
Three-Component Reaction of Benzylamines, Diethyl Phosphite and Triethyl Orthoformate: Dependence of the Reaction Course on the Structural Features of the Substrates and Reaction Conditions


Abstract
:
1. Introduction
2. Results and Discussion
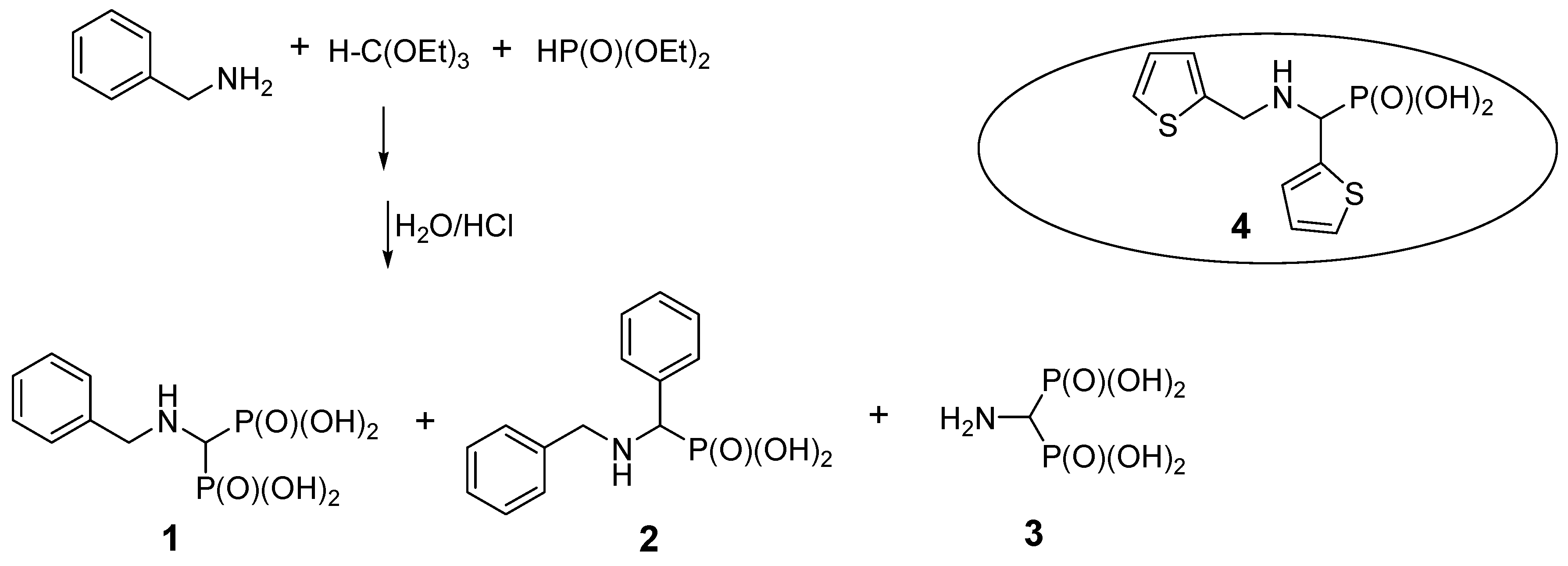
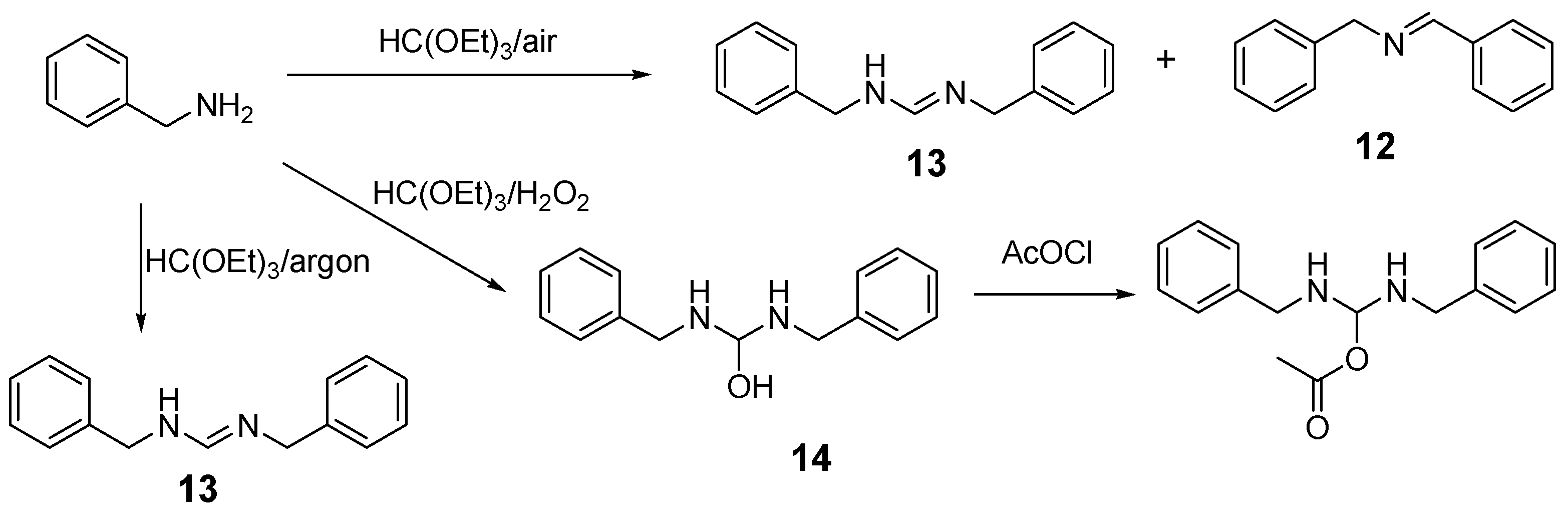
2.1. Benzylamine as a Substrate
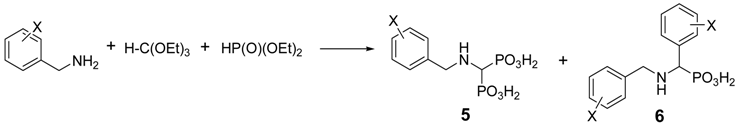
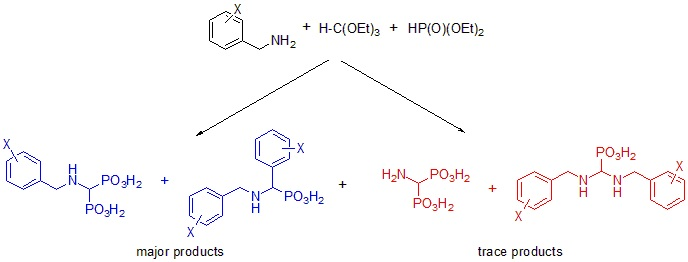
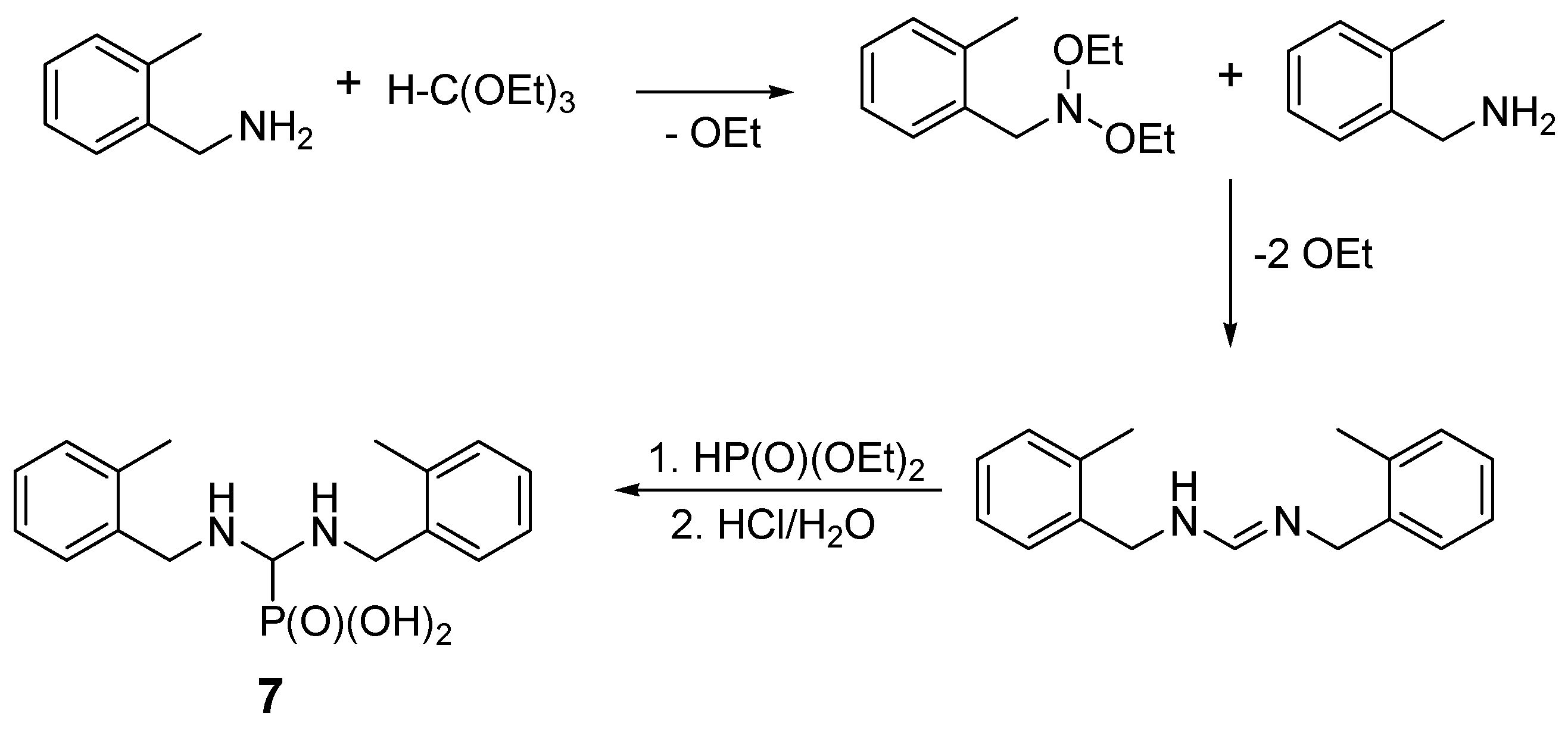
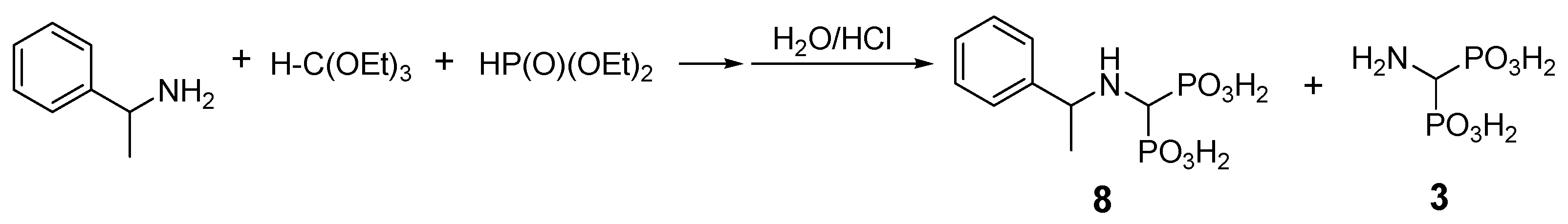
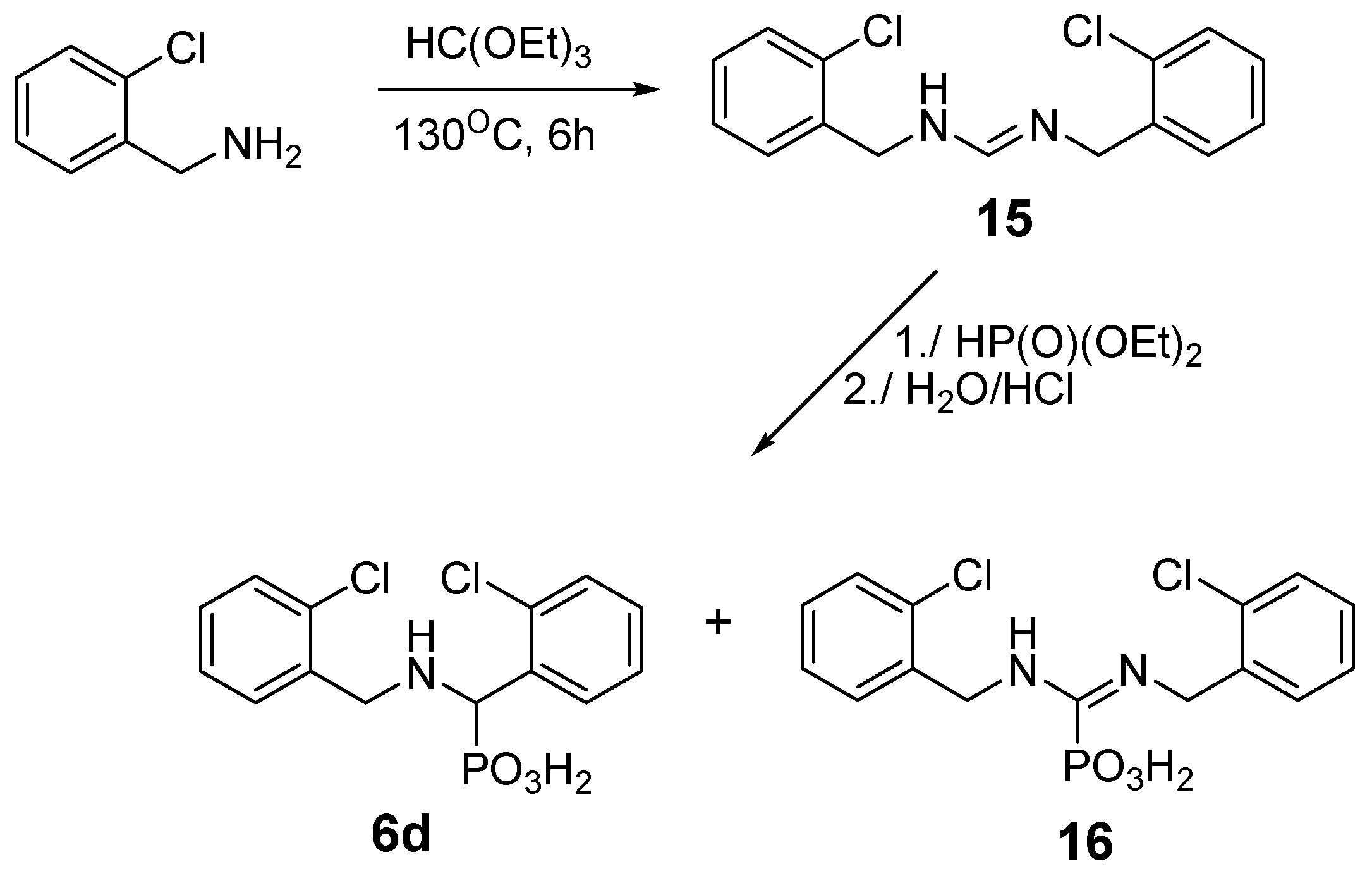
2.2. Reactions of Benzylamines with Substituents on the Phenyl Ring
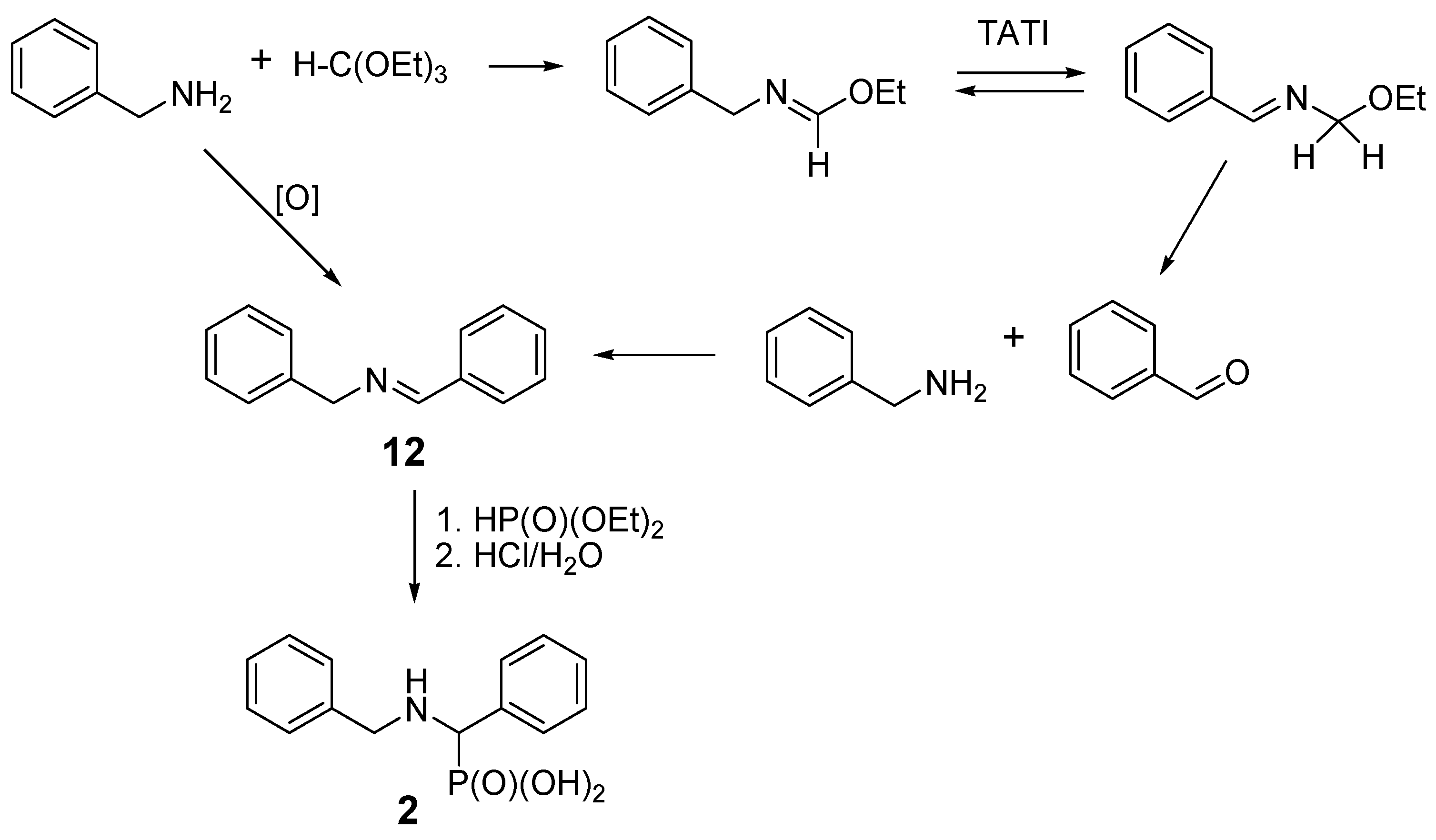
2.3. Presumable Mechanism of Reaction Leading to N-Benzyl Aminobenzylphosphonic Acid (2)
3. Experimental Section
3.1. General Information
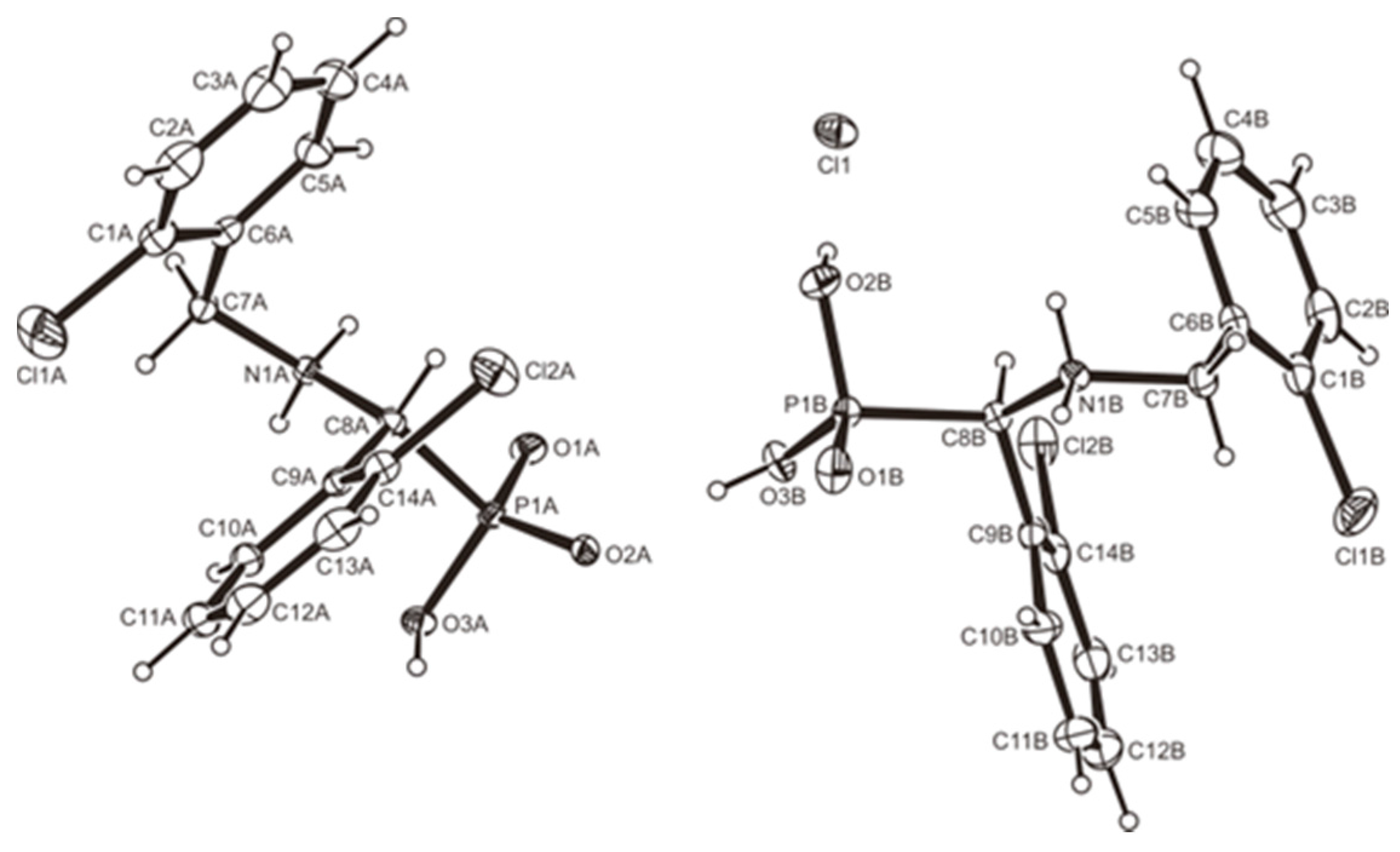
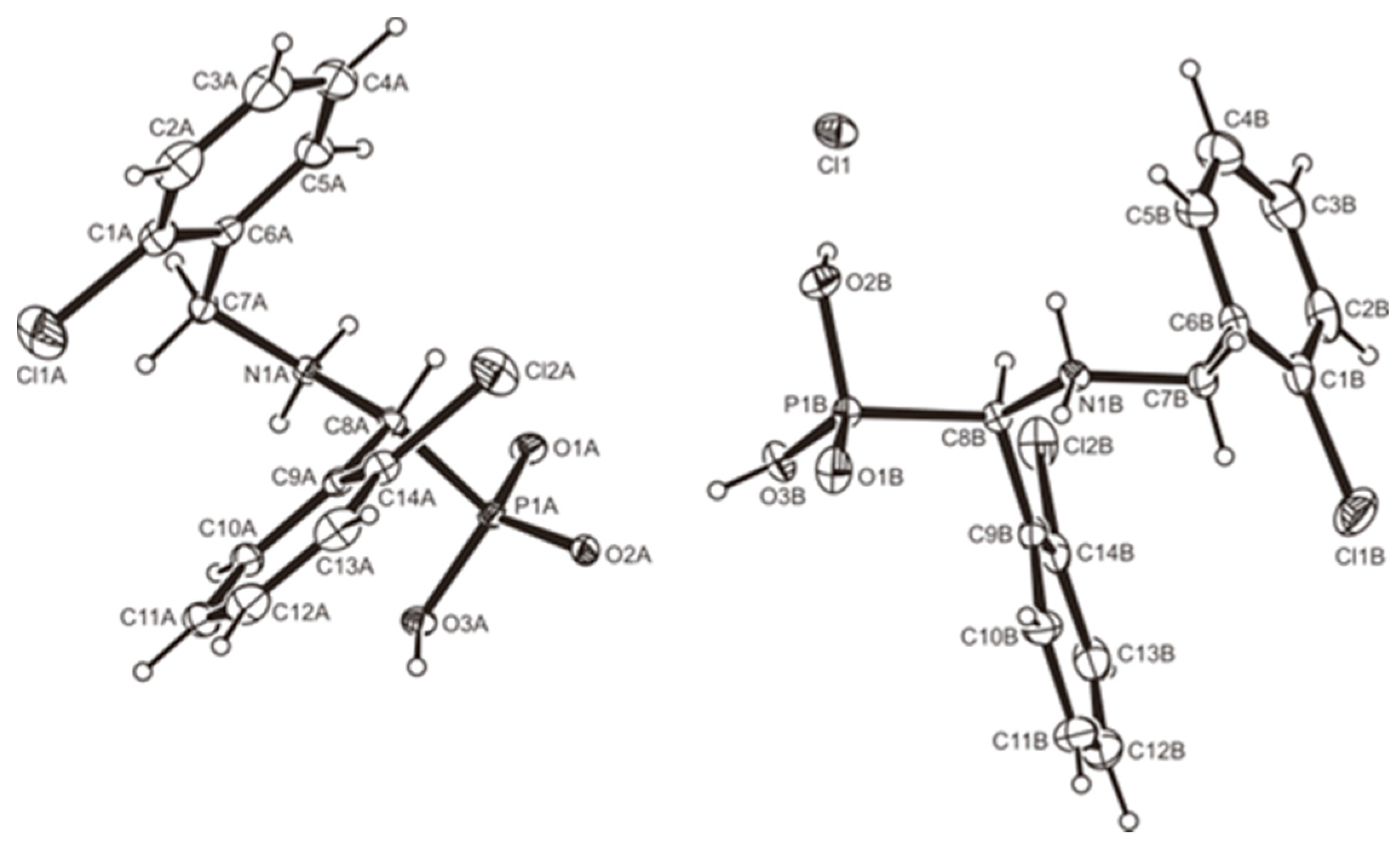
3.2. Crystallography
3.3. General Procedure for the Synthesis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chmielewska, E.; Kafarski, P. Physiologic activity of bisphosphonates—Recent advances. Open J. Pharm. Sci. J. 2016, 3, 56–78. [Google Scholar] [CrossRef]
- Ebetino, F.H.; Hogan, A.M.; Sun, S.; Tsoumpra, M.K.; Duan, X.; Triffitt, J.T.; Kwaasi, A.A.; Dunfort, J.E.; Barnett, B.L.; Oppermann, U.; et al. The relationship between the chemistry and biological activity of the bisphosphonates. Bone 2011, 49, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Demkowicz, S.; Rachoń, J.; Dasko, M.; Kozak, W. Selected organophosphorus compounds with biological activity. Applications in medicine. RSC Adv. 2016, 6, 7101–7112. [Google Scholar] [CrossRef]
- Eriksen, E.F.; Diez-Peres, A.; Boonen, S. Update on long-term treatment with bisphosphonates for postmenopausal osteoporosis: A systematic review. Bone 2014, 58, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Sezer, O.; Croucher, P.I.; Garcia-Sanz, R.; Boccadoro, S.; San Miguel, J.; Ashcroft, J.; Blade, J.; Cavo, M.; Delforge, M.; et al. European Myeloma Network. The use of bisphosphonates in multiple myeloma: Recommendations of an expert panel on behalf of the European Myeloma Network. Ann. Oncol. 2009, 20, 1303–1317. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.S.; Park, J.; de Schutter, J.W.; Huang, X.F.; Berghuis, A.M.; Sebag, M.; Tsantrizos, Y.S. Design and synthesis of active site inhibitors of the human farnesyl pyrophosphate synthase: Apoptosis and inhibition of ERK phosphorylation in multiple myeloma cells. J. Med. Chem. 2012, 55, 3201–3215. [Google Scholar] [CrossRef] [PubMed]
- Ringe, J.D.; Body, J.-J. A review of bone pain relief with ibandronate and other bisphosphonates in disorders of increased bone turnover. Clin. Exp. Rheumatol. 2007, 25, 766–774. [Google Scholar] [PubMed]
- Varenna, M. Bisphosphonates beyond their anti-osteoclastic properties. Rheumatology 2014, 53, 965–967. [Google Scholar] [CrossRef] [PubMed]
- Kosikowska, P.; Bochno, M.; Macegoniuk, K.; Forlani, G.; Kafarski, P.; Berlicki, Ł. Bisphosphonic acids as effective inhibitors of Mycobacterium tuberculosis glutamine synthetase. J. Enzym. Inhib. Med. Chem. 2016, 31, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Hudock, M.P.; Sanz-Rodriques, C.E.; Song, Y.; Chan, J.M.W.; Zhang, Y.; Odeh, S.; Kosztowski, T.; Leon-Rossell, A.; Conceptión, J.L.; Yardley, V.; et al. Inhibition of Trypanosoma cruzi hexokinase by bisphosphonates. J. Med. Chem. 2006, 49, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhang, Y.; Sinko, W.; Hensler, M.E.; Olson, J.; Molohon, K.J.; Lindert, S.; Cao, R.; Li, K.; Wang, K.; et al. Antibacterial drug leads targeting isoprenoid biosynthesis. PNAS 2013, 110, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Wang, Y.; Li, K.; Gao, J.; Huang, C.H.; Chen, C.C.; Ko, T.P.; Zhang, Y.; Guo, R.T.; Oldfield, E. tibacterial Drug leads: DNA and enzyme multitargeting. J. Med. Chem. 2015, 58, 1215–1227. [Google Scholar] [CrossRef] [PubMed]
- Lacbay, C.M.; Mancuso, J.; Lin, Y.S.; Bennett, N.; Gotte, M.; Tsantrizos, Y.S. Modular assembly of purine-like bisphosphonates as inhibitors of HIV-1 reverse transcriptase. J. Med. Chem. 2014, 57, 7435–7449. [Google Scholar] [CrossRef] [PubMed]
- Agapkina, J.; Yanvarev, D.; Anisenko, A.; Korolev, S.; Vepsäläinen, J.; Kochetkov, S.; Gottikh, M. Specific features of HIV-1 integrase inhibition by bisphosphonate derivatives. Eur. J. Med. Chem. 2014, 73, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Maier, L. Organische phosphorverbindungen 75: Herstellung und Eigenschaften von Aminomethylendiphosphinaten und -diphosphonaten, RR1NCH[P(O)R2(OR3)]2 und Derivaten. Phosphorus Sulfur Relat. Elem. 1981, 11, 311–332. [Google Scholar] [CrossRef]
- Kantoci, D.; Denike, J.K.; Wechter, W.J. Synthesis of Aminobisphosphonate. Synth. Commun. 1996, 26, 2037–2043. [Google Scholar] [CrossRef]
- Chmielewska, E.; Kafarski, P. Synthetic procedures leading towards aminobisphosphonates. Molecules 2016, 21, 1474. [Google Scholar] [CrossRef] [PubMed]
- Bálint, E.; Tajti, Á.; Dzielak, A.; Hägele, G.; Keglevich, G. Microwave-assisted synthesis of (aminomethylene)bisphosphine oxides and (aminomethylene)bisphosphonates by a three-component condensation. Beilstein J. Org. Chem. 2016, 12, 1493–1502. [Google Scholar]
- Dąbrowska, E.; Burzyńska, A.; Mucha, A.; Matczak-Jon, E.; Sawka-Dobrowolska, W.; Berlicki, Ł.; Kafarski, P. Insight into the mechanism of three component condensation leading to aminomethylenebisphosphonates. J. Organomet. Chem. 2009, 694, 3806–3813. [Google Scholar]
- Tyka, R. Novel synthesis of α-aminophosphonic acids. Tetrahedron Lett. 1970, 11, 677–680. [Google Scholar] [CrossRef]
- Ziora, Z.; Kafarski, P. Amidoalkylation of phosphorus trichloride with acetamide and alkyl oxocycloalkanecarboxylates. Phosphorus Sulfur Relat. Elem. 2009, 184, 1047–1053. [Google Scholar] [CrossRef]
- Lejczak, B.; Boduszek, B.; Kafarski, P.; Forlani, G.; Wojtasek, H.; Wieczorek, P. Mode of action of herbicidal derivatives of aminomethylenebisphosphonic acid. 1. Physiologic activity and inhibition of anthocyanin biosynthesis. J. Plant Growth Regul. 1996, 15, 109–113. [Google Scholar] [CrossRef]
- Miszczyk, P.; Wieczorek, D.; Gałęzowska, J.; Dziuk, B.; Wietrzyk, J.; Chmielewska, E. Reaction of 3-amino-1,2,4-triazole with diethyl phosphite and triethyl orthoformate: acid-base properties and antiosteoporotic activities of the products. Molecules 2017, 22, 254. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wang, L.; Gao, S. Recent advances in aerobic oxidation of alcohols and amines to imines. ACS Catal. 2015, 5, 5851–5876. [Google Scholar] [CrossRef]
- Qin, J.; Zhang, L.; Lv, J.; Luo, S.; Cheng, J.-P. Bioinspired organocatalytic aerobic C-H oxidation of amines with an ortho-quinone catalyst. Org. Lett. 2015, 17, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Schalfelberger, F.; Hu, L.; Ramström, O. trans-Symmetric dynamic covalent systems: connected transamination and transimination reactions. Chem. Eur. J. 2015, 21, 9776–9783. [Google Scholar] [CrossRef] [PubMed]
- CrysAlis PRO Software System (Version 1.171.37.33); Agilent Technologies: Wrocław, Poland, 2014.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Farrugia, L.J. WinGX and ORTEP for Windows: an update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Chmielewska, E.; Kempińska, K.; Wietrzyk, J.; Piątek, A.; Kuryszko, J.J.; Kiełbowicz, Z.; Kafarski, P. Novel Bisphosphonates and Their Use. Int Pat Appl WO2015159153 (A1), 22 October 2015. [Google Scholar]
- Redmore, D.; Thompson, N.E.S. Alpha-Amino Phosphonic Acids. US Pat. US 4098849 A, 4 July 1978. [Google Scholar]
- Schőllkopf, U.; Hoppe, I.; Thiele, A. Asymmetric synthesis of α-aminophosphonic acids, I. Enantioselective synthesis of l-(1-aminoethyl)phosphonic acid by asymmetric catalytic hydrogenation of N-[1-(dimethoxyphosphoryl)ethenyl]formamide. Liebigs Ann. Chem. 1985, 555–559. [Google Scholar]
- Sample Availability: Samples of the compounds 1–16 are available from the authors.
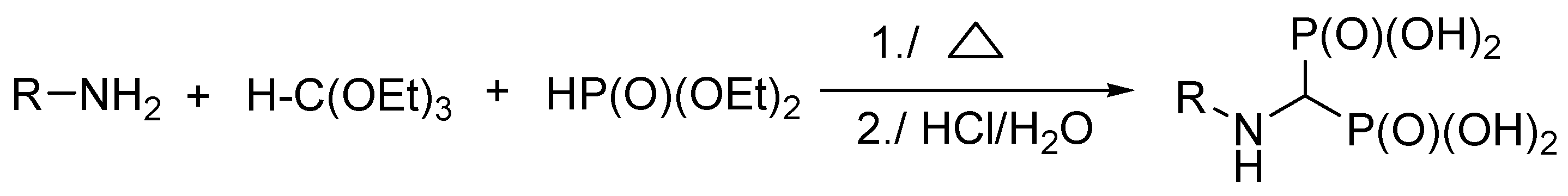
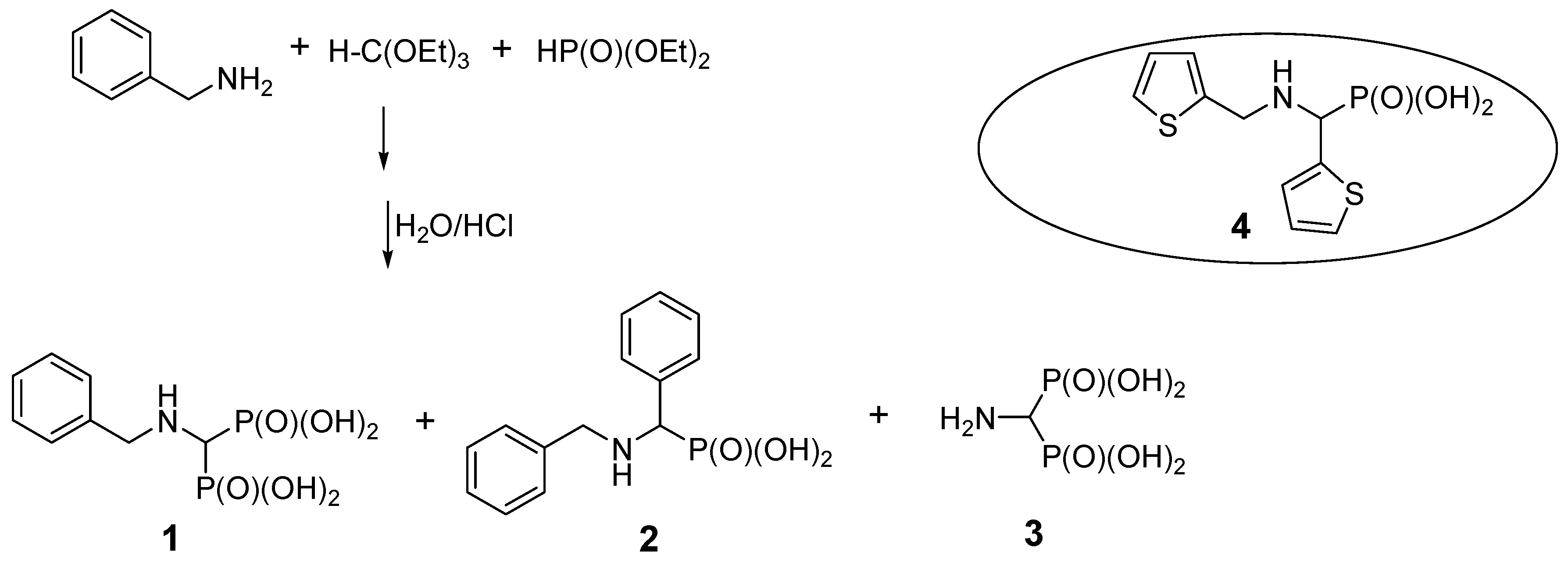
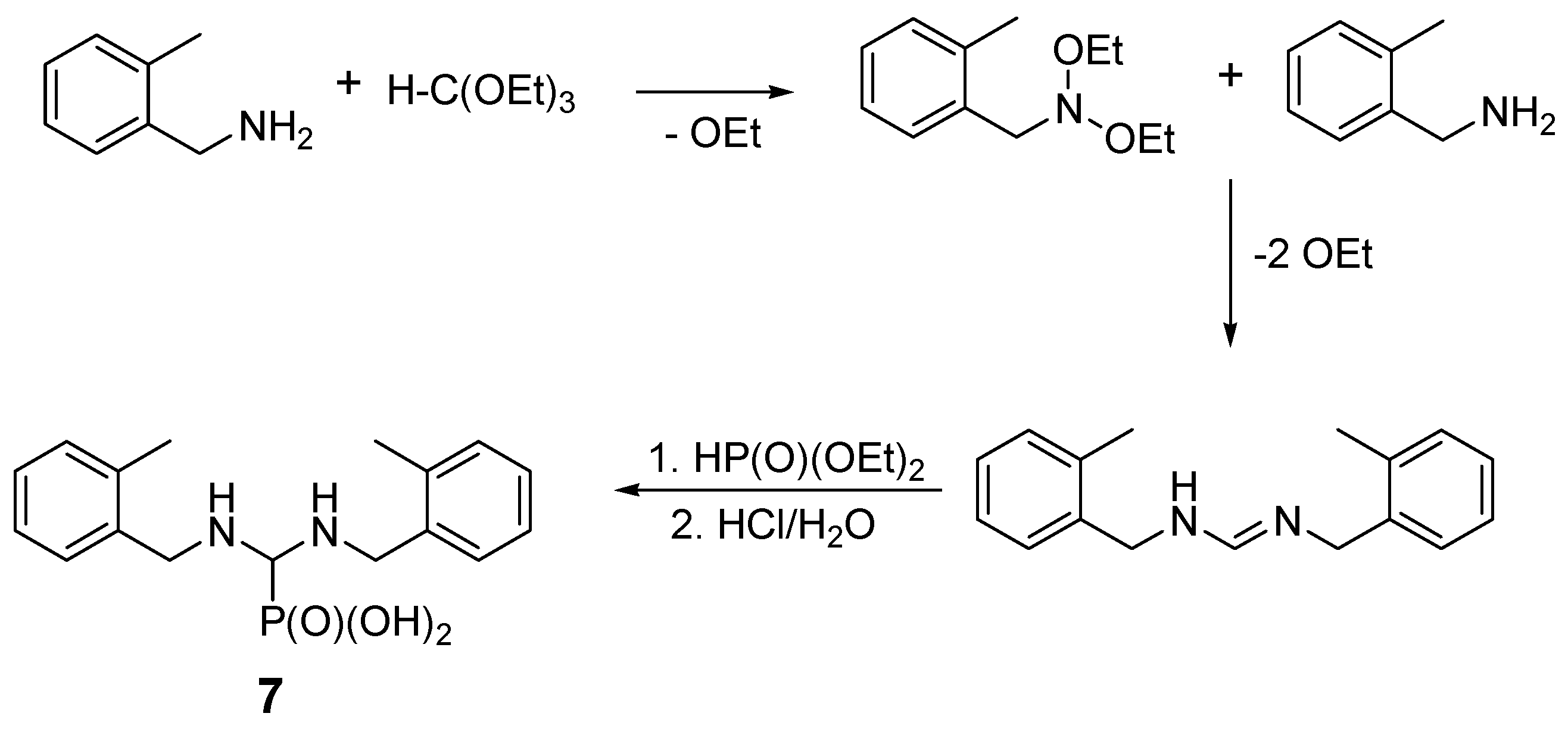
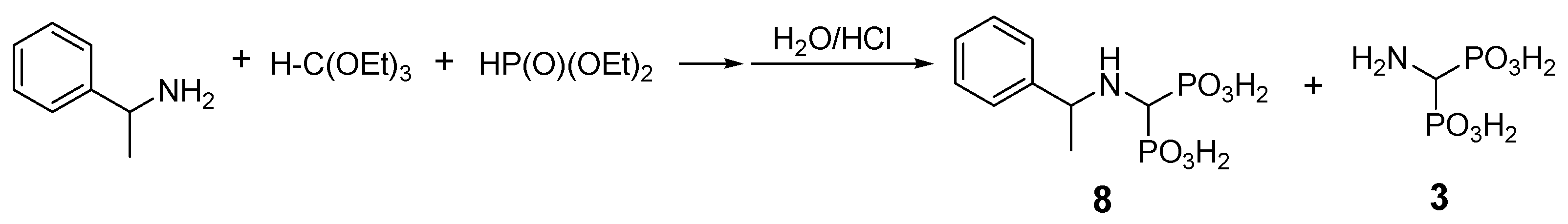

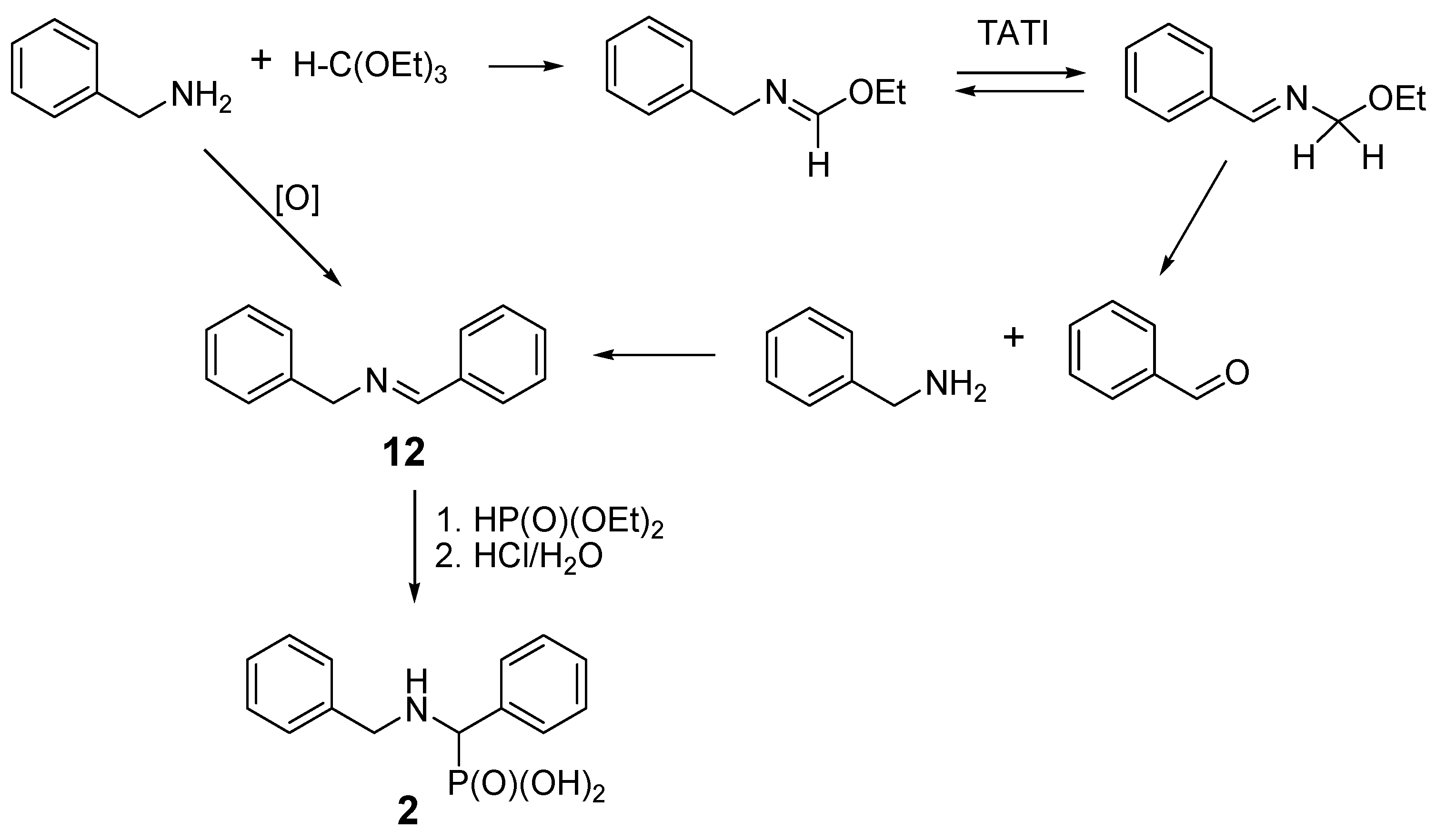
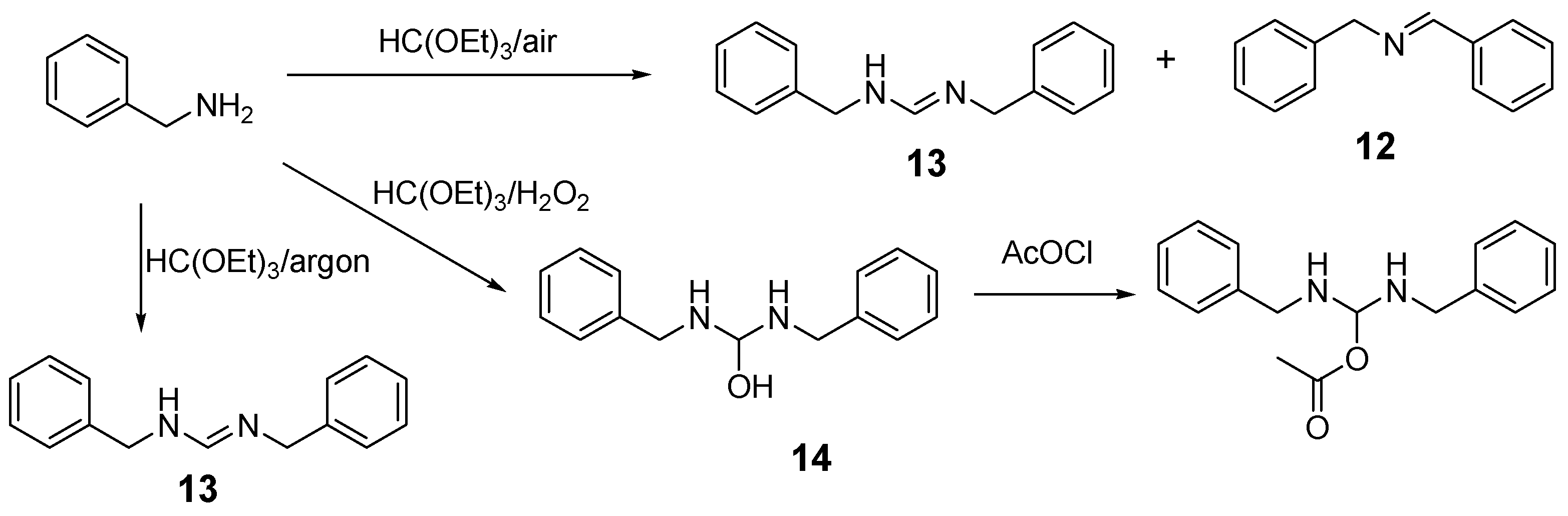
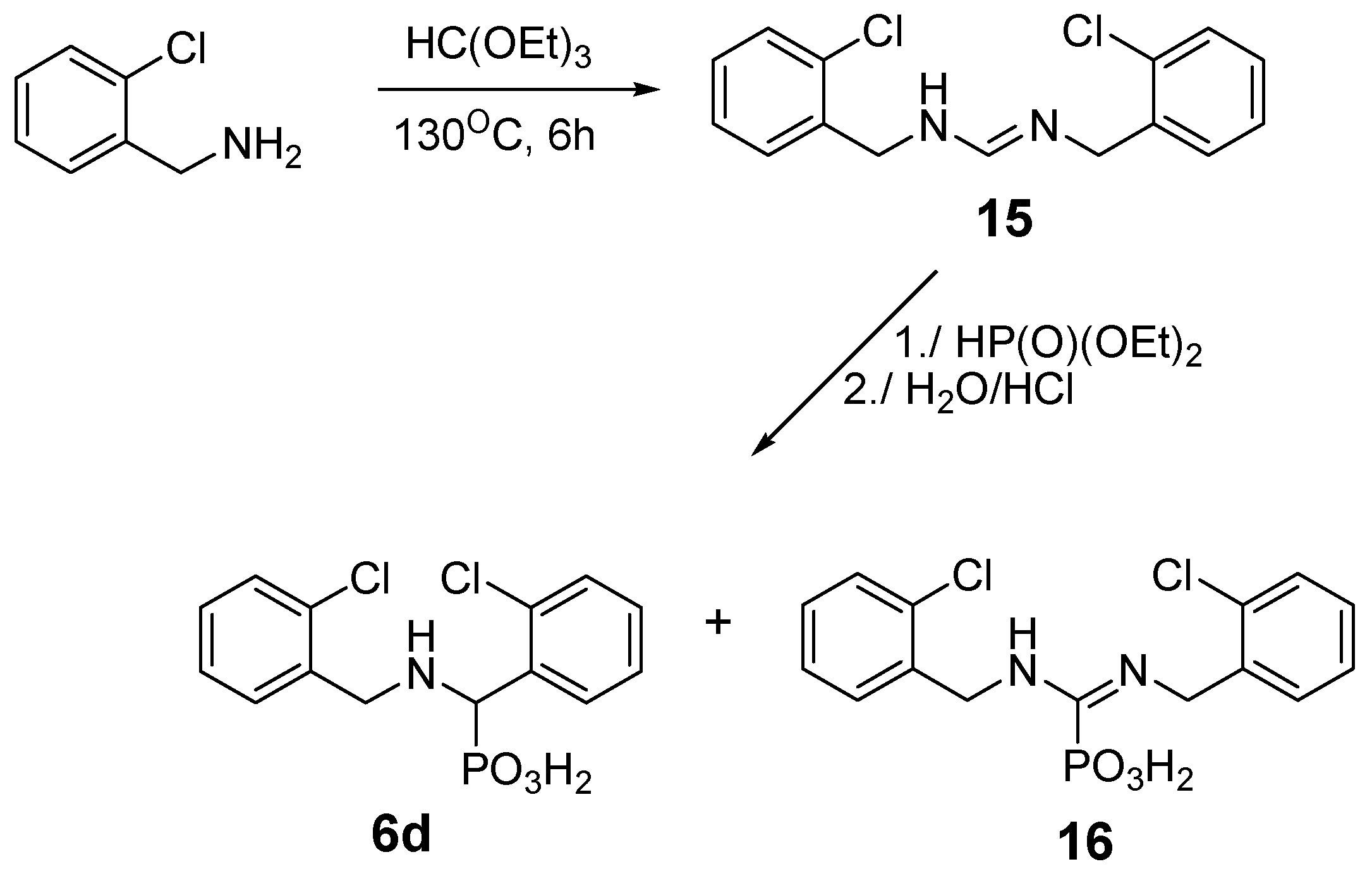

| Entry | Amine: Phosphite: Orthoformate Molar Ratio | Reaction Conditions | Molar Amounts of Products (1:2) * | Total Isolated Yield (%) |
|---|---|---|---|---|
| 1 | 1:2:1 | 80 °C, 12 h | 0:100 | 53 |
| 2 | 1:2:1 | 135 °C, 12 h | 43:57 | 10.5 |
| 3 | 1:2:1 | reflux with removal of ethanol/12 h | 65:35 | 20 |
| 4 | 1:2:1 | 80 °C, 6 min, MW | NR | |
| 5 | 1:2:1 | 80 °C, 12 min, MW | 0:100 | 11.5 |
| 6 | 1:2:1 | 135 °C, 6 min, MW | 0:100 | 5 |
| 7 | 1:2:1 | 135 °C, 12 min, MW | 0:100 | 13.5 |
| 8 | 1:4:2 | 135 °C, 12 h | 100:0 | 68 |
| Entry | Substituent (X) | Product(s) | Amine:Phosphite:Orthoformate Molar Ratio | Molar Amounts of Products (5:6) * | Total Isolated Yield (%) |
|---|---|---|---|---|---|
| 1 | o-CH3 | 6a | 1:2:1 | ** | |
| 2 | o-CH3 | 5a | 1:4:2 | 100:0 | 31 |
| 3 | m-CH3 | 6b | 1:2:1 | 0:100 | 19 |
| 4 | m-CH3 | 5b | 1:4:2 | 100:0 | 36 |
| 5 | p-CH3 | 6c | 1:2:1 | 85:15 | 7 |
| 6 | p-CH3 | 5c | 1:4:2 | 100:0 | 28 |
| 7 | p-CH3 | 5c + 6c | 1:2:1 (80 °C) | 0:100 | 13 |
| 8 | o-Cl | 6d | 1:2:1 | 0:100 | 24 |
| 9 | m-Cl | 6e | 1:2:1 | 0:100 | 31 |
| 10 | p-Cl | 6f | 1:2:1 | 0:100 | 24 |
| 11 | p-Cl | 5f + 6f | 1:4:2 | 57:43 | 75 |
| 12 | p-F | 5g + 6g | 1:2:1 | 26:74 | 53 |
| 13 | m-Br | 5h + 6h | 1:2:1 | 36:64 | 33 |
| 14 | m-Br | 5h | 1:4:2 | 100:0 | 88 |
| 15 | o-NO2 | 6i | 1:2:1 | NR |
{kind=link}
{kind=link}
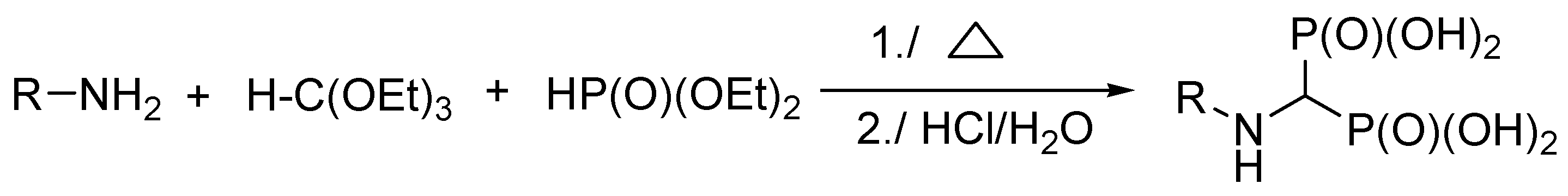{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miszczyk, P.; Turowska-Tyrk, I.; Kafarski, P.; Chmielewska, E. Three-Component Reaction of Benzylamines, Diethyl Phosphite and Triethyl Orthoformate: Dependence of the Reaction Course on the Structural Features of the Substrates and Reaction Conditions. Molecules 2017, 22, 450. https://doi.org/10.3390/molecules22030450
Miszczyk P, Turowska-Tyrk I, Kafarski P, Chmielewska E. Three-Component Reaction of Benzylamines, Diethyl Phosphite and Triethyl Orthoformate: Dependence of the Reaction Course on the Structural Features of the Substrates and Reaction Conditions. Molecules. 2017; 22(3):450. https://doi.org/10.3390/molecules22030450
Chicago/Turabian StyleMiszczyk, Patrycja, Ilona Turowska-Tyrk, Paweł Kafarski, and Ewa Chmielewska. 2017. "Three-Component Reaction of Benzylamines, Diethyl Phosphite and Triethyl Orthoformate: Dependence of the Reaction Course on the Structural Features of the Substrates and Reaction Conditions" Molecules 22, no. 3: 450. https://doi.org/10.3390/molecules22030450