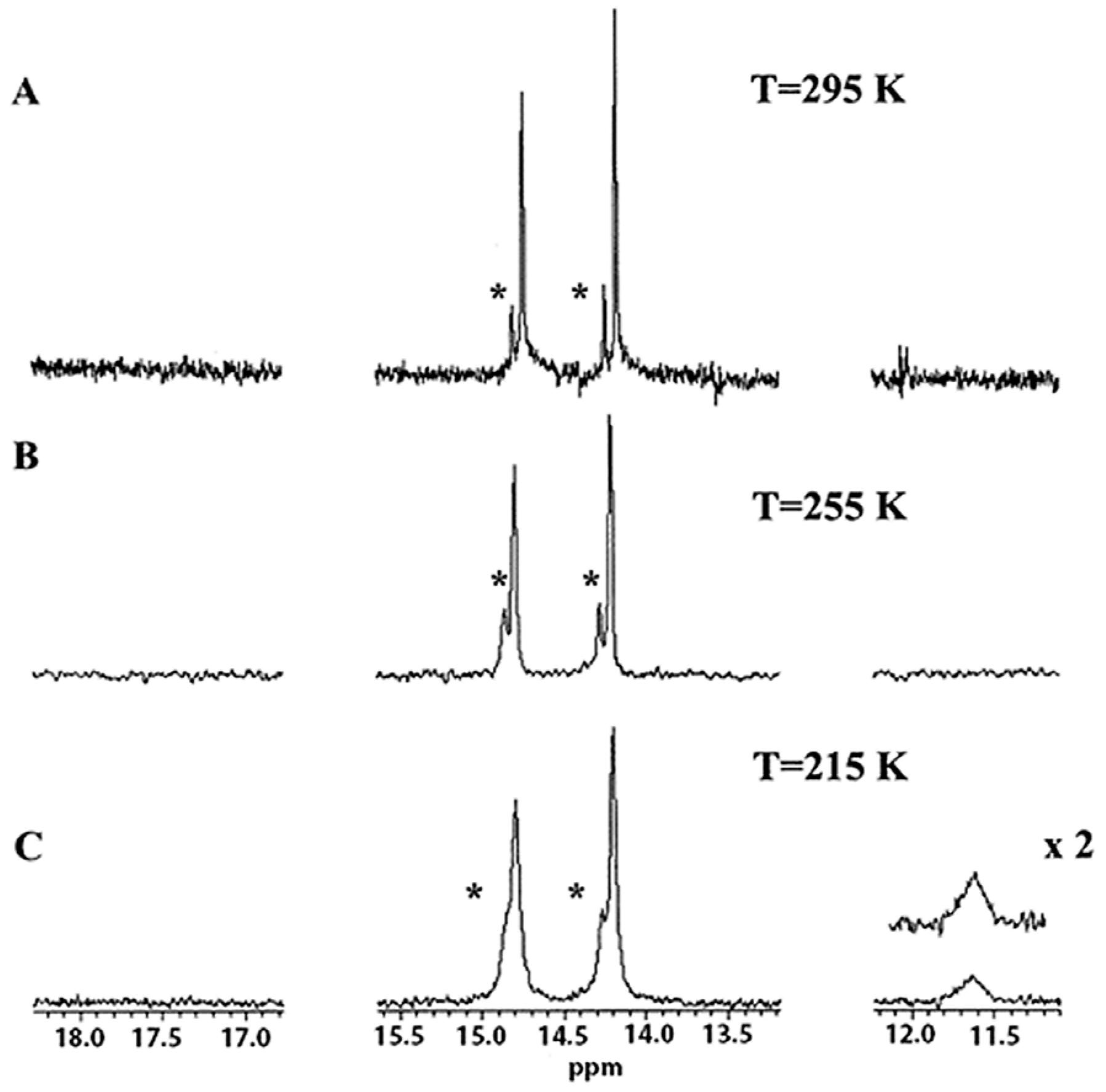
3.4. Comparison of X-ray with NMR Crystallography in Solution—The Location of Labile Hydrogens
4. Conclusions
- (i)
- in resolving conflicting literature data and ambiguities in resonance assignment;
- (ii)
- (iii)
- in investigating the nature of hydrogen bonding.
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jeffrey, G.A.; Saenger, S.W. Hydrogen Bonding in Biological Structures; Springer Verlag: Berlin, Germany, 1991. [Google Scholar]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Scheider, S. Hydrogen Bonding: A Theoretical Perspective; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Perrin, C.L.; Nielson, J.B. “Strong” hydrogen bonds in chemistry and biology. Annu. Rev. Phys. Chem. 1997, 48, 511–544. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Rozas, I.; Elguero, J. Non-conventional hydrogen bonds. Chem. Soc. Rev. 1998, 27, 163–170. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: New York, NY, USA, 1999. [Google Scholar]
- Steiner, T. The hydrogen bond in the solid state. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Grabowski, S.J. (Ed.) Hydrogen bonding—New insights. In Challenges and Advances in Computational Chemistry and Physics; Springer: Dordrecht, The Netherlands, 2005; Volume 3.
- Perrin, C.L. Are short, low-barrier hydrogen bonds unusually strong? Acc. Chem. Res. 2010, 43, 1550–1557. [Google Scholar] [CrossRef] [PubMed]
- Gilli, P.; Pretto, L.; Bertolasi, V.; Gilli, G. Predicting hydrogen-bond strengths from acid-base molecular properties. The pKa slide rule: Toward the solution of a long-lasting problem. Acc. Chem. Res. 2009, 42, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Aakeröy, C.B.; Salmon, D.J.; Smith, M.M.; Desper, J. Cyanooximes as effective and selective co-crystallizing agents. Cryst. Eng. Commun. 2009, 11, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Laurence, C.; Brameld, K.A.; Graton, J.; LeQuestel, J.-Y.; Renault, E. The pKBHX database: Toward a better understanding of hydrogen-bond basicity for medicinal chemists. J. Med. Chem. 2009, 52, 4073–4086. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, D.; Hunter, C.A.; Prohens, R.; Scuderi, S.; McCabe, J.F. Virtual cocrystal screeningists. Chem. Sci. 2011, 2, 883–890. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Epa, K.; Forbes, S.; Schultheiss, N.; Desper, J. Ranking relative hydrogen-bond strengths in hydroxybenzoic acids for crystal-engineering purposes. Chem. Eur. J. 2013, 19, 14998–15003. [Google Scholar] [CrossRef] [PubMed]
- Massa, W. Crystal Structure Determination; Springer: Berlin, Germany, 2004. [Google Scholar]
- Smart, L.E.; Moore, E.A. Solid State Chemistry: An Introduction; Taylor & Francis: London, UK, 2005. [Google Scholar]
- Steiner, T.; Majerz, I.; Wilson, C.C. First O-H-N hydrogen bond with a centered proton obtained by thermally induced proton migration. Angew. Chem. Int. Ed. 2001, 40, 2651–2654. [Google Scholar] [CrossRef]
- Allen, F.H. A systematic pairwise comparison of geometric parameters obtained by X-ray and neutron diffraction. Acta Crystallogr. Sect. B 1986, 42, 515–522. [Google Scholar] [CrossRef]
- Jeffrey, G.A.; Lewis, L. Cooperative aspects of hydrogen bonding in carbohydrates. Carbohydr. Res. 1978, 60, 179–182. [Google Scholar] [CrossRef]
- Evers, J.; Göbel, M.; Krumm, B.; Martin, F.; Medvedyev, S.; Oehlinger, G.; Xaver Steemann, F.; Troyan, I.; Klapötke, J.M.; Eremets, M.I. Molecular structure of hydrazoic acid with hydrogen-bonded tetramers in nearly planar layers. J. Am. Chem. Soc. 2011, 133, 12100–12105. [Google Scholar] [CrossRef] [PubMed]
- Shedrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Allan, D.R.; Clark, S.J. Comparison of the high-pressure and low-temperture structures of ethanol and acetic acid. Phys. Rev. B 1999, 60, 6328–6334. [Google Scholar] [CrossRef]
- Horiuchi, S.; Tokunaga, Y.; Giovannetti, G.; Picozzi, S.; Itoh, H.; Shimano, R.; Kumai, R.; Tokura, Y. Above-room-temperature ferroelectricity in a single-component molecular crystal. Nature 2010, 463, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Deringer, V.L.; Hoepfner, V.; Dronskowski, R. Accurate hydrogen positions in organic crystals: Assessing a quantum-chemical aide. Cryst. Growth Des. 2012, 12, 1014–1021. [Google Scholar] [CrossRef]
- Dračínský, M. The chemical bond: The perspective of NMR spectroscopy. Ann. Rep. NMR Spectrosc. 2016, 90, 1–40. [Google Scholar]
- Brunner, E.; Sternberg, U. Solid-state NMR investigations on the nature of hydrogen bonds. Nucl. Magn. Reson. Spectrosc. 1998, 32, 21–57. [Google Scholar] [CrossRef]
- Aliev, A.E.; Harris, K.D.M. Probing hydrogen bonding in solids using solid state NMR spectroscopy. Struct. Bond. 2004, 108, 1–53. [Google Scholar]
- Jeffrey, G.A.; Yeon, Y. The correlation between hydrogen-bond lengths and proton chemical shifts in crystals. Acta Crystallogr. B 1986, 42, 410–413. [Google Scholar] [CrossRef]
- Sternberg, U.; Brunner, E. The influence of short-range geometry on the chemical shift of protons in hydrogen bonds. J. Magn. Reson. A 1994, 108, 142–150. [Google Scholar] [CrossRef]
- Harris, T.K.; Mildvan, S.A. High-precision measurement of hydrogen bond lengths in proteins by nuclear magnetic resonance methods. Proteins Struct. Funct. Genet. 1999, 35, 275–282. [Google Scholar] [CrossRef]
- Sigala, P.A.; Fafarman, A.T.; Schwans, J.P.; Fried, S.D.; Fenn, T.D.; Caaveiro, J.M.; Pybus, B.; Ringe, D.; Petsko, G.A.; Boxer, S.G.; et al. Quantitative dissection of hydrogen bond-mediated proton transfer in the ketosteroid isomerase active site. Proc. Natl. Acad. Sci. USA 2013, 110, E2552–E2561. [Google Scholar] [CrossRef]
- Baxter, N.J.; Williamson, M.P. Temperature dependence of 1H chemical shifts in proteins. J. Biomol. NMR 1997, 9, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Rance, M.; Houghten, R.A.; Lerner, R.A.; Wright, P.E. Folding of immunogenic peptide fragments of proteins in water solution. I. Sequence requirements for the formation of a reverse turn. J. Mol. Biol. 1988, 201, 161–200. [Google Scholar] [CrossRef]
- Englander, S.W.; Kallenbach, N.R. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys. 1983, 16, 521–655. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, D.; Williamson, M.P. The nuclear Overhauser Effect in Structural and Conformational Analysis, 2nd ed.; Willey–VCH: New York, NY, USA, 2000. [Google Scholar]
- Vögeli, B. The nuclear Overhauser effect from a quantitative perspective. Progr. Nucl. Magn. Reson. Spectrosc. 2014, 78, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Dingley, A.J.; Grzesiek, S. Direct observation of hydrogen bonds in nucleic acid base pairs by internucleotide 2JNN couplings. J. Am. Chem. Soc. 1998, 120, 8293–8297. [Google Scholar] [CrossRef]
- Grzesiek, S.; Cordier, F.; Dingley, A.J. Scalar couplings across hydrogen bonds. Methods Enzymol. 2002, 338, 111–133. [Google Scholar]
- Bolvig, S.; Hansen, P.E. Isotope effects on chemical shifts as an analytical tool in structural studies of intramolecular hydrogen bonded compounds. Curr. Org. Chem. 2000, 4, 19–54. [Google Scholar] [CrossRef]
- Naito, A.; Nishimura, K.; Kimura, S.; Aida, M.; Yasuoka, N.; Tuzi, S.; Saito, H. Determination of the three-dimensional structure of a new crystalline form of N-Acetyl-Pro-Gly-Phe as revealed by 13C REDOR, X-ray diffraction, and molecular dynamics calculation. J. Phys. Chem. 1996, 100, 14995–15004. [Google Scholar] [CrossRef]
- Zhao, X.; Sudmeier, J.I.; Bachovchin, W.W.; Levitt, M.H. Measurement of NH bond lengths by fast magic-angle spinning solid-state NMR spectroscopy: A new method for the quantification of hydrogen bonds. J. Am. Chem. Soc. 2001, 123, 11097–11098. [Google Scholar] [CrossRef] [PubMed]
- Schnell, I.; Saalwächter, K. 15N–H bond length determination in natural abundance by inverse detection in fast-MAS solid-state NMR Spectroscopy. J. Am. Chem. Soc. 2002, 124, 10938–10939. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Improra, R.; Rega, N. Quantum mechanical computations and spectroscopy: From small rigid molecules in the gas phase to large flexible molecules in solution. Acc. Chem. Res. 2008, 41, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Mulder, F.A.A.; Filatov, M. NMR chemical shift data and ab initio shielding calculations: Emerging tools for protein structure determination. Chem. Soc. Rev. 2010, 39, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of 1H-NMR and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef] [PubMed]
- Siskos, M.G.; Kontogianni, V.G.; Tsiafoulis, C.G.; Tzakos, A.G.; Gerothanassis, I.P. Investigation of solute–solvent interactions in phenol compounds: Accurate ab initio calculations of solvent effects on 1H-NMR chemical shifts. Org. Biomol. Chem. 2013, 11, 7400–7411. [Google Scholar] [CrossRef] [PubMed]
- Siskos, M.G.; Tzakos, A.G.; Gerothanassis, I.P. Accurate ab initio calculations of O–H⋯O and O–H⋯−O proton chemical shifts: Towards elucidation of the nature of the hydrogen bond and prediction of hydrogen bond distances. Org. Biomol. Chem. 2015, 13, 8852–8868. [Google Scholar] [CrossRef] [PubMed]
- Lomas, J.S. 1H-NMR spectra of butane-1,4-diol and other 1,4 diols: DFT calculations of shifts and coupling constants. Magn. Reson. Chem. 2014, 52, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.K.; Ghi, P.Y.; Hammond, R.B.; Ma, C.-Y.; Roberts, K.J. Refinement of hydrogen atomic position in a hydrogen bond using a combination of solid-state NMR and computation. Chem. Commun. 2003, 2834–2835. [Google Scholar] [CrossRef]
- Siskos, M.G.; Choudhary, M.C.; Tzakos, A.G.; Gerothanassis, I.P. 1H-NMR chemical shift assignment, structure and conformational elucidation of hypericin with the use of DFT calculations—The challenge of accurate labile hydrogens. Tetrahedron 2016, 72, 8287–8293. [Google Scholar] [CrossRef]
- Harris, R.K.; Wasylishen, R.E.; Duer, M.J. (Eds.) NMR Crystallography; John Willey & Sons: Hoboken, NJ, USA, 2009.
- Appreley, D.; Harris, R.K.; Hodgkinson, P. Solid state NMR: Basic Principles & Practice; Momentum Press LLC: New York, NY, USA, 2012. [Google Scholar]
- Ashbrook, S.E.; McKay, D. Combining solid state NMR spectroscopy with first principles calculations—A guide to NMR crystallography. Chem. Commun. 2016, 52, 7186–7204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Florence, A.J.; Bardin, J.; Johnston, B.; Shankland, N.; Griffin, T.A.N.; Shankland, K. Structure determination from powder data: Mogul and CASTEP. Zeit. Kristallogr. 2009, 30, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.J.; Matthew, I.; Segall, D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Zeit. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burcke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Hilbert, F.; Emsley, J. Hydrogen bonding and chemical reactivity. Adv. Phys. Org. Chem. 1990, 26, 255–379. [Google Scholar]
- Claramunt, R.M.; Lopez, C.; Santa Maria, M.D.; Sanz, D.; Elguero, J. The use of NMR spectroscopy to study tautomerism. Progr. Nucl. Magn. Reson Spectrosc. 2006, 49, 169–206. [Google Scholar] [CrossRef]
- Antonov, L. Tautomerism: Methods and Theories; Willey-VCH Verlag: Weinhein, Germany, 2014. [Google Scholar]
- Thomas, L.H.; Florence, A.J.; Wilson, C.C. Hydrogen atom behaviour imaged in a short intramolecular hydrogen bond using the combined approach of X-ray and neutron diffraction. New J. Chem. 2009, 33, 2486–2490. [Google Scholar] [CrossRef]
- Poppe, L.; van Halbeek, H. NMR spectroscopy of hydroxyl protons in supercooled carbohydrates. Nat. Struct. Mol. Biol. 1994, 1, 215–216. [Google Scholar] [CrossRef]
- Bekiroglou, S.; Sandström, A.; Kenne, L.; Sandtröm, C. Ab initio and NMR studies on the effect of hydration on the chemical shift of hydroxyl protons in carbohydrates using disaccharides and water/methanol/ethers as model systems. Org. Biomol. Chem. 2004, 2, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Charisiadis, P.; Exarchou, V.; Troganis, A.N.; Gerothanassis, I.P. Exploring the “forgotten” –OH 1H-NMR spectral region in natural products. Chem. Commun. 2010, 46, 3589–3591. [Google Scholar] [CrossRef] [PubMed]
- Charisiadis, P.; Primikyri, A.; Exarchou, V.; Tzakos, A.; Gerothanassis, I.P. Unprecedented ultra-high-resolution hydroxy group 1H-NMR spectroscopic analysis of plant extracts. J. Nat. Prod. 2011, 74, 2462–2466. [Google Scholar] [CrossRef] [PubMed]
- Kontogianni, V.G.; Tsiafoulis, C.G.; Tzakos, A.G.; Gerothanassis, I.P. Ηydrogen bonding probes of phenol –OH groups. Org. Biomol. Chem. 2013, 11, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Charisiadis, P.; Kontogianni, V.G.; Tsiafoulis, C.G.; Tzakos, A.G.; Siskos, M.; Gerothanassis, I.P. 1H-NMR as a structural and analytical tool of intra- and intermolecular hydrogen bonds of phenol-containing natural products and model compounds. Molecules 2014, 19, 13643–13682. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Abeygunawardana, C.; Giltis, A.G.; Mildvan, A.S. Hydrogen bonding at the active site of Δ5−3-ketosteroid isomerase. Biochemistry 1997, 36, 14614–14626. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.K.; Zhao, Q.; Mildvan, A.S. NMR studies of strong hydrogen bonds in enzymes and in a model compound. J. Mol. Struct. 2000, 552, 97–109. [Google Scholar] [CrossRef]
- Tolstoy, P.M.; Koeppe, B.; Denisov, G.S.; Limbach, H.-H. Combined NMR and UV-Vis spectroscopy in the solution state: Study of the geometries of strong OHO hydrogen bonds of phenols with carboxylic acids. Angew. Chem. Int. Ed. 2009, 48, 5745–5747. [Google Scholar] [CrossRef] [PubMed]
- Koeppe, B.; Tolstoy, P.M.; Limbach, H.-H. Reactions pathways of proton transfer in hydrogen-bonded phenol-carboxylate complexes explored by combined UV-Vis and NMR spectroscopy. J. Am. Chem. Soc. 2011, 133, 7897–7908. [Google Scholar] [CrossRef] [PubMed]
- Koeppe, B.; Guo, J.; Tolstoy, P.M.; Denisov, G.S.; Limbach, H.-H. Solvent and H/D isotope effects on the proton transfer pathways in heteroconjucated hydrogen-bonded phenol-carboxylic acid anions observed by the combined UV-Vis and NMR spectroscopy. J. Am. Chem. Soc. 2013, 135, 7553–7566. [Google Scholar] [CrossRef] [PubMed]
- Limbach, H.-H.; Tolstoy, P.M.; Perez-Hernandez, N.; Guo, J.; Shenderovich, I.G.; Denisov, G.S. OHO hydrogen bond geometries and NMR chemical shifts: From equilibrium structures to geometric H/D isotope effects, with applications for water, protonated water, and compressed ice. Isr. J. Chem. 2009, 49, 199–216. [Google Scholar] [CrossRef]
- Pylaeva, S.; Allolio, C.; Koeppe, B.; Denisov, G.S.; Limbach, H.-H.; Sebastiani, D.; Tolstoy, P.M. Proton transfer pathways in a short hydrogen bond caused be solvation shell fluctuation: An ab initio MD and NMR/UV study of N(OHO)− bonded system. Phys. Chem. Chem. Phys. 2015, 17, 4634–4644. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBEO model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Berglund, B.; Vaughan, R.W. Correlations between proton chemical shift tensors, deuterium quadrupole couplings, and bond distances for hydrogen bonds in solids. J. Chem. Phys. 1980, 73, 2037–2043. [Google Scholar] [CrossRef]
- Harris, R.K.; Jackson, P.; Merwin, L.H.; Say, B.J.; Hägele, G. Perspectives in high-resolution solid-state nuclear magnetic resonance, with emphasis on combined rotation and multiple-pulse spectroscopy. J. Chem. Soc. Faraday Trans. 1988, 84, 3649–3672. [Google Scholar] [CrossRef]
- Bertolasi, V.; Gilli, P.; Ferretti, V.; Gilli, G. Intramolecular O–H···O hydrogen bonds assisted by resonance. Correlation between crystallographic data and 1H-NMR chemical shifts. J. Chem. Soc. Perkin Trans. 2 1997, 945–952. [Google Scholar] [CrossRef]
- Mariam, Y.H.; Musin, R.N. Transition from moderate to strong hydrogen bonds: Its identification and physical bases in the case of O–H···O intramolecular hydrogen bonds. J. Phys. Chem. A 2008, 112, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.J.; Mobli, M. An NMR, IR and theoretical investigation of 1H chemical shifts and hydrogen bonding in phenols. Magn. Reson. Chem. 2007, 45, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tolstoy, P.M.; Koeppe, B.; Golubev, N.S.; Denisov, G.S.; Smirnov, S.N.; Limbach, H.-H. Hydrogen bond geometries and proton tautomerism of homoconjugated anions of carboxylic acids studied via H/D isotope effects on 13C-NMR chemical shift. J. Phys. Chem. A 2012, 116, 11180–11188. [Google Scholar] [CrossRef] [PubMed]
- Tolstoy, P.M.; Schah-Mohammedi, P.; Smirnov, S.N.; Golubev, N.S.; Denisov, G.S.; Limbach, H.-H. Characterization of fluxional hydrogen-bonded complexes of acetic acid and acetate by NMR: Geometries and isotope and solvent effects. J. Am. Chem. Soc. 2004, 126, 5621–5634. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Masuda, Y. Effect of solvent and proton location and dynamic behavior in short intramolecular hydrogen bonds studied by molecular dynamics simulations and NMR experiments. Chem. Phys. 2015, 458, 18–29. [Google Scholar] [CrossRef]
- Altman, L.J.; Laungani, P.; Gunnarsson, G.; Wennerstrom, H.; Forsen, S. Proton, deuterium, and tritium nuclear magnetic resonance of intramolecular hydrogen bonds. Isotope effects and the shape of the potential energy function. J. Am. Chem. Soc. 1978, 100, 8264–8266. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural bond orbital analysis of near Hartree-Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. [Google Scholar] [CrossRef]
- Weinhold, F.; Klein, R.A. What is hydrogen bond? Mutually consistent theoretical and experimental criteria for characterizing H-bonding interactions. Mol. Phys. 2012, 110, 565–579. [Google Scholar] [CrossRef]
- Grabowski, S.J. What is the covalency of hydrogen bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef] [PubMed]
- Steiner, T.; Saenger, W. Covalent bond lengthening in hydroxyl groups involved in three-center and in cooperative hydrogen bonds. Analysis of low-temperature neutron diffraction data. J. Am. Chem. Soc. 1992, 114, 7123–7126. [Google Scholar] [CrossRef]
- Desiraju, G.R. A bond by any other name. Angew. Chem. Int. Ed. 2011, 50, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Interpretation of spectroscopic methods of hydrogen bonds. Phys. Chem. Chem. Phys. 2016, 17, 2263–2271. [Google Scholar]
- Scheiner, S. Assessment of the presence and strength of H-bonds by means of corrected NMR. Molecules 2016, 21, 1426. [Google Scholar] [CrossRef] [PubMed]
- Charisiadis, P.; Tsiafoulis, C.G.; Exarchou, V.; Tzakos, A.G.; Gerothanassis, I.P. Rapid and direct low micromolecular NMR method for the simultaneous detection of hydrogen peroxide and phenolics in plant extracts. J. Agric. Food Chem. 2012, 60, 4508–4513. [Google Scholar] [CrossRef] [PubMed]
- Neratzaki, A.A.; Tsiafoulis, C.G.; Charisiadis, P.; Kontogianni, V.G.; Gerothanassis, I.P. Novel determination of the total phenolic content in crude plant extracts by the use of 1H NMR of the –OH spectral region. Anal. Chim. Acta 2011, 688, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Charisiadis, P.; Kontogianni, V.G.; Tsiafoulis, C.G.; Tzakos, A.G.; Gerothanassis, I.P. Determination of polyphenolic phytochemicals using highly deshielded –OH 1H-NMR signals. Phytochem. Anal. 2017. [Google Scholar] [CrossRef]
- Falk, H. From the photosensitizer hypericin to the photoreceptor stentorian—The chemistry of phenantroperylene quinines. Angew. Chem. Int. Ed. 1999, 38, 3117–3136. [Google Scholar] [CrossRef]
- Alecu, M.; Ursaciuc, C.; Halalau, F.; Coman, G.; Merlevede, W.; Waelkens, E.; de Witte, P. Photodynamic treatment of basal cell carcinoma and squamous cell carcinoma with hypericin. Anticancer Res. 1998, 18, 4651–4654. [Google Scholar] [PubMed]
- Freeman, D.; Frolow, F.; Kapinus, E.; Lavie, D.; Meruelo, D.; Mazur, Y. Acidic properties of hypericin and its octahydroxy analogue in the ground and excited states. J. Chem. Soc. Chem. Commun. 1994, 891–892. [Google Scholar] [CrossRef]
- Leonhartsberger, J.G.; Falk, H. The protonation and deprotonation equilibria of hypericin revisited. Monatsh. Chem. 2002, 133, 167–172. [Google Scholar] [CrossRef]
- Smirnov, A.; Fulton, D.B.; Andreotti, A.; Petrich, J.W. Exploring ground-state heterogeneity of hypericin and hypocrellin A and B: Dynamic and 2D ROESY NMR study. J. Am. Chem. Soc. 1999, 121, 7979–7988. [Google Scholar] [CrossRef]
- Dax, T.G.; Falk, H.; Kapinus, E.I. Ein nachweis fur die struktur des 1,6-dioxo-tautomeren des hypericin. Monatsh. Chem. 1999, 130, 827–831. [Google Scholar] [CrossRef]
- Skalkos, D.; Tatsis, E.; Gerothanassis, I.P.; Troganis, A. Towards a consensus structure of hypericin in solution: Direct evidence for a single tautomer and different ionization states in protic and non protic solvents by the use of variable temperature gradient 1H-NMR. Tetrahedron 2002, 58, 4925–4929. [Google Scholar] [CrossRef]
- Klein, R.A.; Mennucci, B.; Tomasi, J. Ab initio calculations of 17O-NMR-chemical shifts for water. The limits of PCM theory and the role of hydrogen-bond geometry and cooperativity. J. Phys. Chem. A 2004, 108, 5851–5863. [Google Scholar] [CrossRef]
- Pickard, C.J.; Mauri, F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys. Rev. B 2001, 63, 245101. [Google Scholar] [CrossRef]
- Bonhomme, C.; Gervais, C.; Babonneau, F.; Coelho, C.; Pourpoint, F.; Azaïs, T.; Ashbrook, S.E.; Griffin, J.M.; Yates, J.R.; Mauri, F.; et al. First-principles calculation of NMR parameters using the gauge including projector augmented wave method: A chemist’s point of view. Chem. Rev. 2012, 112, 5733–5779. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R.; Dobbins, S.E.; Pickard, C.J.; Mauri, F.; Chi, P.Y.; Harris, R.K. A combined first principles computational and solid-state NMR study of a molecular crystal: Flurbiprofen. Phys. Chem. Chem. Phys. 2005, 7, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Flippen, J.L.; Gilardi, R.D. (+)-2-(2-Fluoro-4-biphenyl) propionic acid (flurbiprofen). Acta Crystallogr. Sect. B 1975, 31, 926–928. [Google Scholar] [CrossRef]
- Widdifield, C.M.; Robson, H.; Hodgkinson, P. Furosemide’s one little hydrogen atom: NMR crystallography structure verification of powdered molecular organics. Chem. Commun. 2016, 52, 6685–6688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baias, M.; Widdifield, C.M.; Dumez, J.-N.; Thompson, H.P.G.; Cooper, T.G.; Salager, E.; Bassil, S.; Stein, R.S.; Lesage, A.; Day, G.M.; et al. Powder crystallography of pharmaceutical materials by combined crystal structure prediction and solid-state 1H NMR spectroscopy. Phys. Chem. Chem. Phys. 2013, 15, 8069–8080. [Google Scholar] [CrossRef] [PubMed]
- Filip, X.; Grosu, I.-G.; Miclăuş, M.; Filip, C. NMR crystallography methods to probe complex hydrogen bonding networks: Application to structure elucidation of anhydrous quercetin. Cryst. Eng. Commun. 2013, 15, 4131–4142. [Google Scholar] [CrossRef]
- Filip, X.; Filip, C. Can the conformation of flexible hydroxyl groups be constrained by simple NMR crystallography approaches? The case of the quercetin solid forms. Solid State NMR 2015, 65, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.-Z.; Yamagata, Y.; Tomita, K. Structure of quercetin dihydrate. Acta Cryst. C 1990, 46, 310–313. [Google Scholar] [CrossRef]
- Pickard, C.J.; Salager, E.; Pintacuda, G.; Elena, B.; Emsley, L. Resolving structures from powders by NMR crystallography using combined proton spin diffusion and plane wave DFT calculations. J. Am. Chem. Soc. 2007, 129, 8932–8933. [Google Scholar] [CrossRef] [PubMed]
- Vila, A.J.; Lagier, C.M.; Olivieri, A.C. 13C-NMR and AM1 study of the intramolecular proton transfer in solid 1,3-diphenylpropane-1,3-dione. J. Chem. Soc. Perkin Trans. 2 1990, 9, 1615–1618. [Google Scholar] [CrossRef]
- Gerothanassis, I.P. Oxygen-17 NMR spectroscopy: Basic principles and applications (Part I). Progr. NMR Spectrosc. 2010, 56, 96–197. [Google Scholar] [CrossRef] [PubMed]
- Gerothanassis, I.P. Oxygen-17 NMR spectroscopy: Basic principles and applications (Part II). Progr. NMR Spectrosc. 2010, 27, 1–110. [Google Scholar] [CrossRef] [PubMed]
- Dziembowska, T.; Hansen, P.E.; Rozwadowski, Z. Studies based on deuterium isotope effect on 13C chemical shifts. Progr. NMR Spectrosc. 2004, 45, 1–29. [Google Scholar] [CrossRef]
- Borisov, E.Y.; Zhang, W.; Bolvig, S.; Hansen, P.E. nJ(13C,O1H) coupling constants of intramolecularly hydrogen-bonded compounds. Magn. Reson. Chem. 1998, 36, S104–S110. [Google Scholar] [CrossRef]
- Bogle, X.S.; Singleton, D.A. Isotope-induced desymmetrization can mimic isotopic perturbation of equilibria. On the symmetry of bromonium ions and hydrogen bonds. J. Am. Chem. Soc. 2011, 133, 17172–17175. [Google Scholar] [CrossRef] [PubMed]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
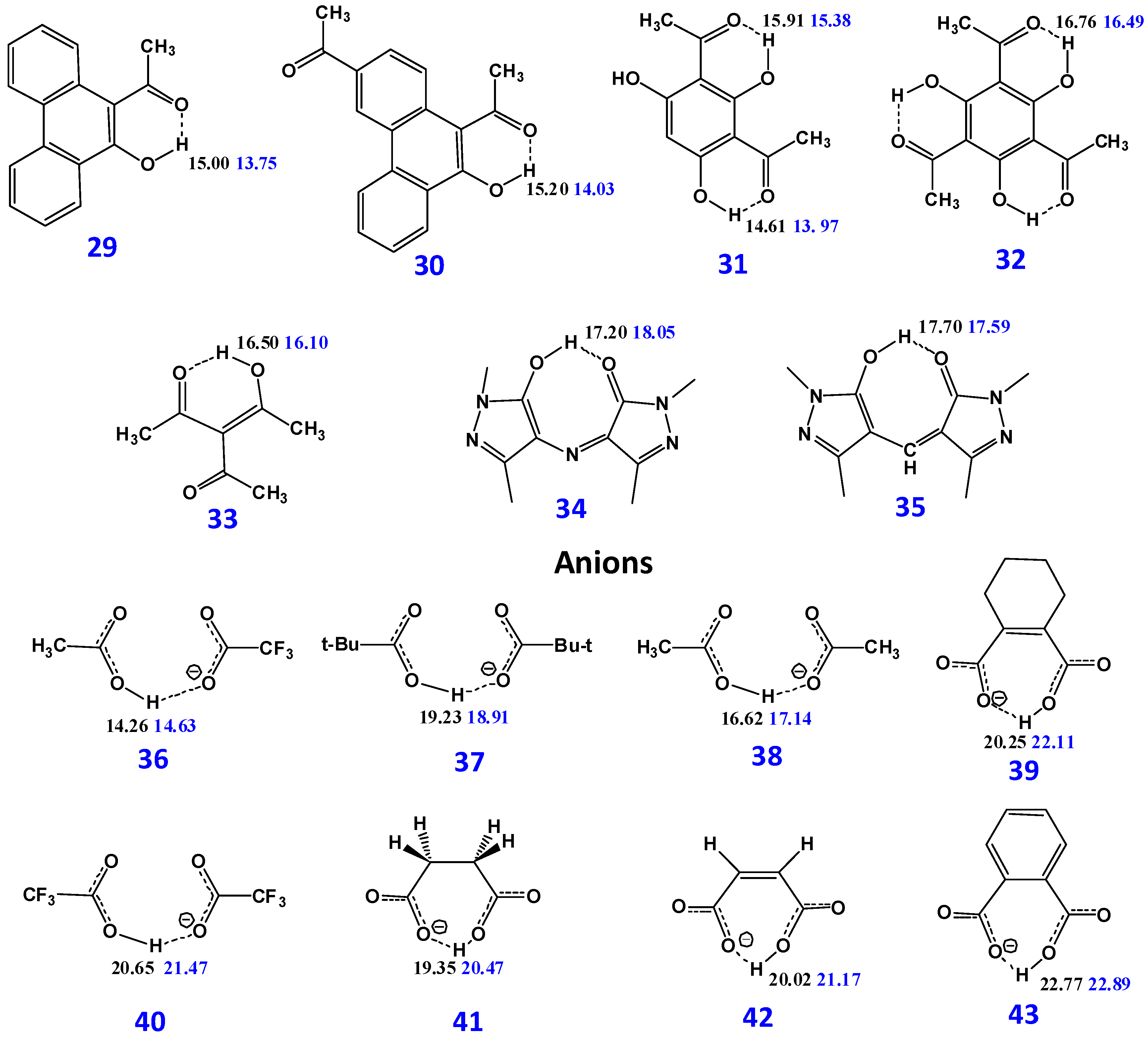{kind=link}
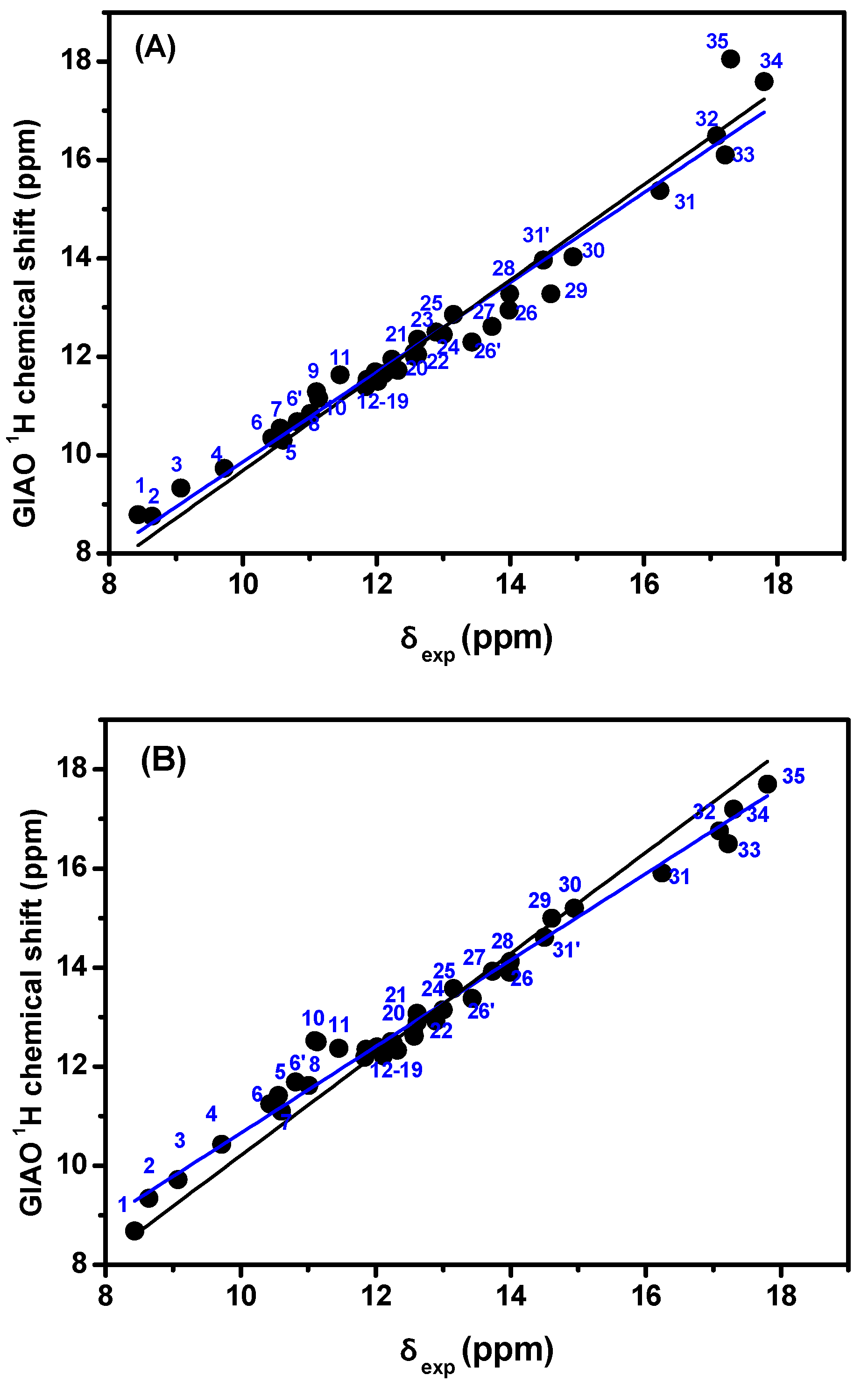{kind=link}
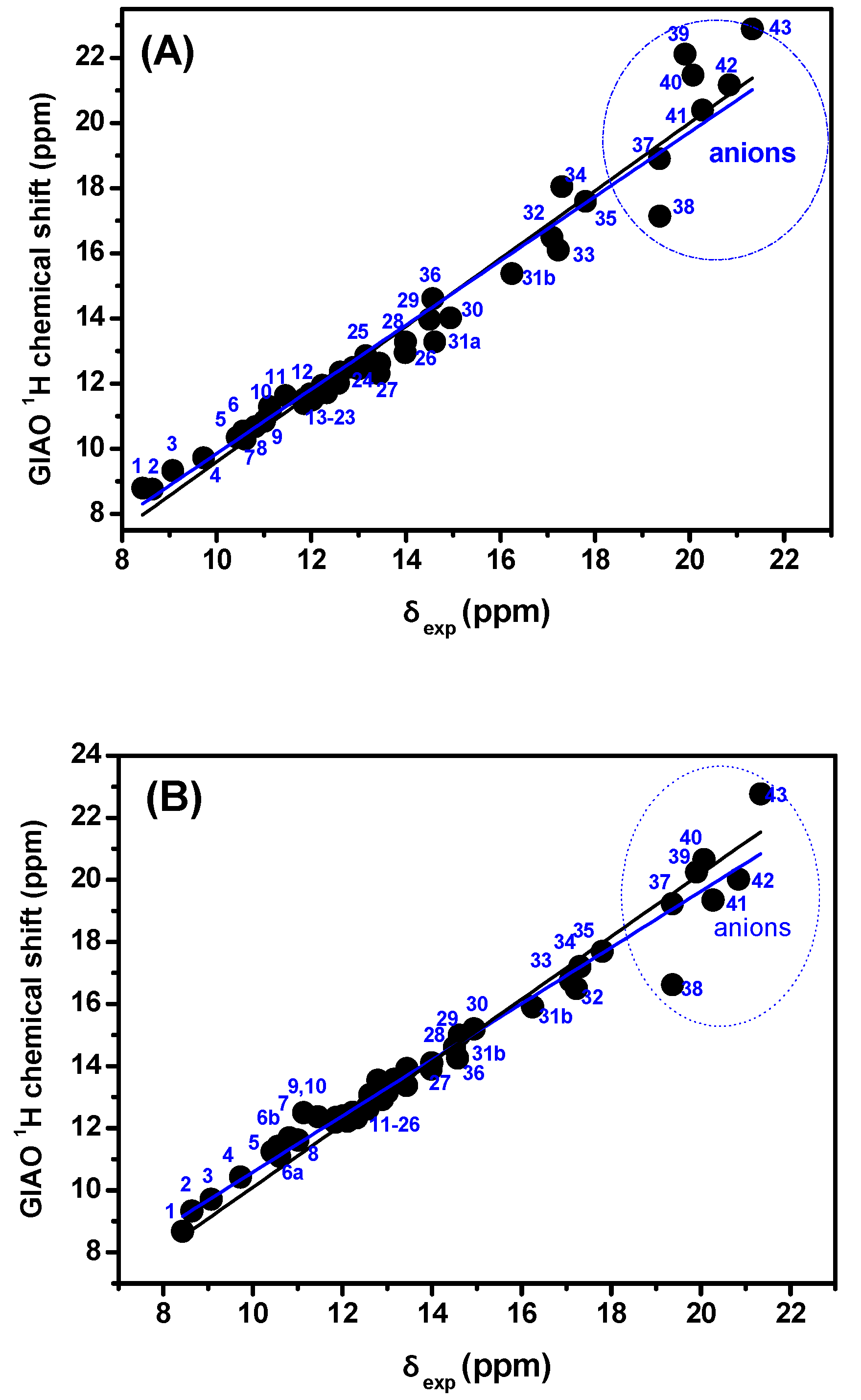{kind=link}
{kind=link}
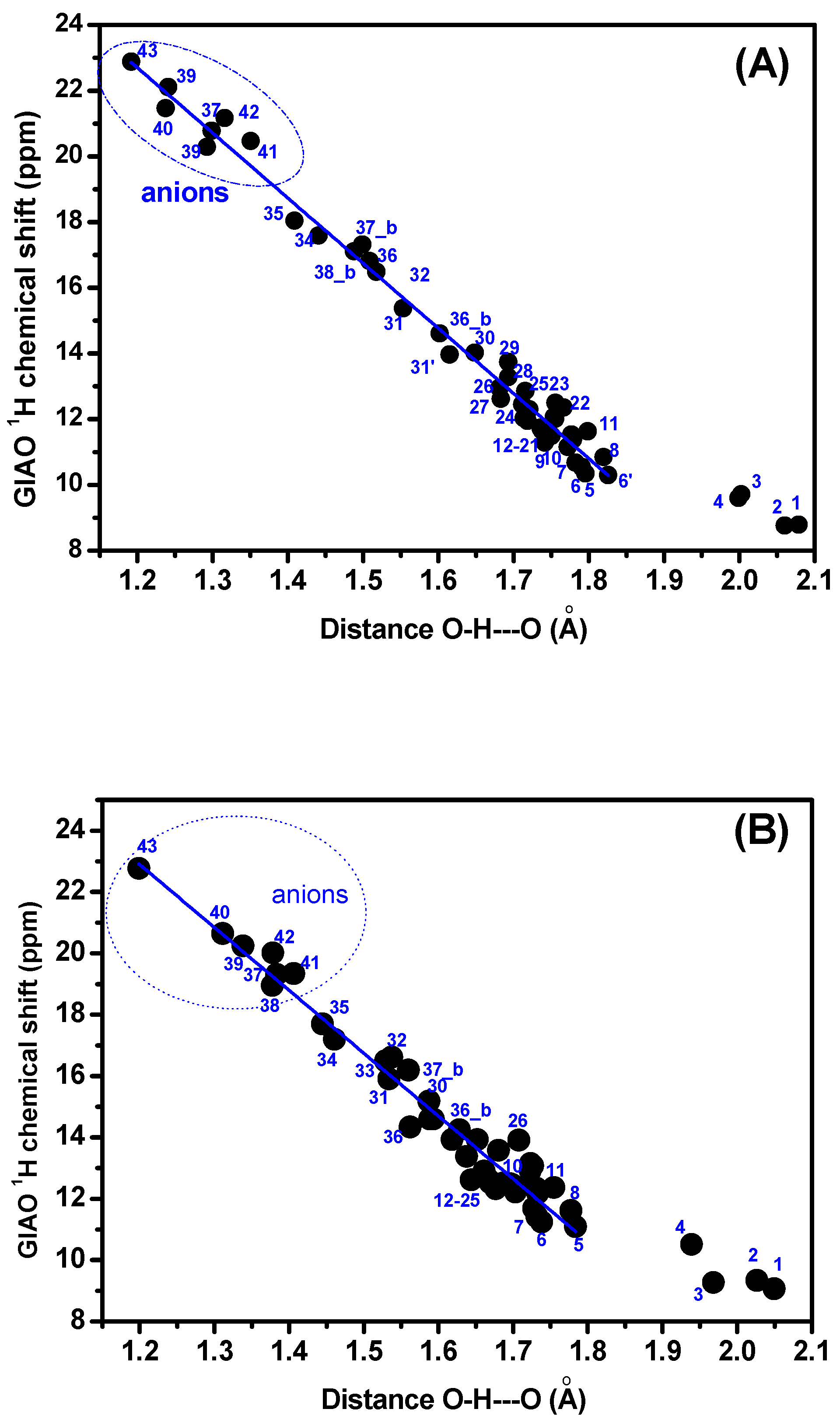{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
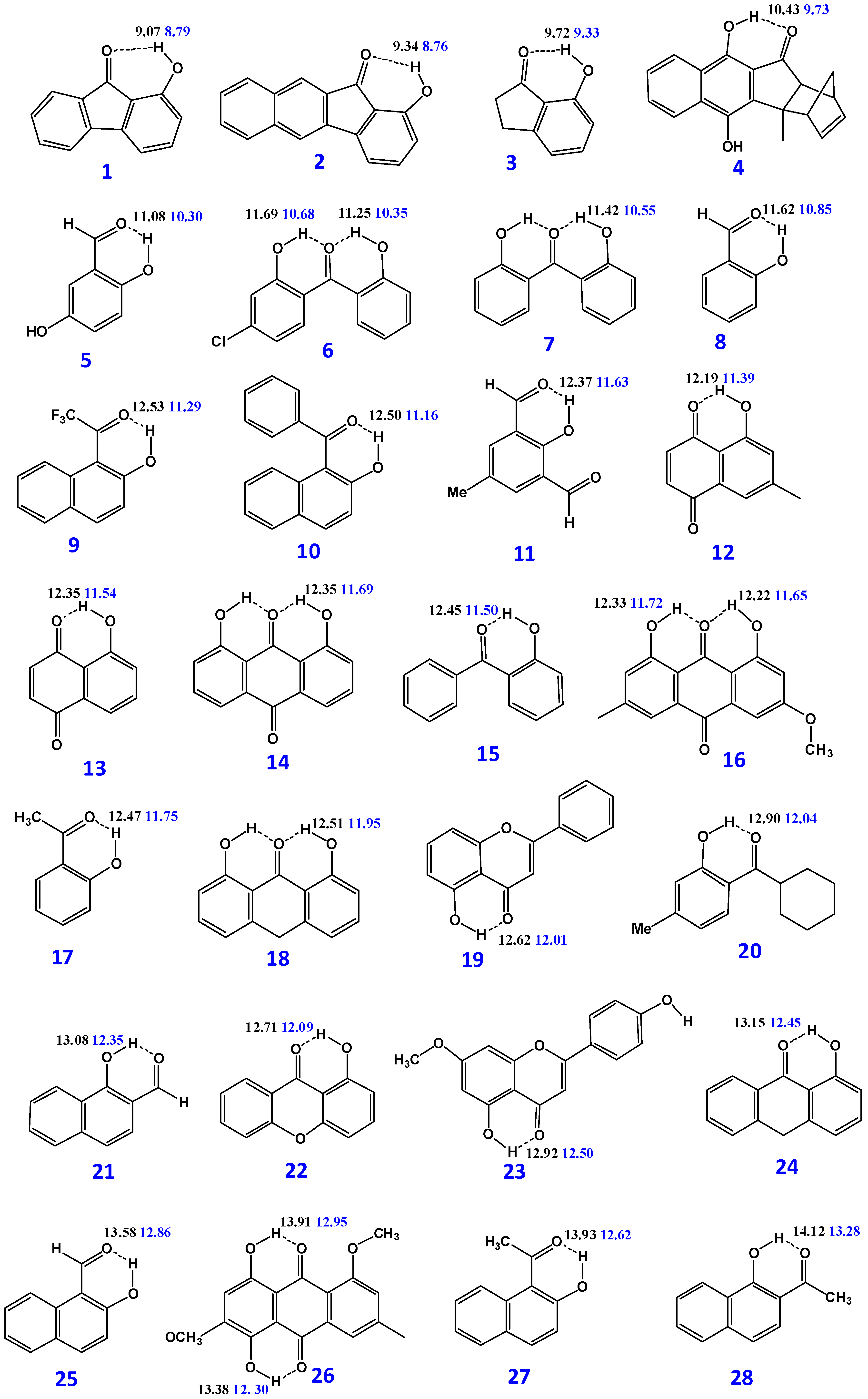
| Compounds | B3LYP/6-31 + G(d) δ (ppm) | M06-2X/6-31 + G(d) δ (ppm) | δexp (ppm) | (O)H···O (Å) | O–H (Å) | NBO O···H-O | ΔQ × 103 |
|---|---|---|---|---|---|---|---|
| 1 | 9.07 | 8.79 | 8.76 | 2.04923 | 0.98315 | (−0.578, +0.533, −0.690) | 112 |
| 2.07842 | 0.97824 | (−0.572, +0.541, −0.706) | 134 | ||||
| 2 | 9.34 | 8.76 | 8.64 | 2.02634 | 0.98385 | (−0.588, +0.533, −0.691) | 103 |
| 2.05999 | 0.97875 | (−0.579, +0.543, −0.706) | 127 | ||||
| 3 | 9.72 | 9.33 | 9.07, 9.04 | 1.96789 | 0.98563 | (−0.604, +0.533, −0.693) | 89 |
| 2.00225 | 0.97982 | (−0.601, +0.543, −0.709) | 108 | ||||
| 4 | 10.43 | 9.73 | 9.72 | 1.93876 | 0.98692 | (−0.612, +0.537, −0.695) | 83 |
| 1.99843 | 0.98047 | (−0.608, +0.547, −0.711) | 103 | ||||
| 5 | 11.01(49.65%) 11.15(50.35%) 11.08 | 10.08 (48.5%) 10.47(51.5%) 10.30 | 10.6 | 1.78342 | 0.98868 | (−0.593, +0.533, −0.699) | 106 |
| 1.78486 | 0.98866 | (−0.595, +0.533, −0.697) | 102 | ||||
| 1.82655 | 0.97982 | (−0.590, +0.544, −0.718) | 128 | ||||
| 1.82531 | 0.98079 | (−0.592, +0.545, −0.716) | 124 | ||||
| 6 | 11.25 11.69 | 10.35 10.68 | 10.43 10.81 | 1.73815 | 0.98692 | (−0.672, +0.536, −0.696) | 24 |
| 1.72708 | 0.98862 | (−0.672, +0.538, −0.692) | 20 | ||||
| 1.79525 | 0.97878 | (−0.667, +0.547, −0.714) | 47 | ||||
| 1.78196 | 0.98027 | (−0.667, +0.549, −0.710) | 43 | ||||
| 7 | 11.42 | 10.55 | 10.54 | 1.73187 | 0.98741 | (−0.673, +0.536, −0.698) | 25 |
| 10.59 | 1.79045 | 0.97921 | (−0.668, +0.547, −0.716) | 48 | |||
| 8 | 11.62 | 10.85 | 11.01 | 1.77701 | 0.98985 | (−0.596, +0.534, −0.693) | 97 |
| 1.81916 | 0.98173 | (−0.595, +0.546, −0.712) | 117 | ||||
| 9 | 12.53 | 11.29 | 11.10 | 1.66898 | 0.99237 | (−0582, +0.536, −0.678) | 96 |
| 1.74125 | 0.98160 | (−0.576, +0.548, −0.699) | 123 | ||||
| 10 | 12.50 | 11.16 | 11.13 | 1.69508 | 0.99237 | (−0.617, +0.537, −0.693) | 76 |
| 1.77197 | 0.98155 | (−0.611, +0.548, −0.712) | 101 | ||||
| 11 | 12.37 | 11.63 | 11.45 | 1.75446 | 0.99152 | (−0.588, +0.534, −0.657) | 69 |
| 1.79817 | 0.98306 | (−0.587, +0.549, −0.679) | 92 | ||||
| 12 | 12.19 | 11.39 | 11.84 | 1.73269 | 0.99280 | (−0.591, +0.536, −0.690) | 99 |
| 1.77877 | 0.98324 | (−0.589, +0.548, −0.710) | 121 | ||||
| 13 | 12.35 | 11.54 | 11.86 | 1.73107 | 0.99232 | (−0.586, +0.536, −0.689) | 103 |
| 1.77694 | 0.98291 | (−0.584, +0.548, −0.710) | 126 | ||||
| 14 | 12.35 | 11.69 | 11.98 | 1.70550 | 0.99079 | (−0.666, +0.538, −0.690) | 24 |
| 1.74172 | 0.98252 | (−0.669, +0.549, −0.709) | 40 | ||||
| 15 | 12.45 | 11.50 | 12.02 | 1.69731 | 0.99279 | (−0.611, +0.535, −0.698) | 87 |
| 1.75040 | 0.98318 | (−0.608, +0.548, −0.717) | 109 | ||||
| 16 | 12.22 12.33 | 11.65 11.72 | 12.11 12.32 | 1.70733 | 0.98130 | (−0.674, +0.538, −0.694) | 20 |
| 1.73743 | 0.98316 | (−0.678, +0.549, −0.711) | 33 | ||||
| 1.70310 | 0.99197 | (−0.674, +0.538, −0.693) | 19 | ||||
| 1.73576 | 0.98362 | (−0.678, +0.549, −0.709) | 31 | ||||
| 17 | 12.47 | 11.75 | 12.26 | 1.69790 | 0.99348 | (−0.610, +0.534, −0.698) | 85 |
| 1.73631 | 0.98459 | (−0.609, +0.547, −0.717) | 108 | ||||
| 18 | 12.51 | 11.95 | 12.23 | 1.68655 | 0.99150 | (−0.679, +0.536, −0.697) | 18 |
| 1.71821 | 0.98338 | (−0.585, +0.548, −0.714) | 29 | ||||
| 19 | 12.62 | 12.01 | 12.57 | 1.72324 | 0.99693 | (−0.637, +0.536, −0.699) | 42 |
| 1.75500 | 0.98794 | (−0.639, +0.549, −0.716) | 77 | ||||
| 20 | 12.90 | 12.04 | 12.61 | 1.66975 | 0.99577 | (−0.624, +0.535, −0.700) | 76 |
| 1.71307 | 0.98607 | (−0.621, +0.548, −0.718) | 97 | ||||
| 21 | 13.08 | 12.35 | 12.61 | 1.72608 | 0.99522 | (−0.608, +0.538, −0.692) | 84 |
| 1.76602 | 0.98619 | (−0.607, +0.552, −0.711) | 104 | ||||
| 22 | 12.71 | 12.09 | 12.56 | 1.71940 | 0.99448 | (−0.631, +0.535, −0.694) | 63 |
| 12.63 | 1.75323 | 0.98626 | (−0.631, +0.548, −0.712) | 81 | |||
| 23 | 12.92 | 12.50 | 12.89 | 1.72271 | 0.99755 | (−0.631, +0.535, −0.694) | 63 |
| 1.75509 | 0.98830 | (−0.644, +0.549, −0.716) | 72 | ||||
| 24 | 13.15 | 12.45 | 13.00 | 1.67646 | 0.99547 | (−0.627, +0.534, −0.700) | 73 |
| 1.71115 | 0.98643 | (−0.626, +0.547, −0.717) | 91 | ||||
| 25 | 13.58 | 12.86 | 13.15 | 1.68007 | 0.99749 | (−0.610, +0.535, −0.687) | 77 |
| 1.71520 | 0.98807 | (−0.610, +0.549, −0.708) | 98 | ||||
| 26 | 13.38 13.91 | 12.30 12.95 | 13.43 13.98 | 1.66846 | 0.99736 | (−0.622, +0.536, −0.676) | 54 |
| 1.72076 | 0.98570 | (−0.617, +0.549, −0.699) | 82 | ||||
| 1.63738 | 1.00210 | (−0.610, +0.537, −0.701) | 89 | ||||
| 1.68097 | 0.98996 | (−0.606, +0.551, −0.721) | 115 | ||||
| 27 | 13.93 | 12.62 | 13.93 | 1.61806 | 1.00022 | (−0.629, +0.536, −0.689) | 60 |
| 13.44 | 1.68256 | 0.98808 | (−0.627, +0.550, −0.710) | 83 | |||
| 28 | 14.12 | 13.28 | 14.11 | 1.65192 | 0.99990 | (−0.621, +0.538, −0.696) | 75 |
| 13.99 | 1.69262 | 0.98997 | (−0.621, +0.553, −0.716) | 95 | |||
| 29 | 15.00 | 13.75 | 14.61 | 1.59292 | 1.00480 | (−0.634, +0.539 −0.692) | 58 |
| 1.64796 | 0.99260 | ||||||
| 30 | 15.20 | 14.03 | 14.94 | 1.58712 | 1.01611 | (−0.630, +0.539, −0.686) | 56 |
| 1.63945 | 0.99403 | (−0.631, +0.555, −0.708) | 77 | ||||
| 31 | 14.61 15.91 | 13.97 15.38 | 14.5 | 1.58832 | 1.00792 | (−0.629, +0.538, −0.689) | 60 |
| 1.61450 | 0.99800 | (−0.632, +0.553, −0.710) | 78 | ||||
| 16.24 | 1.53351 | 1.01518 | (−0.630, +0.539, −0.701) | 71 | |||
| 1.55290 | 1.00622 | (−0.634, +0.555, −0.725) | 91 | ||||
| 32 | 16.76 | 16.49 | 17.09 | 1.5068 | 1.02233 | (−0.629, +0.540, −0.692) | 63 |
| 1.51752 | 1.01495 | (−0.635, +0.556, −0.717) | 82 | ||||
| 33 | 16.50 | 16.10 | 17.22 | 1.52904 | 1.02208 | (−0.640, +0.538, −0.669) | 29 |
| 1.54311 | 1.01478 | (−0.642, +0.554, −0.702) | 60 | ||||
| 34 | 17.20 | 18.05 | 17.3 | 1.46069 | 1.04128 | (−0.693, +0.542, −0.696) | 3 |
| 1.44083 | 1.04060 | (−0.707, +0.557, −0.723) | 16 | ||||
| 35 | 17.70 | 17.59 | 17.8 | 1.44455 | 1.04669 | (−0.703, +0.542, −0.701) | −2 |
| 1.40867 | 1.05278 | (−0.722, +0.557, −0.729) | 7 | ||||
| Anions | |||||||
| 36 | 14.34 a(8.2%) c 14.25 b(91.8%) 14.26 d | 16.82 a(1%) c 14.61 b(99%) c 14.63 d | 14.57 | 1.56178 a | 1.03503 a | (−0.754, +0.541, −0.755) | −1 |
| 1.6277 b | 1.01526 b | (−0.758, +0.535, −0.748) | 10 | ||||
| 1.50817 a | 1.04133 a | (−0.772, +0.551, −0.781) | −9 | ||||
| 1.60161 b | 1.01290 b | (−0.781, +0.548, −0.776) | 5 | ||||
| 37 | 19.32 a(97.18%) c | 20.78 a(46.01%) c | 19.36 | 1.38308 | 1.10012 | (−0.793, +0.535, −0.762) | −31 |
| 16.11 b(2.82%) c | 17.32 b(53.99%) c | 1.55928 b | 1.03303 b | (−0.817, +0.540, −0.748) b | −69 | ||
| 1.29850 | 1.13567 | (−0.810, +0.549, −0.792) | −18 | ||||
| 19.23 d | 18.91 d | 1.49879 b | 1.04125b | (−0.837, +0.551, −0.776) b | −51 | ||
| 38 | 18.96 a(11.87%) c | 20.29(1.0%) | 19.37 | 1.37708 b | 1.10516 | (−0.795, +0.538, −0.764) | −31 |
| 16.30 b(88.13%) c | 17.11 b(99%) c | 1.53724 b | 1.03931 b | (−0.808, +0.537, −0.755) b | −53 | ||
| 1.29212 | 1.14240 | (−0.810, +0.550, −0.793) | −17 | ||||
| 16.62 d | 17.14 d | 1.48775 b | 1.04413 b | (−0.827, +0.548, −0.784) b | −43 | ||
| 39 | 20.25 | 22.11 | 19.90 | 1.40593 | 1.08017 | (−0.743, +0.525, −0.764) | −22 |
| 1.24084 | 1.13673 | (−0.779, +0.540, −0.771) | −8 | ||||
| 40 | 20.65 | 21.47 | 20.07 | 1.31074 | 1.13759 | (−0.744, +0.530, −0.723) | −21 |
| 1.23737 | 1.17881 | (−0.764, +0.544, −0.758) | −6 | ||||
| 41 | 19.35 | 20.47 | 20.27 | 1.40593 | 1.08017 | (−0.792, +0.528, −0.762) | −30 |
| 1.35039 | 1.09709 | (−0.811, +0.543, −0.790) | −21 | ||||
| 42 | 20.02 | 21.17 | 20.84 | 1.37790 | 1.08291 | (−0.771, +0.526, −0.748) | −23 |
| 1.31591 | 1.10506 | (−0.790, +0.542, −0.774) | −16 | ||||
| 43 | 22.77 | 22.89 | 21.33 | 1.19860 | 1.18586 | (−0.746, +0.522, −0.747) | 1 |
| 1.19173 | 1.17940 | (−0.772, +0.540, −0.770) | −2 |
| Bond | Bond Length (Å) | ||
|---|---|---|---|
| X-ray | Calculated: Protons Relaxed | Calculated: All Atoms Relaxed | |
| O–H | 1.286 | 1.023 | 1.036 |
| O···H | 1.358 | 1.618 | 1.522 |
| O–H···O | 2.641 | 2.641 | 2.559 |
| Methyl C–H | 1.128 | 1.091 | 1.091 |
| Aromatic C–H | 1.132 | 1.083 | 1.084 |
| C7–H | 1.149 | 1.090 | 1.092 |
| Interatomic Distances (Å) | Crystal Structure a | B3LYP/6-31+G(d) (IEF-PCM in DMSO) | TPSS/TZVP (IEF-PCM in DMSO) |
|---|---|---|---|
| O(1)···O(14) | 2.497(5) | 2.526 | 2.497 |
| O(13)···O(14) | 2.494(4) | 2.543 | 2.514 |
| O(6)···O(7) | 2.495(5) | 2.528 | 2.497 |
| O(8)···O(7) | 2.529(5) | 2.549 | 2.517 |
| O(3)···O(4) | 2.362(4) | 2.384 | 2.365 |
| O(1)–H(1) | 0.89(6) | 1.002 | 1.009 |
| O(1)–H(1)···O(14) | 1.74(6) | 1.611 | 1.564 |
| O(13)–H(13) | 1.10(5) | 1.001 | 1.006 |
| O(13)–H(13)···O(14) | 1.52(6) | 1.633 | 1.591 |
| O(6)–H(6) | 1.13(6) | 1.002 | 1.009 |
| O(6)–H(6)···O(7) | 1.50(6) | 1.615 | 1.563 |
| O(8)–H(8) | 0.81(6) | 1.000 | 1.007 |
| O(8)–H(8)···O(7) | 1.79(6) | 1.643 | 1.585 |
| O(3)···H(intra) | 1.17(5) | 1.081 | 1.125 |
| O(3)–H(intra)···O(4) | 1.20(5) | 1.312 | 1.245 |
| Dihedral Angles (°) | |||
| C(10)–C(10a)–C(10b)–C(11) | 32.4 | 34.0 | 33.9 |
| C(3)–C(3a)–C(3b)–C(4) | 19.2 | 19.8 | 18.5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siskos, M.G.; Choudhary, M.I.; Gerothanassis, I.P. Hydrogen Atomic Positions of O–H···O Hydrogen Bonds in Solution and in the Solid State: The Synergy of Quantum Chemical Calculations with 1H-NMR Chemical Shifts and X-ray Diffraction Methods. Molecules 2017, 22, 415. https://doi.org/10.3390/molecules22030415
Siskos MG, Choudhary MI, Gerothanassis IP. Hydrogen Atomic Positions of O–H···O Hydrogen Bonds in Solution and in the Solid State: The Synergy of Quantum Chemical Calculations with 1H-NMR Chemical Shifts and X-ray Diffraction Methods. Molecules. 2017; 22(3):415. https://doi.org/10.3390/molecules22030415
Chicago/Turabian StyleSiskos, Michael G., M. Iqbal Choudhary, and Ioannis P. Gerothanassis. 2017. "Hydrogen Atomic Positions of O–H···O Hydrogen Bonds in Solution and in the Solid State: The Synergy of Quantum Chemical Calculations with 1H-NMR Chemical Shifts and X-ray Diffraction Methods" Molecules 22, no. 3: 415. https://doi.org/10.3390/molecules22030415